Job Results:
Ligand
Structure
Job ID
f5a9e34d4d6e0fad4e04e6dd0d3ae3aa
Job name
NA
Time
2026-01-10 22:50:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Tyrosine-protein kinase Kit (KIT) | 1T46 | 7.15 | |
Target general information Gen name KIT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; p145 c-kit; Proto-oncogene tyrosine-protein kinase Kit; Proto-oncogene c-Kit; Piebald trait protein; PBT; Mast/stem cell growth factor re Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function In response to KITLG/SCF binding, KIT can activate several signaling pathways. Phosphorylates PIK3R1, PLCG1, SH2B2/APS and CBL. Activates the AKT1 signaling pathway by phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase. Activated KIT also transmits signals via GRB2 and activation of RAS, RAF1 and the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3, STAT5A and STAT5B. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate. KIT signaling is modulated by protein phosphatases, and by rapid internalization and degradation of the receptor. Activated KIT promotes phosphorylation of the protein phosphatases PTPN6/SHP-1 and PTPRU, and of the transcription factors STAT1, STAT3, STAT5A and STAT5B. Promotes phosphorylation of PIK3R1, CBL, CRK (isoform Crk-II), LYN, MAPK1/ERK2 and/or MAPK3/ERK1, PLCG1, SRC and SHC1. Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine KITLG/SCF and plays an essential role in the regulation of cell survival and proliferation, hematopoiesis, stem cell maintenance, gametogenesis, mast cell development, migration and function, and in melanogenesis. Related diseases Piebald trait (PBT) [MIM:172800]: Autosomal dominant genetic developmental abnormality of pigmentation characterized by congenital patches of white skin and hair that lack melanocytes. {ECO:0000269|PubMed:11074500, ECO:0000269|PubMed:1370874, ECO:0000269|PubMed:1376329, ECO:0000269|PubMed:1717985, ECO:0000269|PubMed:7687267, ECO:0000269|PubMed:8680409, ECO:0000269|PubMed:9450866, ECO:0000269|PubMed:9699740}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:11505412, ECO:0000269|PubMed:15824741, ECO:0000269|PubMed:9438854, ECO:0000269|PubMed:9697690}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Testicular germ cell tumor (TGCT) [MIM:273300]: A common malignancy in males representing 95% of all testicular neoplasms. TGCTs have various pathologic subtypes including: unclassified intratubular germ cell neoplasia, seminoma (including cases with syncytiotrophoblastic cells), spermatocytic seminoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma, and teratoma. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. The gene represented in this entry is involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of KIT are detected in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the kinase domain can result in a constitutively activated kinase.; DISEASE: Mastocytosis, cutaneous (MASTC) [MIM:154800]: A form of mastocytosis, a heterogeneous group of disorders associated with abnormal proliferation and accumulation of mast cells in various tissues, especially in the skin and hematopoietic organs. MASTC is an autosomal dominant form characterized by macules, papules, nodules, or diffuse infiltration of the skin, often associated with localized hyperpigmentation. Gentle rubbing of the lesions induces histamine release from mechanically activated mast cells, causing local wheals, erythema, and often pruritus, a phenomenon termed Darier sign. {ECO:0000269|PubMed:15173254, ECO:0000269|PubMed:19865100, ECO:0000269|PubMed:21689725, ECO:0000269|PubMed:24289326, ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mastocytosis, systemic (MASTSYS) [MIM:154800]: A severe form of mastocytosis characterized by abnormal proliferation and accumulation of mast cells in several organs, resulting in a systemic disease that may affect bone, gastrointestinal tract, lymphatics, spleen, and liver. In some cases, it is associated with a clonal hematologic non-mast-cell lineage disease, such as a myelodysplastic or myeloproliferative disorder. It can also lead to mast cell leukemia, which carries a high risk of mortality. {ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB09103; DB15233; DB01254; DB12147; DB12010; DB00619; DB09078; DB06080; DB06595; DB04868; DB05913; DB06589; DB12978; DB01962; DB08901; DB08896; DB14840; DB00398; DB01268; DB11800; DB05146 Interacts with P00519; P42684; O75815; P51451; Q8WV28; P46108; P07332; P09769; O75791; P62993; Q14451; P08631; Q96JZ2; P21583; P06239; P07948; P16333; O43639; P27986; O00459; Q92569; P19174; P16885; Q13882; Q06124; Q92729; P20936; Q9UQQ2; O14796; Q9NP31; Q8N5H7; P78314; Q15464; P29353; P98077; Q92529; Q9H6Q3; O14508; O14543; O14544; P12931; Q9ULZ2; Q9HBL0; Q63HR2; Q68CZ2; P42681; P07947; P43403; Q8VBX6; P35235 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33575.6 Length 297 Aromaticity 0.11 Instability index 45.37 Isoelectric point 8.37 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -9.75  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GNNYVYIDPTQLPYDHKWEFPRNRLSFGKTLGAGAFGKVVEATAYGLIKSDAAMTVAVKMLKPSAHLTEREALMSELKVLSYLGNHMNIVNLLGACTIGGPTLVITEYCCYGDLLNFLRRKRDSFLALDLEDLLSFSYQVAKGMAFLASKNCIHRDLAARNILLTHGRITKICDFGLARDIKNDSNYVVKGNARLPVKWMAPESIFNCVYTFESDVWSYGIFLWELFSLGSSPYPGMPVDSKFYKMIKEGFRMLSPEHAPAEMYDIMKTCWDADPLKRPTFKQIVQLIEKQISESTN Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Nicotinamide N-methyltransferase | 2IIP | 7.15 | |
Target general information Gen name NNMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class Transferase Function Nicotinamide N-methyltransferase activity.Pyridine N-methyltransferase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00627 Interacts with NA EC number 2.1.1.1 Uniprot keywords 3D-structure; Acetylation; Citrullination; Cytoplasm; Direct protein sequencing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27886.8 Length 251 Aromaticity 0.1 Instability index 40.66 Isoelectric point 5.23 Charge (pH=7) -5.11 2D Binding mode Binding energy (Kcal/mol) -9.75 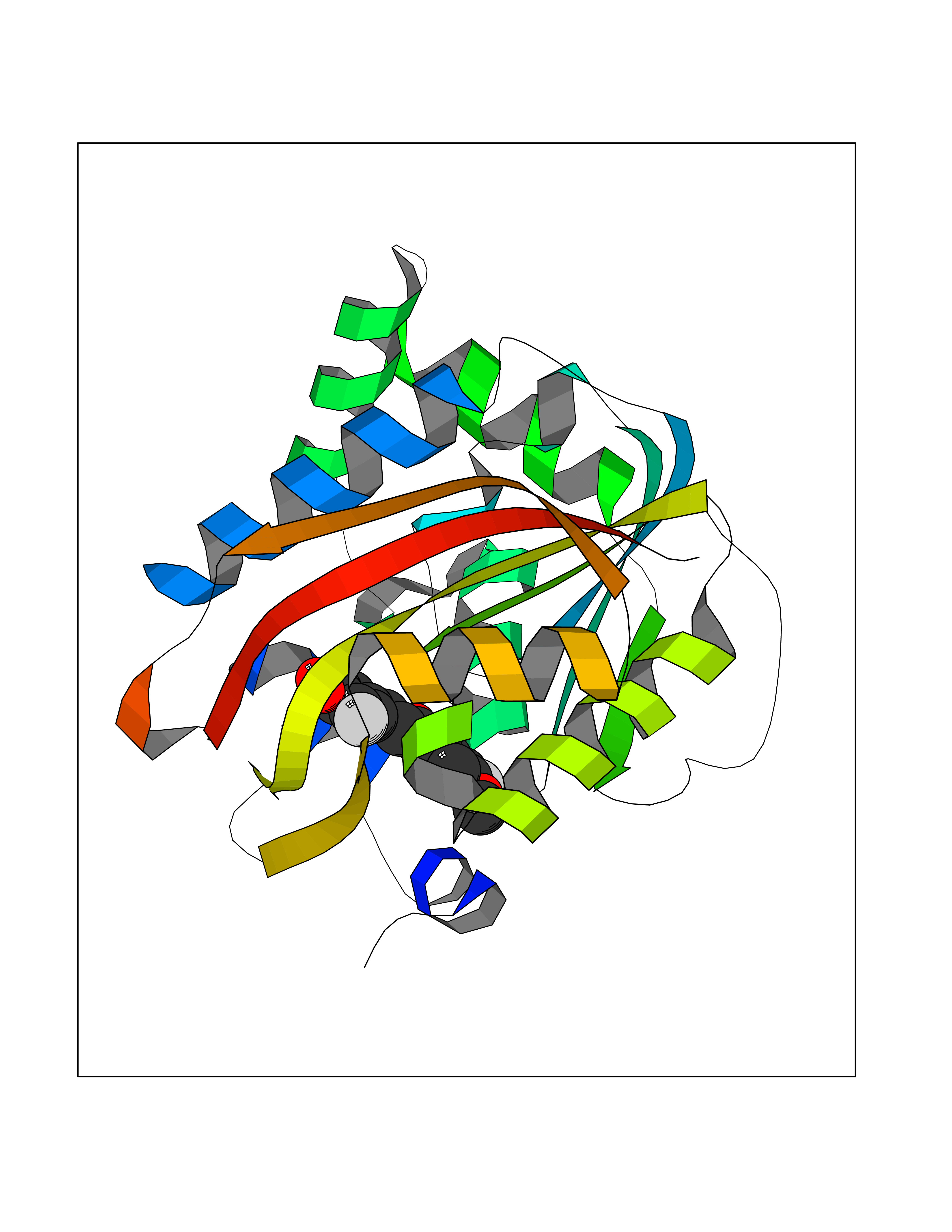 Molscript Map 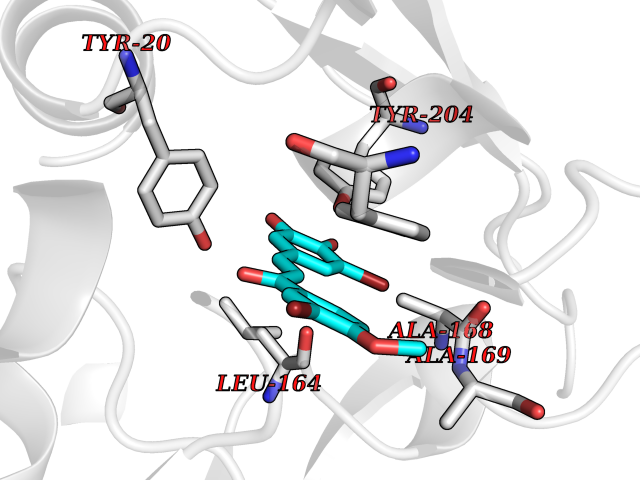 Pymol Map 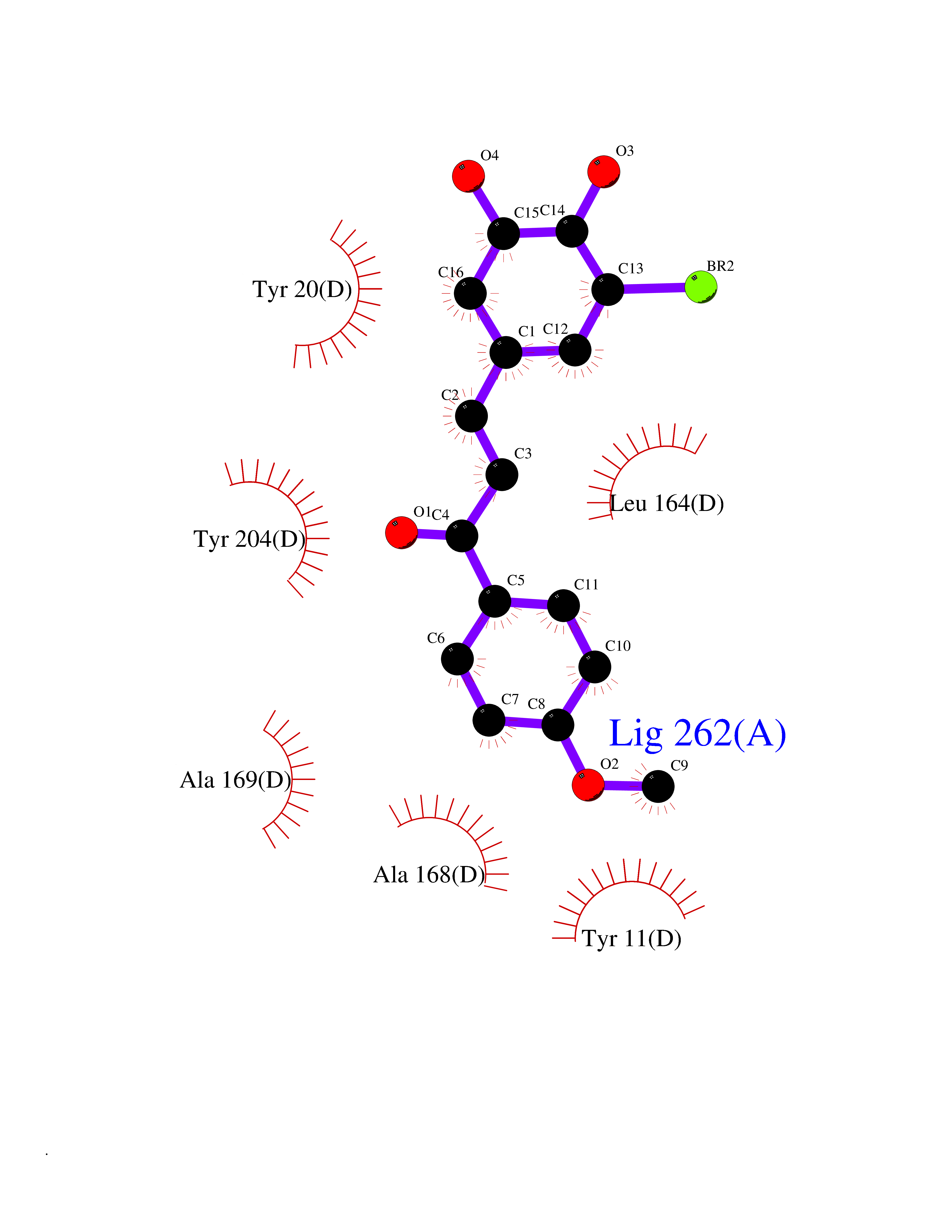 Ligplot Map 3D Binding mode Sequence GFTSKDTYLSHFNPRDYLEKYYSAESQILKHLLKNLFKIFCLDGVKGDLLIDIGSGPTIYQLLSACESFKEIVVTDYSDQNLQELEKWLKAAPAAFDWSPVVTYVCDLEGNRVKGPEKEEKLRQAVKQVLKCDVTQSQPLGAVPLPPADCVLSTLCLDAACPDLPTYCRALRNLGSLLKPGGFLVIMDALKSSYYMIGEQKFSSLPLGREAVEAAVKEAGYTIEWFEVISQSYSSTMANNEGLFSLVARKL Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Lysine-specific histone demethylase 1B (KDM1B) | 4HSU | 7.15 | |
Target general information Gen name KDM1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 2; LSD2; Flavin-containing amine oxidase domain-containing protein 1; C6orf193; AOF1 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Required for de novo DNA methylation of a subset of imprinted genes during oogenesis. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Demethylates both mono- and di-methylated 'Lys-4' of histone H3. Has no effect on tri-methylated 'Lys-4', mono-, di- or tri-methylated 'Lys-9', mono-, di- or tri-methylated 'Lys-27', mono-, di- or tri-methylated 'Lys-36' of histone H3, or on mono-, di- or tri-methylated 'Lys-20' of histone H4. Histone demethylase that demethylates 'Lys-4' of histone H3, a specific tag for epigenetic transcriptional activation, thereby acting as a corepressor. Related diseases Angioedema, hereditary, 1 (HAE1) [MIM:106100]: An autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema due to C1 esterase inhibitor deficiency is comprised of two clinically indistinguishable forms. In hereditary angioedema type 1, serum levels of C1 esterase inhibitor are decreased, while in type 2, the levels are normal or elevated, but the protein is non-functional. {ECO:0000269|PubMed:12773530, ECO:0000269|PubMed:1363816, ECO:0000269|PubMed:1451784, ECO:0000269|PubMed:14635117, ECO:0000269|PubMed:16409206, ECO:0000269|PubMed:2118657, ECO:0000269|PubMed:2296585, ECO:0000269|PubMed:22994404, ECO:0000269|PubMed:2365061, ECO:0000269|PubMed:24456027, ECO:0000269|PubMed:3178731, ECO:0000269|PubMed:7814636, ECO:0000269|PubMed:7883978, ECO:0000269|PubMed:8172583, ECO:0000269|PubMed:8529136, ECO:0000269|PubMed:8755917, ECO:0000269|Ref.41}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96L03 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Developmental protein; FAD; Flavoprotein; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85795.5 Length 763 Aromaticity 0.1 Instability index 37.87 Isoelectric point 8.41 Charge (pH=7) 9.16 2D Binding mode Binding energy (Kcal/mol) -9.76 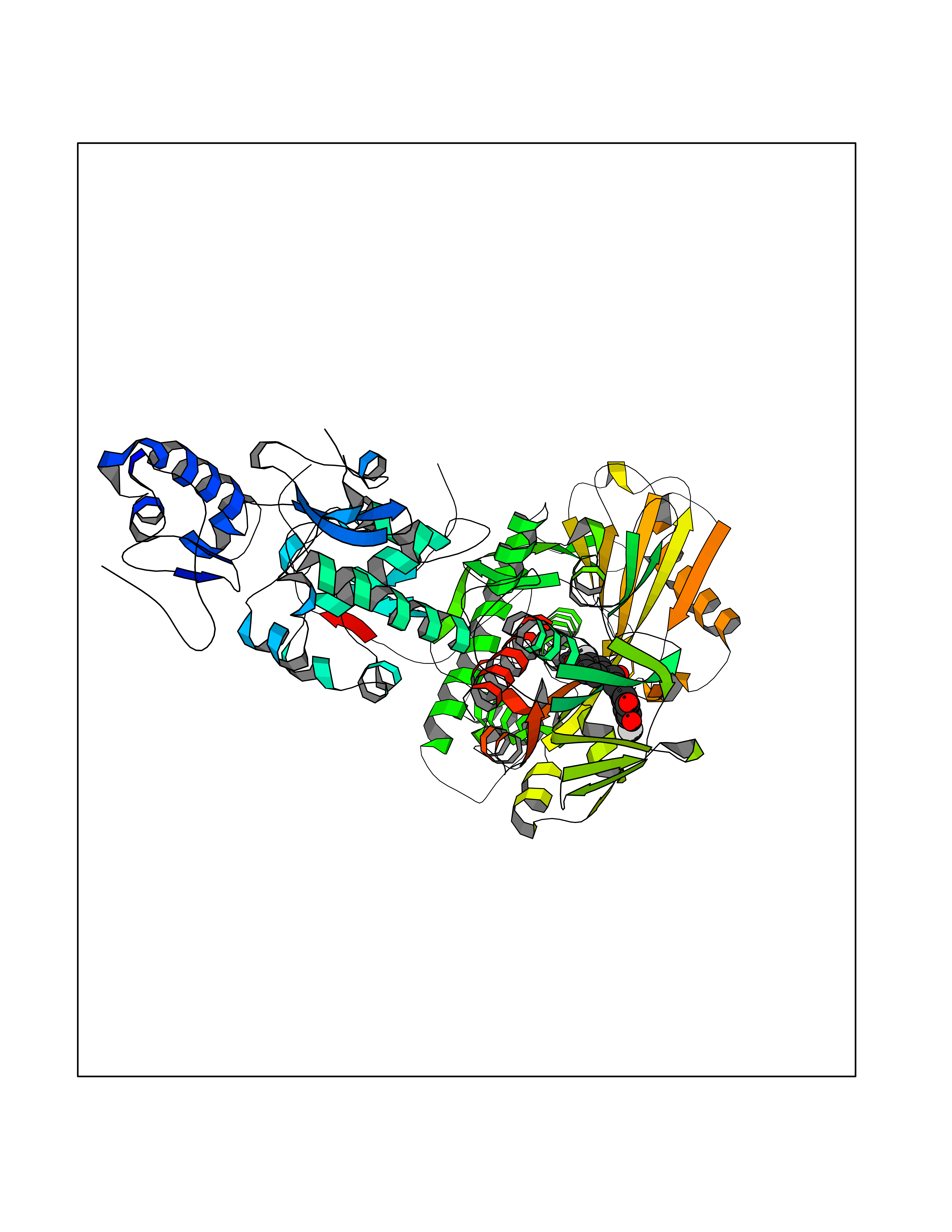 Molscript Map 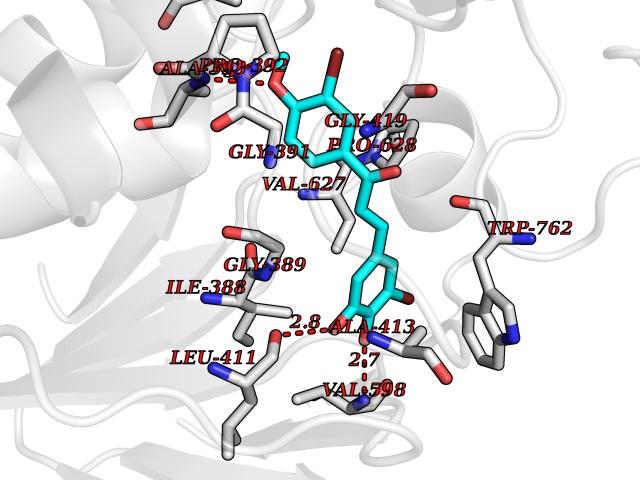 Pymol Map 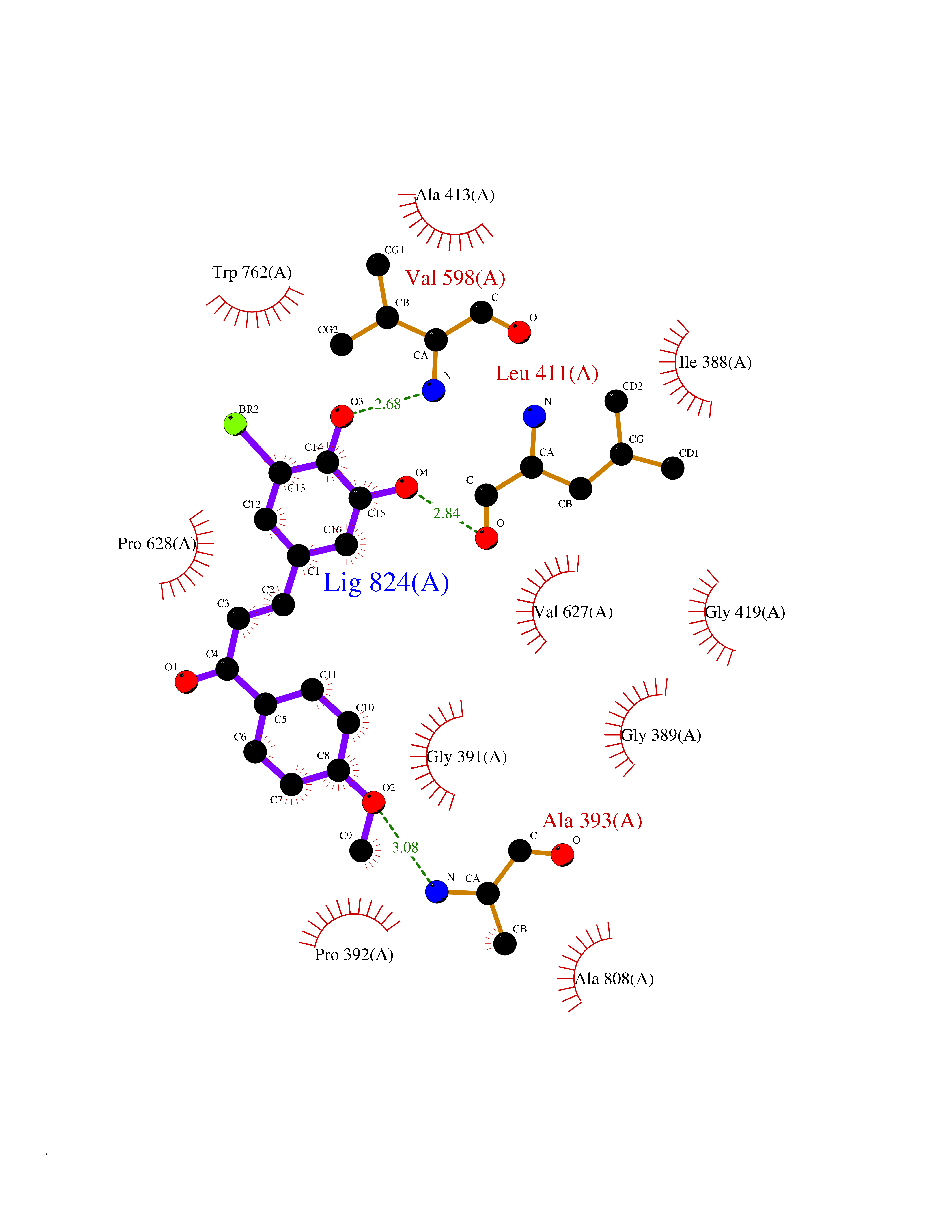 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQLATKAAR Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | p53-binding protein Mdm4 (MDM4) | 6Q9Y | 7.15 | |
Target general information Gen name MDM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Mdmx; Mdm2-like p53-binding protein; Double minute 4 protein Protein family MDM2/MDM4 family Biochemical class MDM2/MDM4 family Function Inhibits p53/TP53- and TP73/p73-mediated cell cycle arrest and apoptosis by binding its transcriptional activation domain. Inhibits degradation of MDM2. Can reverse MDM2-targeted degradation of TP53 while maintaining suppression of TP53 transactivation and apoptotic functions. Related diseases Bone marrow failure syndrome 6 (BMFS6) [MIM:618849]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS6 is an autosomal dominant form characterized by intermittent neutropenia, lymphopenia, or anemia associated with hypocellular bone marrow, and increased susceptibility to cancer. {ECO:0000269|PubMed:32300648}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NX04; P10415; Q7Z479; O95971; P48729; Q00987; Q13064; P41227; P06400; Q9Y4L5; P23297; P29034; P33763; P04271; P31947; P04637; P62837; Q93009; O14972; P61964; P62258; P61981; P63104; Q9BRR0; A0A0S2Z6X0; Q3YBA8; P03255-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19722 Length 173 Aromaticity 0.08 Instability index 50.78 Isoelectric point 8.48 Charge (pH=7) 2.27 2D Binding mode Binding energy (Kcal/mol) -9.76 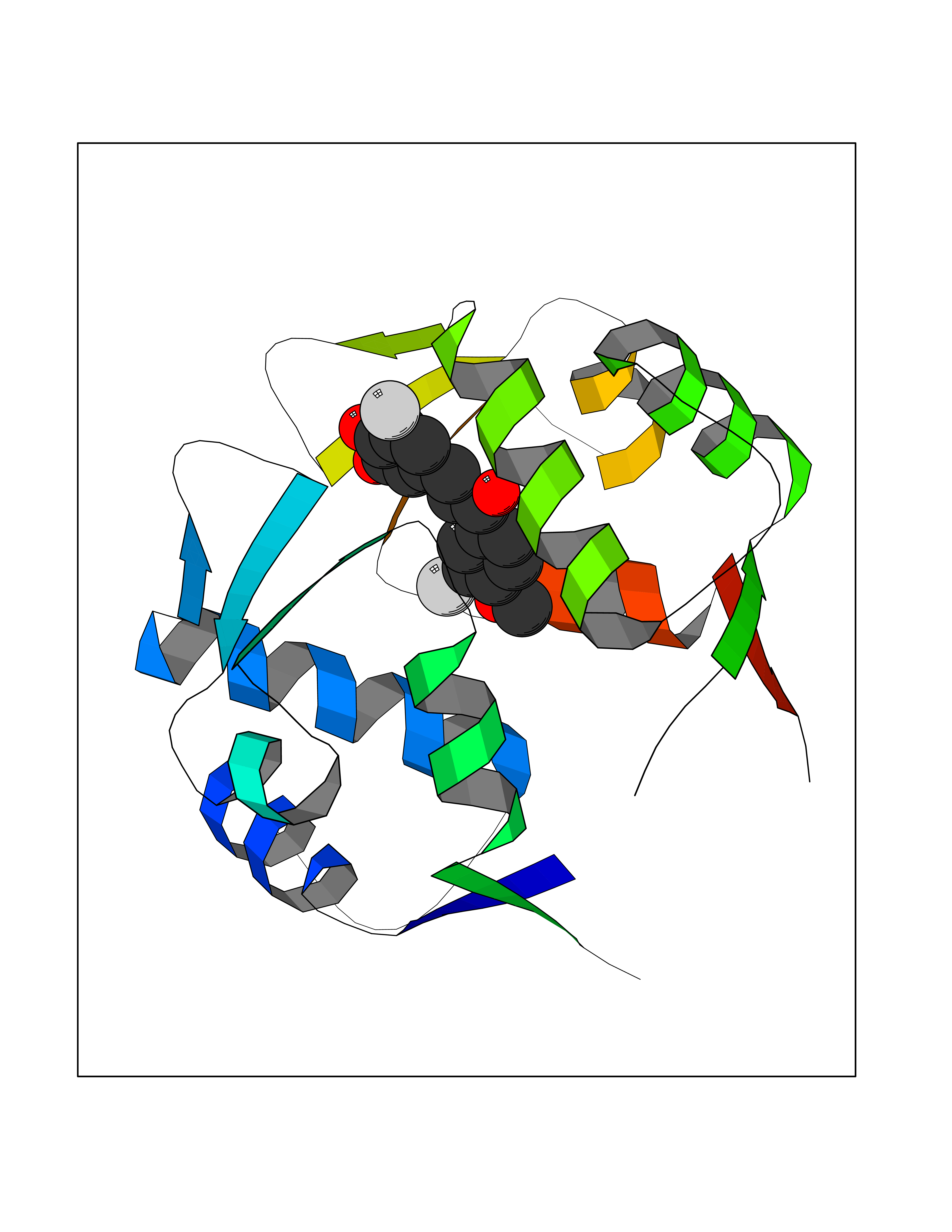 Molscript Map 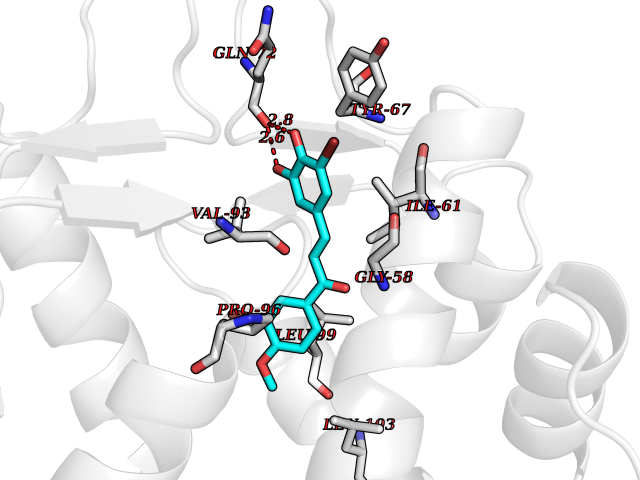 Pymol Map 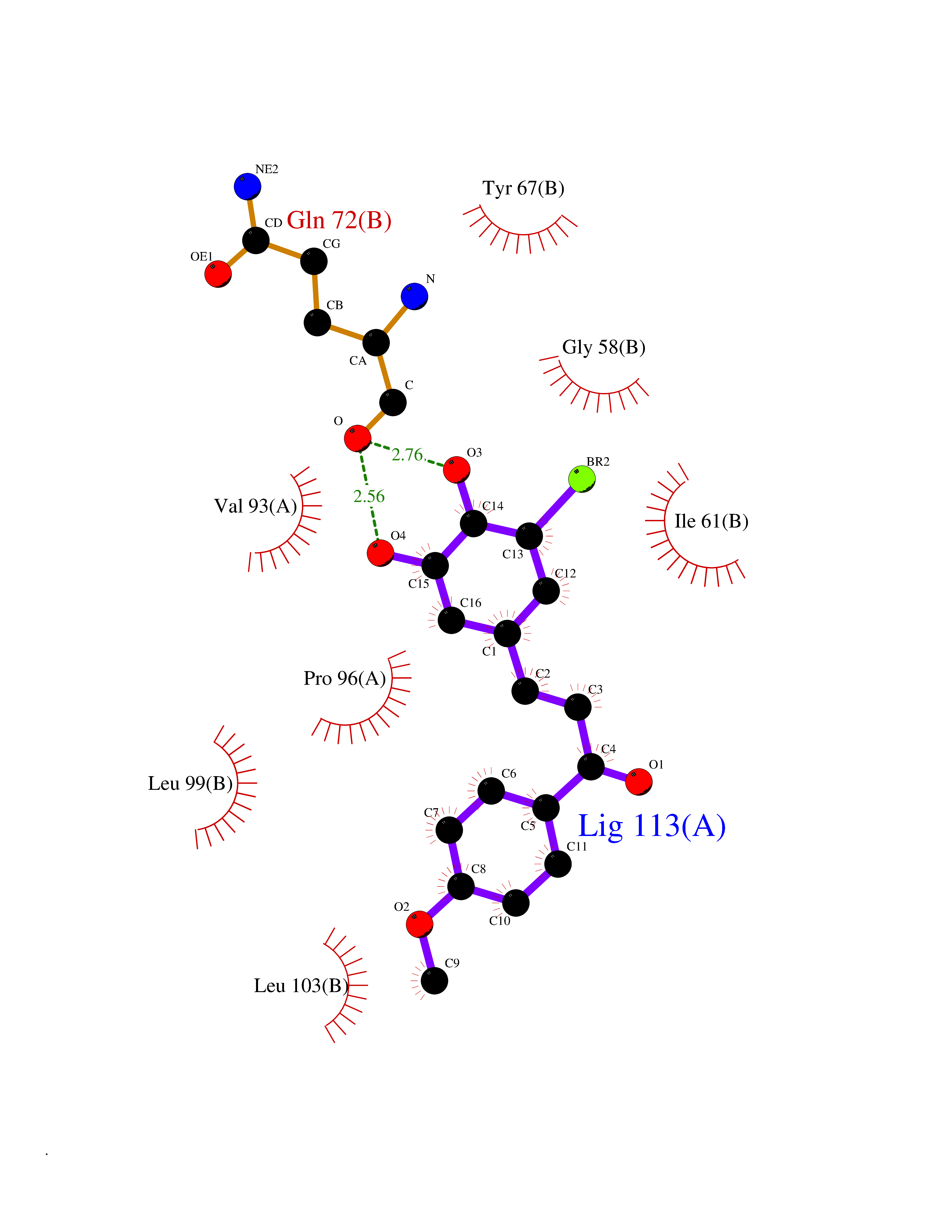 Ligplot Map 3D Binding mode Sequence QVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLAQINQVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLA Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Mutated Histone H3.3 (H3F3A) | 4GUS | 7.15 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -9.75  Molscript Map 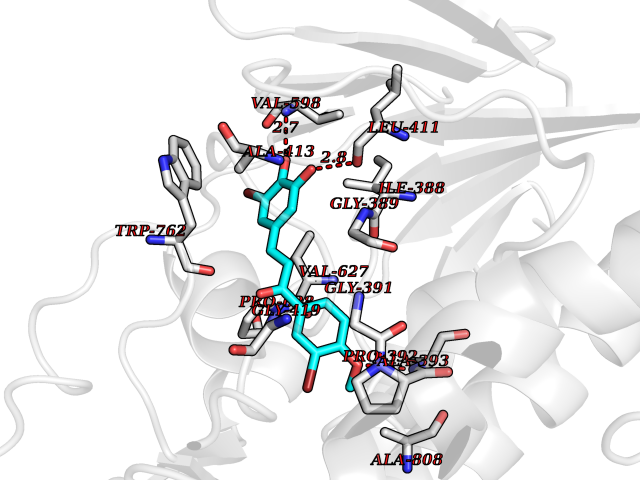 Pymol Map 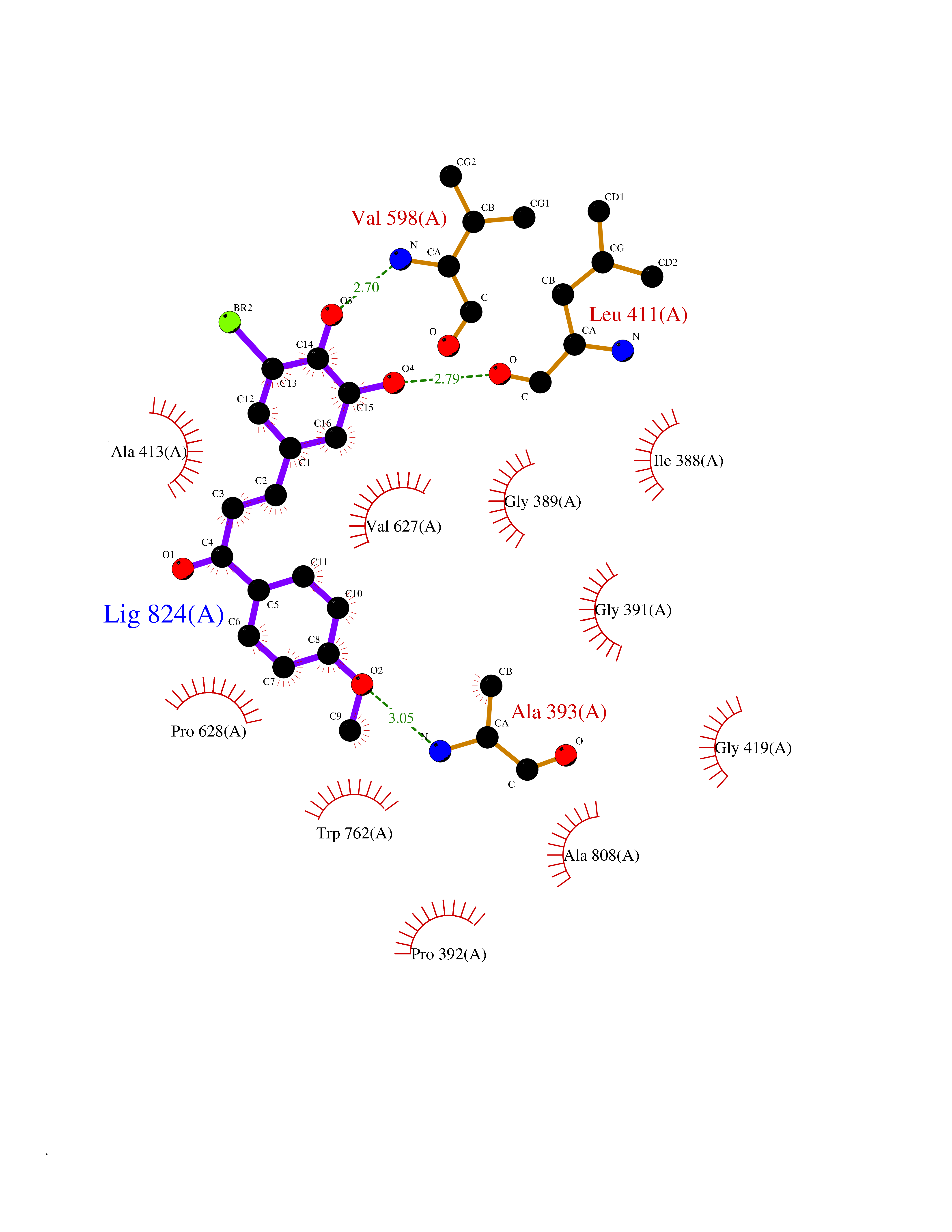 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 7.15 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -9.76 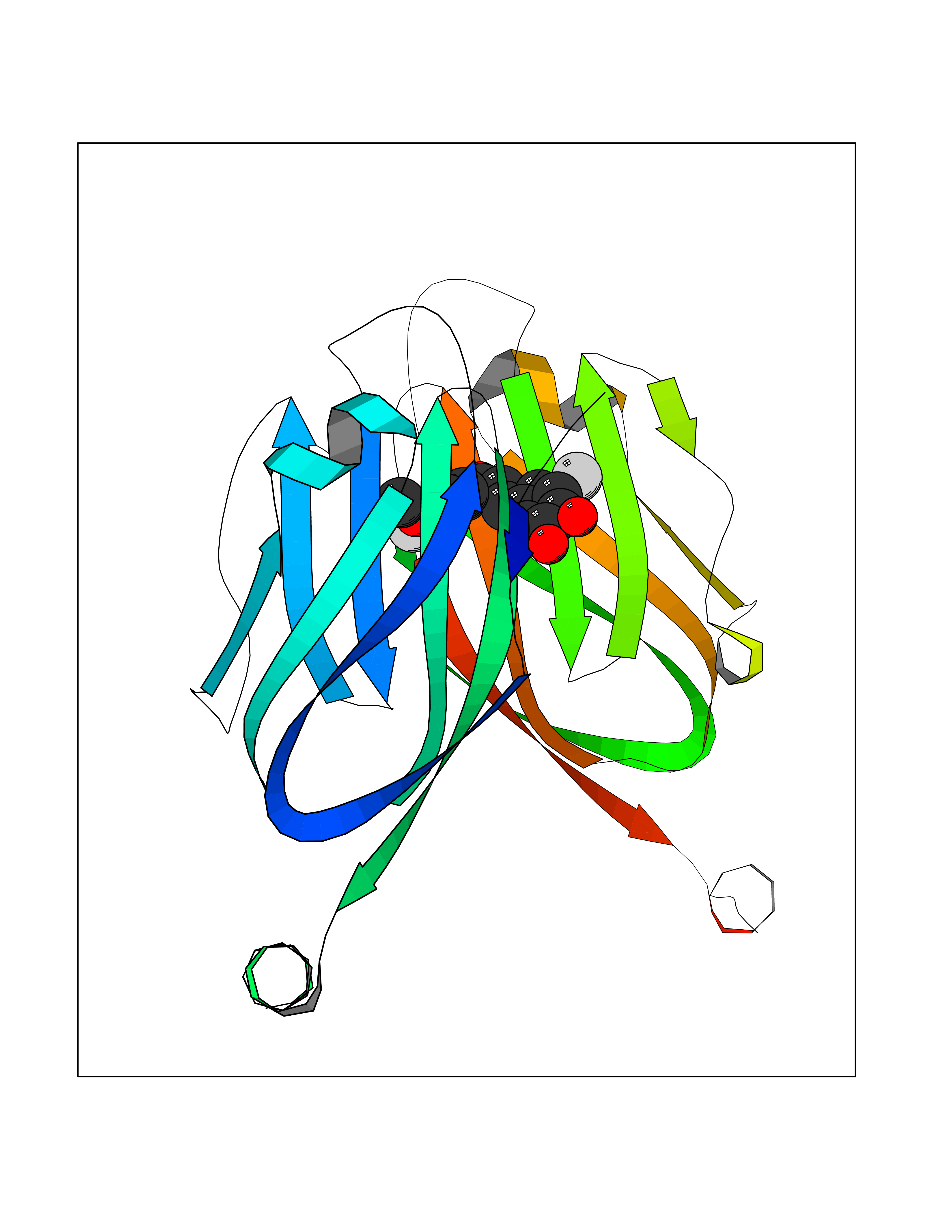 Molscript Map 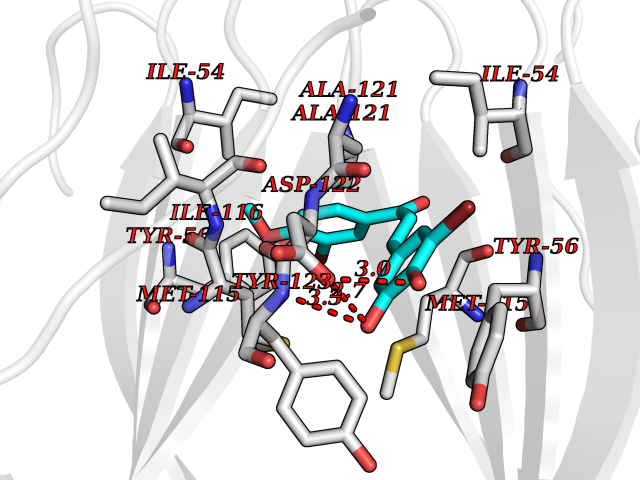 Pymol Map 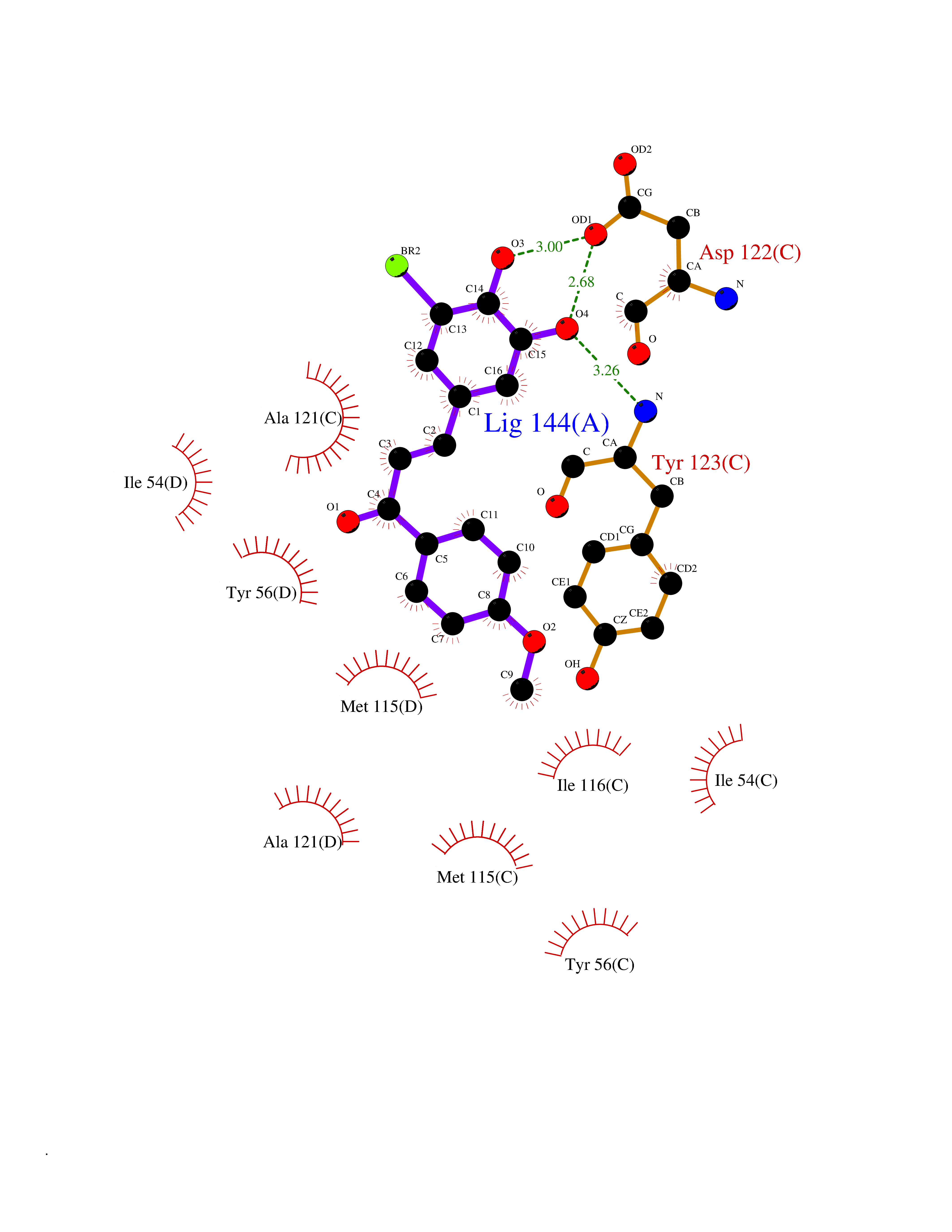 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 7.14 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -9.74  Molscript Map 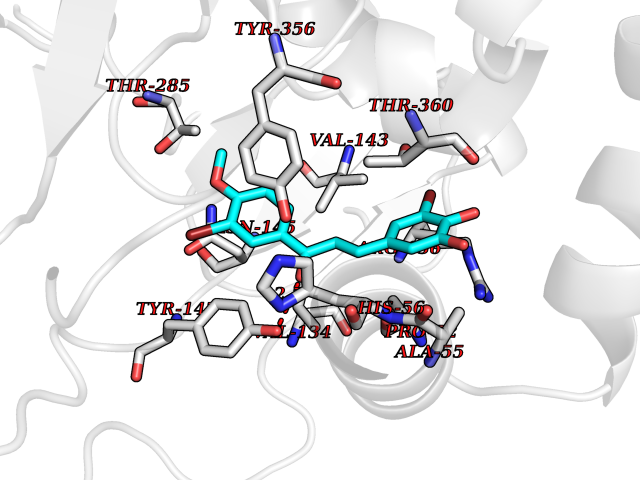 Pymol Map  Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Serine/threonine-protein kinase cot (COT) | 4Y85 | 7.14 | |
Target general information Gen name MAP3K8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor progression locus 2; TPL-2; Proto-oncogene c-Cot; Mitogen-activated protein kinase kinase kinase 8; ESTF; Cancer Osaka thyroid oncogene; COT Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class NA Function Required for lipopolysaccharide (LPS)-induced, TLR4-mediated activation of the MAPK/ERK pathway in macrophages, thus being critical for production of the proinflammatory cytokine TNF-alpha (TNF) during immune responses. Involved in the regulation of T-helper cell differentiation and IFNG expression in T-cells. Involved in mediating host resistance to bacterial infection through negative regulation of type I interferon (IFN) production. In vitro, activates MAPK/ERK pathway in response to IL1 in an IRAK1-independent manner, leading to up-regulation of IL8 and CCL4. Transduces CD40 and TNFRSF1A signals that activate ERK in B-cells and macrophages, and thus may play a role in the regulation of immunoglobulin production. May also play a role in the transduction of TNF signals that activate JNK and NF-kappa-B in some cell types. In adipocytes, activates MAPK/ERK pathway in an IKBKB-dependent manner in response to IL1B and TNF, but not insulin, leading to induction of lipolysis. Plays a role in the cell cycle. Isoform 1 shows some transforming activity, although it is much weaker than that of the activated oncogenic variant. Related diseases Hyperinsulinemic hypoglycemia, familial, 2 (HHF2) [MIM:601820]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF2 is a common cause of persistent hypoglycemia in infancy. Unless early and aggressive intervention is undertaken, brain damage from recurrent episodes of hypoglycemia may occur. HHF2 inheritance can be autosomal dominant or autosomal recessive. {ECO:0000269|PubMed:10204114, ECO:0000269|PubMed:12364426, ECO:0000269|PubMed:15562009, ECO:0000269|PubMed:15579781, ECO:0000269|PubMed:15807877, ECO:0000269|PubMed:15998776, ECO:0000269|PubMed:16332676, ECO:0000269|PubMed:16357843, ECO:0000269|PubMed:18596924, ECO:0000269|PubMed:19357197, ECO:0000269|PubMed:7847376, ECO:0000269|PubMed:8923010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Diabetes mellitus, permanent neonatal, 2 (PNDM2) [MIM:618856]: A form of permanent neonatal diabetes mellitus, a type of diabetes characterized by onset of persistent hyperglycemia within the first six months of life. Initial clinical manifestations include intrauterine growth retardation, hyperglycemia, glycosuria, osmotic polyuria, severe dehydration, and failure to thrive. Some PNDM2 patients may also have developmental delay, muscle weakness, epilepsy and dysmorphic features. PNDM2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:15115830, ECO:0000269|PubMed:15292329, ECO:0000269|PubMed:15448106, ECO:0000269|PubMed:15448107, ECO:0000269|PubMed:15580558, ECO:0000269|PubMed:15583126, ECO:0000269|PubMed:16609879, ECO:0000269|PubMed:16731833, ECO:0000269|PubMed:17213273, ECO:0000269|PubMed:17652641, ECO:0000269|PubMed:17855752, ECO:0000269|PubMed:20022885, ECO:0000269|PubMed:28842488}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Transient neonatal diabetes mellitus 3 (TNDM3) [MIM:610582]: Neonatal diabetes mellitus, defined as insulin-requiring hyperglycemia within the first month of life, is a rare entity. In about half of the neonates, diabetes is transient and resolves at a median age of 3 months, whereas the rest have a permanent form of diabetes. In a significant number of patients with transient neonatal diabetes mellitus, diabetes type 2 appears later in life. The onset and severity of TNDM3 is variable with childhood-onset diabetes, gestational diabetes or adult-onset diabetes described. {ECO:0000269|PubMed:15718250, ECO:0000269|PubMed:15784703}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KCNJ11 may contribute to non-insulin-dependent diabetes mellitus (NIDDM), also known as diabetes mellitus type 2.; DISEASE: Maturity-onset diabetes of the young 13 (MODY13) [MIM:616329]: A form of diabetes that is characterized by an autosomal dominant mode of inheritance, onset in childhood or early adulthood (usually before 25 years of age), a primary defect in insulin secretion and frequent insulin-independence at the beginning of the disease. {ECO:0000269|PubMed:22701567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P08238; P19838; Q00653; Q13526; Q8NFZ5 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell cycle; Cytoplasm; Immunity; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 34773.8 Length 307 Aromaticity 0.09 Instability index 39.24 Isoelectric point 6.68 Charge (pH=7) -1.2 2D Binding mode Binding energy (Kcal/mol) -9.74  Molscript Map 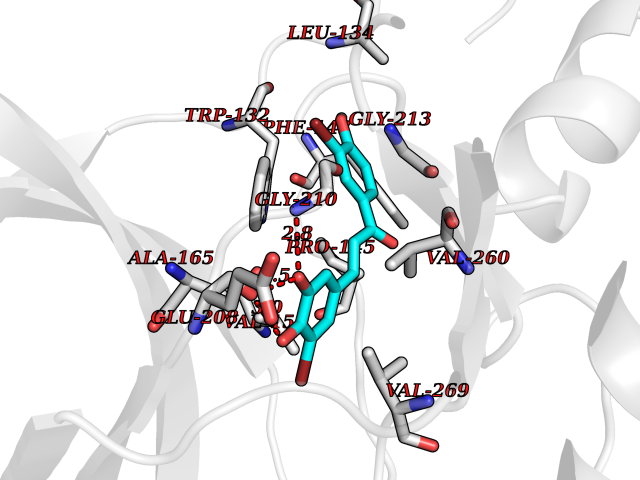 Pymol Map 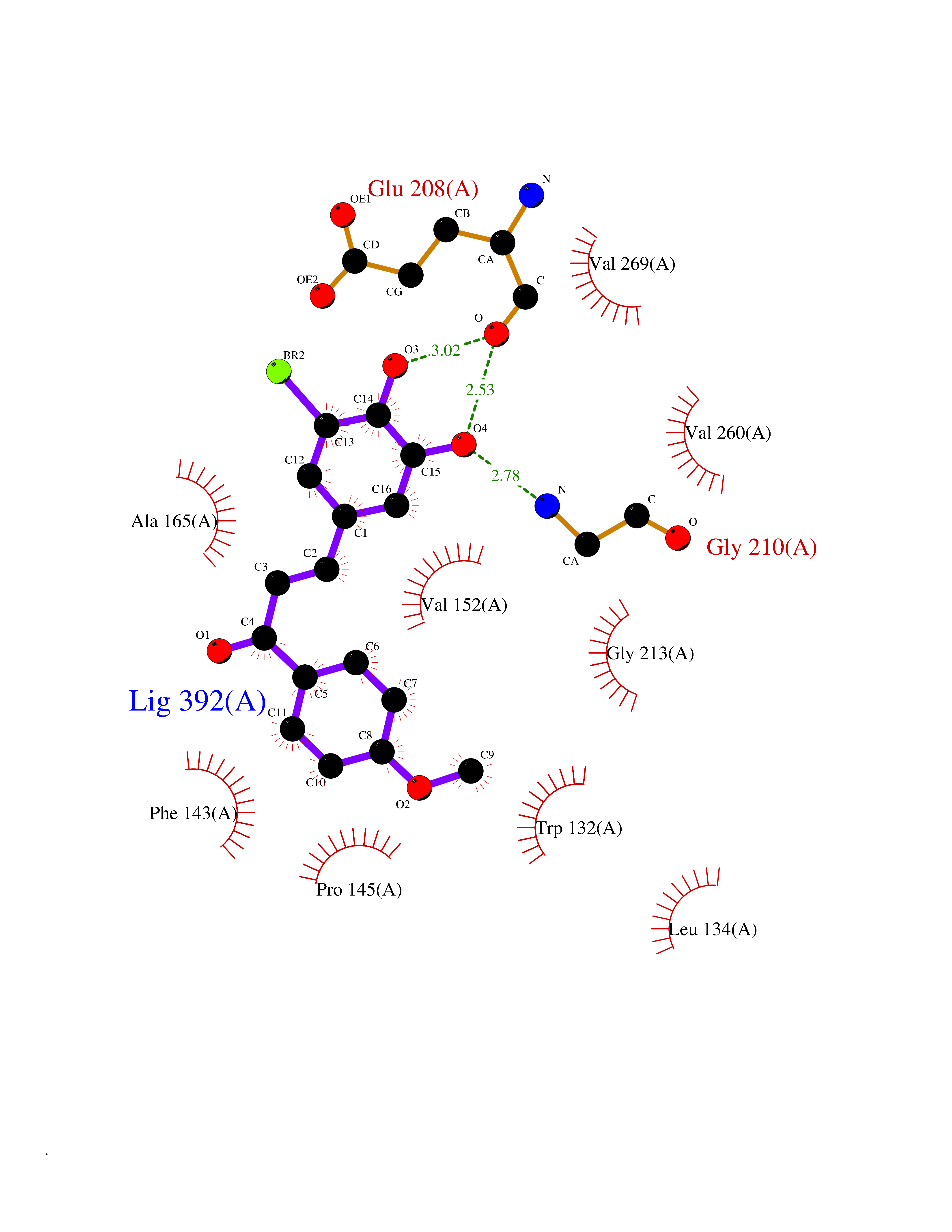 Ligplot Map 3D Binding mode Sequence LSSVRYGTVEDLLAFANHISNTPQESGILLNMVITPQNGRYQIDSDVLLIPWKLTYRNIFIPRGAFGKVYLAQDIKTKKRMACKLIPVDQFKPSDVEIQACFRHENIAELYGAVLWGETVHLFMEAGEGGSVLEKLESCGPMREFEIIWVTKHVLKGLDFLHSKKVIHHDIKPSNIVFMSTKAVLVDFGLSVQMTEDVYFPKDLRGTEIYMSPEVILCRGHSTKADIYSLGATLIHMQTGTPPWVKRYPRSAYPSYLYIIHKQAPPLEDIADDCSPGMRELIEASLERNPNHRPRAADLLKHEALNP Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Retinoic acid receptor gamma (RARG) | 1FCY | 7.11 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -9.7 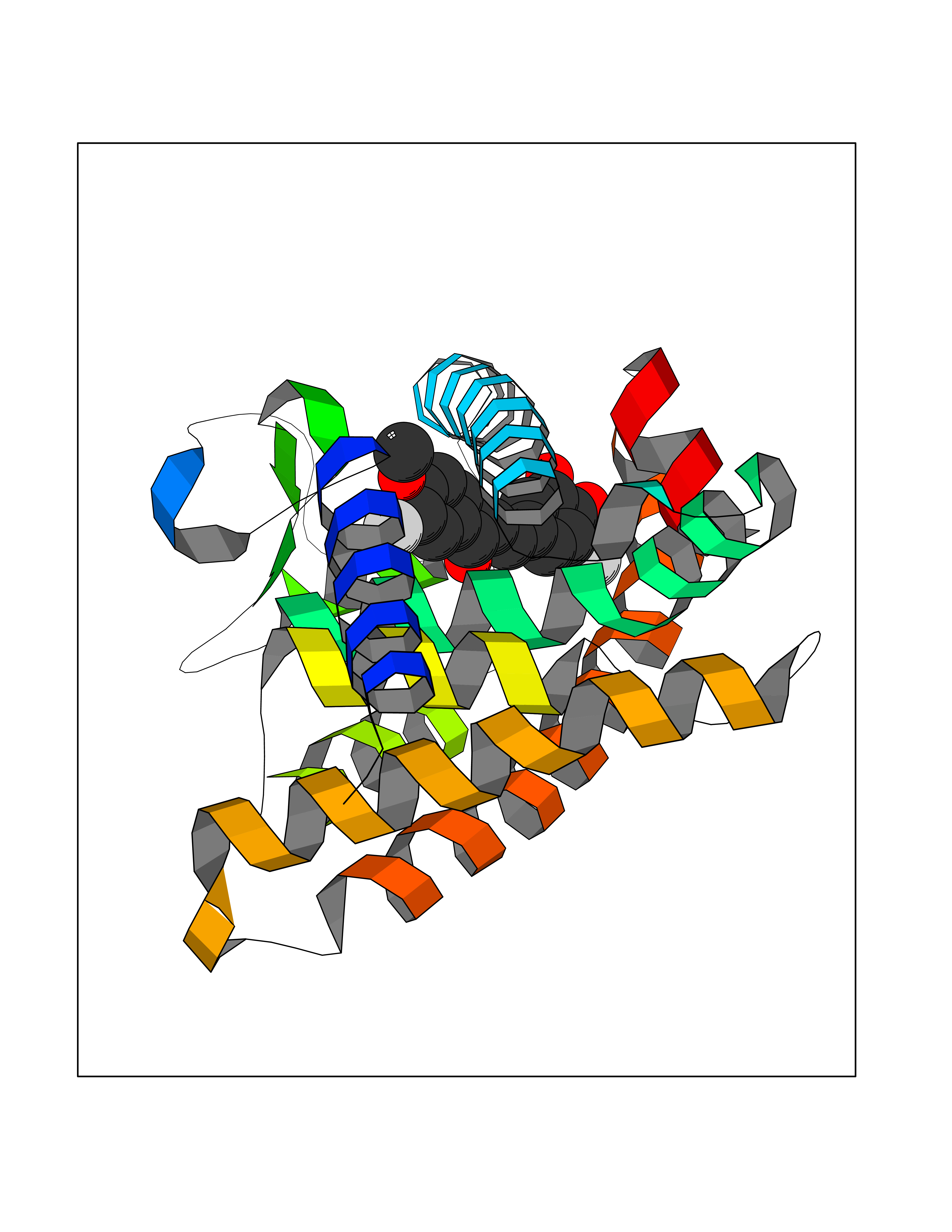 Molscript Map 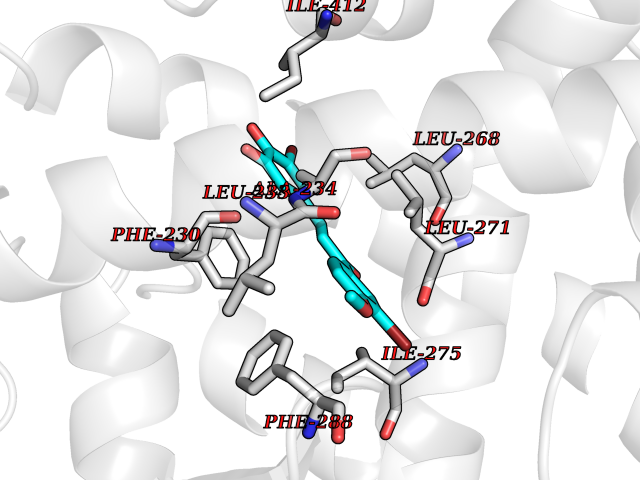 Pymol Map 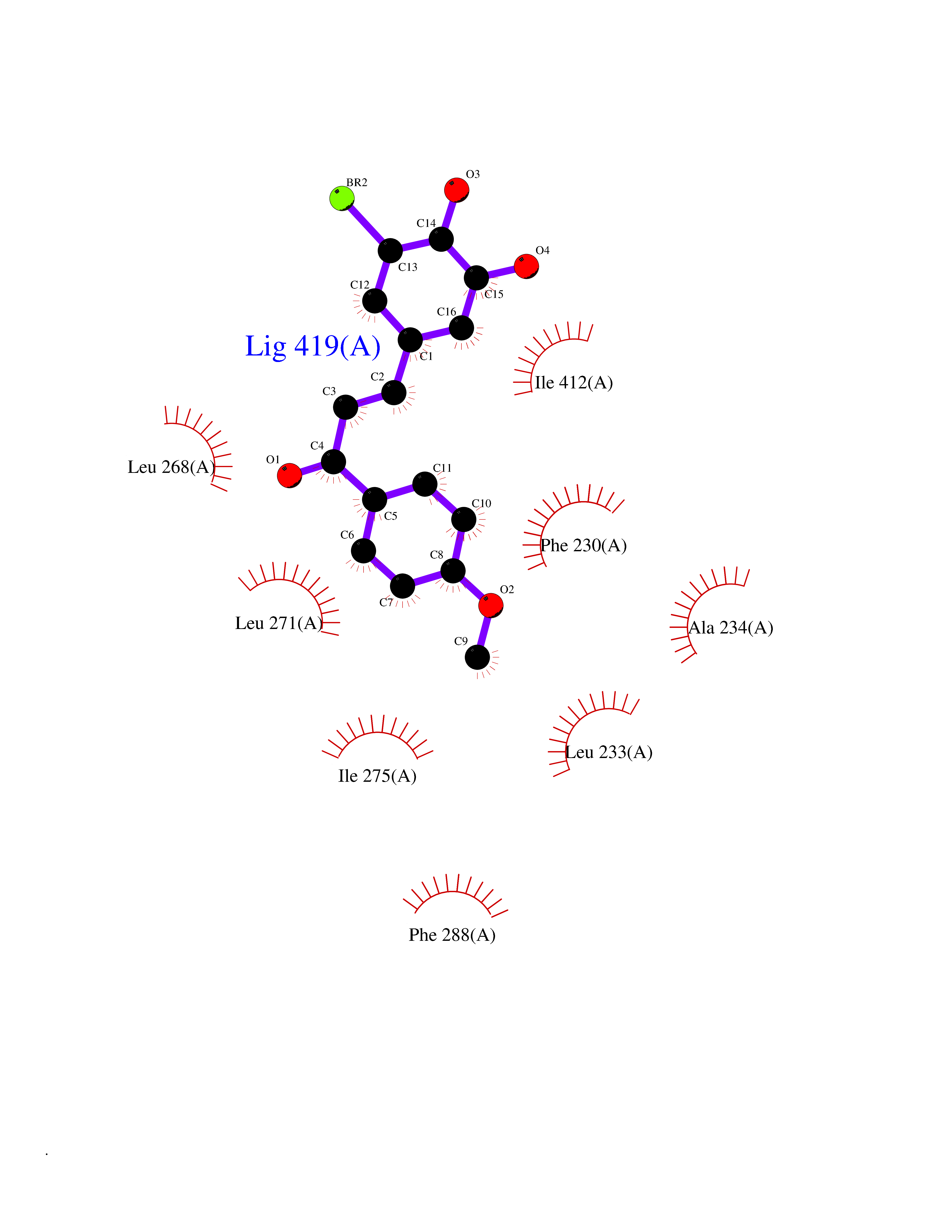 Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Caspase-7 (CASP7) | 1SHJ | 7.11 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -9.7 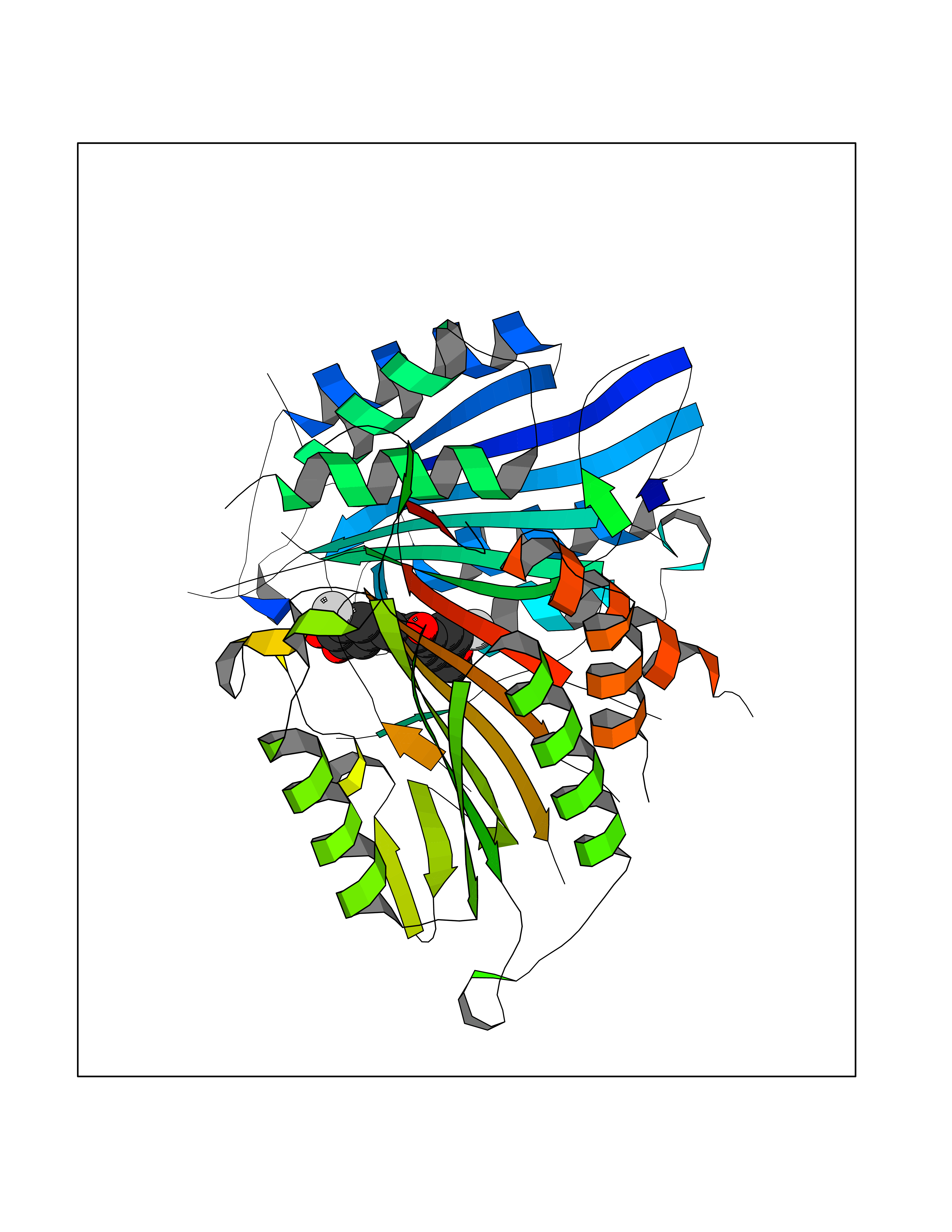 Molscript Map 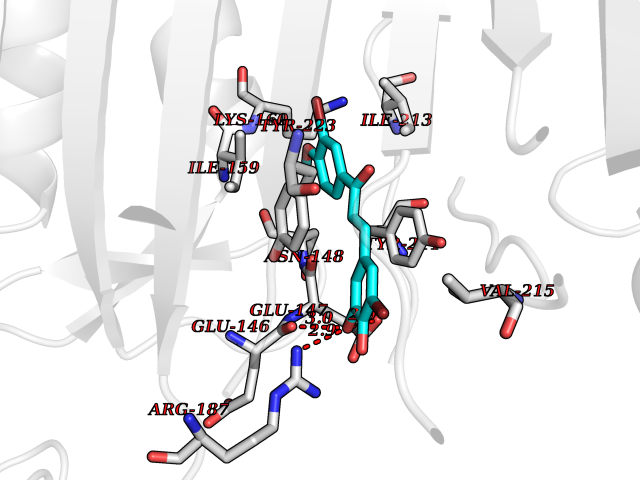 Pymol Map 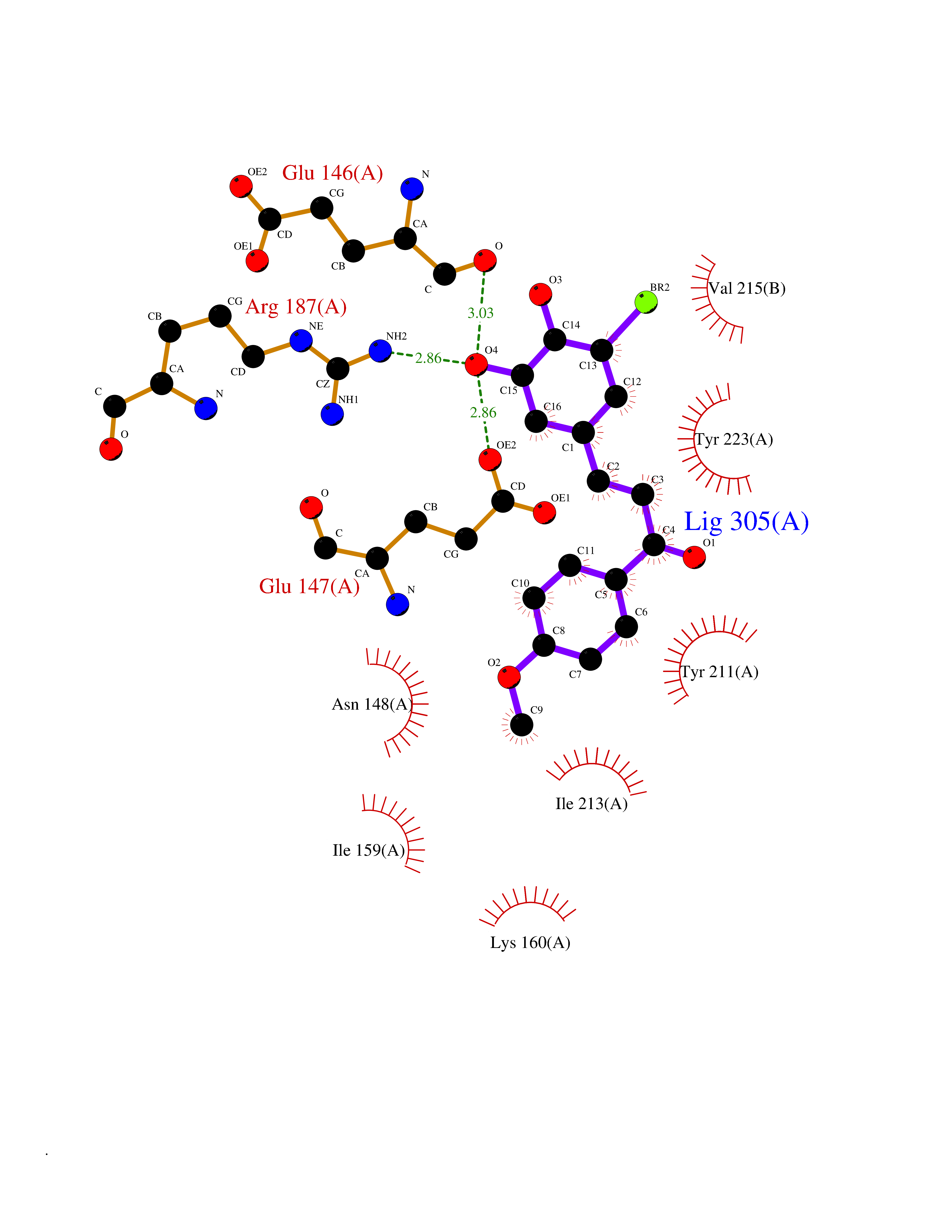 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | BUB1 mitotic checkpoint serine/threonine kinase (BUB1) | 6F7B | 7.10 | |
Target general information Gen name BUB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hBUB1; Mitotic checkpoint serine/threonine-protein kinase BUB1; BUB1L; BUB1A Protein family Protein kinase superfamily, Ser/Thr protein kinase family, BUB1 subfamily Biochemical class Kinase Function Has a key role in the assembly of checkpoint proteins at the kinetochore, being required for the subsequent localization of CENPF, BUB1B, CENPE and MAD2L1. Required for the kinetochore localization of PLK1. Required for centromeric enrichment of AUKRB in prometaphase. Plays an important role in defining SGO1 localization and thereby affects sister chromatid cohesion. Acts as a substrate for anaphase-promoting complex or cyclosome (APC/C) in complex with its activator CDH1 (APC/C-Cdh1). Necessary for ensuring proper chromosome segregation and binding to BUB3 is essential for this function. Can regulate chromosome segregation in a kinetochore-independent manner. Can phosphorylate BUB3. The BUB1-BUB3 complex plays a role in the inhibition of APC/C when spindle-assembly checkpoint is activated and inhibits the ubiquitin ligase activity of APC/C by phosphorylating its activator CDC20. This complex can also phosphorylate MAD1L1. Kinase activity is essential for inhibition of APC/CCDC20 and for chromosome alignment but does not play a major role in the spindle-assembly checkpoint activity. Mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. Serine/threonine-protein kinase that performs 2 crucial functions during mitosis: it is essential for spindle-assembly checkpoint signaling and for correct chromosome alignment. Related diseases Microcephaly 30, primary, autosomal recessive (MCPH30) [MIM:620183]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age, sex and ethnically matched mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. MCPH30 is characterized by small head, poor overall growth, and global developmental delay with variably impaired intellectual development. Affected individuals may also have variable congenital anomalies, including atrial septal defect, dysmorphic facial features, tracheal stenosis, and anomalies of the skin and teeth. {ECO:0000269|PubMed:35044816}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95376; O60566; O43684; P46108; Q8NG31; Q8NG31-2; Q9GZQ8; P03070 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell division; Centromere; Chromosome; Chromosome partition; Host-virus interaction; Intellectual disability; Kinase; Kinetochore; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39564.8 Length 343 Aromaticity 0.12 Instability index 31.17 Isoelectric point 8.49 Charge (pH=7) 4.7 2D Binding mode Binding energy (Kcal/mol) -9.69 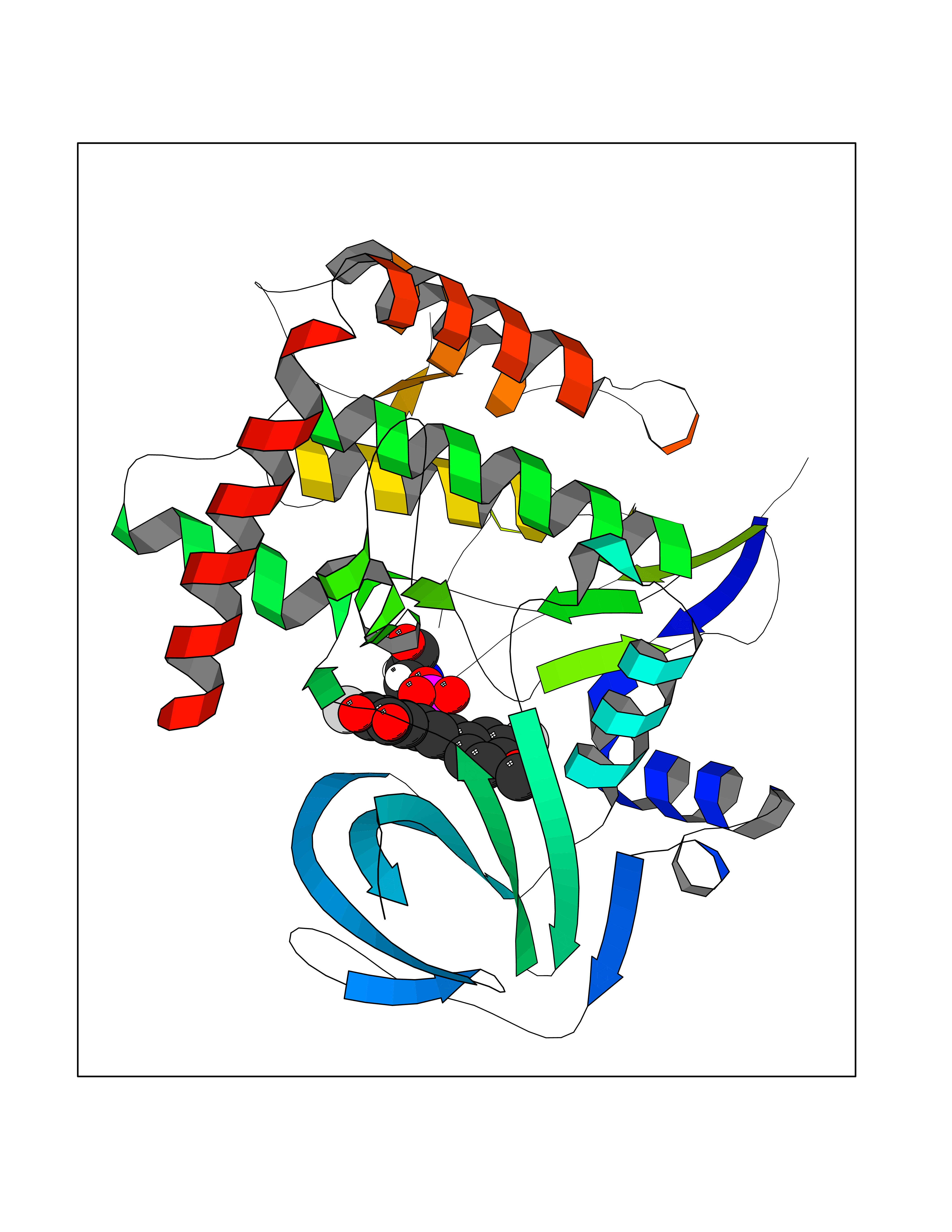 Molscript Map 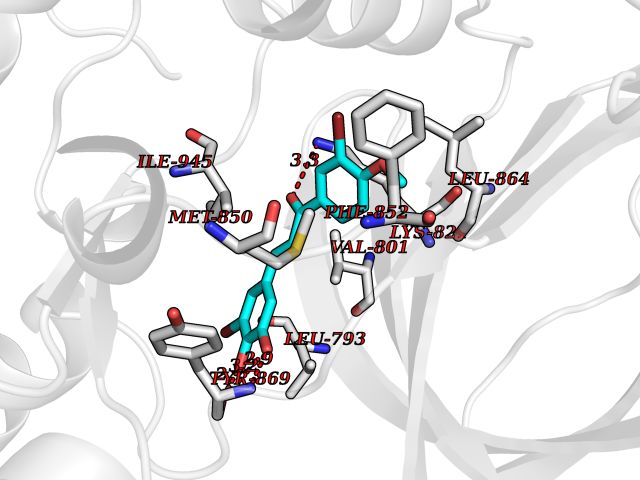 Pymol Map 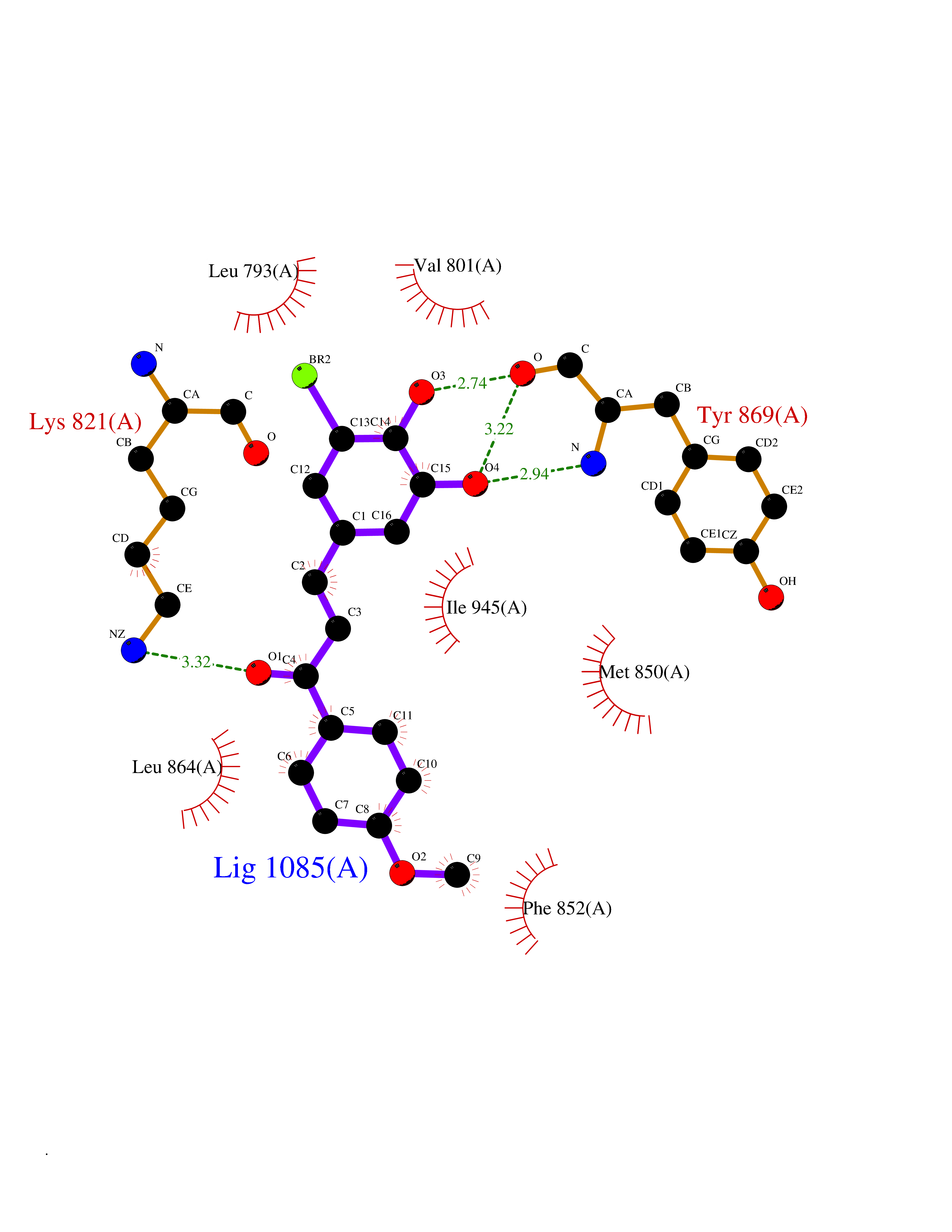 Ligplot Map 3D Binding mode Sequence APNFIVGNPWDDKLIFKLLSGLSKPVSSYPNTFEWQCKLPAIKPKTEFQLGSKLVYVHHLLGEGAFAQVYEATQKNKQKFVLKVQKPANPWEFYIGTQLMERLKPSMQHMFMKFYSAHLFQNGSVLVGELYSYGTLLNAINLYKNTPEKVMPQGLVISFAMRMLYMIEQVHDCEIIHGDIKPDNFILGNGFLEQDDEDDLSAGLALIDLGQSIDMKLFPKGTIFTAKCETXGFQCVEMLSNKPWNYQIDYFGVAATVYCMLFGTYMKVKNEECKPEGLFRRLPHLDMWNEFFHVMLNIPDCHHLPSLDLLRQKLKKVFQQHYTNKIRALRNRLIVLLLECKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Matrix metalloproteinase-13 (MMP-13) | 2OW9 | 7.10 | |
Target general information Gen name MMP13 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Matrix metalloproteinase 13; Collagenase-3; Collagenase 3 Protein family Peptidase M10A family Biochemical class Peptidase Function Cleaves triple helical collagens, including type I, type II and type III collagen, but has the highest activity with soluble type II collagen. Can also degrade collagen type IV, type XIV and type X. May also function by activating or degrading key regulatory proteins, such as TGFB1 and CTGF. Plays a role in wound healing, tissue remodeling, cartilage degradation, bone development, bone mineralization and ossification. Required for normal embryonic bone development and ossification. Plays a role in the healing of bone fractures via endochondral ossification. Plays a role in wound healing, probably by a mechanism that involves proteolytic activation of TGFB1 and degradation of CTGF. Plays a role in keratinocyte migration during wound healing. May play a role in cell migration and in tumor cell invasion. Plays a role in the degradation of extracellular matrix proteins including fibrillar collagen, fibronectin, TNC and ACAN. Related diseases Spondyloepimetaphyseal dysplasia, Missouri type (SEMDM) [MIM:602111]: A bone disease characterized by moderate to severe metaphyseal changes, mild epiphyseal involvement, rhizomelic shortening of the lower limbs with bowing of the femora and/or tibiae, coxa vara, genu varum and pear-shaped vertebrae in childhood. Epimetaphyseal changes improve with age. {ECO:0000269|PubMed:16167086}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metaphyseal anadysplasia 1 (MANDP1) [MIM:602111]: A bone development disorder characterized by skeletal anomalies that resolve spontaneously with age. Clinical characteristics are evident from the first months of life and include slight shortness of stature and a mild varus deformity of the legs. Patients attain a normal stature in adolescence and show improvement or complete resolution of varus deformity of the legs and rhizomelic micromelia. {ECO:0000269|PubMed:19615667}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metaphyseal dysplasia, Spahr type (MDST) [MIM:250400]: An autosomal recessive, rare disease characterized by moderate short stature, mild genua vara, and radiographic signs of metaphyseal dysplasia, but no biochemical signs of rickets. {ECO:0000269|PubMed:24648384, ECO:0000269|PubMed:24781753}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02049; DB01996; DB03033; DB07827; DB08388; DB08561; DB08490; DB06423; DB02697; DB00786; DB04759; DB04760; DB04761; DB07013; DB02071 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Calcium; Collagen degradation; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18757.8 Length 167 Aromaticity 0.15 Instability index 9.97 Isoelectric point 5.47 Charge (pH=7) -7.44 2D Binding mode Binding energy (Kcal/mol) -9.69  Molscript Map 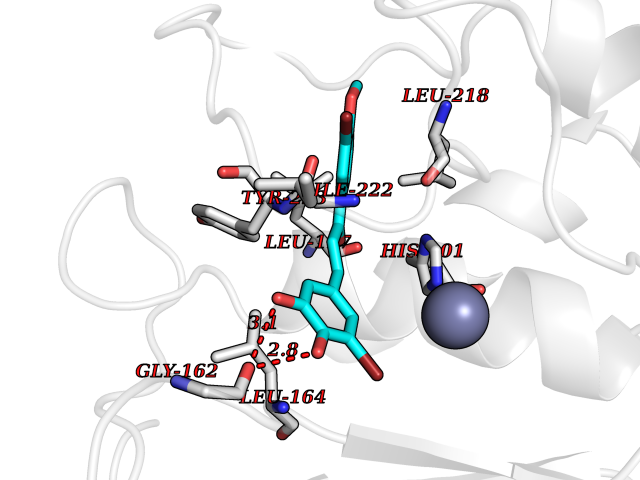 Pymol Map 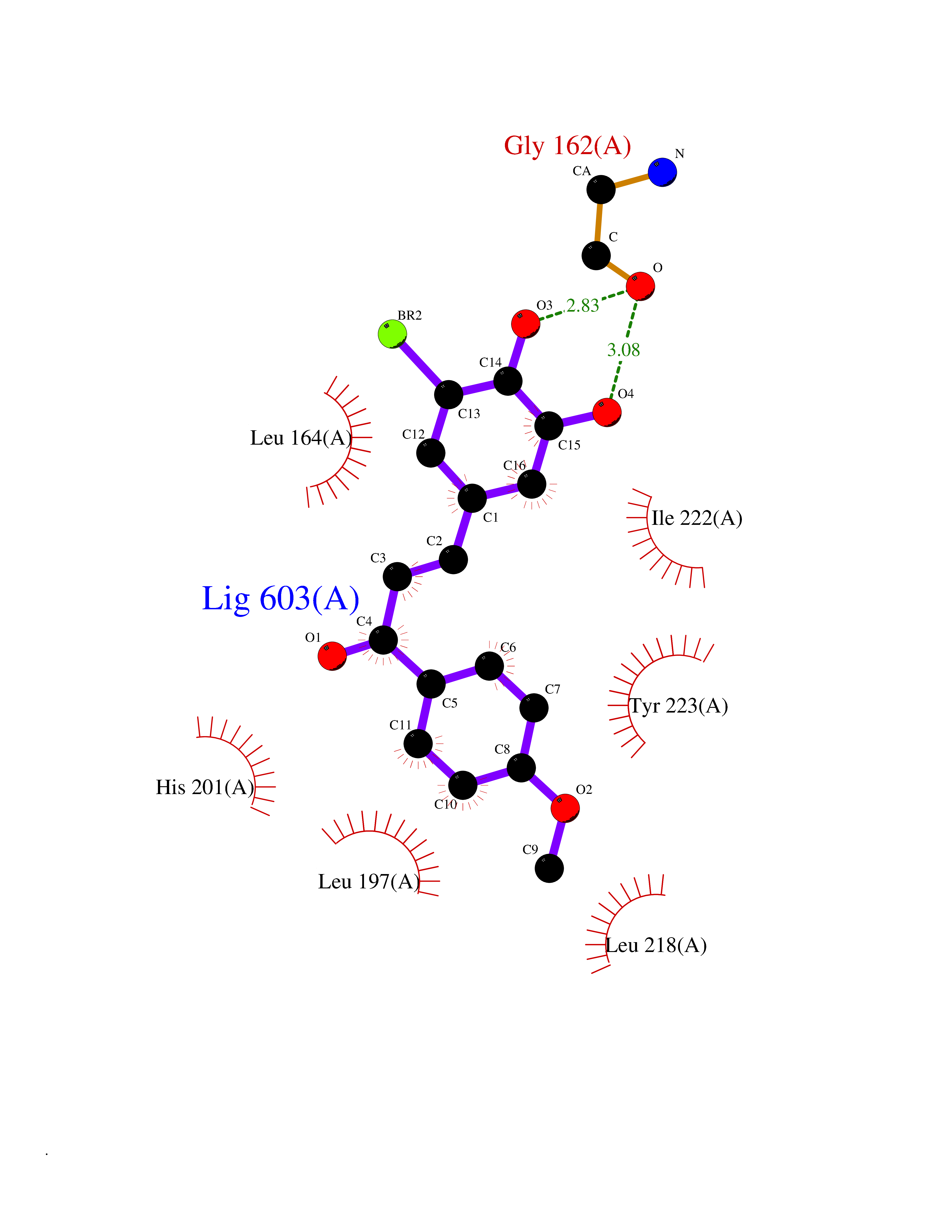 Ligplot Map 3D Binding mode Sequence YNVFPRTLKWSKMNLTYRIVNYTPDMTHSEVEKAFKKAFKVWSDVTPLNFTRLHDGIADIMISFGIKEHGDFYPFDGPSGLLAHAFPPGPNYGGDAHFDDDETWTSSSKGYNLFLVAAHEFGHSLGLDHSKDPGALMFPIYTYTGKSHFMLPDDDVQGIQSLYGPGD Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Nicotinamide phosphoribosyltransferase (NAMPT) | 2E5D | 7.10 | |
Target general information Gen name NAMPT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Visfatin; PreBcell colonyenhancing factor 1; PreB cellenhancing factor; Pre-B-cell colony-enhancing factor 1; Pre-B cell-enhancing factor; PBEF1; PBEF; Nampt; NAmPRTase Protein family NAPRTase family Biochemical class Glycosyltransferases Function It is the rate limiting component in the mammalian NAD biosynthesis pathway. The secreted form behaves both as a cytokine with immunomodulating properties and an adipokine with anti-diabetic properties, it has no enzymatic activity, partly because of lack of activation by ATP, which has a low level in extracellular space and plasma. Plays a role in the modulation of circadian clock function. NAMPT-dependent oscillatory production of NAD regulates oscillation of clock target gene expression by releasing the core clock component: CLOCK-ARNTL/BMAL1 heterodimer from NAD-dependent SIRT1-mediated suppression. Catalyzes the condensation of nicotinamide with 5-phosphoribosyl-1-pyrophosphate to yield nicotinamide mononucleotide, an intermediate in the biosynthesis of NAD. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12980; DB12731; DB05217 Interacts with P02792; Q01628; P03886; P43490; Q70CQ1-2 EC number EC 2.4.2.12 Uniprot keywords 3D-structure; Acetylation; Biological rhythms; Cytokine; Cytoplasm; Glycosyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Pyridine nucleotide biosynthesis; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105483 Length 932 Aromaticity 0.11 Instability index 34.4 Isoelectric point 6.68 Charge (pH=7) -2.24 2D Binding mode Binding energy (Kcal/mol) -9.69 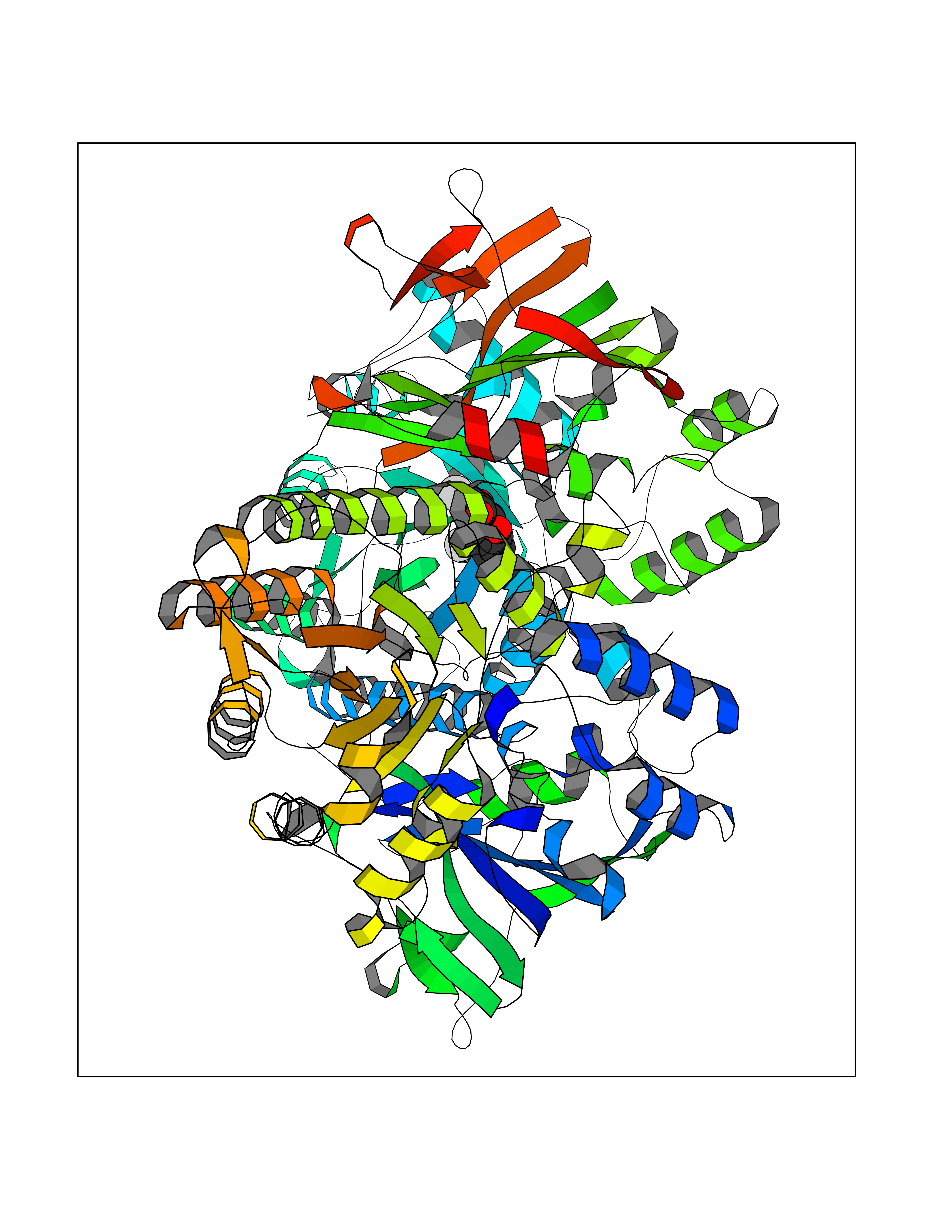 Molscript Map 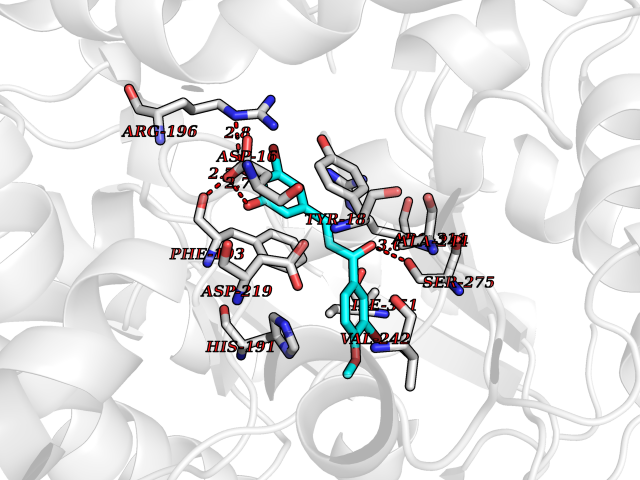 Pymol Map 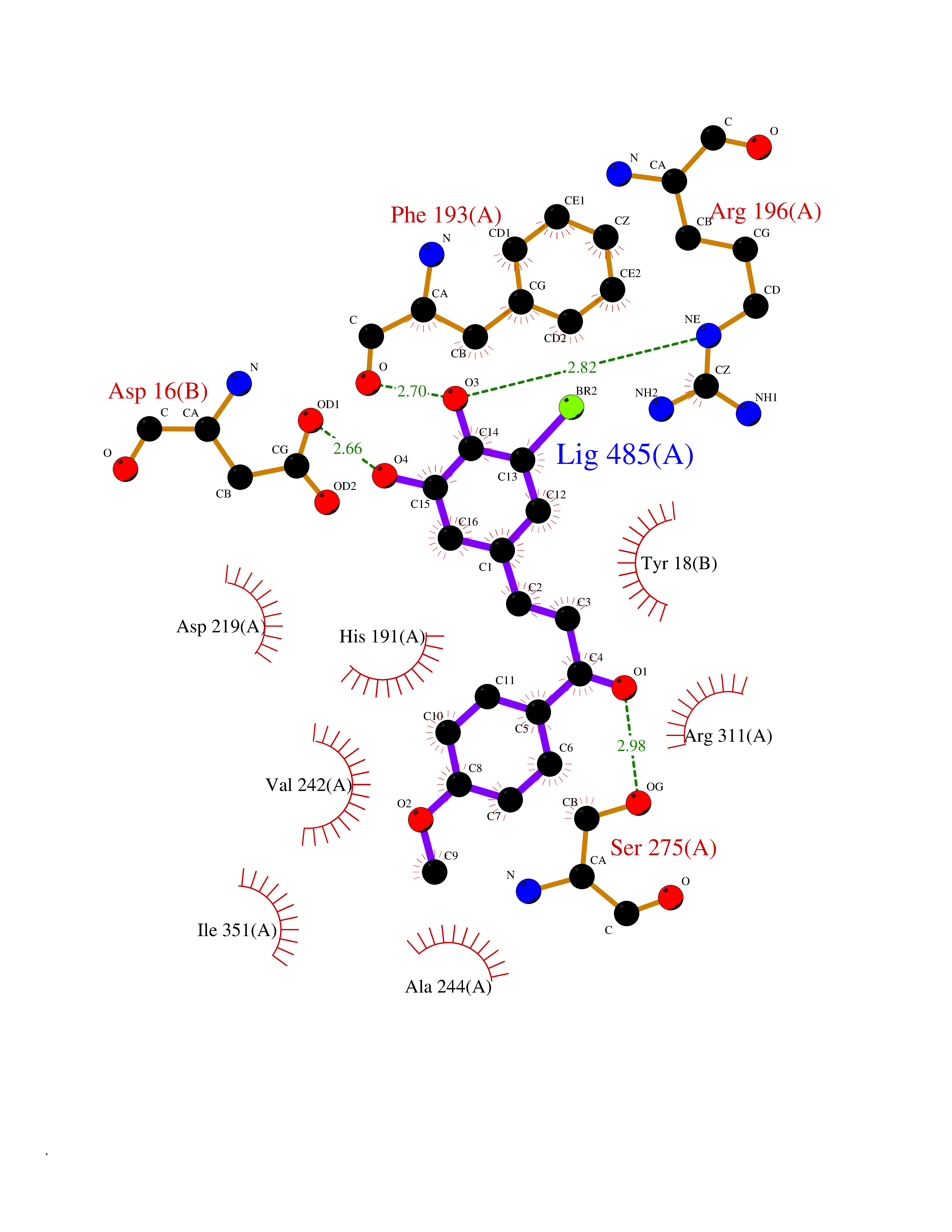 Ligplot Map 3D Binding mode Sequence EFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLNEFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLN Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Fatty acid synthase (FASN) | 3TJM | 7.09 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -9.68 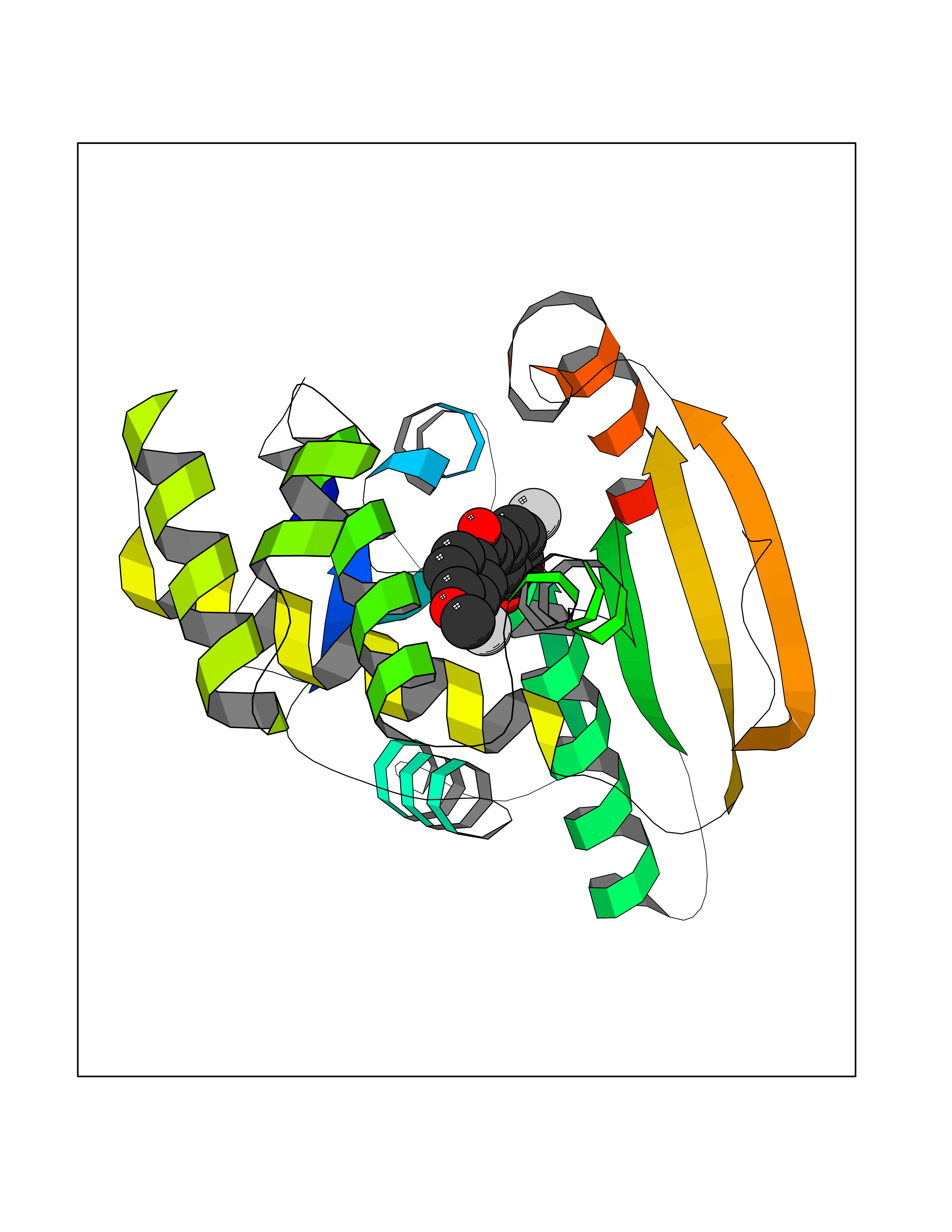 Molscript Map 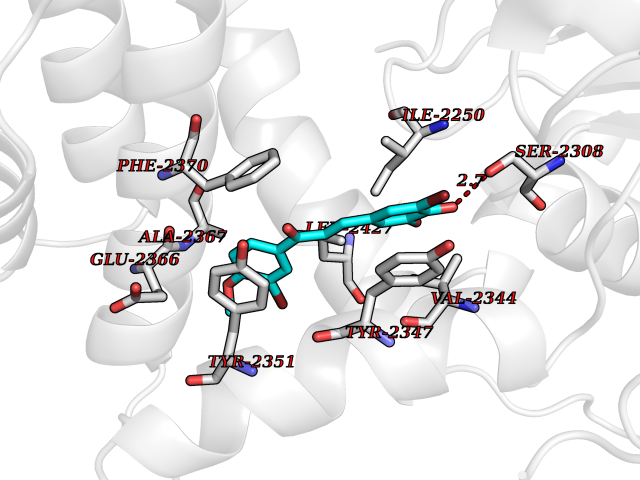 Pymol Map 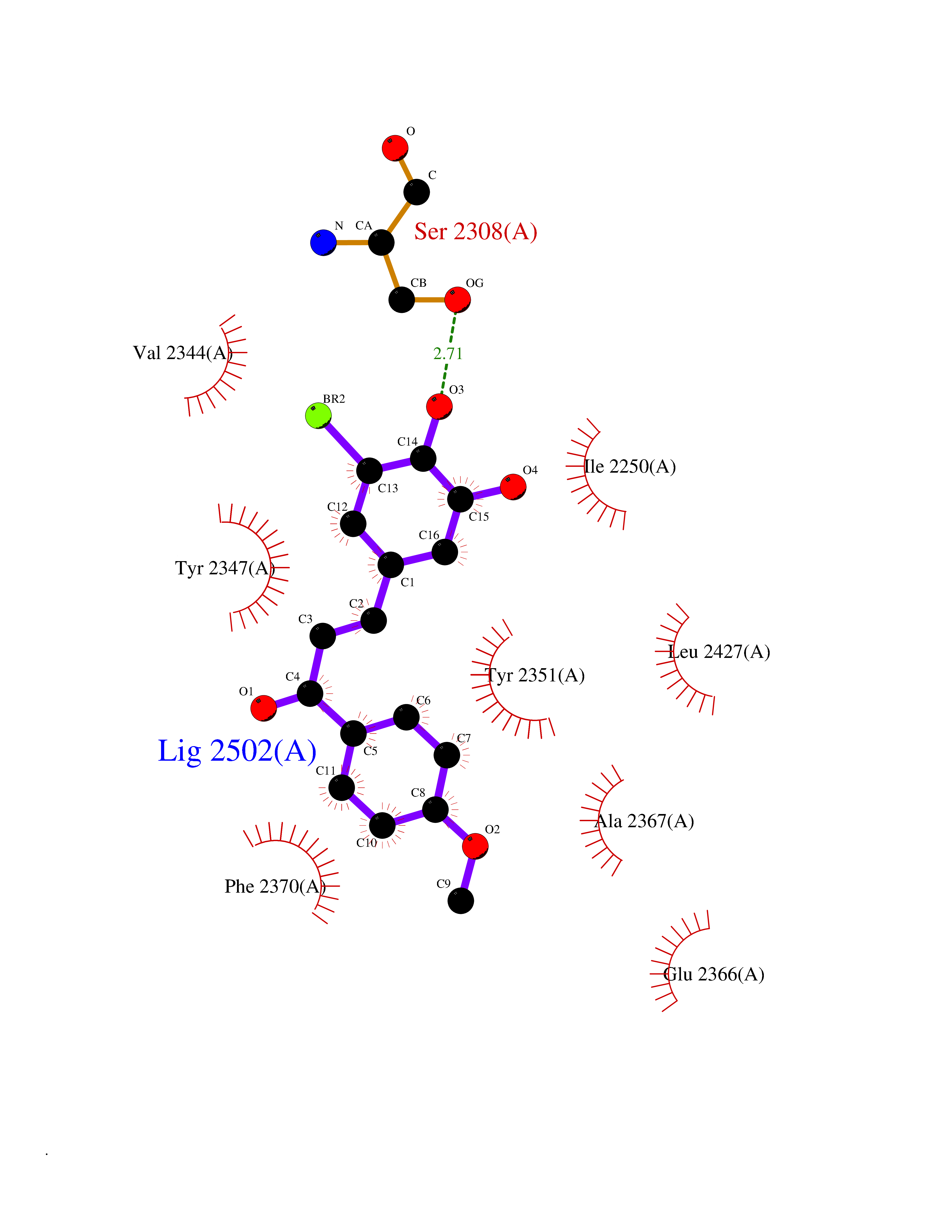 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | SEC14-like protein 4 | 4TLG | 7.09 | |
Target general information Gen name SEC14L4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP3 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB11635; DB11251; DB00163 Interacts with Q96LC9; O43186; P78358; Q9NYQ3; Q0VD86; Q15323; O76011; P50221; Q6FHY5; Q02548; P26367; Q9H8W4; Q04864; Q04864-2; Q9UHV2; P15884; P15884-3; Q96N21; Q9BYV2; Q8N6Y0; Q9H0C1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 23947.6 Length 210 Aromaticity 0.1 Instability index 50.84 Isoelectric point 5.55 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -9.67  Molscript Map 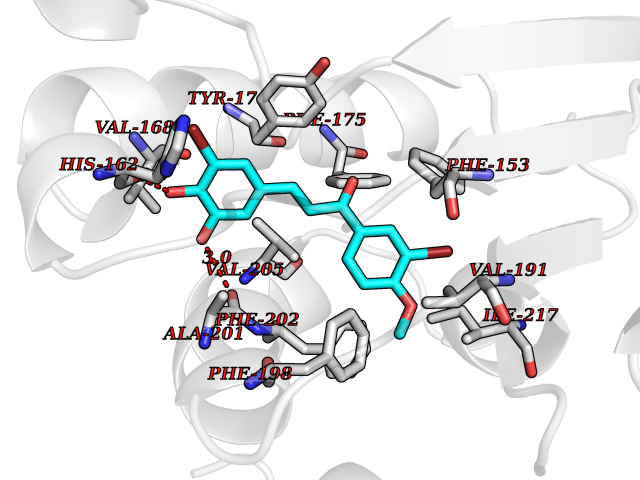 Pymol Map 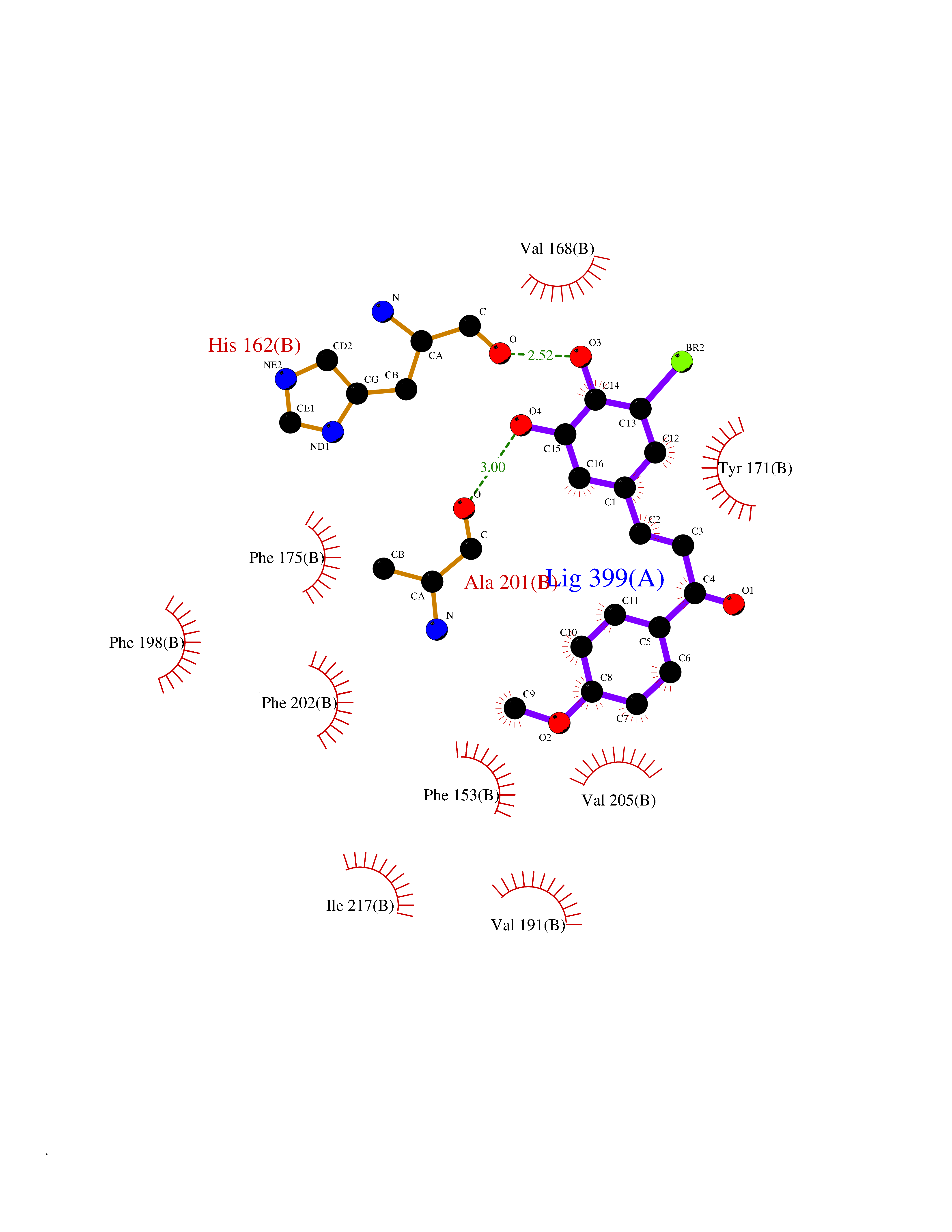 Ligplot Map 3D Binding mode Sequence VTWQPPEVIQLYDSGGLCGYDYEGCPVYFNIIGSLDPKGLLLSASKQDMIRKRIKVCELLLHECELQTQKLGRKIEMALMVFDMEGLSLKHLWKPAVEVYQQFFSILEANYPETLKNLIVIRAPKLFPVAFNLVKSFMSEETRRKIVILGDNWKQELTKFISPDQLPVEFGGTMTDPDGNPKCLTKINYGGEVPKSYYPDKASEETLQSM Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 7.09 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -9.67 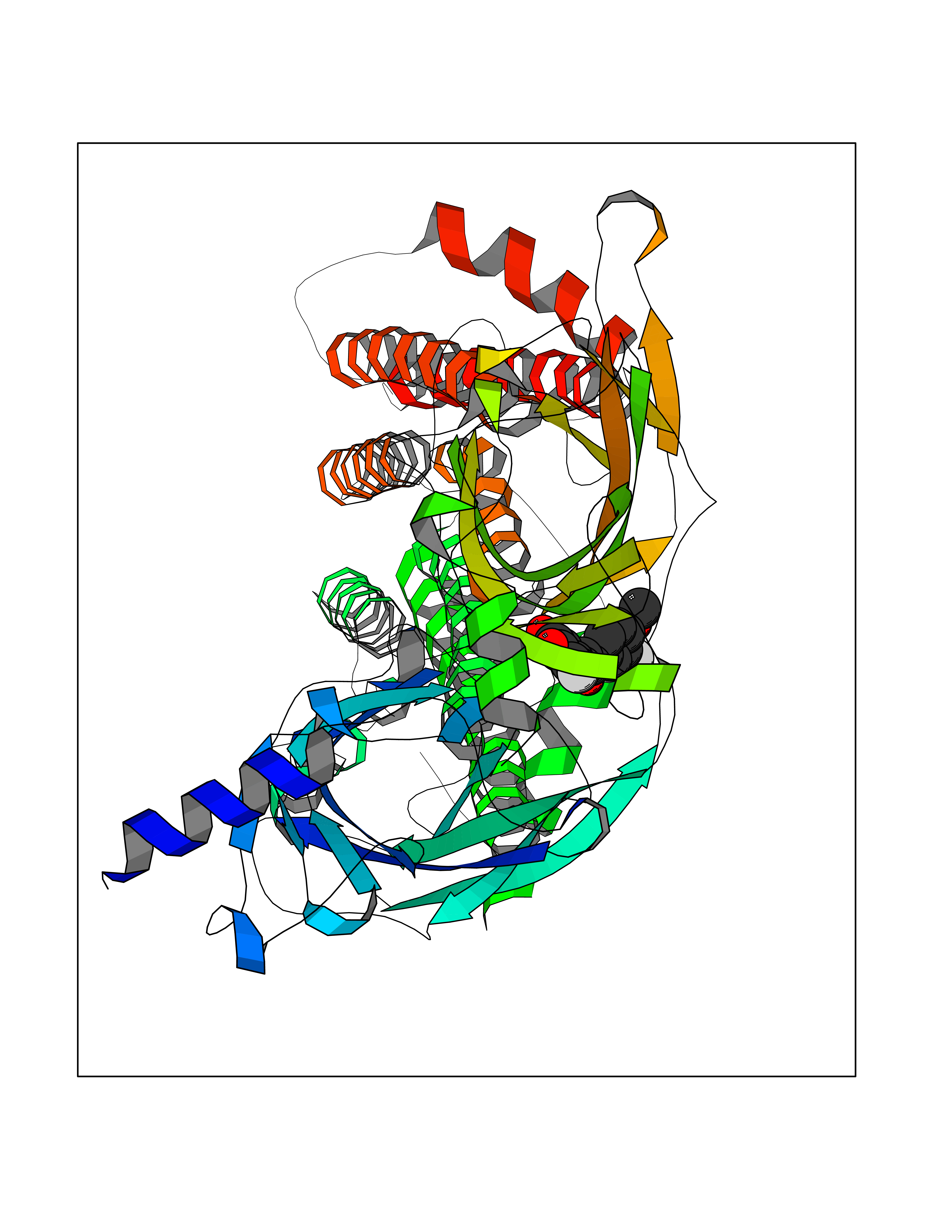 Molscript Map  Pymol Map 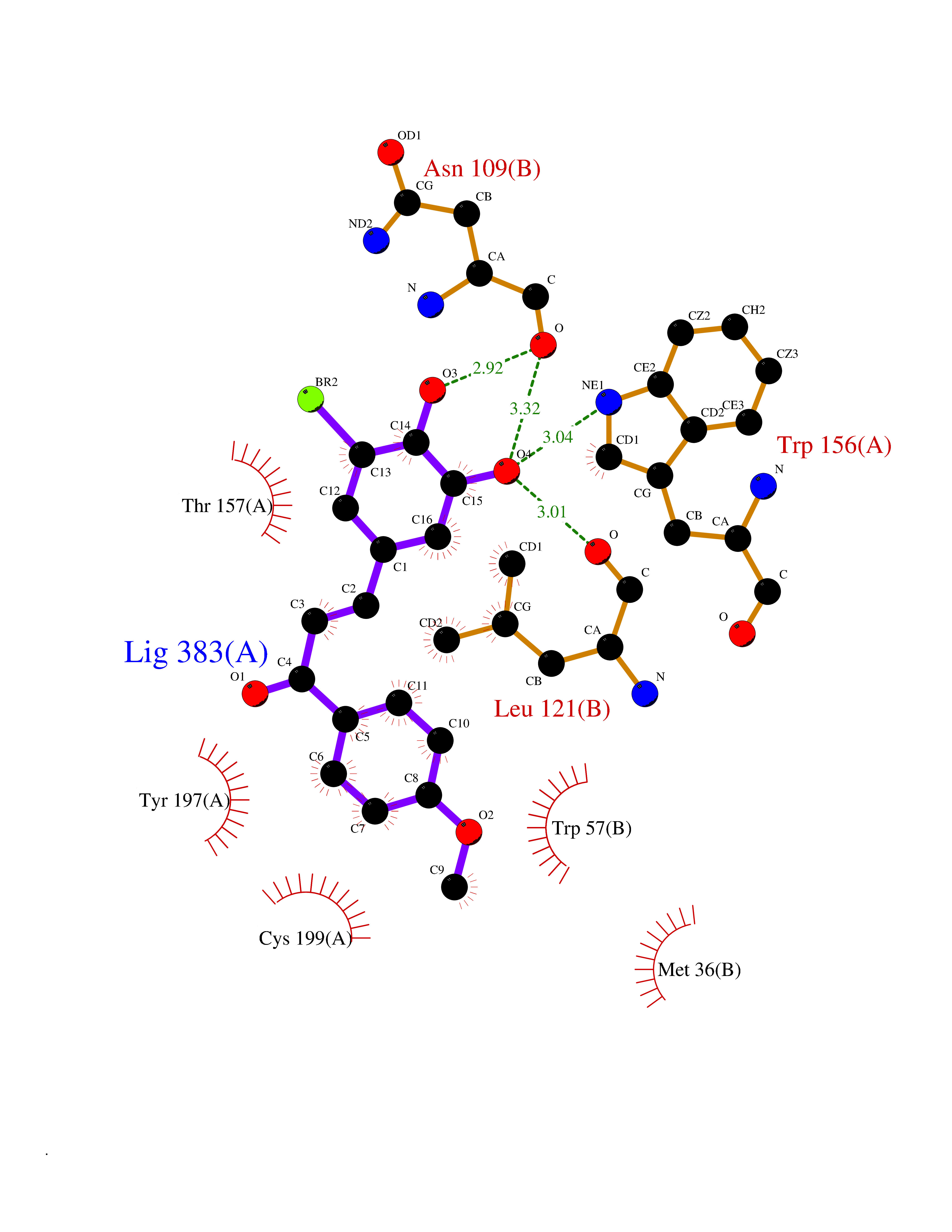 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 7.09 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -9.67 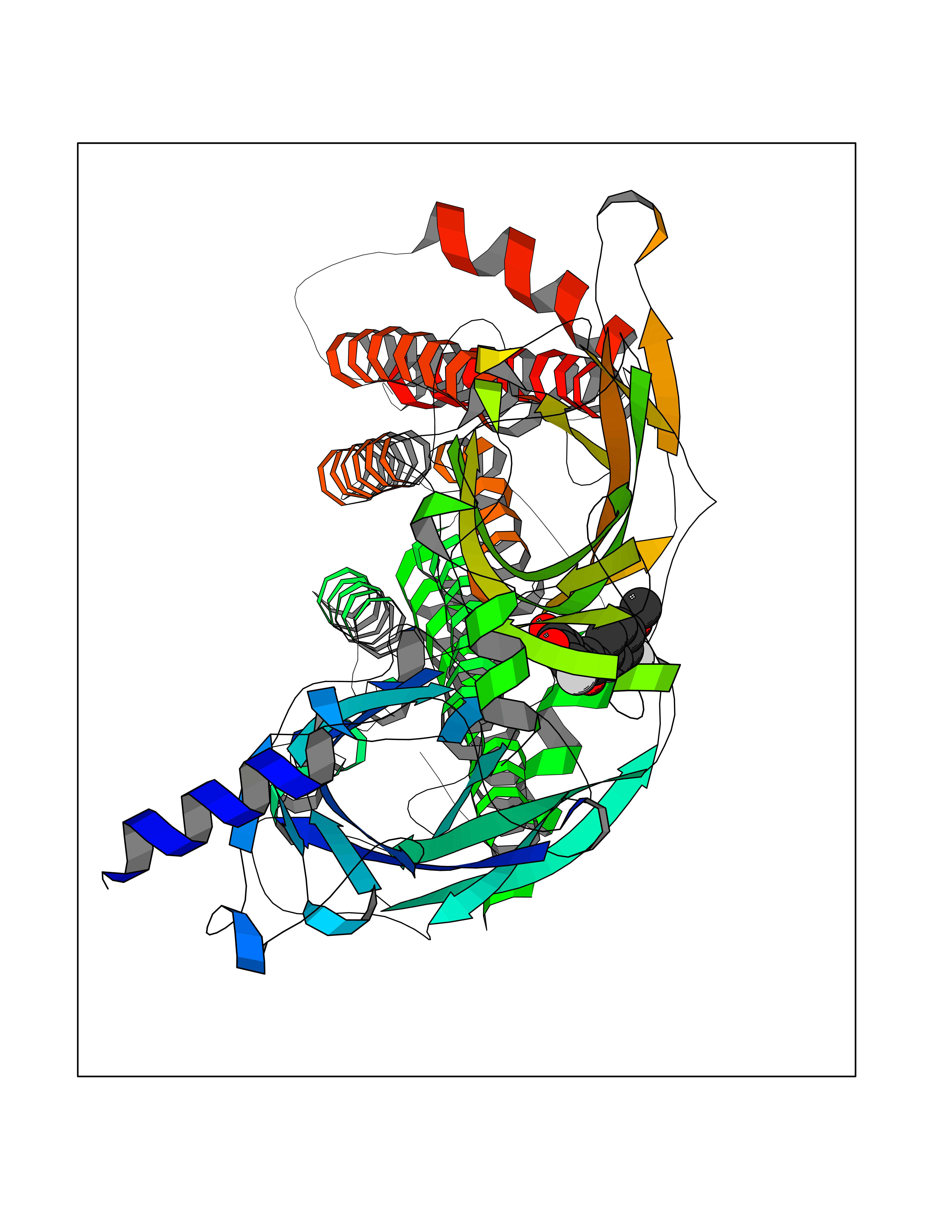 Molscript Map 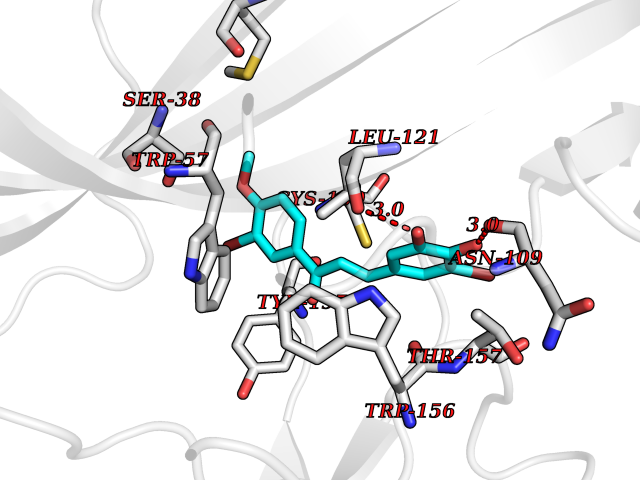 Pymol Map 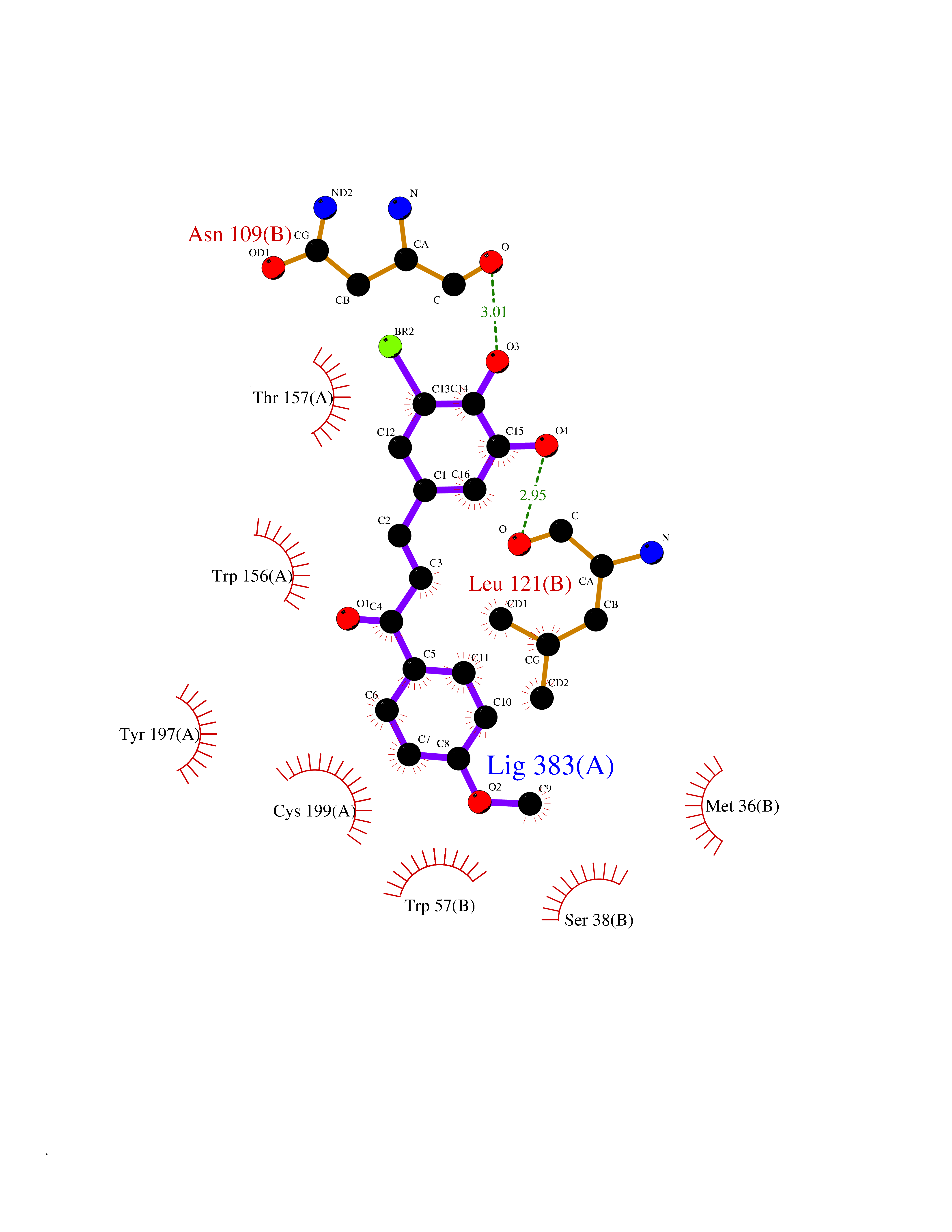 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Retinoic acid receptor beta (RARB) | 4DM6 | 7.08 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -9.66  Molscript Map 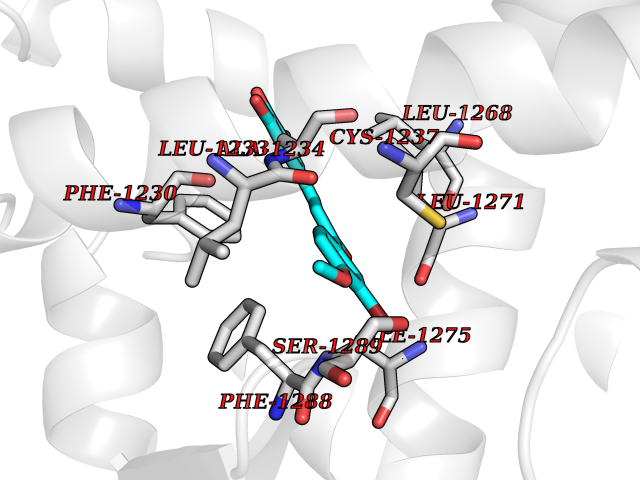 Pymol Map  Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Cytochrome c oxidase subunit 1 | 3DTU | 7.07 | |
Target general information Gen name ctaD Organism Cereibacter sphaeroides (Rhodobacter sphaeroides) Uniprot ID TTD ID NA Synonyms NA Protein family Heme-copper respiratory oxidase family Biochemical class Oxidoreductase Function Copper ion binding.Cytochrome-c oxidase activity.Heme binding.Iron ion binding. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03619 Interacts with Q03736 EC number 7.1.1.9 Uniprot keywords 3D-structure; Cell membrane; Copper; Electron transport; Heme; Hydrogen ion transport; Ion transport; Iron; Membrane; Metal-binding; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,C Molecular weight (Da) 88419.8 Length 794 Aromaticity 0.15 Instability index 40.59 Isoelectric point 6.09 Charge (pH=7) -10.78 2D Binding mode Binding energy (Kcal/mol) -9.64  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence FTRWFMSTNHKDIGVLYLFTGGLVGLISVAFTVYMRMELMAPGVQFMCAEHLESGLVKGFFQSLWPSAVENCTPNGHLWNVMITGHGILMMFFVVIPALFGGFGNYFMPLHIGAPDMAFPRMNNLSYWLYVAGTSLAVASLFAPGGNGQLGSGIGWVLYPPLSTSESGYSTDLAIFAVHLSGASSILGAINMITTFLNMRAPGMTMHKVPLFAWSIFVTAWLILLALPVLAGAITMLLTDRNFGTTFFQPSGGGDPVLYQHILWFFGHPEVYIIVLPAFGIVSHVIATFAKKPIFGYLPMVYAMVAIGVLGFVVWAHHMYTAGLSLTQQSYFMMATMVIAVPTGIKIFSWIATMWGGSIELKTPMLWALGFLFLFTVGGVTGIVLSQASVDRYYHDTYYVVAHFHYVMSLGAVFGIFAGIYFWIGKMSGRQYPEWAGKLHFWMMFVGANLTFFPQHFLGRQGMPRRYIDYPEAFATWNFVSSLGAFLSFASFLFFLGVIFYTLTRGARVTANNYWNEHADTLEWTLTSPPPEHTFEQSLEIIGRPQPGGTGFQPSASPVATQIHWLDGFILVIIAAITIFVTLLILYAVWRFHEKRNKVPARFTHNSPLEIAWTIVPIVILVAIGAFSLPVLFNQQEIPEADVTVKVTGYQWYWGYEYPDEEISFESYMIGSPATGGDNRMSPEVEQQLIEAGYSRDEFLLATDTAMVVPVNKTVVVQVTGADVIHSWTVPAFGVKQDAVPGRLAQLWFRAEREGIFFGQCSELCGISHAYMPITVKVVSEEAYAAWLEQHHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 7.07 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -9.65  Molscript Map 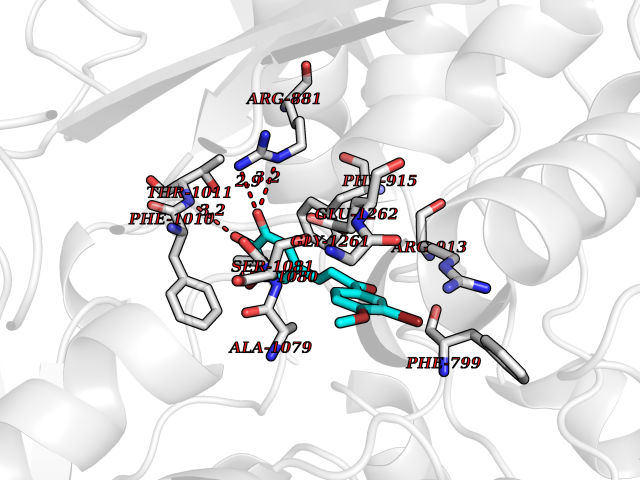 Pymol Map 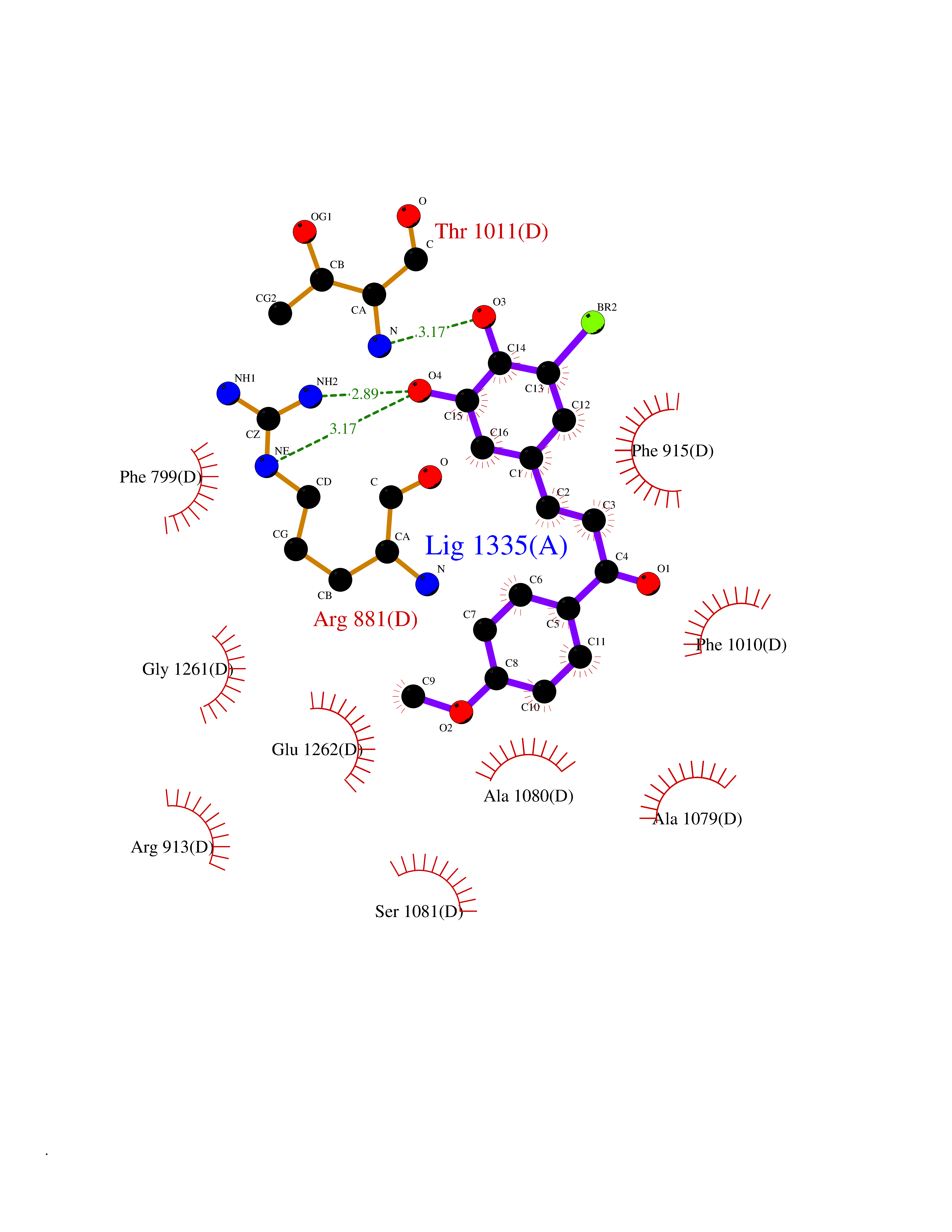 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||