Job Results:
Ligand
Structure
Job ID
a99a6085d597d3b02a857114a6fe159a
Job name
NA
Time
2026-01-11 00:01:47
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | p53-binding protein Mdm4 (MDM4) | 6Q9Y | 7.62 | |
Target general information Gen name MDM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Mdmx; Mdm2-like p53-binding protein; Double minute 4 protein Protein family MDM2/MDM4 family Biochemical class MDM2/MDM4 family Function Inhibits p53/TP53- and TP73/p73-mediated cell cycle arrest and apoptosis by binding its transcriptional activation domain. Inhibits degradation of MDM2. Can reverse MDM2-targeted degradation of TP53 while maintaining suppression of TP53 transactivation and apoptotic functions. Related diseases Bone marrow failure syndrome 6 (BMFS6) [MIM:618849]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS6 is an autosomal dominant form characterized by intermittent neutropenia, lymphopenia, or anemia associated with hypocellular bone marrow, and increased susceptibility to cancer. {ECO:0000269|PubMed:32300648}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NX04; P10415; Q7Z479; O95971; P48729; Q00987; Q13064; P41227; P06400; Q9Y4L5; P23297; P29034; P33763; P04271; P31947; P04637; P62837; Q93009; O14972; P61964; P62258; P61981; P63104; Q9BRR0; A0A0S2Z6X0; Q3YBA8; P03255-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19722 Length 173 Aromaticity 0.08 Instability index 50.78 Isoelectric point 8.48 Charge (pH=7) 2.27 2D Binding mode Binding energy (Kcal/mol) -10.39  Molscript Map 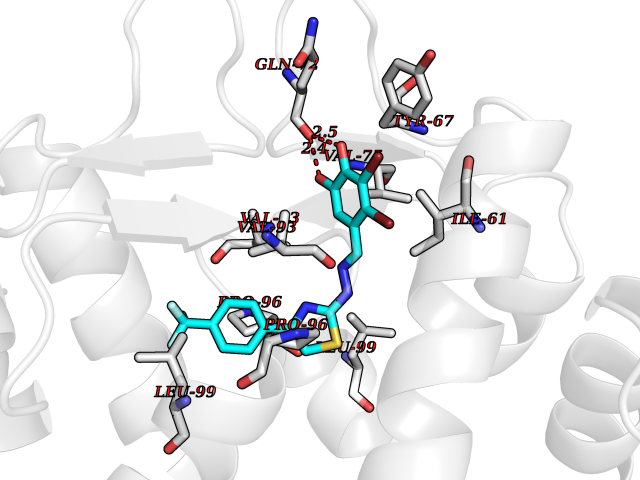 Pymol Map 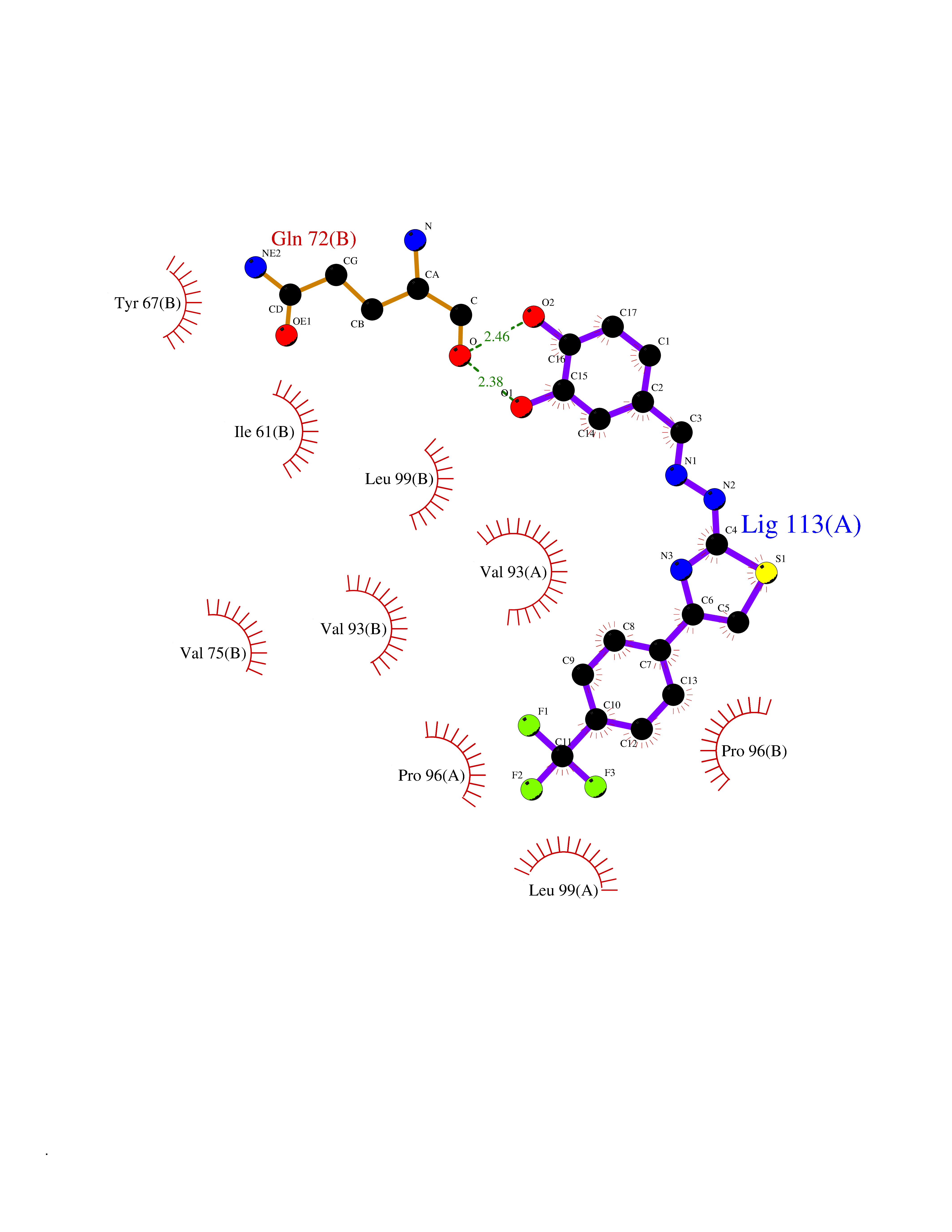 Ligplot Map 3D Binding mode Sequence QVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLAQINQVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLA Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 7.61 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -10.38 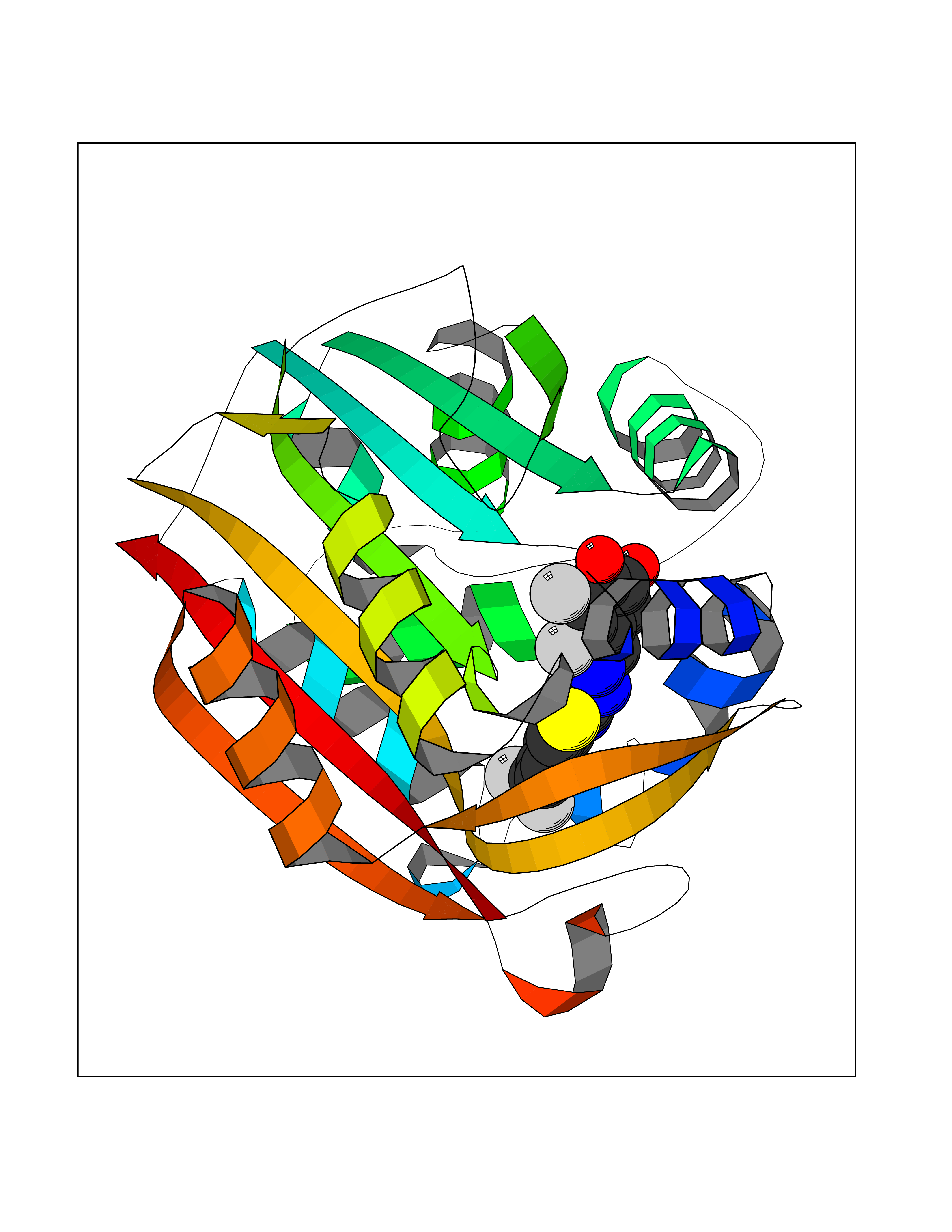 Molscript Map 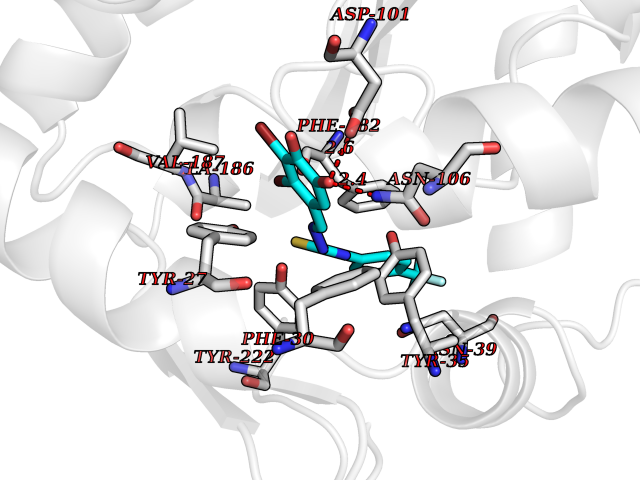 Pymol Map 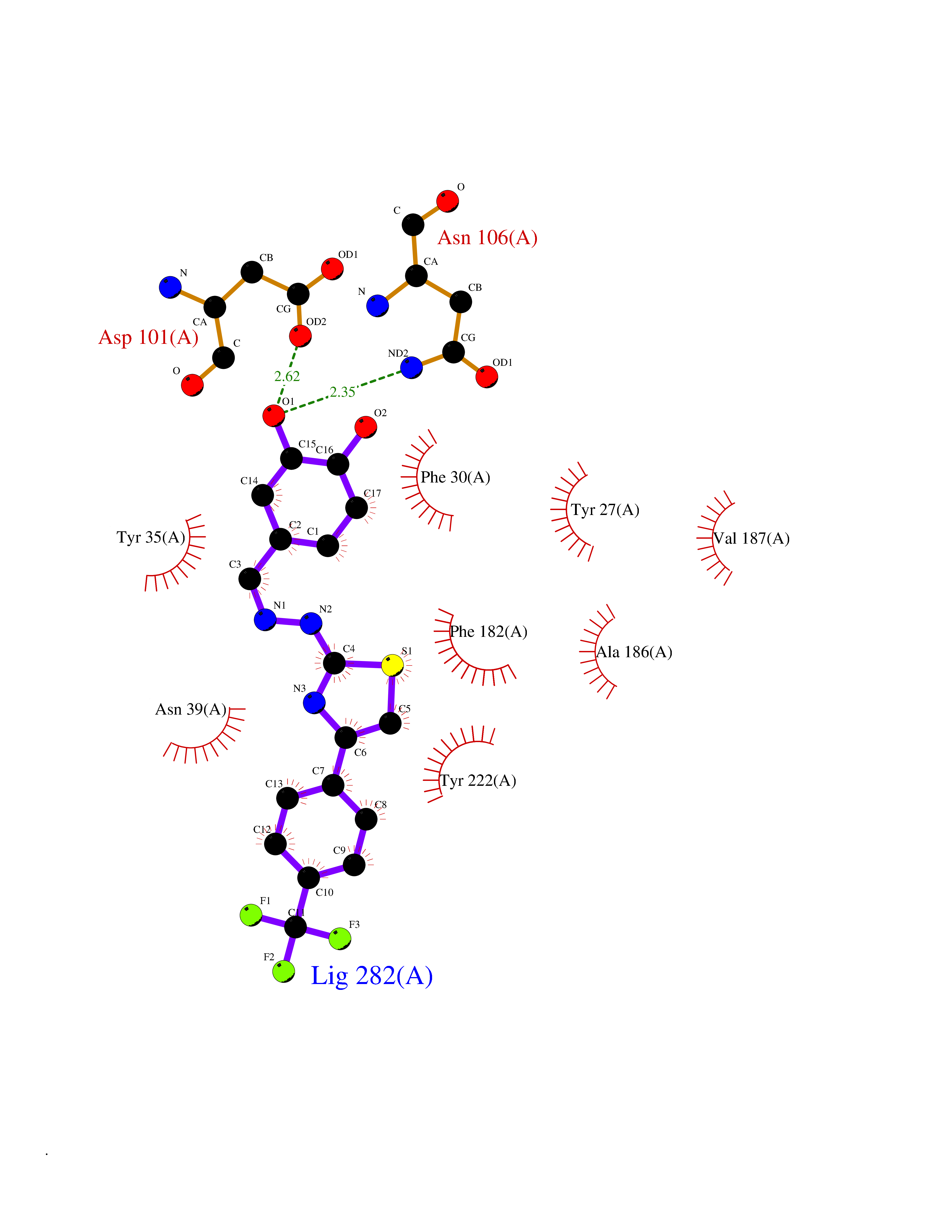 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Beta-arrestin-1 (ARRB1) | 6TKO | 7.60 | |
Target general information Gen name ARRB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Non-visual arrestin-2; Betaarrestin1; Arrestin beta1; Arrestin beta-1; ARR1 Protein family Arrestin family Biochemical class Arrestin protein Function During homologous desensitization, beta-arrestins bind to the GPRK-phosphorylated receptor and sterically preclude its coupling to the cognate G-protein; the binding appears to require additional receptor determinants exposed only in the active receptor conformation. The beta-arrestins target many receptors for internalization by acting as endocytic adapters (CLASPs, clathrin-associated sorting proteins) and recruiting the GPRCs to the adapter protein 2 complex 2 (AP-2) in clathrin-coated pits (CCPs). However, the extent of beta-arrestin involvement appears to vary significantly depending on the receptor, agonist and cell type. Internalized arrestin-receptor complexes traffic to intracellular endosomes, where they remain uncoupled from G-proteins. Two different modes of arrestin-mediated internalization occur. Class A receptors, like ADRB2, OPRM1, ENDRA, D1AR and ADRA1B dissociate from beta-arrestin at or near the plasma membrane and undergo rapid recycling. Class B receptors, like AVPR2, AGTR1, NTSR1, TRHR and TACR1 internalize as a complex with arrestin and traffic with it to endosomal vesicles, presumably as desensitized receptors, for extended periods of time. Receptor resensitization then requires that receptor-bound arrestin is removed so that the receptor can be dephosphorylated and returned to the plasma membrane. Involved in internalization of P2RY4 and UTP-stimulated internalization of P2RY2. Involved in phosphorylation-dependent internalization of OPRD1 ands subsequent recycling. Involved in the degradation of cAMP by recruiting cAMP phosphodiesterases to ligand-activated receptors. Beta-arrestins function as multivalent adapter proteins that can switch the GPCR from a G-protein signaling mode that transmits short-lived signals from the plasma membrane via small molecule second messengers and ion channels to a beta-arrestin signaling mode that transmits a distinct set of signals that are initiated as the receptor internalizes and transits the intracellular compartment. Acts as signaling scaffold for MAPK pathways such as MAPK1/3 (ERK1/2). ERK1/2 activated by the beta-arrestin scaffold is largely excluded from the nucleus and confined to cytoplasmic locations such as endocytic vesicles, also called beta-arrestin signalosomes. Recruits c-Src/SRC to ADRB2 resulting in ERK activation. GPCRs for which the beta-arrestin-mediated signaling relies on both ARRB1 and ARRB2 (codependent regulation) include ADRB2, F2RL1 and PTH1R. For some GPCRs the beta-arrestin-mediated signaling relies on either ARRB1 or ARRB2 and is inhibited by the other respective beta-arrestin form (reciprocal regulation). Inhibits ERK1/2 signaling in AGTR1- and AVPR2-mediated activation (reciprocal regulation). Is required for SP-stimulated endocytosis of NK1R and recruits c-Src/SRC to internalized NK1R resulting in ERK1/2 activation, which is required for the antiapoptotic effects of SP. Is involved in proteinase-activated F2RL1-mediated ERK activity. Acts as signaling scaffold for the AKT1 pathway. Is involved in alpha-thrombin-stimulated AKT1 signaling. Is involved in IGF1-stimulated AKT1 signaling leading to increased protection from apoptosis. Involved in activation of the p38 MAPK signaling pathway and in actin bundle formation. Involved in F2RL1-mediated cytoskeletal rearrangement and chemotaxis. Involved in AGTR1-mediated stress fiber formation by acting together with GNAQ to activate RHOA. Appears to function as signaling scaffold involved in regulation of MIP-1-beta-stimulated CCR5-dependent chemotaxis. Involved in attenuation of NF-kappa-B-dependent transcription in response to GPCR or cytokine stimulation by interacting with and stabilizing CHUK. May serve as nuclear messenger for GPCRs. Involved in OPRD1-stimulated transcriptional regulation by translocating to CDKN1B and FOS promoter regions and recruiting EP300 resulting in acetylation of histone H4. Involved in regulation of LEF1 transcriptional activity via interaction with DVL1 and/or DVL2 Also involved in regulation of receptors other than GPCRs. Involved in Toll-like receptor and IL-1 receptor signaling through the interaction with TRAF6 which prevents TRAF6 autoubiquitination and oligomerization required for activation of NF-kappa-B and JUN. Binds phosphoinositides. Binds inositolhexakisphosphate (InsP6). Involved in IL8-mediated granule release in neutrophils. Required for atypical chemokine receptor ACKR2-induced RAC1-LIMK1-PAK1-dependent phosphorylation of cofilin (CFL1) and for the up-regulation of ACKR2 from endosomal compartment to cell membrane, increasing its efficiency in chemokine uptake and degradation. Involved in the internalization of the atypical chemokine receptor ACKR3. Negatively regulates the NOTCH signaling pathway by mediating the ubiquitination and degradation of NOTCH1 by ITCH. Participates to the recruitment of the ubiquitin-protein ligase to the receptor. Functions in regulating agonist-mediated G-protein coupled receptor (GPCR) signaling by mediating both receptor desensitization and resensitization processes. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P63010-2; O15169; P0DP25; P20963; P25101; P50148; Q5JWF2; Q14749; P06396; Q16665; P11142; Q99683; P53779; P45984; Q00987; P19338; Q14978; P14618; P14859-6; P35813; O75688; Q13523; P06702; P12931; Q15208; Q13428; P04637; P27348; P25490; O43298; O95218; Q7DB77 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Coated pit; Cytoplasm; Cytoplasmic vesicle; Membrane; Nucleus; Phosphoprotein; Protein transport; Proteomics identification; Reference proteome; Signal transduction inhibitor; Transcription; Transcription regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32455.6 Length 293 Aromaticity 0.12 Instability index 28.99 Isoelectric point 9.12 Charge (pH=7) 9.86 2D Binding mode Binding energy (Kcal/mol) -10.36  Molscript Map 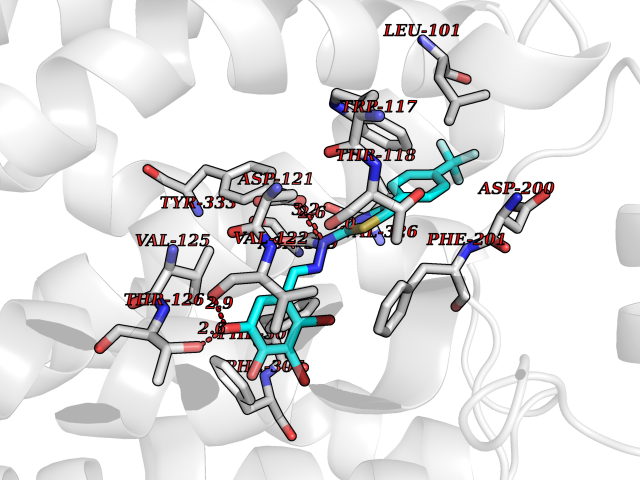 Pymol Map 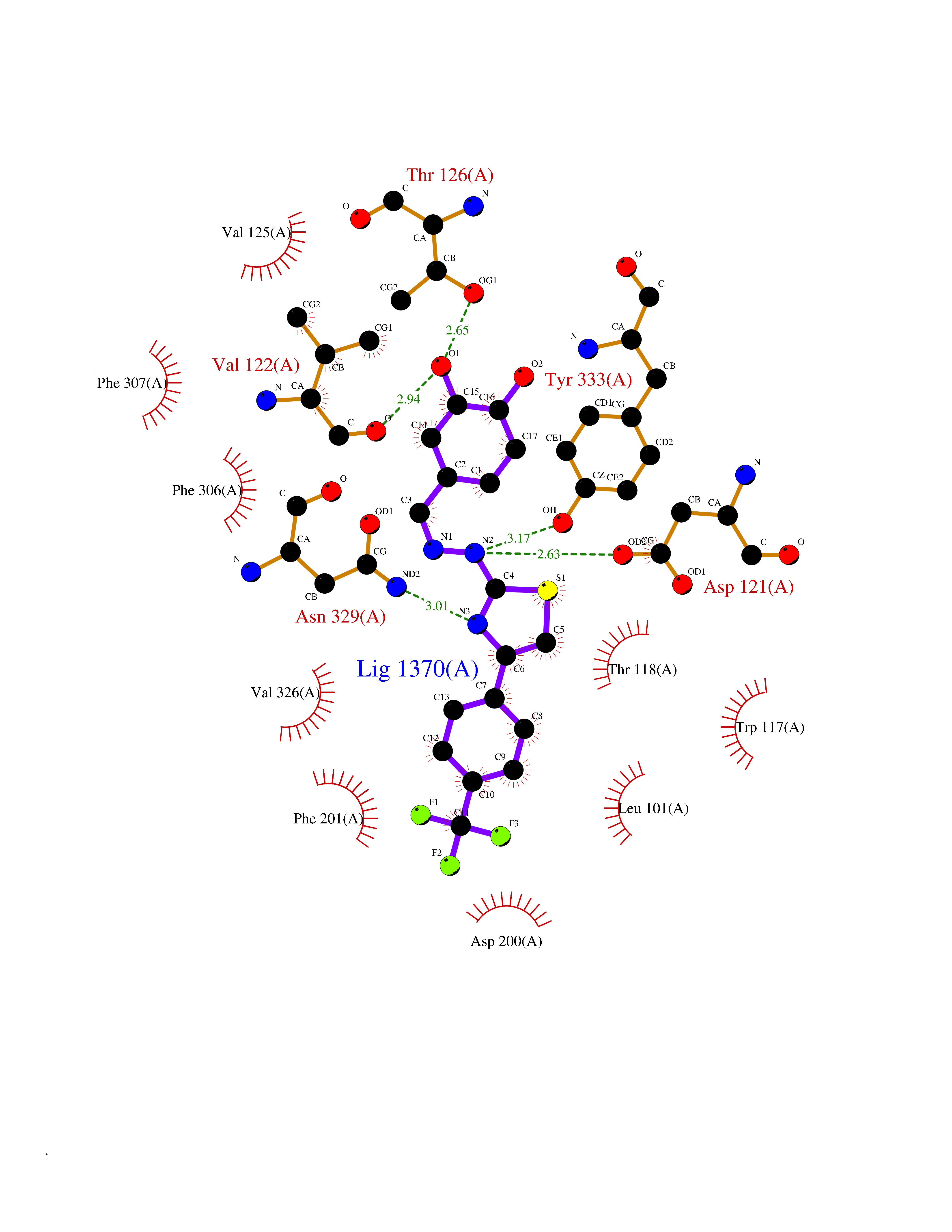 Ligplot Map 3D Binding mode Sequence AGCSLLMALVVLLIVAGNVLVIAAIGRTQRLQTLTNLFITSLACADLVVGLLVVPFGATLVCRGTWLWGSFLCELWTSLDVLCVTASIWTLCVIAIDRYLAITSPFRYQSLMTRARAKVIICTVWAISALVSFLPIMMHWWRDEDPQALKCYQDPGCCDFVTNRAYAIASSIISFYIPLLIMIFVYLRVYREAKEQIRKIDVMAMREHKALKTLGIIMGVFTLCWLPFFLVNIVNVFNRDLVPKWLFVAFNWLGYANSAMNPIIYCRSPDFRKAFKRLLAEXAXXAXXXLAKD Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Aldehyde oxidoreductase | 4USA | 7.59 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -10.36  Molscript Map  Pymol Map 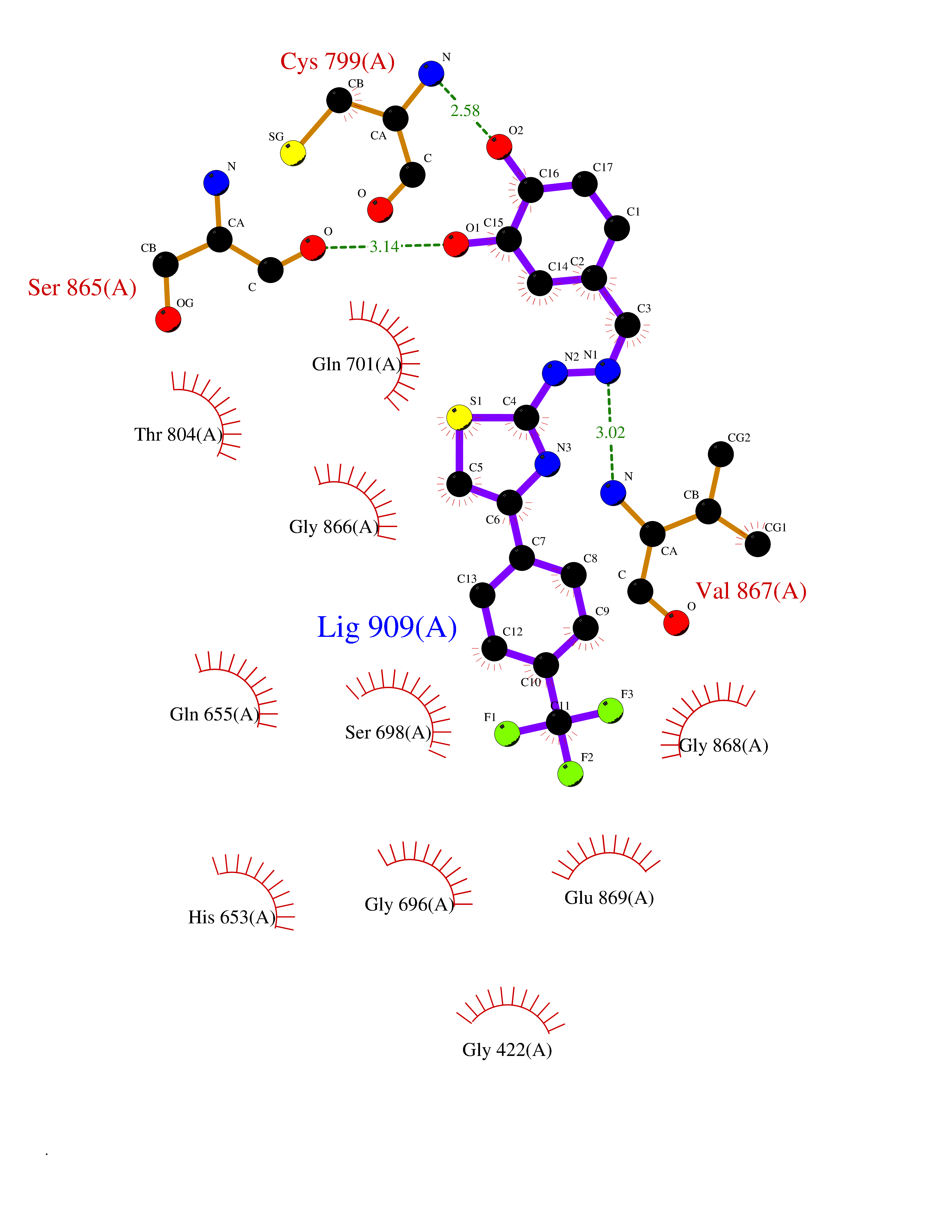 Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Monoglyceride lipase (MAGL) | 3PE6 | 7.59 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -10.35 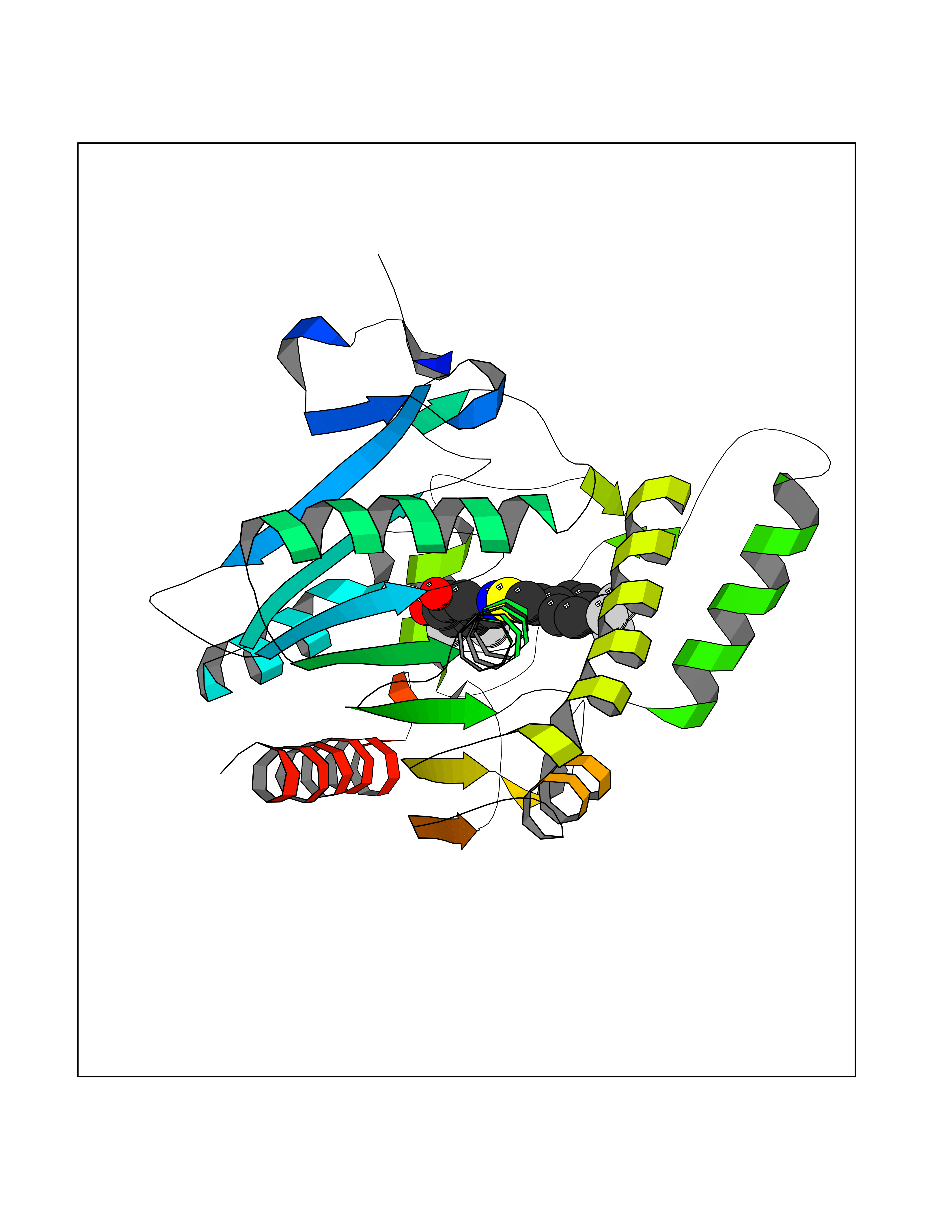 Molscript Map 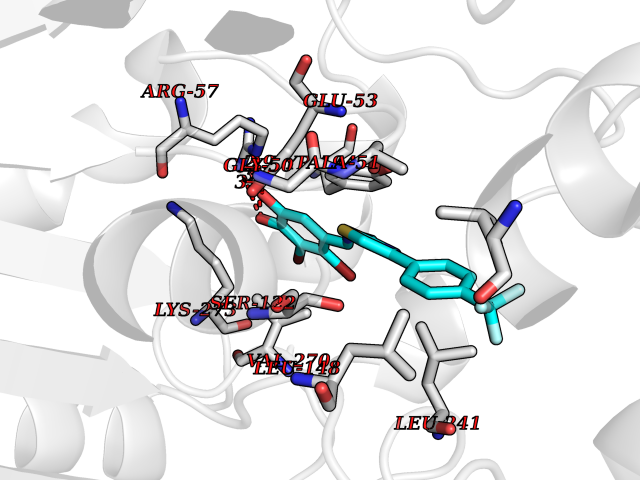 Pymol Map 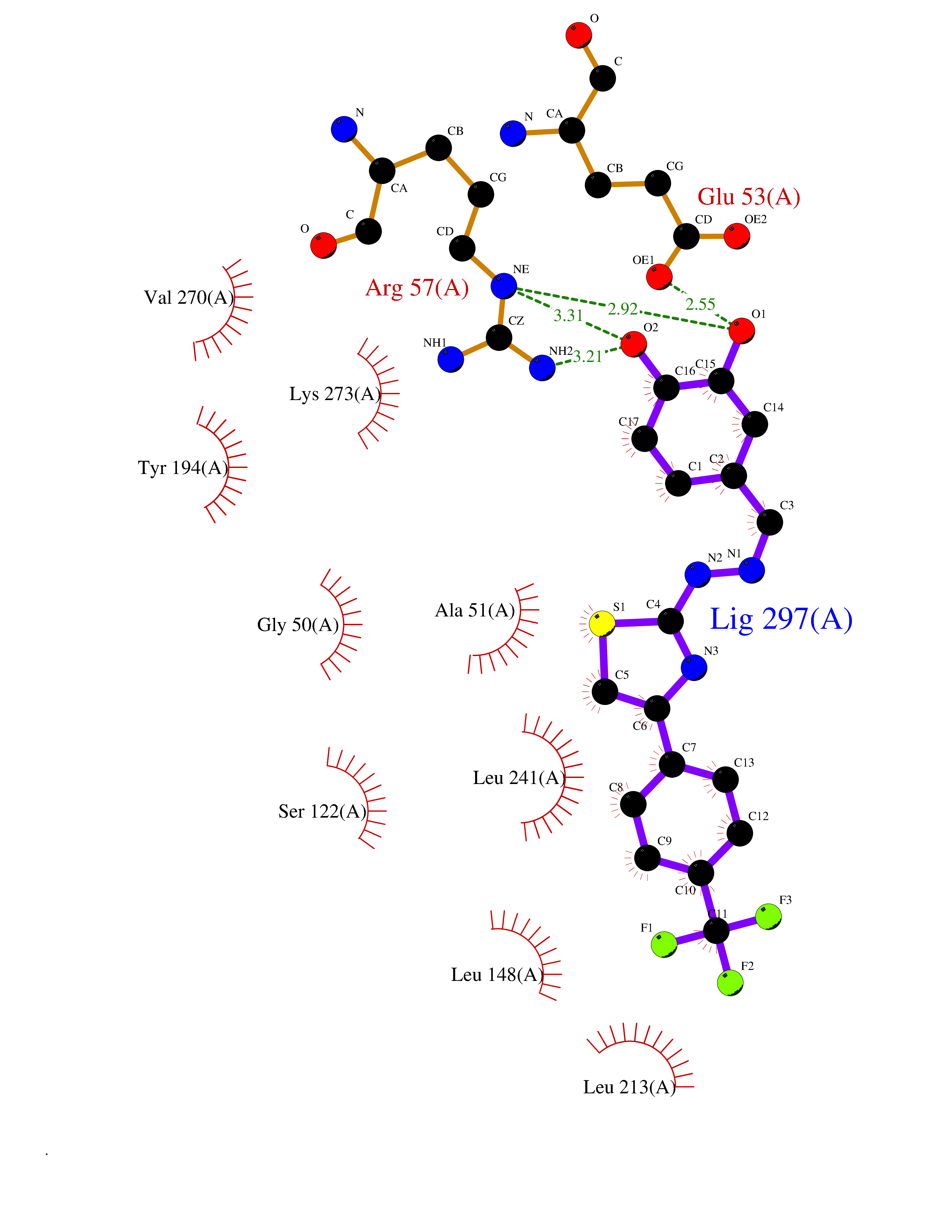 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Monoamine oxidase type A (MAO-A) | 2Z5Y | 7.58 | |
Target general information Gen name MAOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoamine oxidase A; Amine oxidase [flavin-containing] A Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function MAOA preferentially oxidizes biogenic amines such as 5-hydroxytryptamine (5-HT), norepinephrine and epinephrine. Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. Related diseases Brunner syndrome (BRNRS) [MIM:300615]: A form of X-linked non-dysmorphic mild intellectual disability. Male patients are affected by borderline intellectual deficit and exhibit abnormal behavior, including disturbed regulation of impulsive aggression. Obligate female carriers have normal intelligence and behavior. {ECO:0000269|PubMed:8211186}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00918; DB00182; DB06698; DB04889; DB13876; DB01445; DB06774; DB00215; DB04017; DB09130; DB05205; DB07641; DB00988; DB01363; DB00668; DB12329; DB01175; DB03147; DB14914; DB00614; DB01381; DB07919; DB04818; DB01247; DB00601; DB01577; DB00805; DB01442; DB01171; DB08804; DB00952; DB04820; DB00184; DB04821; DB06412; DB01626; DB00780; DB00191; DB00388; DB00397; DB09244; DB04850; DB00721; DB01168; DB00571; DB00852; DB09363; DB00140; DB00953; DB06654; DB01037; DB01104; DB00669; DB14569; DB09042; DB00624; DB13943; DB13944; DB13946; DB09245; DB00752; DB15328; DB09185; DB04832; DB00315; DB00909 Interacts with P27338 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine metabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Intellectual disability; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurotransmitter degradation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 58195.3 Length 513 Aromaticity 0.11 Instability index 34.97 Isoelectric point 7.98 Charge (pH=7) 2.87 2D Binding mode Binding energy (Kcal/mol) -10.34 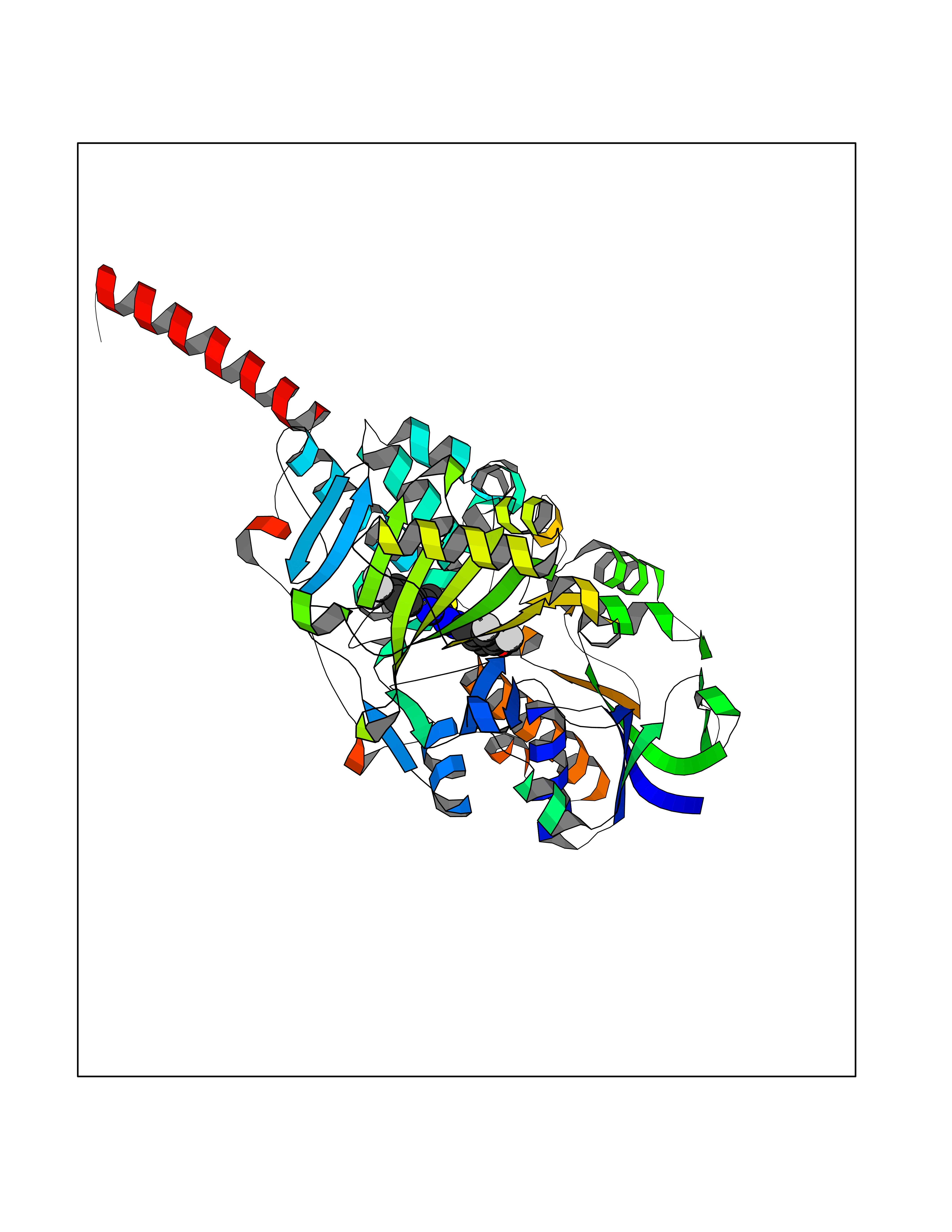 Molscript Map 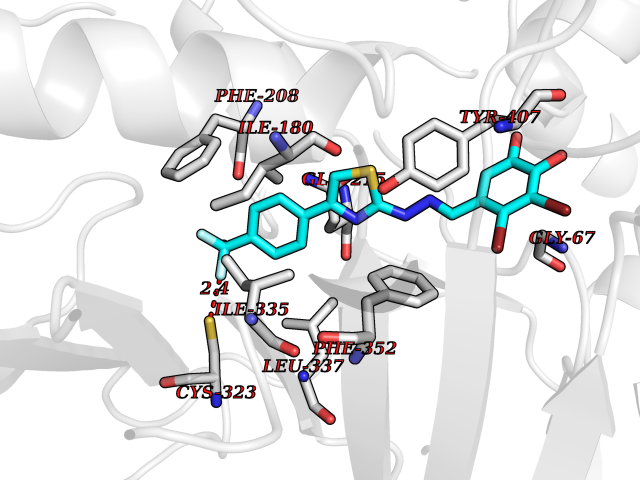 Pymol Map 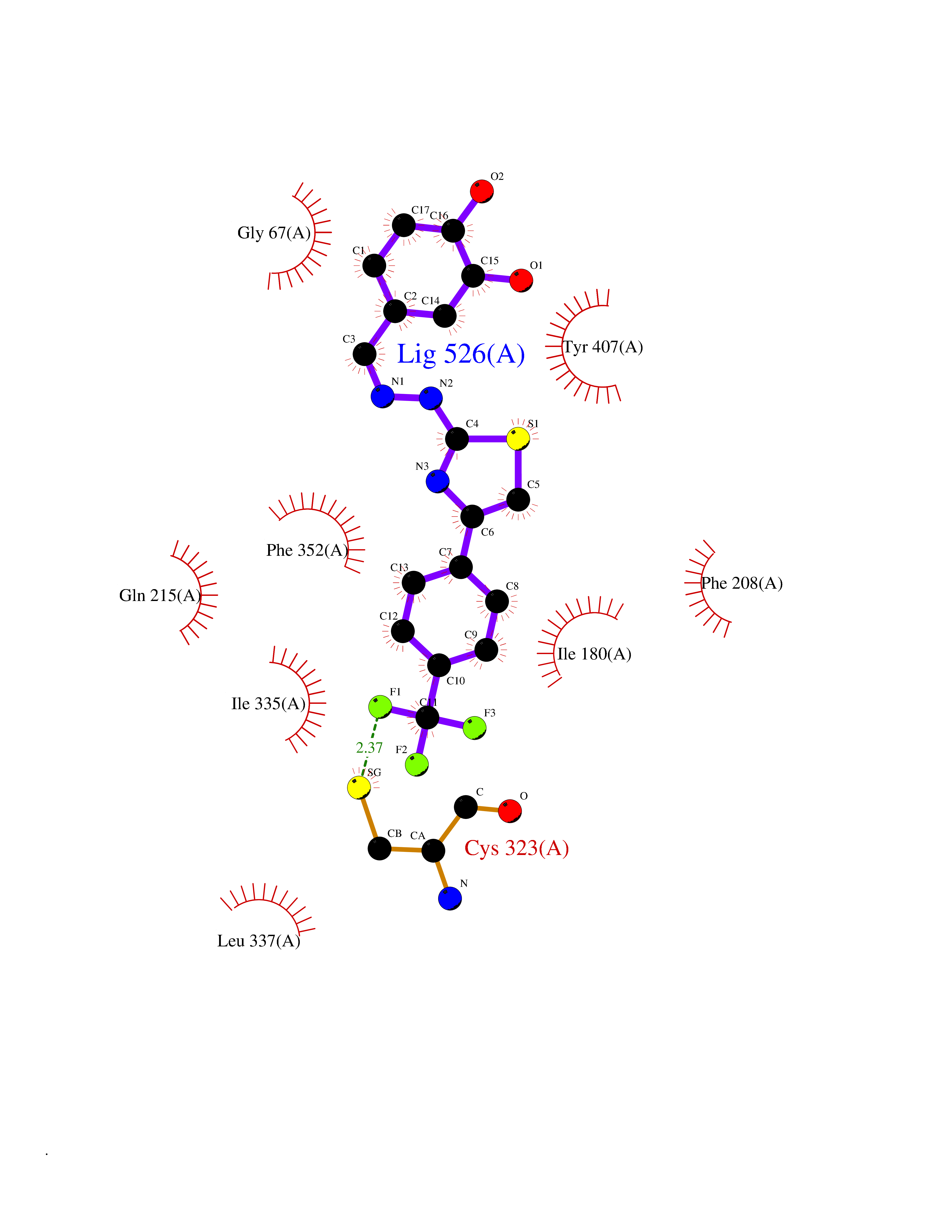 Ligplot Map 3D Binding mode Sequence HMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYVGPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRAAFPPVWNPIAYLDYNNLWRTIDNMGKEIPTDAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSVTNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAKIHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIMGFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYGRVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTFWERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLL Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Monoamine oxidase type B (MAO-B) | 2V5Z | 7.56 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -10.31 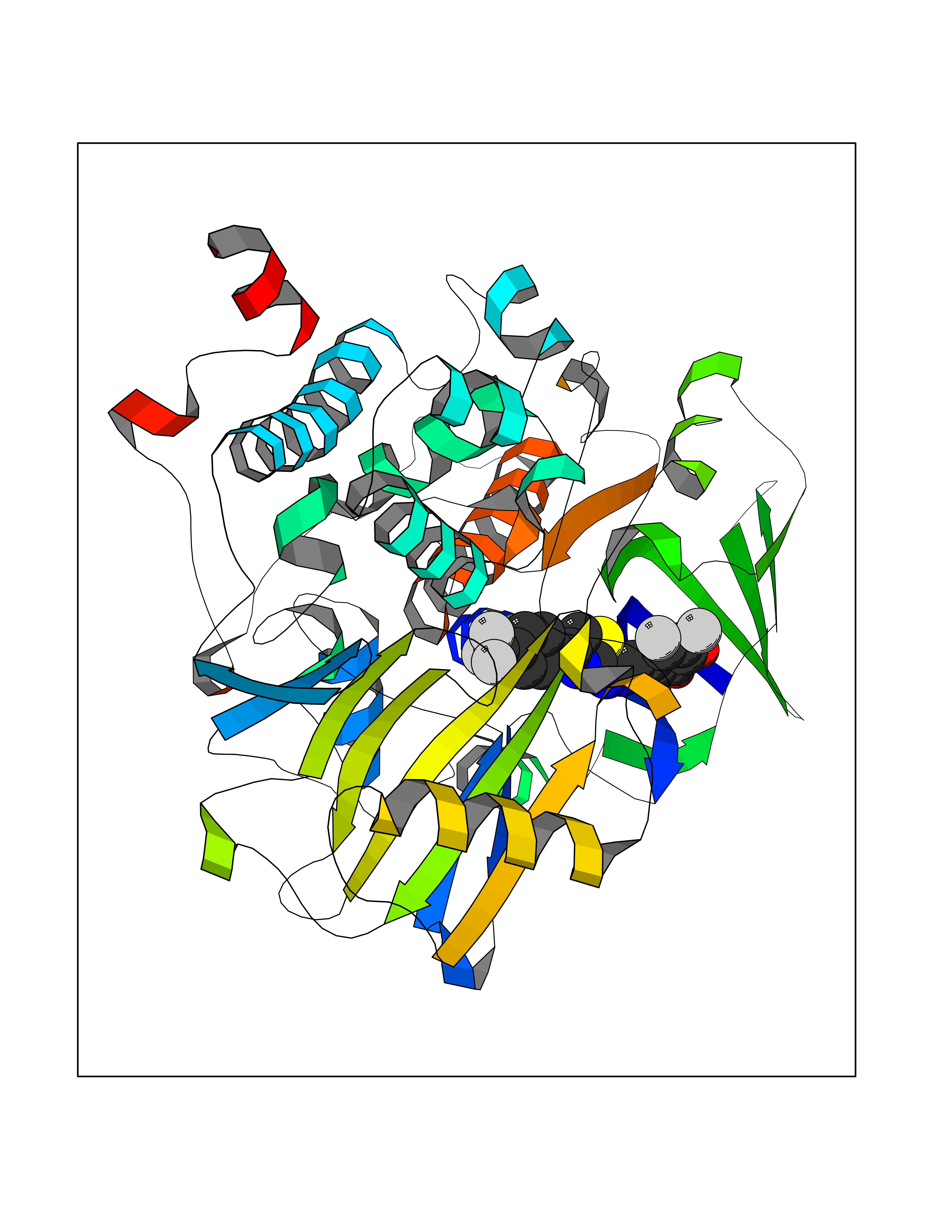 Molscript Map 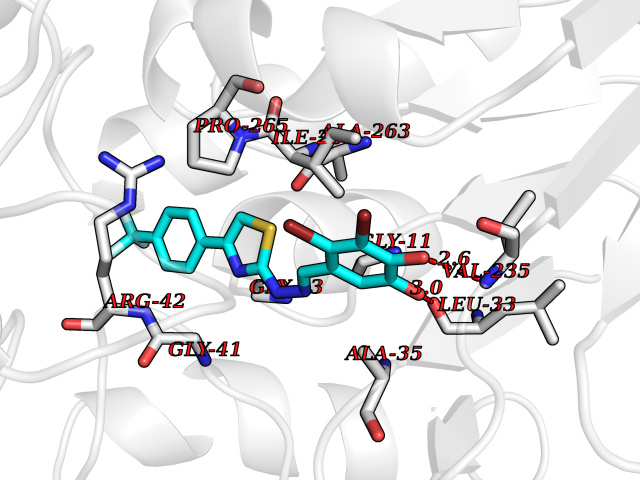 Pymol Map 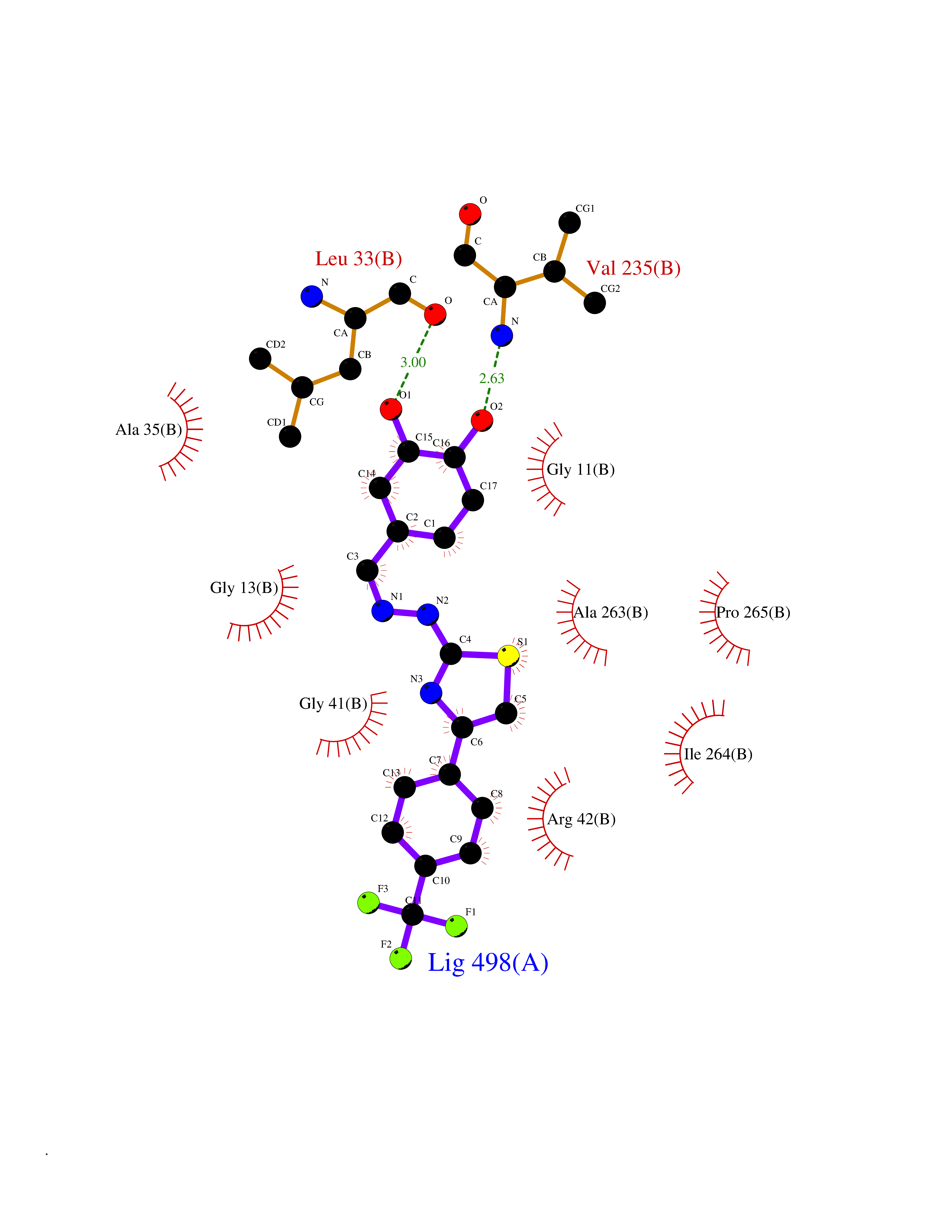 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Guanidinoacetate N-methyltransferase | 3ORH | 7.56 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -10.31 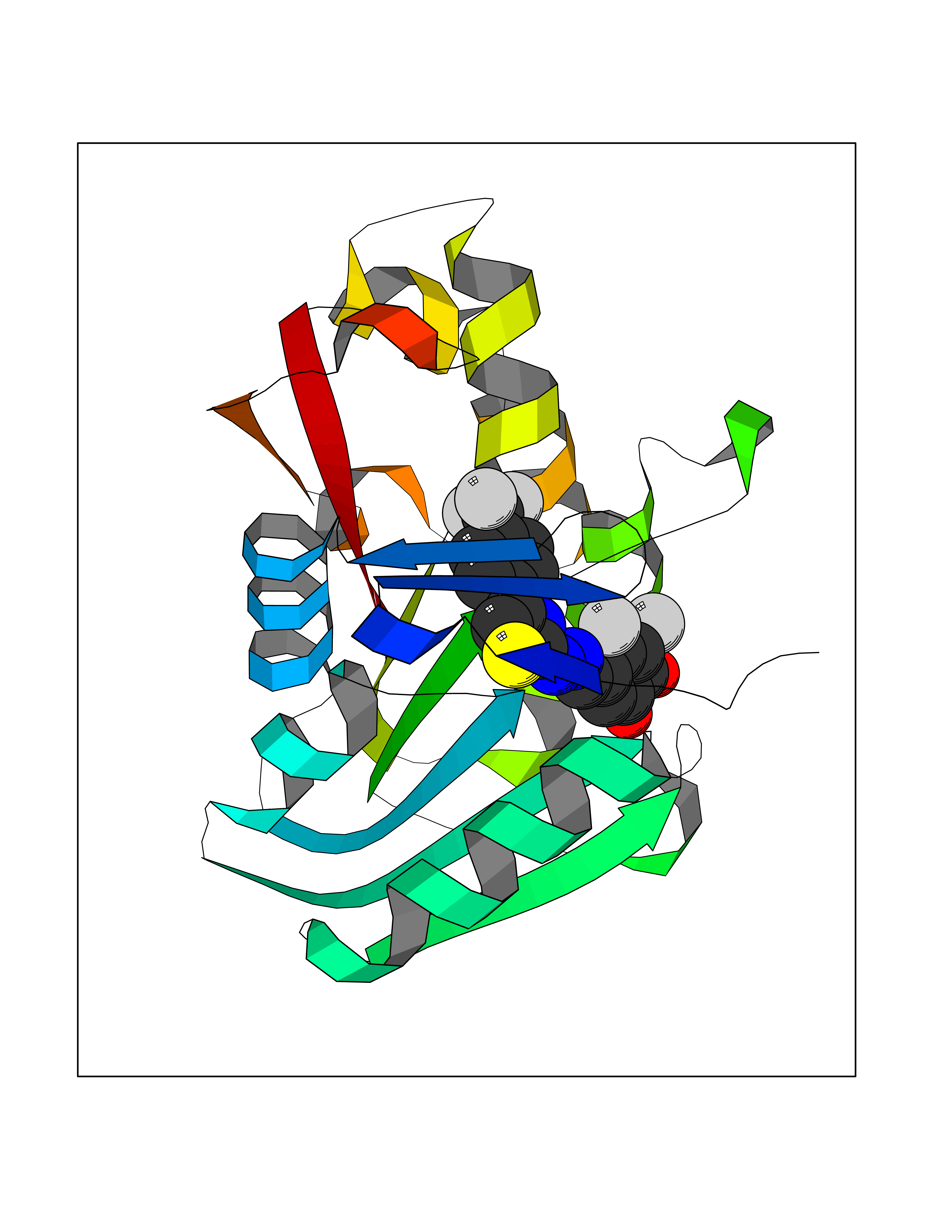 Molscript Map 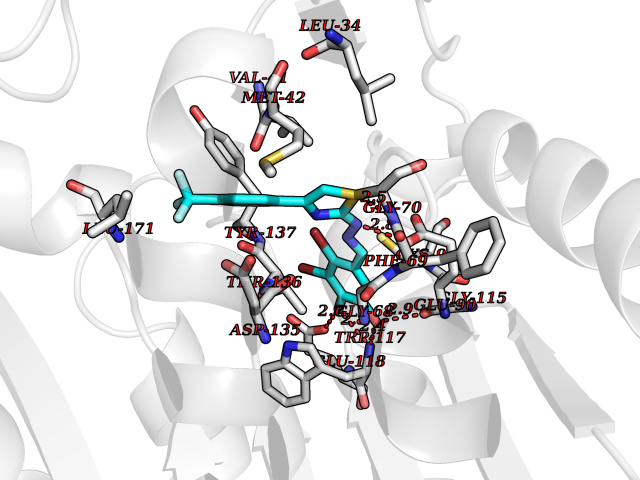 Pymol Map 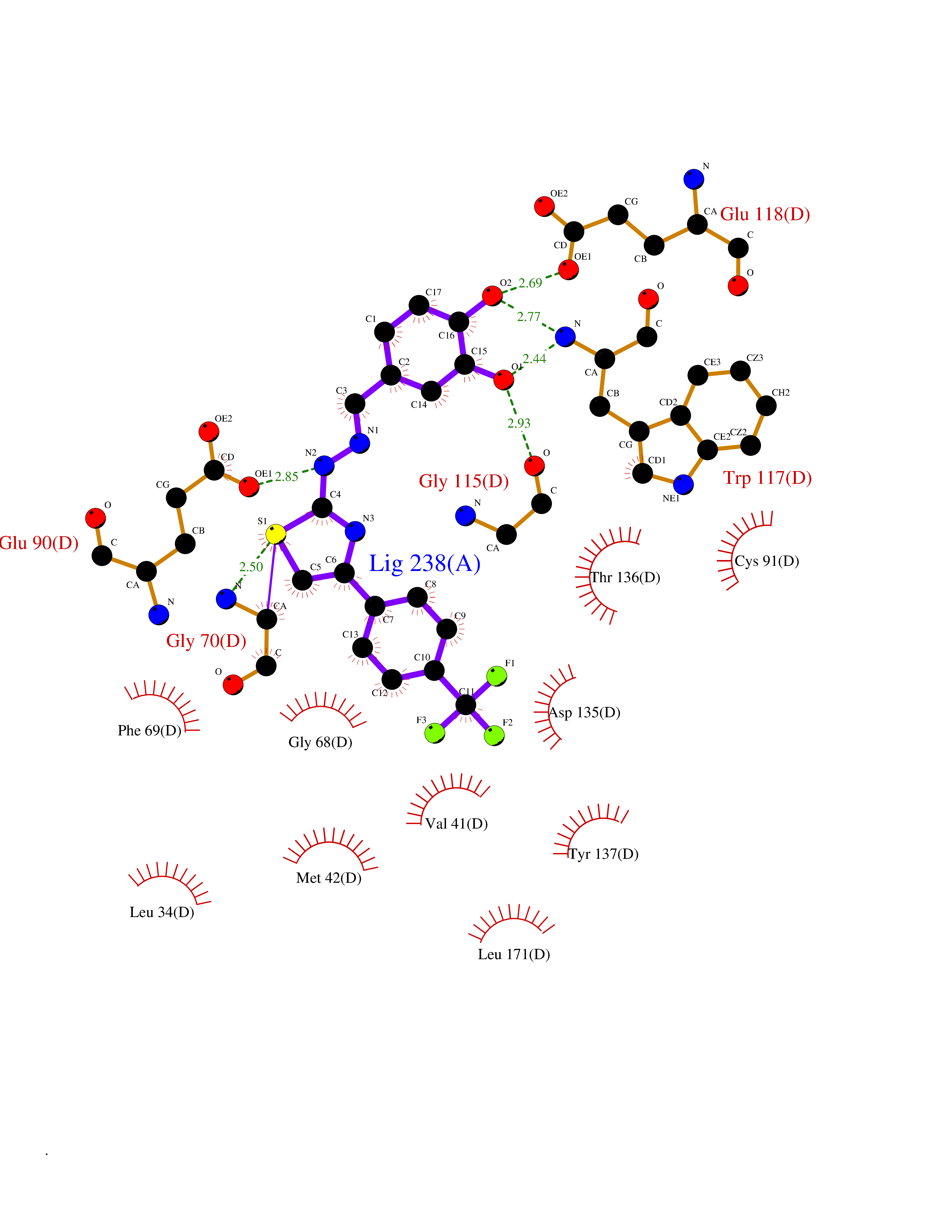 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Oxysterols receptor LXR-alpha (NR1H3) | 3IPQ | 7.56 | |
Target general information Gen name NR1H3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 1 group H member 3; Nuclear receptor LXRalpha; Nuclear orphan receptor LXR-alpha; Liver X receptor alpha; LXRalpha; LXRA Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Interaction with retinoic acid receptor (RXR) shifts RXR from its role as a silent DNA-binding partner to an active ligand-binding subunit in mediating retinoid responses through target genes defined by LXRES. LXRES are DR4-type response elements characterized by direct repeats of two similar hexanuclotide half-sites spaced by four nucleotides. Plays an important role in the regulation of cholesterol homeostasis, regulating cholesterol uptake through MYLIP-dependent ubiquitination of LDLR, VLDLR and LRP8. Interplays functionally with RORA for the regulation of genes involved in liver metabolism. Nuclear receptor that exhibits a ligand-dependent transcriptional activation activity. Related diseases Okur-Chung neurodevelopmental syndrome (OCNDS) [MIM:617062]: An autosomal dominant neurodevelopmental disorder characterized by developmental delay, intellectual disability, behavioral problems, hypotonia, speech problems, microcephaly, pachygyria and variable dysmorphic features. {ECO:0000269|PubMed:27048600}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB08063; DB11994; DB07929; DB13174; DB07080 Interacts with O60869; O60341; Q99750; Q15788; O75376; Q07869; Q07869-1; Q03181; P37231; P19793; P28702; P48443; O43463; P42858; Q99750; O95817; G5E9A7; O95872; P02545; Q99750; P28702; P28702-3; P48443; Q7Z699 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 25389.9 Length 220 Aromaticity 0.09 Instability index 46.42 Isoelectric point 5.51 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -10.31  Molscript Map 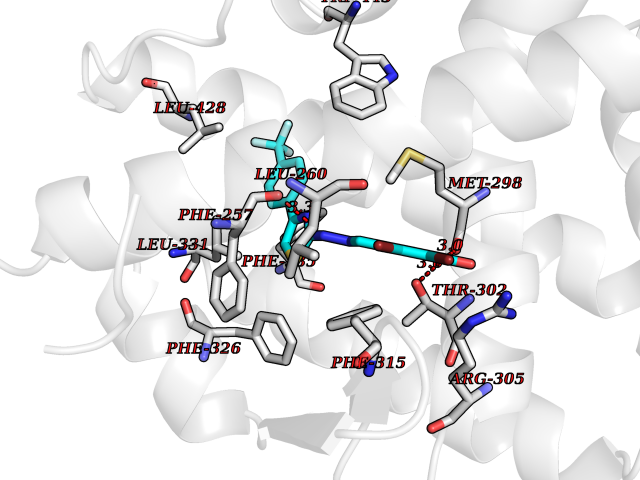 Pymol Map 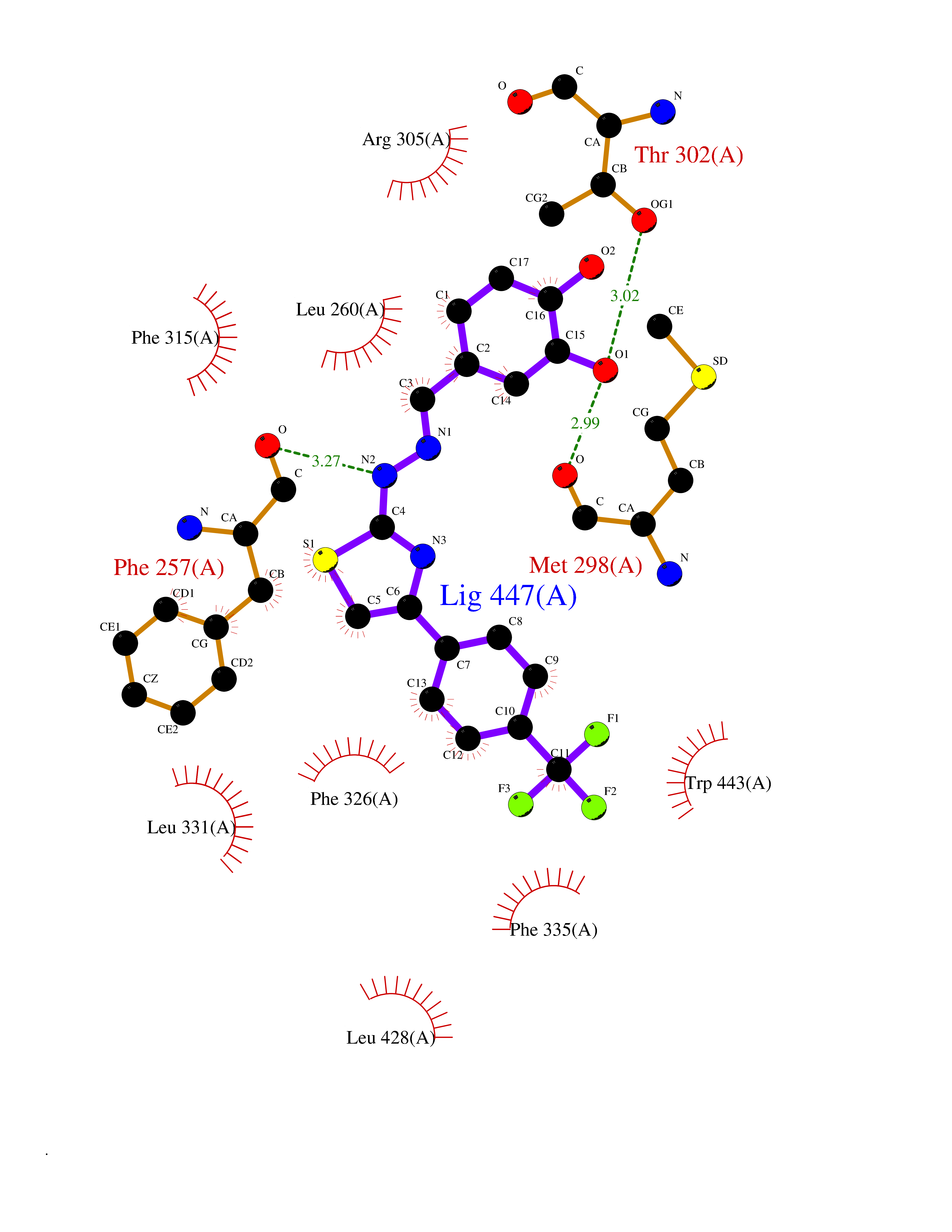 Ligplot Map 3D Binding mode Sequence QLSPEQLGMIEKLVAAQQTPWPEARQQRFAHFTELAIVSVQEIVDFAKQLPGFLQLSREDQIALLKTSAIEVMLLETSRRYNPGSESITFLKDFSYNREDFAKAGLQVEFINPIFEFSRAMNELQLNDAEFALLIAISIFSADRPNVQDQLQVERLQHTYVEALHAYVSIHHPHDRLMFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDV Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 7.55 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -10.3  Molscript Map 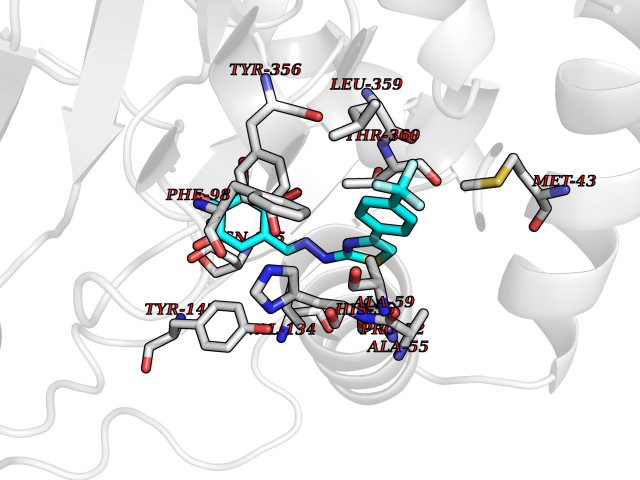 Pymol Map 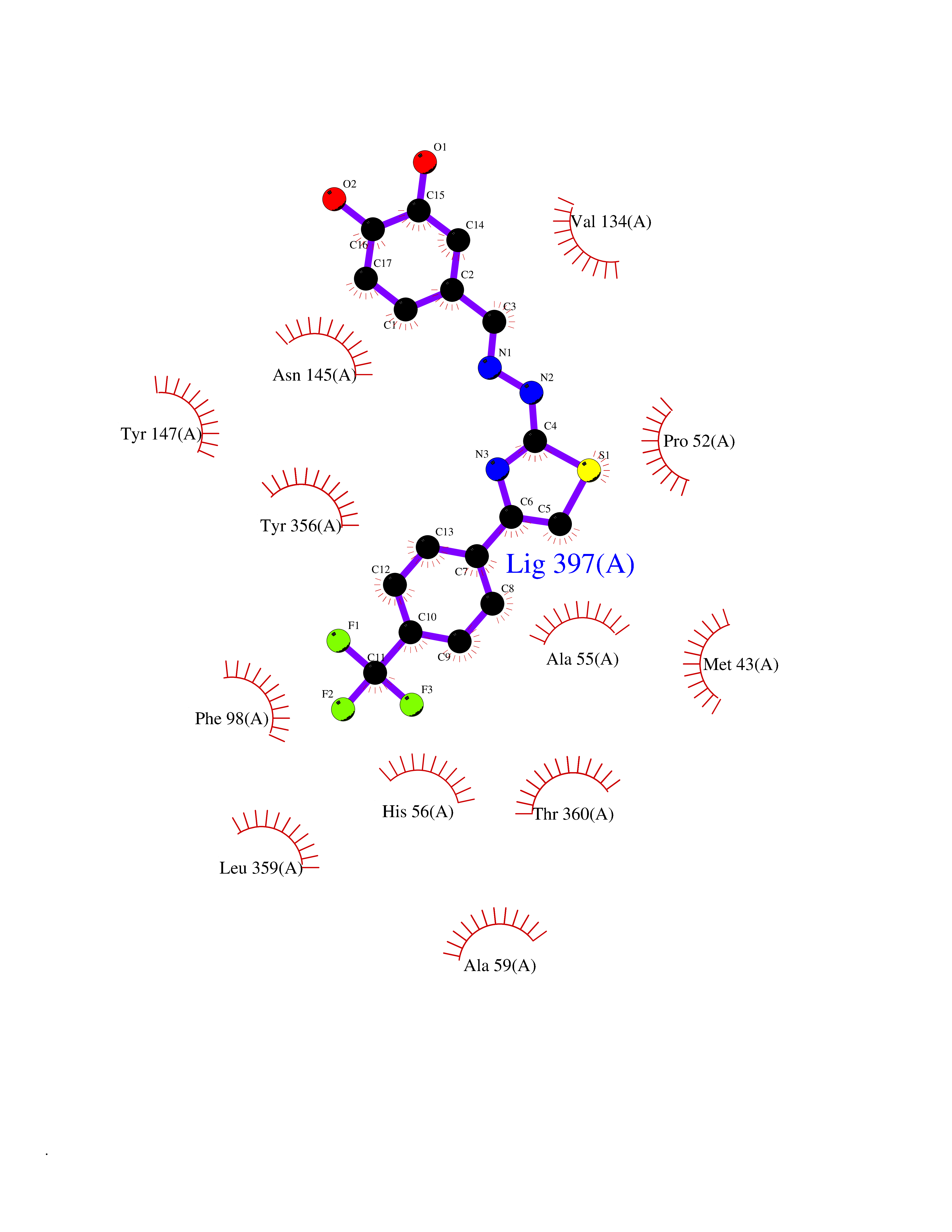 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | NAD-dependent deacetylase sirtuin-2 (SIRT2) | 4RMJ | 7.55 | |
Target general information Gen name SIRT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SIR2like protein 2; SIR2L2; SIR2L; SIR2-like protein 2; Regulatory protein SIR2 homolog 2; NADdependent protein deacetylase sirtuin2; NAD-dependent protein deacetylase sirtuin-2 Protein family Sirtuin family, Class I subfamily Biochemical class Carbon-nitrogen hydrolase Function Participates in the modulation of multiple and diverse biological processes such as cell cycle control, genomic integrity, microtubule dynamics, cell differentiation, metabolic networks, and autophagy. Plays a major role in the control of cell cycle progression and genomic stability. Functions in the antephase checkpoint preventing precocious mitotic entry in response to microtubule stress agents, and hence allowing proper inheritance of chromosomes. Positively regulates the anaphase promoting complex/cyclosome (APC/C) ubiquitin ligase complex activity by deacetylating CDC20 and FZR1, then allowing progression through mitosis. Associates both with chromatin at transcriptional start sites (TSSs) and enhancers of active genes. Plays a role in cell cycle and chromatin compaction through epigenetic modulation of the regulation of histone H4 'Lys-20' methylation (H4K20me1) during early mitosis. Specifically deacetylates histone H4 at 'Lys-16' (H4K16ac) between the G2/M transition and metaphase enabling H4K20me1 deposition by KMT5A leading to ulterior levels of H4K20me2 and H4K20me3 deposition throughout cell cycle, and mitotic S-phase progression. Deacetylates KMT5A modulating KMT5A chromatin localization during the mitotic stress response. Deacetylates also histone H3 at 'Lys-57' (H3K56ac) during the mitotic G2/M transition. Upon bacterium Listeria monocytogenes infection, deacetylates 'Lys-18' of histone H3 in a receptor tyrosine kinase MET- and PI3K/Akt-dependent manner, thereby inhibiting transcriptional activity and promoting late stages of listeria infection. During oocyte meiosis progression, may deacetylate histone H4 at 'Lys-16' (H4K16ac) and alpha-tubulin, regulating spindle assembly and chromosome alignment by influencing microtubule dynamics and kinetochore function. Deacetylates histone H4 at 'Lys-16' (H4K16ac) at the VEGFA promoter and thereby contributes to regulate expression of VEGFA, a key regulator of angiogenesis. Deacetylates alpha-tubulin at 'Lys-40' and hence controls neuronal motility, oligodendroglial cell arbor projection processes and proliferation of non-neuronal cells. Phosphorylation at Ser-368 by a G1/S-specific cyclin E-CDK2 complex inactivates SIRT2-mediated alpha-tubulin deacetylation, negatively regulating cell adhesion, cell migration and neurite outgrowth during neuronal differentiation. Deacetylates PARD3 and participates in the regulation of Schwann cell peripheral myelination formation during early postnatal development and during postinjury remyelination. Involved in several cellular metabolic pathways. Plays a role in the regulation of blood glucose homeostasis by deacetylating and stabilizing phosphoenolpyruvate carboxykinase PCK1 activity in response to low nutrient availability. Acts as a key regulator in the pentose phosphate pathway (PPP) by deacetylating and activating the glucose-6-phosphate G6PD enzyme, and therefore, stimulates the production of cytosolic NADPH to counteract oxidative damage. Maintains energy homeostasis in response to nutrient deprivation as well as energy expenditure by inhibiting adipogenesis and promoting lipolysis. Attenuates adipocyte differentiation by deacetylating and promoting FOXO1 interaction to PPARG and subsequent repression of PPARG-dependent transcriptional activity. Plays a role in the regulation of lysosome-mediated degradation of protein aggregates by autophagy in neuronal cells. Deacetylates FOXO1 in response to oxidative stress or serum deprivation, thereby negatively regulating FOXO1-mediated autophagy. Deacetylates a broad range of transcription factors and co-regulators regulating target gene expression. Deacetylates transcriptional factor FOXO3 stimulating the ubiquitin ligase SCF(SKP2)-mediated FOXO3 ubiquitination and degradation. Deacetylates HIF1A and therefore promotes HIF1A degradation and inhibition of HIF1A transcriptional activity in tumor cells in response to hypoxia. Deacetylates RELA in the cytoplasm inhibiting NF-kappaB-dependent transcription activation upon TNF-alpha stimulation. Inhibits transcriptional activation by deacetylating p53/TP53 and EP300. Deacetylates also EIF5A. Functions as a negative regulator on oxidative stress-tolerance in response to anoxia-reoxygenation conditions. Plays a role as tumor suppressor. NAD-dependent protein deacetylase, which deacetylates internal lysines on histone and alpha-tubulin as well as many other proteins such as key transcription factors. Related diseases Deafness, autosomal recessive, 39 (DFNB39) [MIM:608265]: A form of profound prelingual sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:19576567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15493 Interacts with O60566; O60729; P11413; Q92831; Q04206; Q9BYB0; Q12834; Q9UM11 EC number EC 3.5.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Autophagy; Cell cycle; Cell division; Cell membrane; Cell projection; Chromosome; Cytoplasm; Cytoskeleton; Differentiation; Immunity; Innate immunity; Meiosis; Membrane; Metal-binding; Microtubule; Mitosis; NAD; Neurodegeneration; Neurogenesis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 34093.1 Length 301 Aromaticity 0.1 Instability index 45.2 Isoelectric point 5.59 Charge (pH=7) -6.94 2D Binding mode Binding energy (Kcal/mol) -10.3  Molscript Map 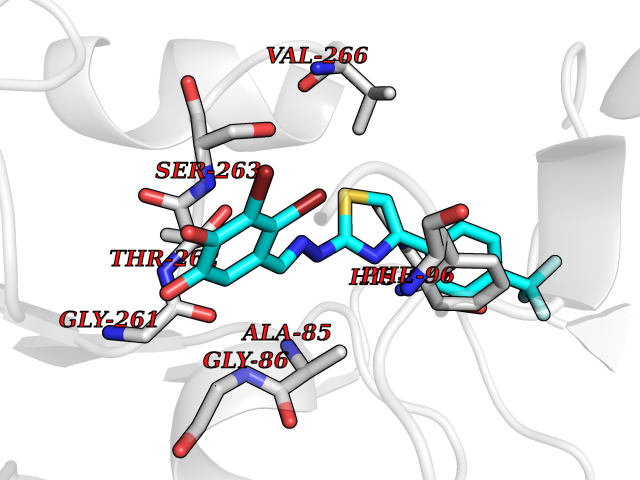 Pymol Map 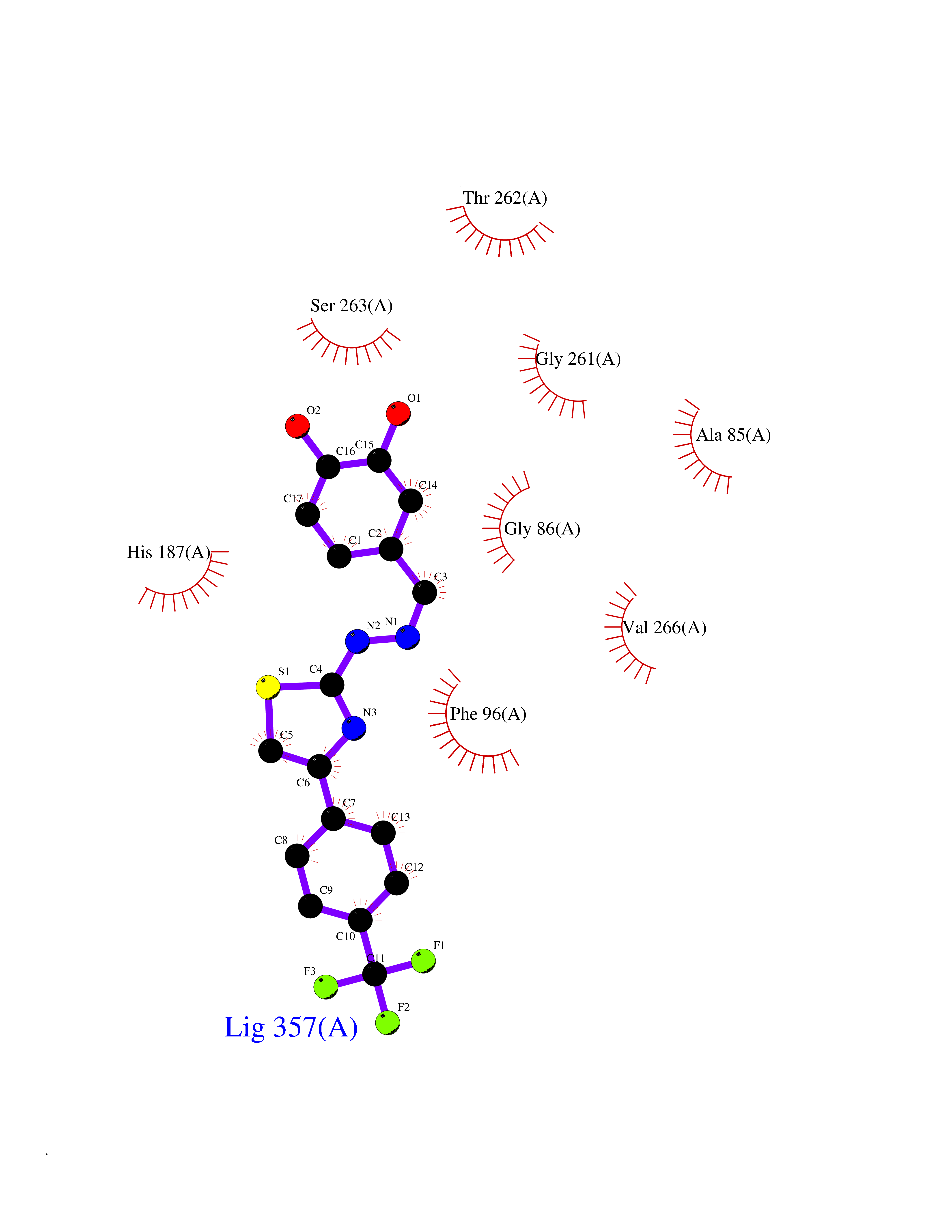 Ligplot Map 3D Binding mode Sequence MERLLDELTLEGVARYMQSERCRRVICLVGAGISTSAGIPDFRSPSTGLYDNLEKYHLPYPEAIFEISYFKKHPEPFFALAKELYPGQFKPTICHYFMRLLKDKGLLLRCYTQNIDTLERIAGLEQEDLVEAHGTFYTSHCVSASCRHEYPLSWMKEKIFSEVTPKCEDCQSLVKPDIVFFGESLPARFFSCMQSDFLKVDLLLVMGTSLQVQPFASLISKAPLSTPRLLINKEKAGQSDPFLGMIMGLGGGMDFDSKKAYRDVAWLGECDQGCLALAELLGWKKELEDLVRREHASIDAQ Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Mutated Histone H3.3 (H3F3A) | 4GUS | 7.54 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -10.29  Molscript Map  Pymol Map 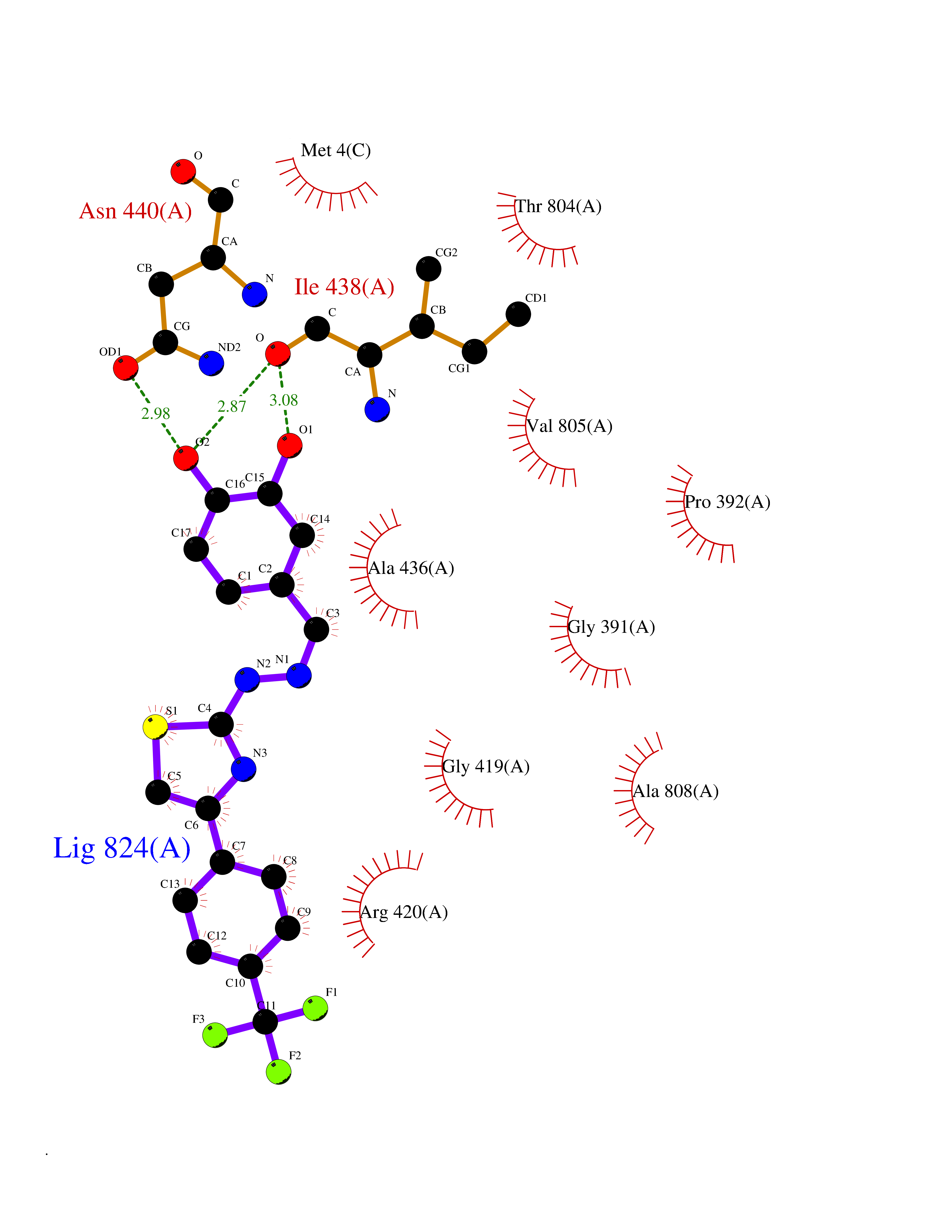 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | SET domain containing 8 (KMT5A) | 5TEG | 7.53 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -10.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Sodium/glucose cotransporter 1 (SGLT1) | 7WMV | 7.53 | |
Target general information Gen name SLC5A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 5 member 1; Na(+)/glucose cotransporter 1; NAGT; High affinity sodium-glucose cotransporter Protein family Sodium:solute symporter (SSF) (TC 2.A.21) family Biochemical class Solute:sodium symporter Function Efficient substrate transport in mammalian kidney is provided by the concerted action of a low affinity high capacity and a high affinity low capacity Na(+)/glucose cotransporter arranged in series along kidney proximal tubules. Actively transports glucose into cells by Na(+) cotransport with a Na(+) to glucose coupling ratio of 2:1. Related diseases Congenital glucose/galactose malabsorption (GGM) [MIM:606824]: Intestinal monosaccharide transporter deficiency. It is an autosomal recessive disorder manifesting itself within the first weeks of life. It is characterized by severe diarrhea and dehydration which are usually fatal unless glucose and galactose are eliminated from the diet. {ECO:0000269|PubMed:10036327, ECO:0000269|PubMed:11406349, ECO:0000269|PubMed:2008213, ECO:0000269|PubMed:8195156, ECO:0000269|PubMed:8563765}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00766; DB01914; DB09341; DB05018; DB12713 Interacts with P00533 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Sodium; Sodium transport; Sugar transport; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 66451.3 Length 602 Aromaticity 0.12 Instability index 35.84 Isoelectric point 8.3 Charge (pH=7) 4.43 2D Binding mode Binding energy (Kcal/mol) -10.28  Molscript Map 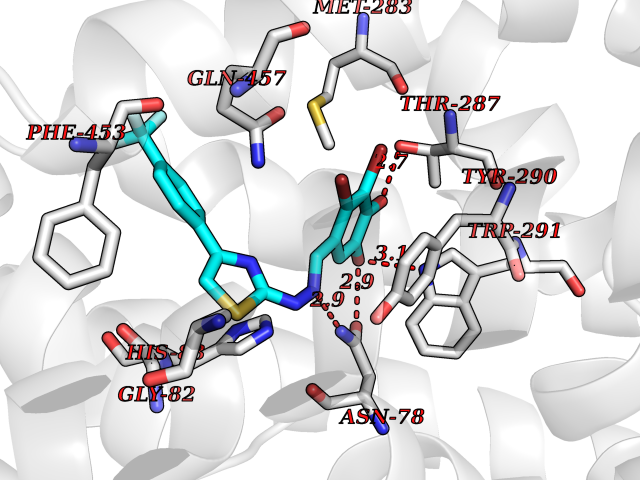 Pymol Map 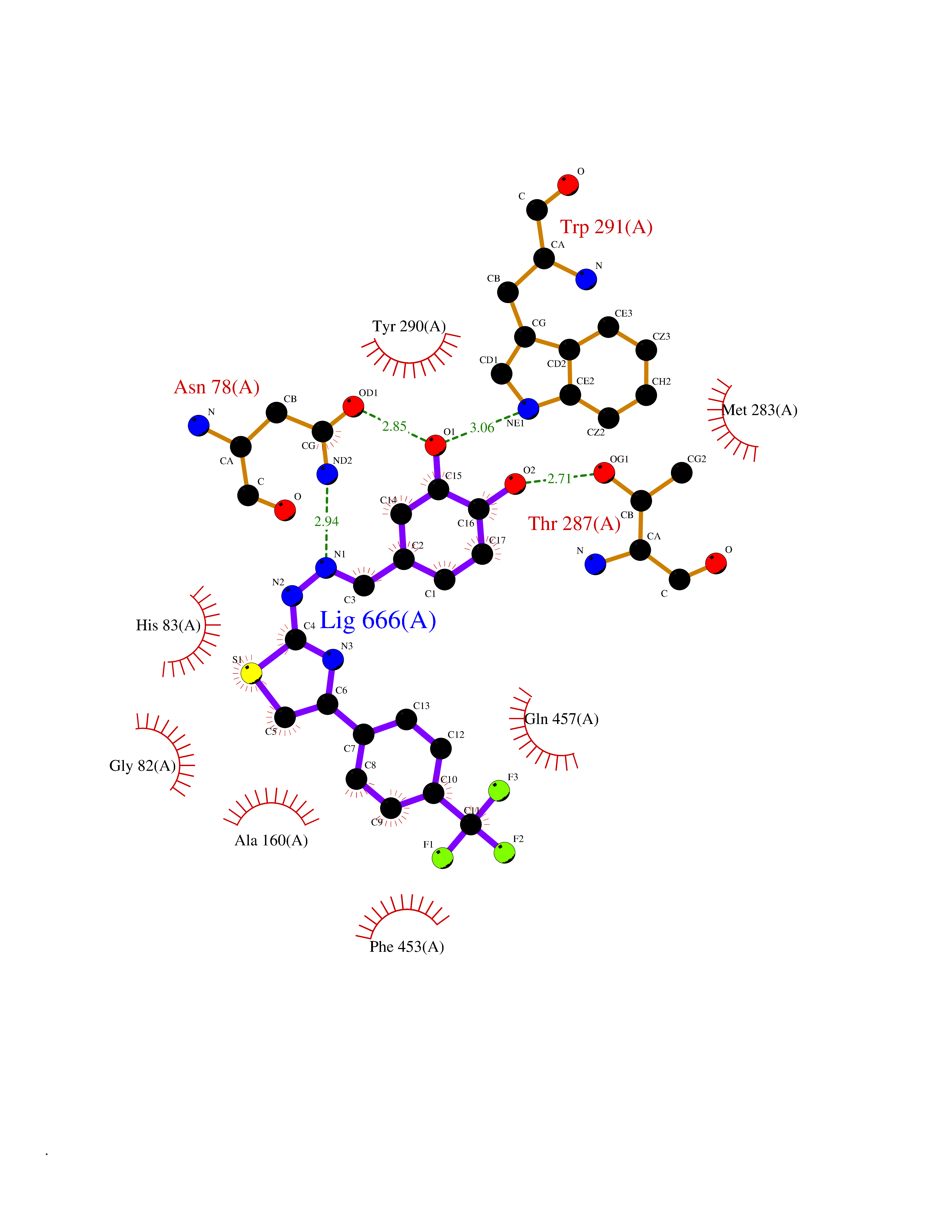 Ligplot Map 3D Binding mode Sequence ETHELIRNAADISIIVIYFVVVMAVGLWAMFSTNRGTVGGFFLAGRSMVWWPIGASLFASNIGSGHFVGLAGTGAASGIAIGGFEWNALVLVVVLGWLFVPIYIKAGVVTMPEYLRKRFGGQRIQVYLSLLSLLLYIFTKISADIFSGAIFINLALGLNLYLAIFLLLAITALYTITGGLAAVIYTDTLQTVIMLVGSLILTGFAFHEVGGYDAFMEKYMKAIPTIVSDGNTTFQEKCYTPRADSFHIFRDPLTGDLPWPGFIFGMSILTLWYWCTDQVIVQRCLSAKNMSHVKGGCILCGYLKLMPMFIMVMPGMISRILYTEKIACVVPSECEKYCGTKVGCTNIAYPTLVVELMPNGLRGLMLSVMLASLMSSLTSIFNSASTLFTMDIYAKVRKRASEKELMIAGRLFILVLIGISIAWVPIVQSAQSGQLFDYIQSITSYLGPPIAAVFLLAIFWKRVNEPGAFWGLILGLLIGISRMITEFAYGTGSCMEPSNCPTIICGVHYLYFAIILFAISFITIVVISLLTKPIPDVHLYRLCWSLRNSKEERIDLDATEEEEKAMKMKMTDTSEKPLWRTVLNVNGIILVTVAVFCHAYFA Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 7.53 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -10.27  Molscript Map 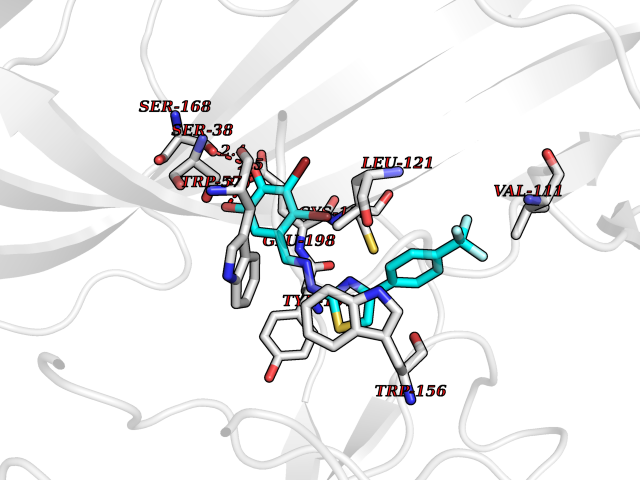 Pymol Map 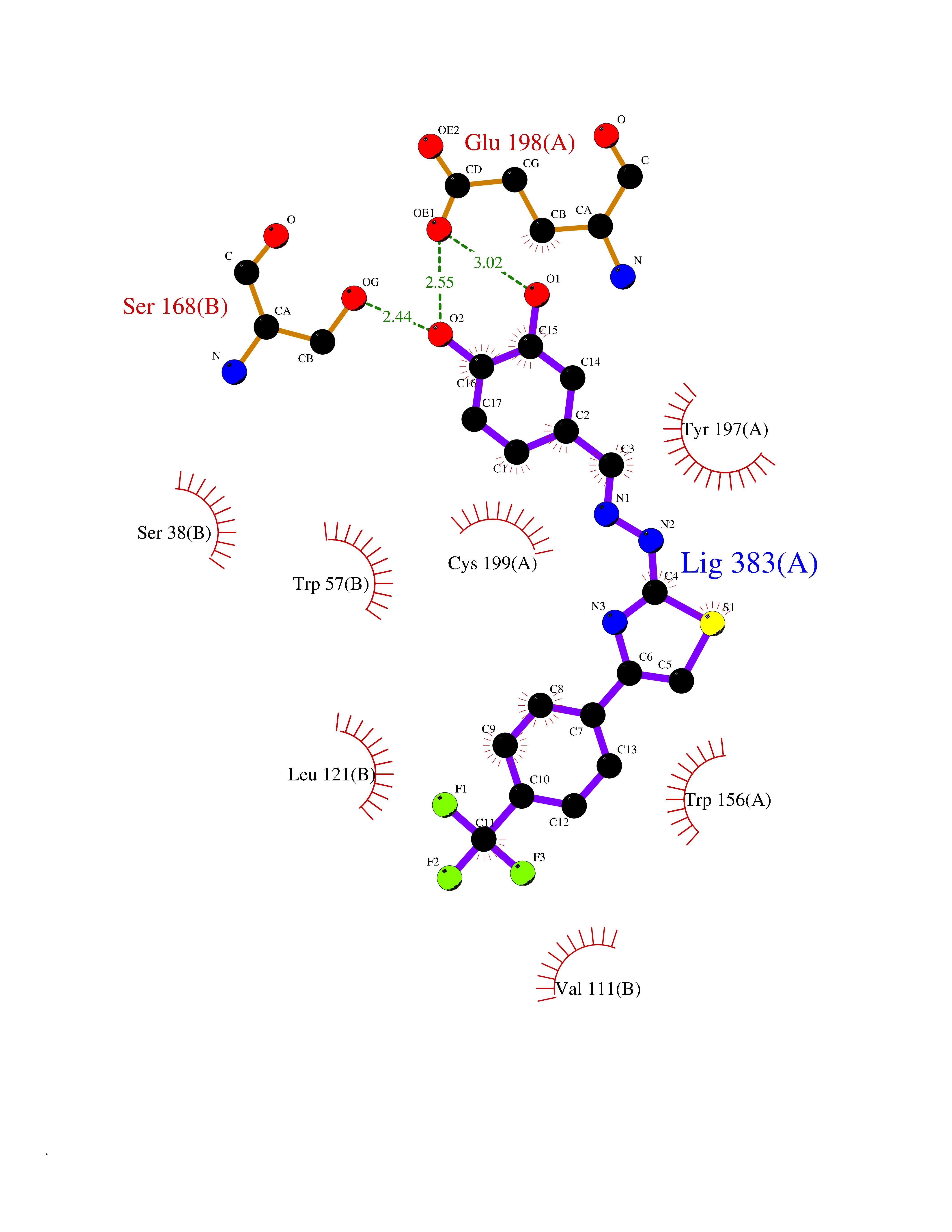 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Cytoplasmic aspartate aminotransferase (GOT1) | 3II0 | 7.53 | |
Target general information Gen name GOT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate oxaloacetate transaminase-1; GOT1 Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Biosynthesis of L-glutamate from L-aspartate or L- cysteine. Important regulator of levels of glutamate, the major excitatory neurotransmitter of the vertebrate central nervous system. Acts as a scavenger of glutamate in brain neuroprotection. The aspartate aminotransferase activity is involved in hepatic glucose synthesis during development and in adipocyte glyceroneogenesis. Using L-cysteine as substrate, regulates levels of mercaptopyruvate, an important source of hydrogen sulfide. Mercaptopyruvate is converted into H(2)S via the action of 3- mercaptopyruvate sulfurtransferase (3MST). Hydrogen sulfide is an important synaptic modulator and neuroprotectant in the brain. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00210; DB00128; DB09130; DB00151; DB00142; DB04299; DB00114 Interacts with NA EC number EC 2.6.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Cytoplasm; Direct protein sequencing; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51732.3 Length 462 Aromaticity 0.1 Instability index 31.83 Isoelectric point 7.48 Charge (pH=7) 0.86 2D Binding mode Binding energy (Kcal/mol) -10.27  Molscript Map 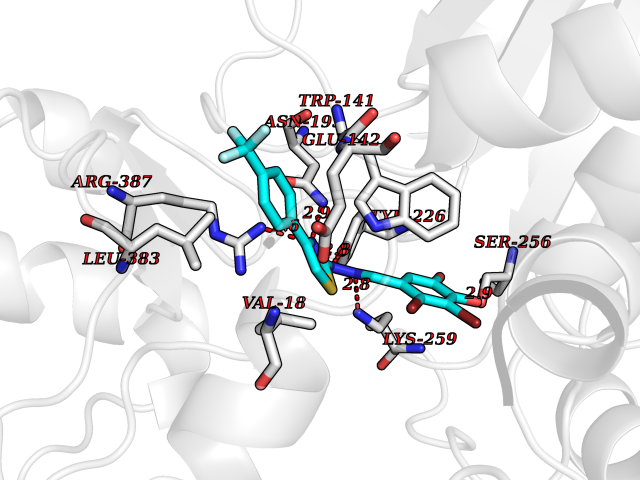 Pymol Map 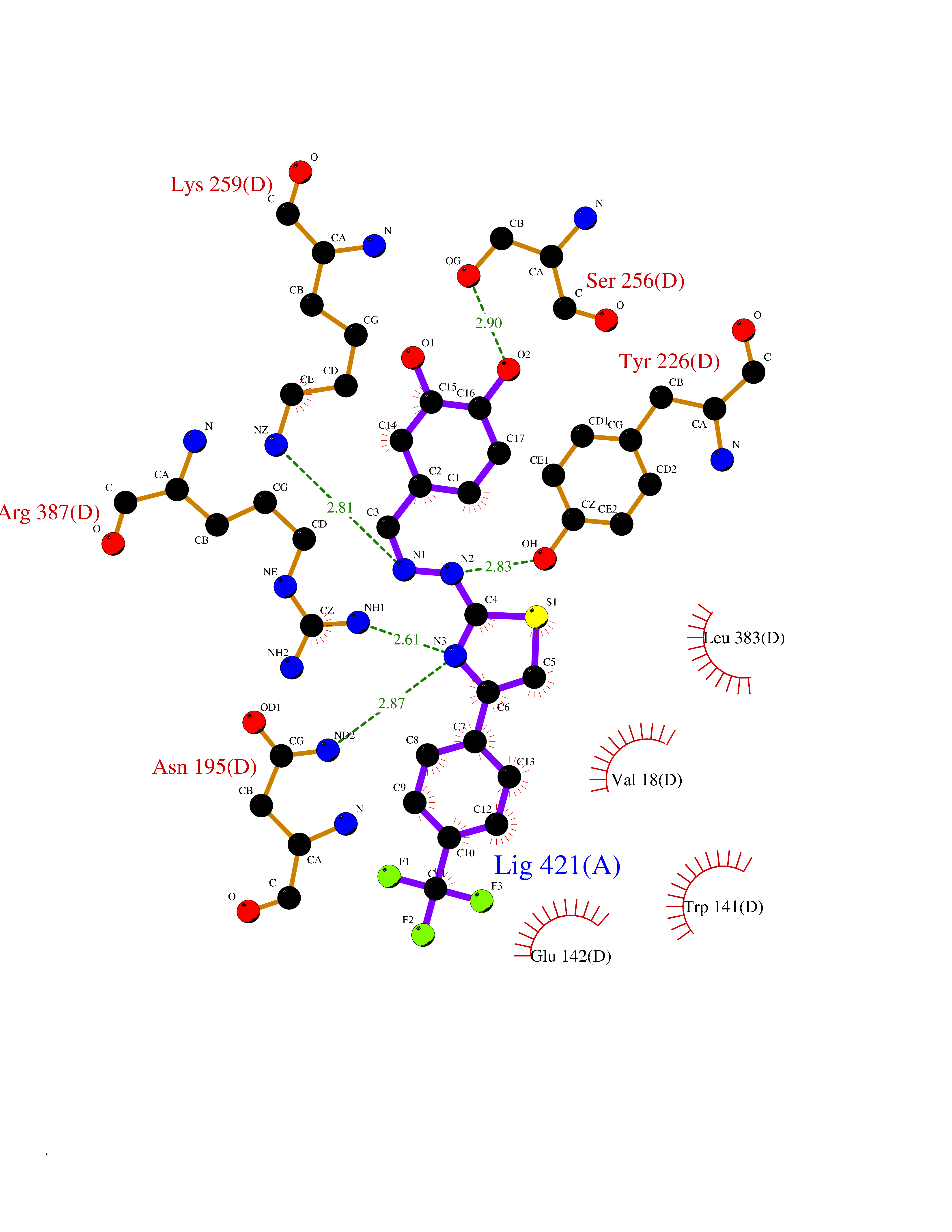 Ligplot Map 3D Binding mode Sequence PVLVFKLTVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRVQSLGGTGALRIGADEKIVRITWSNPMQPVLVFKLTADFREDPDPRKVNLGVGAYRTDDCHPWVLPVVKKVEQKIANDNSLNHEYLPILGLAEFRSCASRLALGDDSPALKEKRVGGVQSLGGTGALRIGADFLARWYNGTNNKNTPVYVSSPTWENHNAVFSAAGFKDIRSYRYWDAEKRGLDLQGFLNDLENAPEFSIVVLHACAHNPTGIDPTPEQWKQIASVMKHRFLFPFFDSAYQGFASGNLERDAWAIRYFVSEGFEFFCAQSFSKNFGLYNERVGNLTVVGKEPESILQVLSQMEKIVRITWSNPPAQGARIVASTLSNPELFEEWTGNVKTMADRILTMRSELRARLEALKTPGTWNHITDQIGMFSFTGLNPKQVEYLVNEKHIYLLPSGRINVSGLTTKNLDYVATSIHEA Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Gamma-butyrobetaine dioxygenase | 4C5W | 7.52 | |
Target general information Gen name BBOX1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms BBH;BBOX Protein family Gamma-BBH/TMLD family Biochemical class Oxidoreductase Function Gamma-butyrobetaine dioxygenase activity.Identical protein binding.Iron ion binding.Zinc ion binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126 Interacts with O75936; A0MZ66-7 EC number 1.14.11.1 Uniprot keywords 3D-structure; Carnitine biosynthesis; Cytoplasm; Dioxygenase; Iron; Metal-binding; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31642.5 Length 275 Aromaticity 0.12 Instability index 35.68 Isoelectric point 6.33 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -10.26  Molscript Map 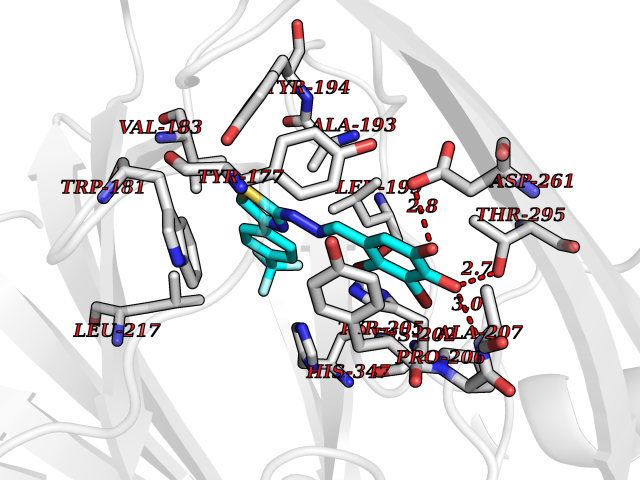 Pymol Map 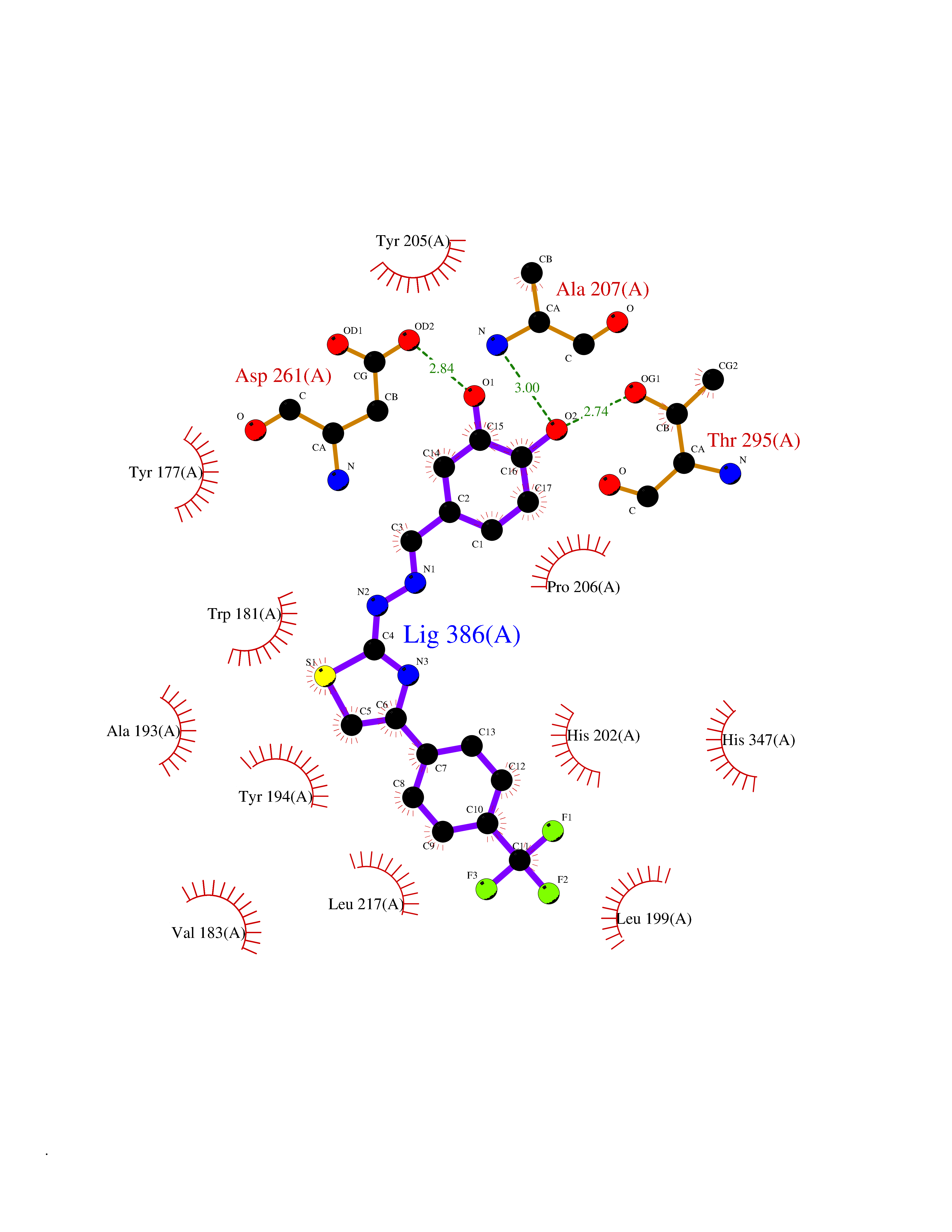 Ligplot Map 3D Binding mode Sequence FPECQYWGSELQLPTLDFEDVLRYDEHAYKWLSTLKKVGIVRLTGASDKPGEVSKLGKRMGFLYLTFYGHTWQVQDKIDANNVAYTTGKLSFHTDYPALHHPPGVQLLHCIKQTVTGGDSEIVDGFNVCQKLKKNNPQAFQILSSTFVDFTDIGVDYCDFSVQSKHKIIELDDKGQVVRINFNNATRDTIFDVPVERVQPFYAALKEFVDLMNSKESKFTFKMNPGDVITFDNWRLLHGRRSYEAGTEISRHLEGAYADWDVVMSRLRILRQRVE Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Leucine carboxyl methyltransferase 1 | 3IEI | 7.52 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -10.26  Molscript Map 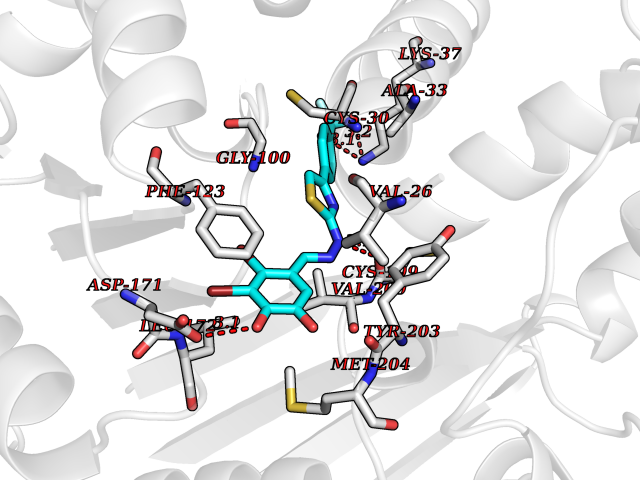 Pymol Map  Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | SET and MYND domain-containing protein 2 (SMYD2) | 5ARF | 7.52 | |
Target general information Gen name SMYD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nlysine methyltransferase SMYD2; N-lysine methyltransferase SMYD2; Lysine Nmethyltransferase 3C; Lysine N-methyltransferase 3C; KMT3C; Histone methyltransferase SMYD2; HSKMB; HSKM-B Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class Methyltransferase Function Specifically methylates histone H3 'Lys-4' (H3K4me) and dimethylates histone H3 'Lys-36' (H3K36me2). Shows even higher methyltransferase activity on p53/TP53. Monomethylates 'Lys-370' of p53/TP53, leading to decreased DNA-binding activity and subsequent transcriptional regulation activity of p53/TP53. Monomethylates RB1 at 'Lys-860'. Protein-lysine N-methyltransferase that methylates both histones and non-histone proteins, including p53/TP53 and RB1. Related diseases Pulmonary hypertension, primary, 4 (PPH4) [MIM:615344]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:23883380}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in this gene may cause developmental delay with sleep apnea (DDSA). A disorder characterized by developmental neurologic, skeletal and respiratory anomalies including microcephaly, arthrogryposis, scoliosis, cleft palate, facial dysmorphology, bilateral talipes, feeding difficulties and central and/or obstructive sleep apnea. Malformations are detected as early as 21 weeks post gestation. Severely affected patients require ongoing treatment with nocturnal O2 or pressure-controlled ventilation. The disease is associated with recurrent de novo gain of function variants. {ECO:0000269|PubMed:36195757}. Drugs (DrugBank ID) NA Interacts with P20290-2; Q96K17; Q9UPZ9; Q9Y5W9; P04637 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48789.6 Length 425 Aromaticity 0.1 Instability index 51.93 Isoelectric point 6.35 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -10.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LGGLERFCSPGKGRGLRALQPFQVGDLLFSCPAYAYVLTVNERGNHCEYCFTRKEGLSKCGRCKQAFYCNVECQKEDWPMHKLECSPMVVFGENWNPSETVRLTARILAKQKIHPERTPSEKLLAVKEFESHLDKLDNEKKDLIQSDIAALHHFYSKHLGFPDNDSLVVLFAQVNCNGFTIEDEELSHLGSAIFPDVALMNHSCCPNVIVTYKGTLAEVRAVQEIKPGEEVFTSYIDLLYPTEDRNDRLRDSYFFTCECQECTTKDKDKAKVEIRKLSDPPKAEAIRDMVRYARNVIEEFRRAKHYKSPSELLEICELSQEKMSSVFEDSNVYMLHMMYQAMGVCLYMQDWEGALQYGQKIIKPYSKHYPLYSLNVASMWLKLGRLYMGLEHKAAGEKALKKAIAIMEVAHGKDHPYISEIKQEI Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 7.51 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -10.25  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||