Job Results:
Ligand
Structure
Job ID
bb998bd8d55e750bf9fe315d9dd71eb1
Job name
NA
Time
2025-12-26 14:34:43
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Pyruvate dehydrogenase [ubiquinone] | 3EYA | 5.80 | |
Target general information Gen name poxB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0871;JW0855 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Identical protein binding.Lipid binding.Magnesium ion binding.Pyruvate dehydrogenase (quinone) activity.Thiamine pyrophosphate binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07003 EC number 1.2.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Lipid-binding; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate; Ubiquinone Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 113027 Length 1046 Aromaticity 0.07 Instability index 35.99 Isoelectric point 5.75 Charge (pH=7) -24.38 2D Binding mode Binding energy (Kcal/mol) -7.91  Molscript Map 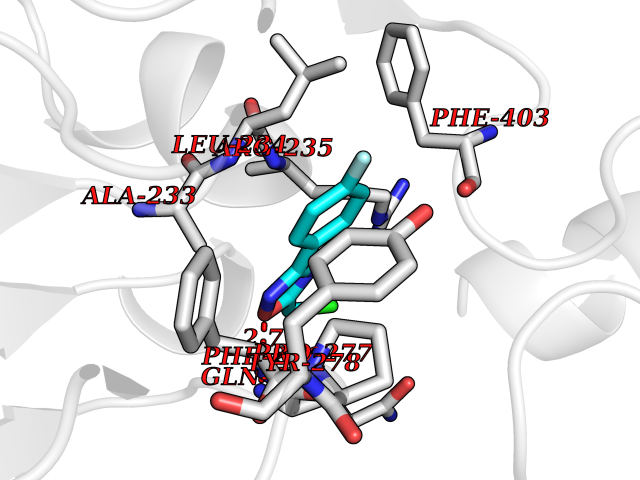 Pymol Map 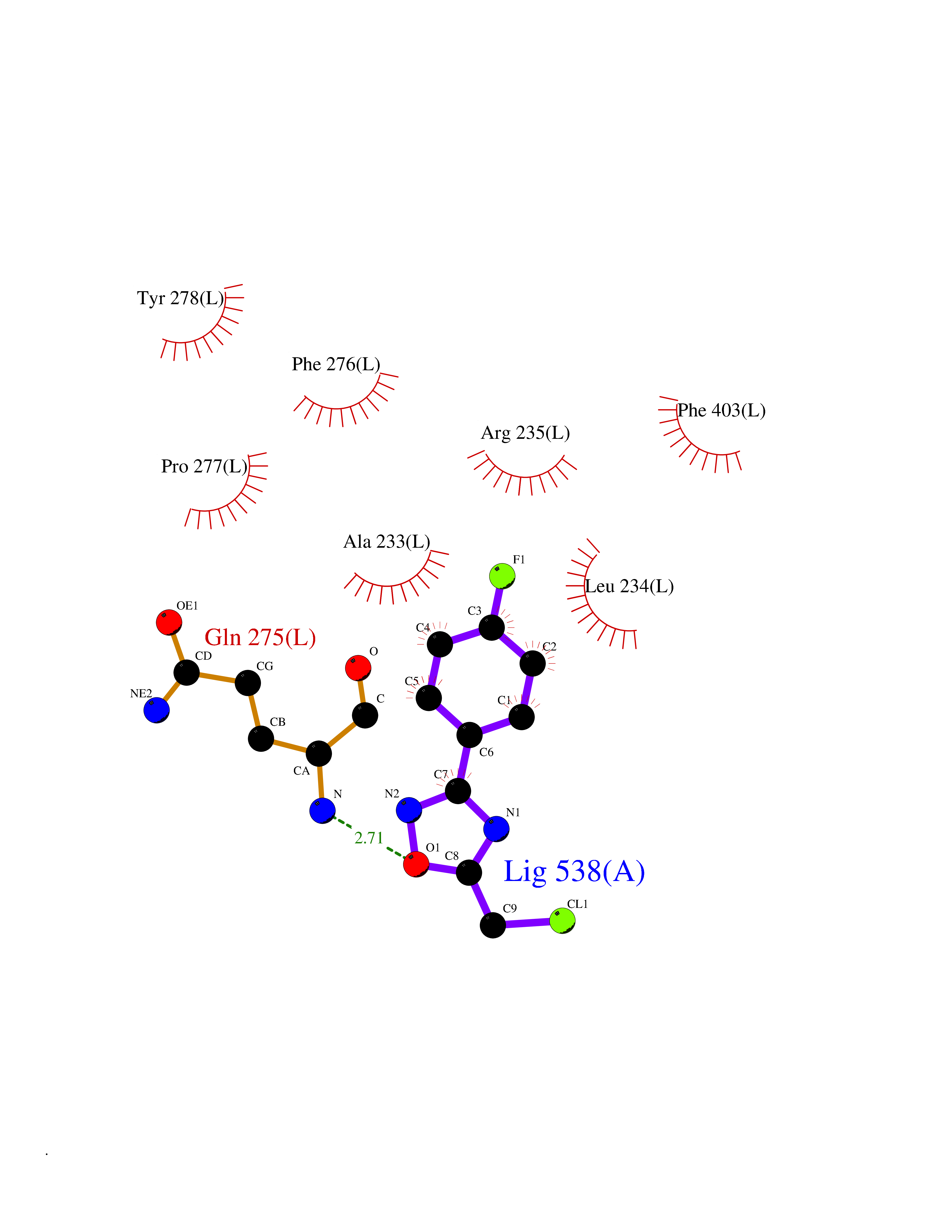 Ligplot Map 3D Binding mode Sequence MKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFDGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIPMKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFVGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIP Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Monomeric sarcosine oxidase | 2GF3 | 5.80 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -7.91  Molscript Map 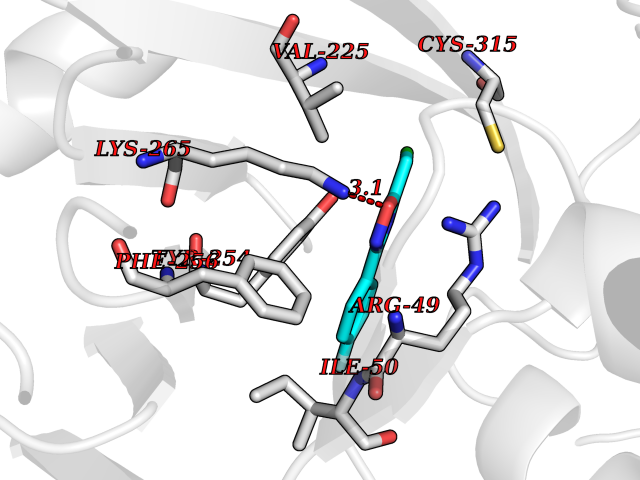 Pymol Map 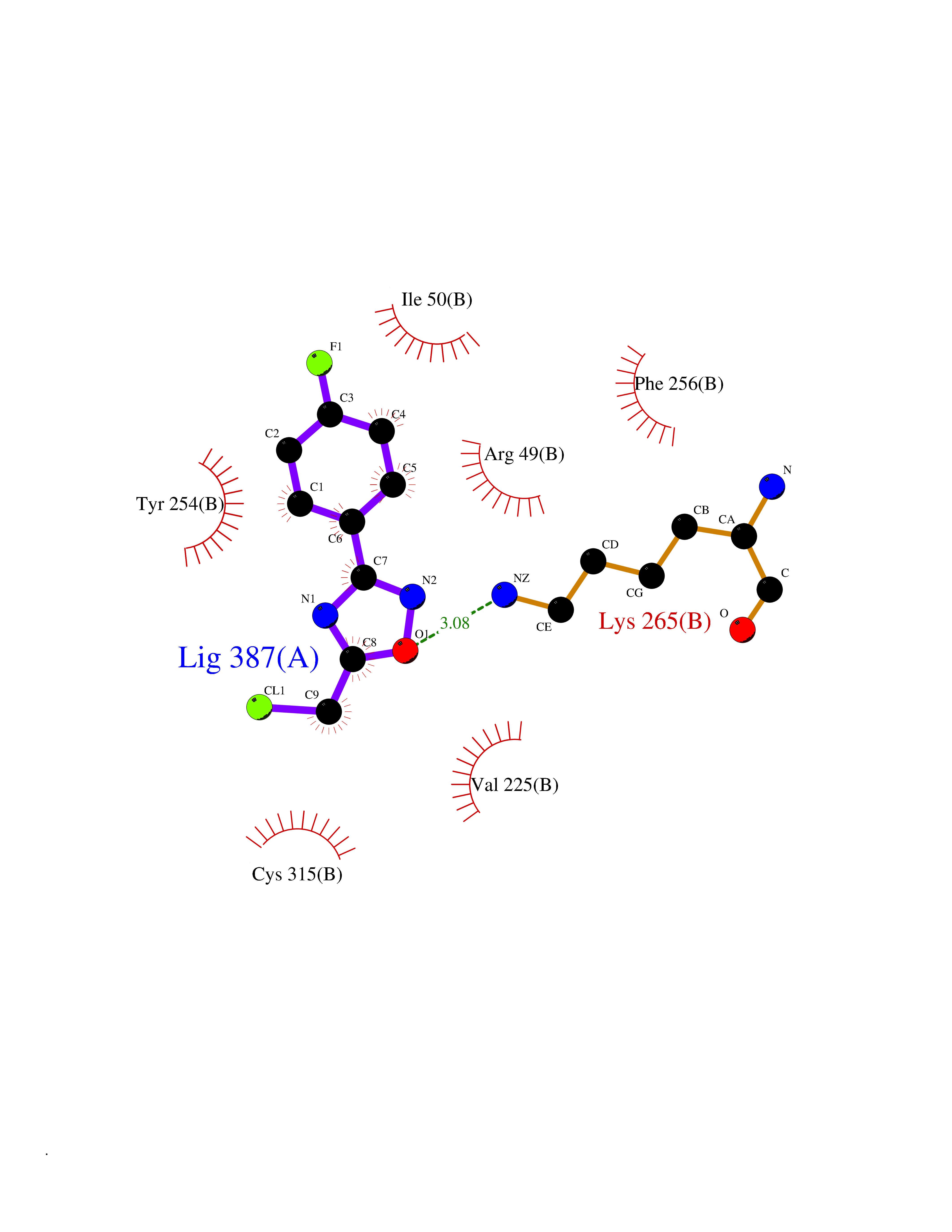 Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Estrogen sulfotransferase | 1G3M | 5.80 | |
Target general information Gen name SULT1E1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms STE Protein family Sulfotransferase 1 family Biochemical class Transferase Function Aryl sulfotransferase activity.Estrone sulfotransferase activity.Flavonol 3-sulfotransferase activity.Steroid binding.Steroid sulfotransferase activity.Sulfotransferase activity. Related diseases Neuromyotonia and axonal neuropathy, autosomal recessive (NMAN) [MIM:137200]: An autosomal recessive neurologic disorder characterized by onset in the first or second decade of a peripheral axonal neuropathy predominantly affecting motor more than sensory nerves. The axonal neuropathy is reminiscent of Charcot-Marie-Tooth disease type 2 and distal hereditary motor neuropathy. Individuals with NMAN also have delayed muscle relaxation and action myotonia associated with neuromyotonic discharges on needle EMG resulting from hyperexcitability of the peripheral nerves. {ECO:0000269|PubMed:16835243, ECO:0000269|PubMed:22961002, ECO:0000269|PubMed:28691797, ECO:0000269|PubMed:29787766, ECO:0000269|PubMed:31088288}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02902; DB03346; DB01812; DB00714; DB14635; DB01176; DB00977; DB09288; DB00675; DB09100 Interacts with O76083; O76083-2 EC number 2.8.2.4 Uniprot keywords 3D-structure; Cytoplasm; Lipid metabolism; Lipid-binding; Proteomics identification; Reference proteome; Steroid-binding; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34022.7 Length 285 Aromaticity 0.15 Instability index 32.76 Isoelectric point 6.33 Charge (pH=7) -2.75 2D Binding mode Binding energy (Kcal/mol) -7.9  Molscript Map 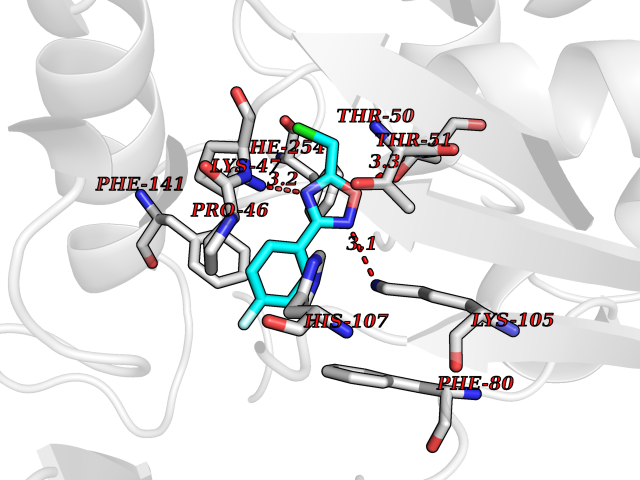 Pymol Map 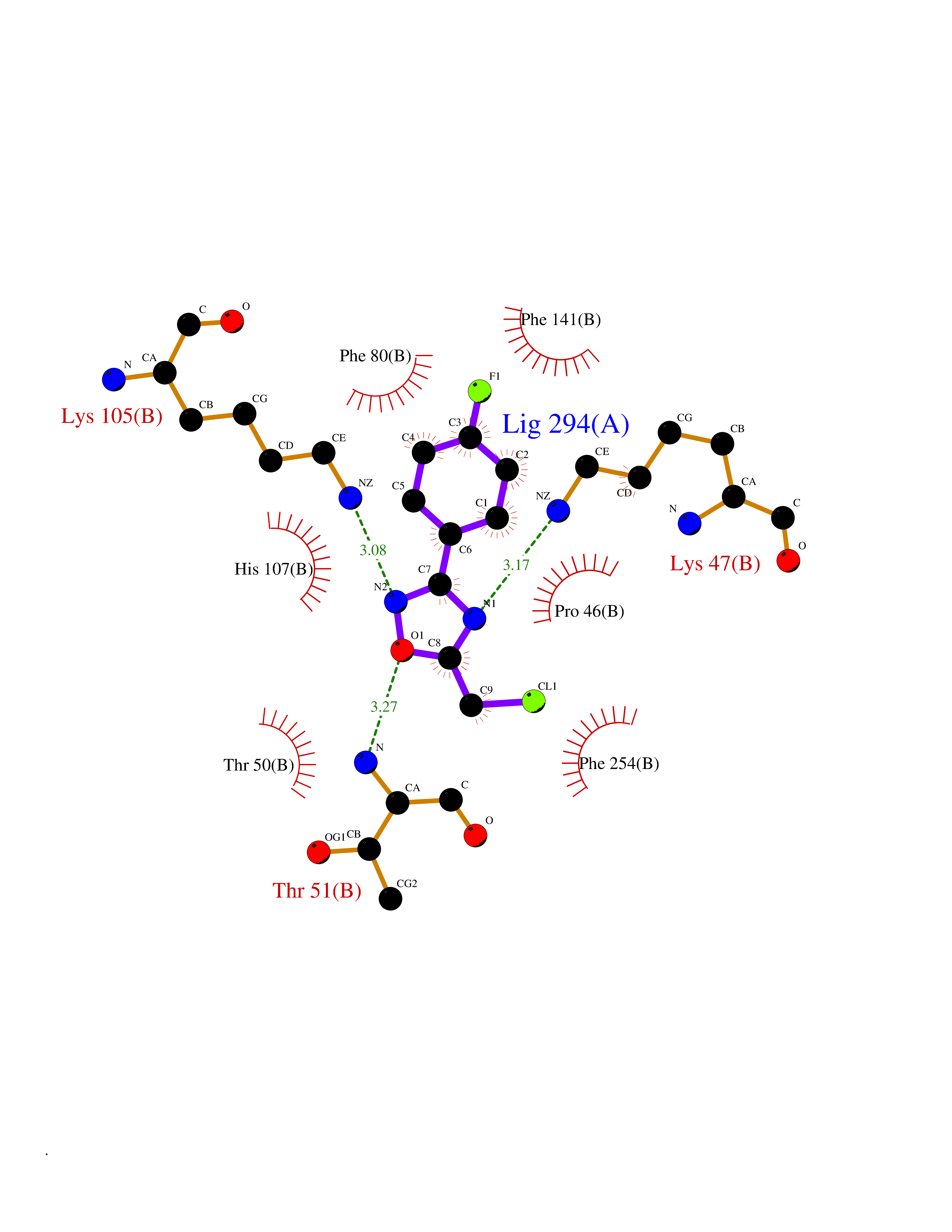 Ligplot Map 3D Binding mode Sequence SELDYYEKFEEVHGILMYKDFVKYWDNVEAFQARPDDLVIATYPKSGTTWVSEIVYMIYKEGDVEKCKEDVIFNRIPFLECRNGVKQLDEMNSPRIVKTHLPPELLPASFWEKDCKIIYLCRNAKDVAVSFYYFFLMVAGHPNPGSFPEFVEKFMQGQVPYGSWYKHVKSWWEKGKSPRVLFLFYEDLKEDIRKEVIKLIHFLERKPSEELVDRIIHHTSFQEMKNNPSTNYTTLPDEIMNQKLSPFMRKGITGDWKNHFTVALNEKFDKHYEQQMKESTLKFRT Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Receptor-type protein-tyrosine phosphatase zeta (PTPRZ1) | 5H08 | 5.80 | |
Target general information Gen name PTPRZ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor protein tyrosine phosphatase zeta; R-PTP-zeta; PTPRZ1 Protein family Protein-tyrosine phosphatase family, Receptor class 5 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein tyrosine phosphatase that negatively regulates oligodendrocyte precursor proliferation in the embryonic spinal cord. Required for normal differentiation of the precursor cells into mature, fully myelinating oligodendrocytes. May play a role in protecting oligondendrocytes against apoptosis. May play a role in the establishment of contextual memory, probably via the dephosphorylation of proteins that are part of important signaling cascades. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UM73; Q12860 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Phosphoprotein; Protein phosphatase; Proteoglycan; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32208.1 Length 282 Aromaticity 0.11 Instability index 38.98 Isoelectric point 7.35 Charge (pH=7) 0.89 2D Binding mode Binding energy (Kcal/mol) -7.91  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GPAIPIKHFPKHVADLHASSGFTEEFEEVQSCTVDLGITADSSNHPDNKHKNRYINIVAYDHSRVKLAQLAEKDGKLTDYINANYVDGYNRPKAYIAAQGPLKSTAEDFWRMIWEHNVEVIVMITNLVEKGRRKCDQYWPADGSEEYGNFLVTQKSVQVLAYYTVRNFTLRNTKIRVVTQYHYTQWPDMGVPEYSLPVLTFVRKAAYAKRHAVGPVVVHCSAGVGRTGTYIVLDSMLQQIQHEGTVNIFGFLKHIRSQRNYLVQTEEQYVFIHDTLVEAILS Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 1SQB | 5.79 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID C,D,F,G,H Molecular weight (Da) 99281.2 Length 866 Aromaticity 0.12 Instability index 43.81 Isoelectric point 8.32 Charge (pH=7) 7.12 2D Binding mode Binding energy (Kcal/mol) -7.89  Molscript Map 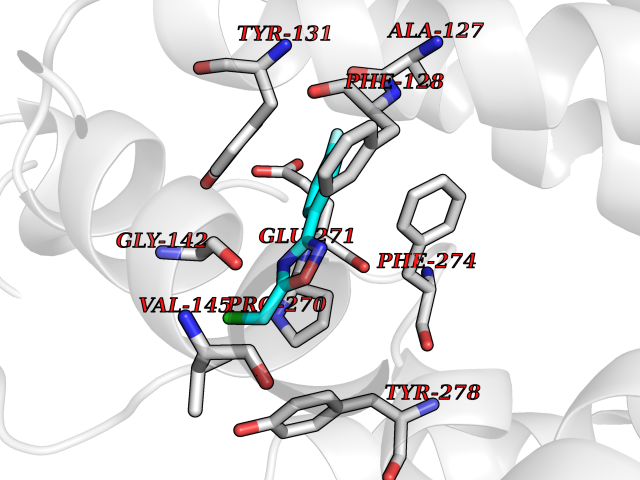 Pymol Map 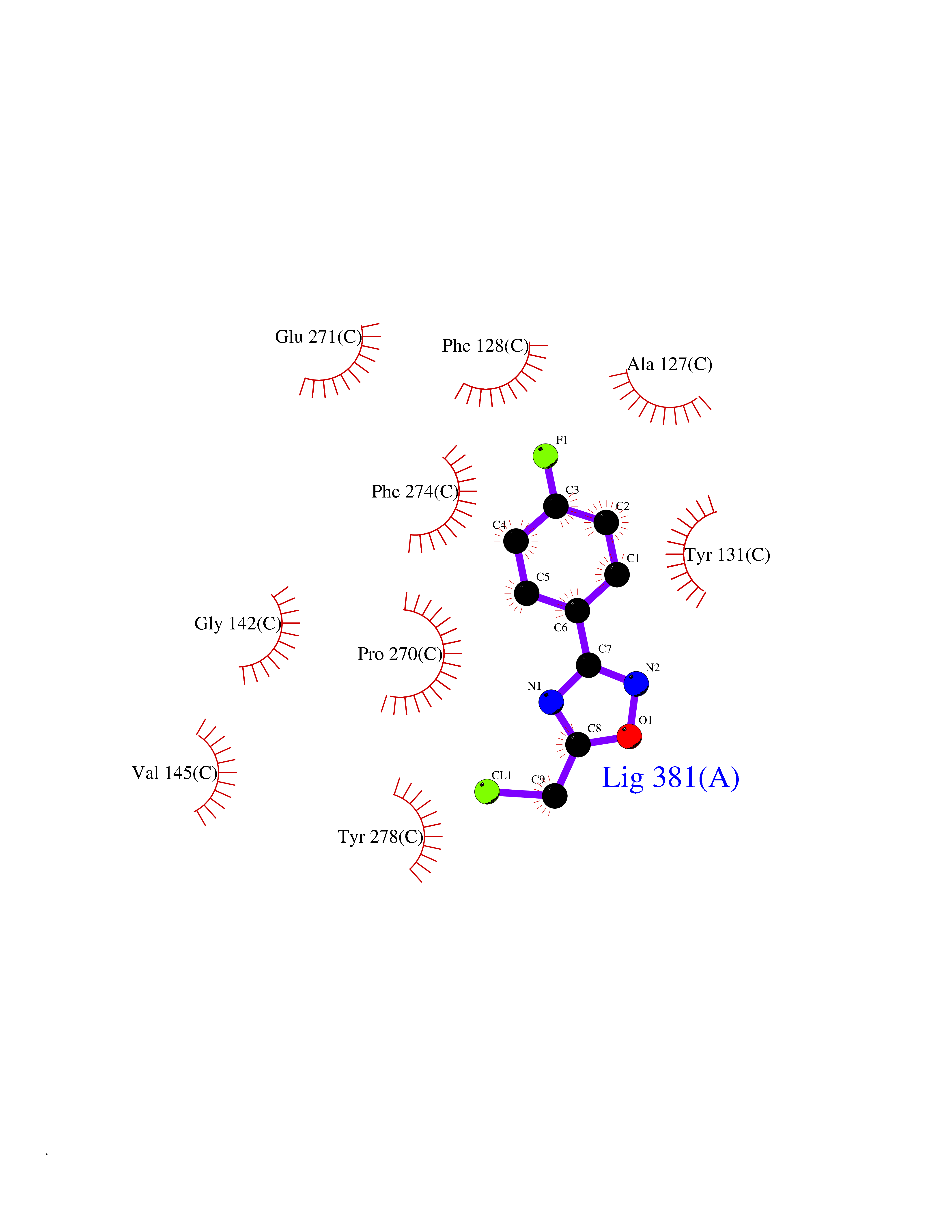 Ligplot Map 3D Binding mode Sequence VSASSRWLEGIRKWYYNAAGFNKLGLMRDDTIHENDDVKEAIRRLPENLYDDRVFRIKRALDLSMRQQILPKEQWTKYEEDKSYLEPYLKEVIRERKEREEWAKKELVDPLTTVREQCEQLEKCVKARERLELCDERVSSRSQTEEDCTEELLDFLHARDHCVAHKLFNSLKTNIRKSHPLMKIVNNAFIDLPAPSNISSWWNFGSLLGICLILQILTGLFLAMHYTSDTTTAFSSVTHICRDVNYGWIIRYMHANGASMFFICLYMHVGRGLYYGSYTFLETWNIGVILLLTVMATAFMGYVLPWGQMSFWGATVITNLLSAIPYIGTNLVEWIWGGFSVDKATLTRFFAFHFILPFIIMAIAMVHLLFLHETGSNNPTGISSDVDKIPFHPYYTIKDILGALLLILALMLLVLFAPDLLGDPDNYTPANPLNTPPHIKPEWYFLFAYAILRSIPNKLGGVLALAFSILILALIPLLHTSKQRSMMFRPLSQCLFWALVADLLTLTWIGGQPVEHPYITIGQLASVLYFLLILVLMPTAGTIENKLLKWSDLELHPPSYPWSHRGLLSSLDHTSIRRGFQVYKQVCSSCHSMDYVAYRHLVGVCYTEDEAKALAEEVEVQDGPNEDGEMFMRPGKLSDYFPKPYPNPEAARAANNGALPPDLSYIVRARHGGEDYVFSLLTGYCEPPTGVSLREGLYFNPYFPGQAIGMAPPIYNEVLEFDDGTPATMSQVAKDVCTFLRWAAEPEHDHRKRMGLKMLLMMGLLLPLVYAMKRHKWSVLKSRKLAYRPPKGRQFGHLTRVRHVITYSLSPFEQRAFPHYFSKGIPNVLRRTRACILRVAPPFVAFYLVYTWGTQEFEKSKRKNPA Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 5.79 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -7.9  Molscript Map 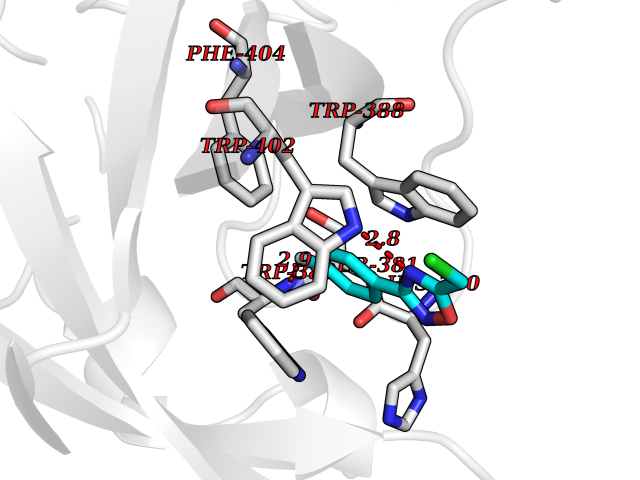 Pymol Map 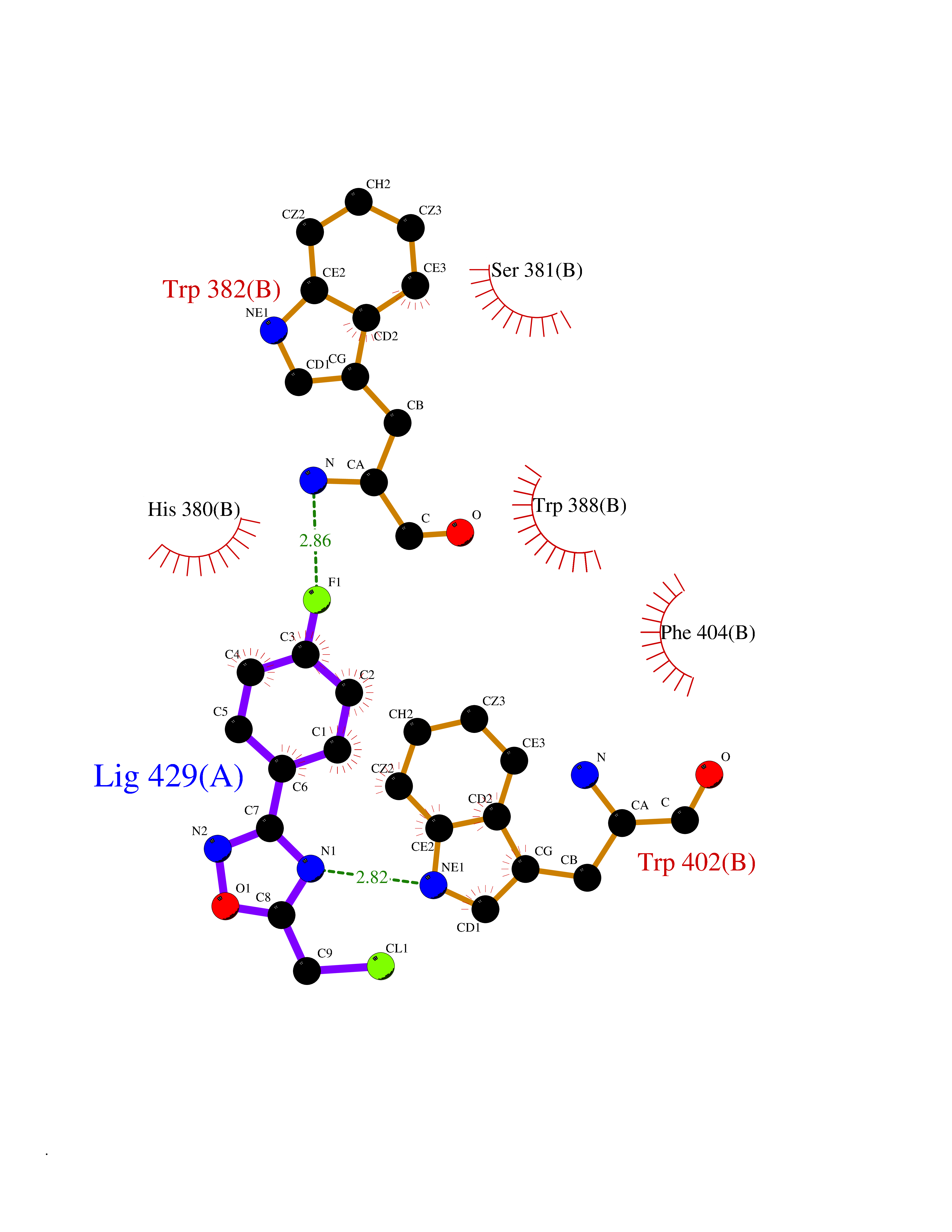 Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 5.79 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -7.9  Molscript Map 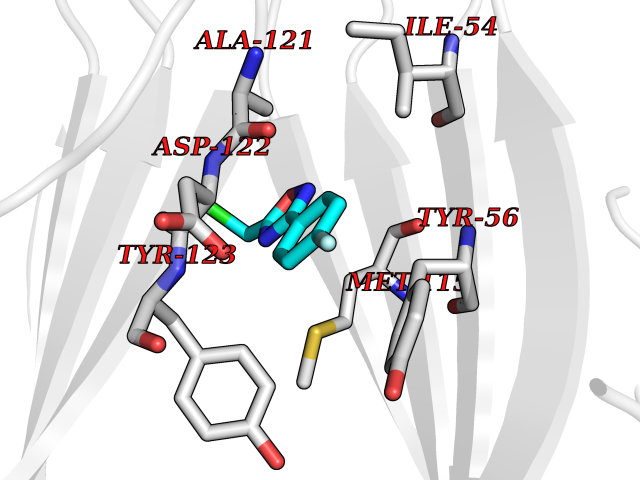 Pymol Map 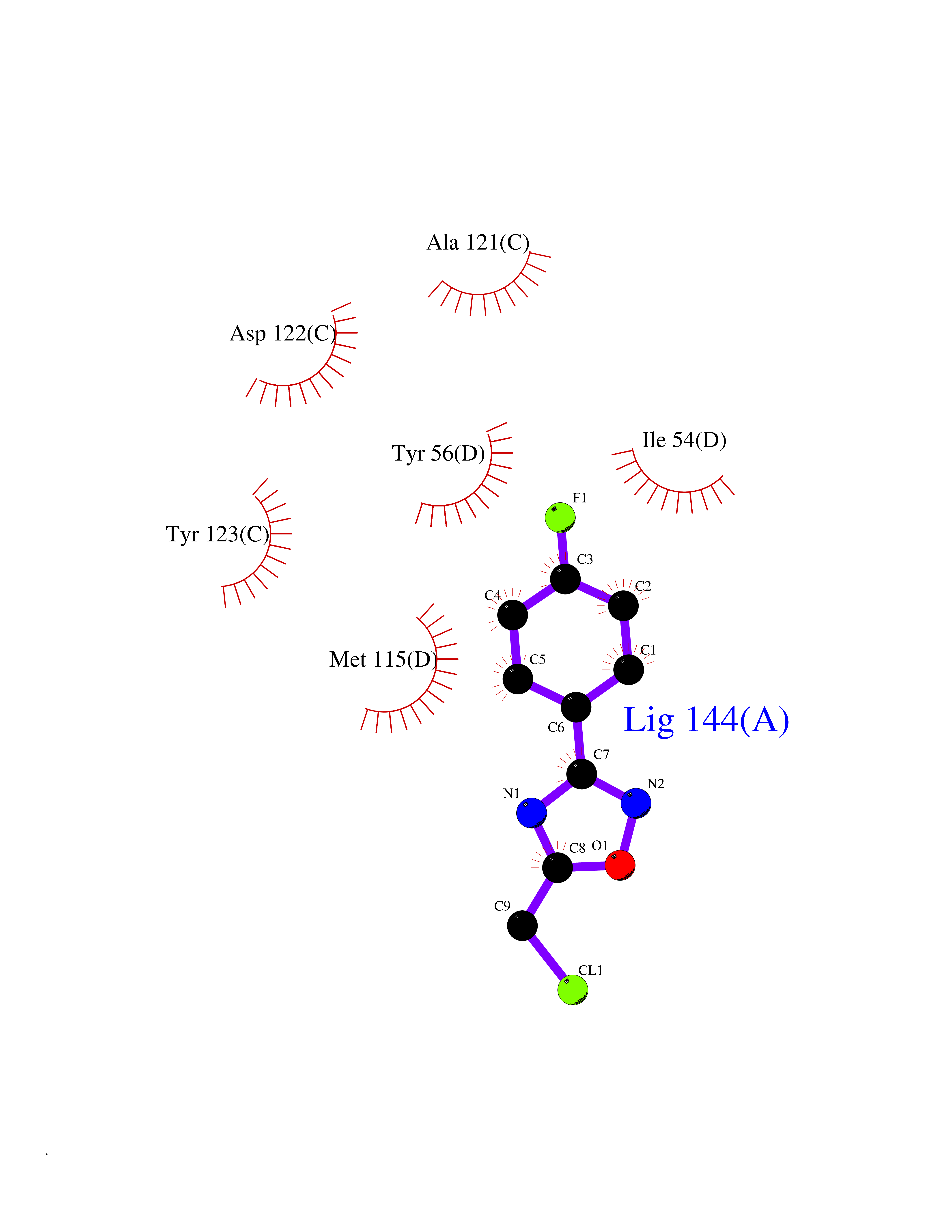 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Macrophage migration inhibitory factor (MIF) | 3IJJ | 5.78 | |
Target general information Gen name MIF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phenylpyruvate tautomerase; MMIF; L-dopachrome tautomerase; L-dopachrome isomerase; Glycosylation-inhibiting factor; GLIF; GIF Protein family MIF family Biochemical class Intramolecular oxidoreductase Function Involved in the innate immune response to bacterial pathogens. The expression of MIF at sites of inflammation suggests a role as mediator in regulating the function of macrophages in host defense. Counteracts the anti-inflammatory activity of glucocorticoids. Has phenylpyruvate tautomerase and dopachrome tautomerase activity (in vitro), but the physiological substrate is not known. It is not clear whether the tautomerase activity has any physiological relevance, and whether it is important for cytokine activity. Pro-inflammatory cytokine. Related diseases Rheumatoid arthritis systemic juvenile (RASJ) [MIM:604302]: An inflammatory articular disorder with systemic onset beginning before the age of 16. It represents a subgroup of juvenile arthritis associated with severe extraarticular features and occasionally fatal complications. During active phases of the disorder, patients display a typical daily spiking fever, an evanescent macular rash, lymphadenopathy, hepatosplenomegaly, serositis, myalgia and arthritis. {ECO:0000269|PubMed:11508429}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01880; DB07888; DB08334; DB08335; DB08333; DB07718; DB08765; DB02728 Interacts with O43521-2; P00533; Q92743; P14174; Q96HA8 EC number EC 5.3.2.1 Uniprot keywords 3D-structure; Acetylation; Cytokine; Cytoplasm; Direct protein sequencing; Immunity; Inflammatory response; Innate immunity; Isomerase; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,C Molecular weight (Da) 24671.9 Length 228 Aromaticity 0.09 Instability index 31.45 Isoelectric point 8.37 Charge (pH=7) 2.26 2D Binding mode Binding energy (Kcal/mol) -7.89  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFAPMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFA Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | p53-binding protein Mdm4 (MDM4) | 6Q9Y | 5.78 | |
Target general information Gen name MDM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Mdmx; Mdm2-like p53-binding protein; Double minute 4 protein Protein family MDM2/MDM4 family Biochemical class MDM2/MDM4 family Function Inhibits p53/TP53- and TP73/p73-mediated cell cycle arrest and apoptosis by binding its transcriptional activation domain. Inhibits degradation of MDM2. Can reverse MDM2-targeted degradation of TP53 while maintaining suppression of TP53 transactivation and apoptotic functions. Related diseases Bone marrow failure syndrome 6 (BMFS6) [MIM:618849]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS6 is an autosomal dominant form characterized by intermittent neutropenia, lymphopenia, or anemia associated with hypocellular bone marrow, and increased susceptibility to cancer. {ECO:0000269|PubMed:32300648}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NX04; P10415; Q7Z479; O95971; P48729; Q00987; Q13064; P41227; P06400; Q9Y4L5; P23297; P29034; P33763; P04271; P31947; P04637; P62837; Q93009; O14972; P61964; P62258; P61981; P63104; Q9BRR0; A0A0S2Z6X0; Q3YBA8; P03255-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19722 Length 173 Aromaticity 0.08 Instability index 50.78 Isoelectric point 8.48 Charge (pH=7) 2.27 2D Binding mode Binding energy (Kcal/mol) -7.89  Molscript Map 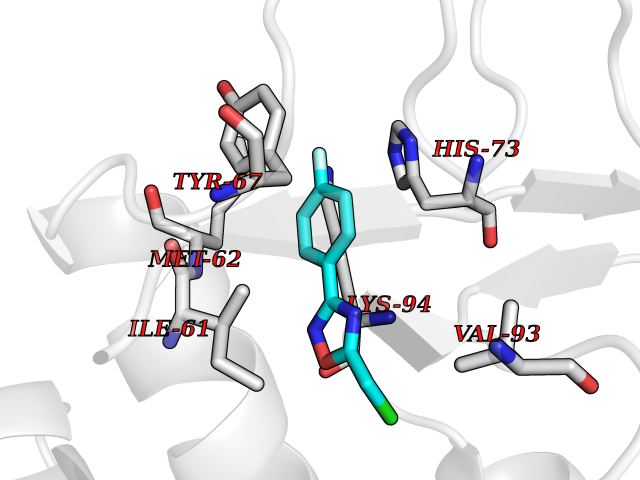 Pymol Map 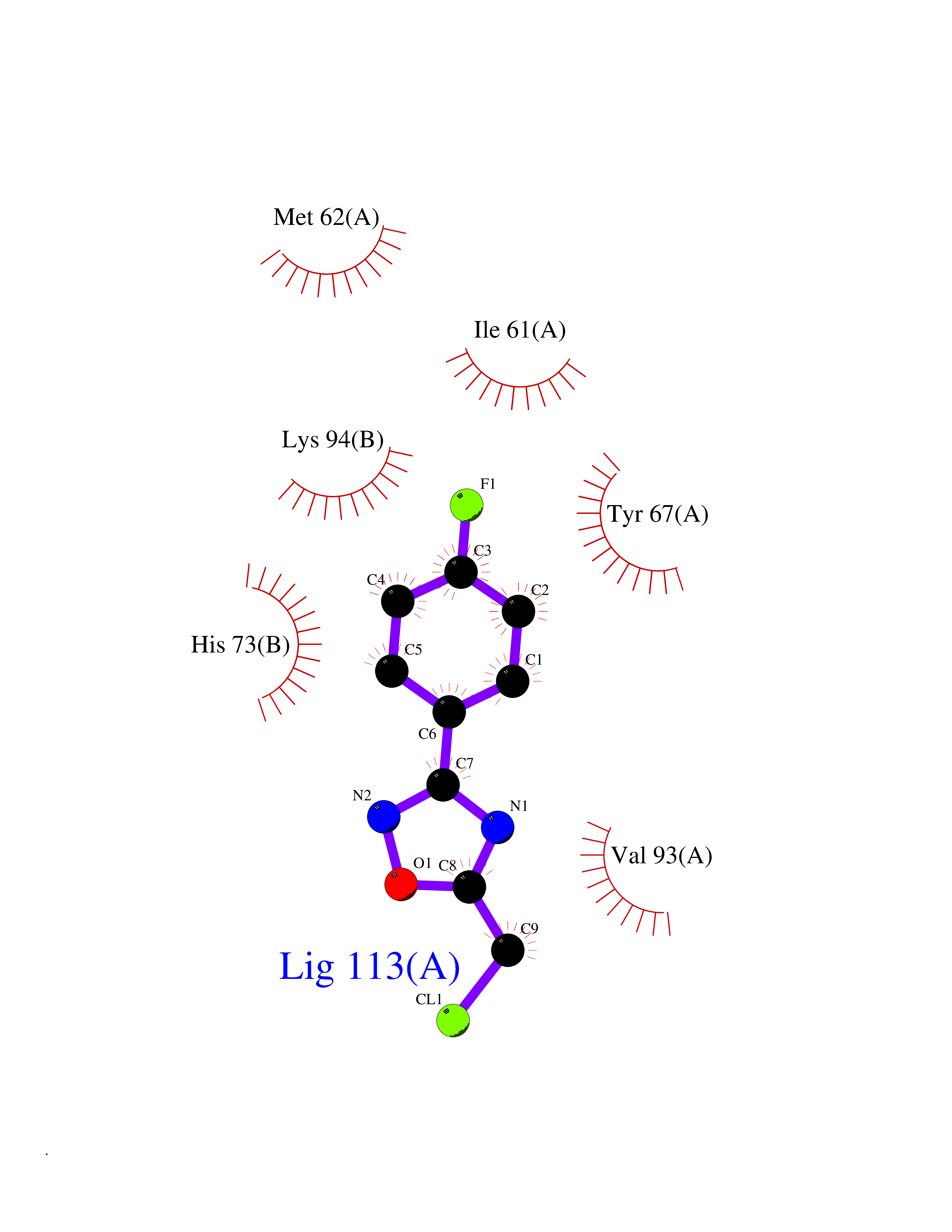 Ligplot Map 3D Binding mode Sequence QVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLAQINQVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLA Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Histamine H1 receptor (H1R) | 7DFL | 5.78 | |
Target general information Gen name HRH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HH1R; H1R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function In peripheral tissues, the H1 subclass of histamine receptors mediates the contraction of smooth muscles, increase in capillary permeability due to contraction of terminal venules, and catecholamine release from adrenal medulla, as well as mediating neurotransmission in the central nervous system. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01615; DB09488; DB06766; DB01246; DB00321; DB00543; DB08799; DB01238; DB14185; DB06216; DB00637; DB00719; DB00972; DB00245; DB00767; DB04890; DB06698; DB11591; DB09128; DB01237; DB00835; DB00354; DB09016; DB00748; DB06016; DB00341; DB08936; DB08800; DB01114; DB00477; DB01239; DB00568; DB00215; DB00283; DB04837; DB00363; DB01176; DB00434; DB01151; DB00967; DB00405; DB09555; DB00985; DB08801; DB01075; DB01146; DB09167; DB01142; DB00366; DB01084; DB05492; DB00751; DB01175; DB06678; DB00950; DB04841; DB00502; DB05381; DB05079; DB00557; DB04946; DB00458; DB08802; DB00920; DB00555; DB01106; DB06282; DB00455; DB09195; DB00408; DB00934; DB00737; DB06691; DB01071; DB00902; DB01403; DB06148; DB00370; DB00540; DB05080; DB06229; DB00334; DB00768; DB01173; DB01267; DB00715; DB08922; DB01619; DB01620; DB06153; DB00433; DB00420; DB01069; DB00777; DB01224; DB00912; DB00734; DB11614; DB05345; DB00342; DB04905; DB11235; DB00797; DB00656; DB00726; DB00792; DB00427; DB09185; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32298.5 Length 275 Aromaticity 0.15 Instability index 36.59 Isoelectric point 9.54 Charge (pH=7) 16.01 2D Binding mode Binding energy (Kcal/mol) -7.88 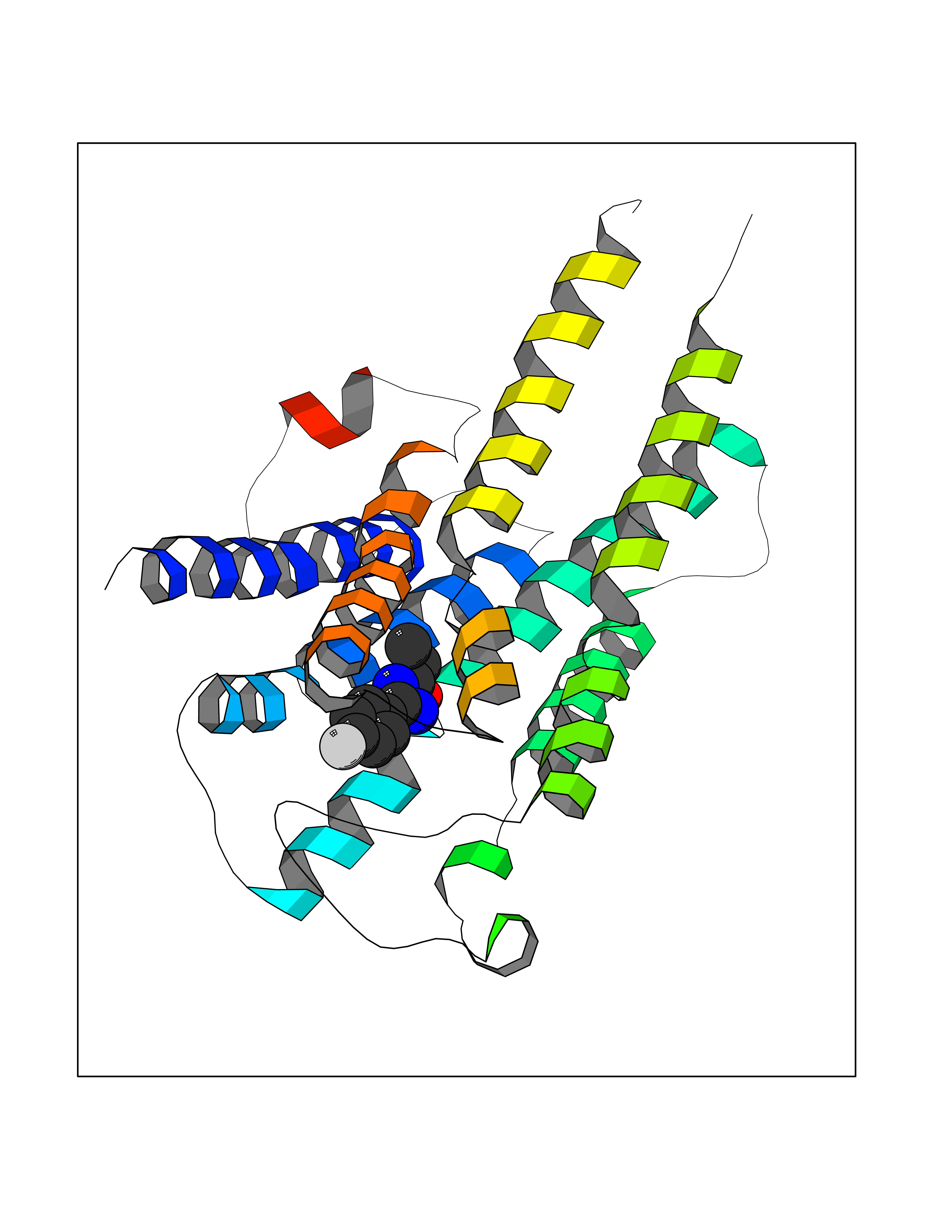 Molscript Map 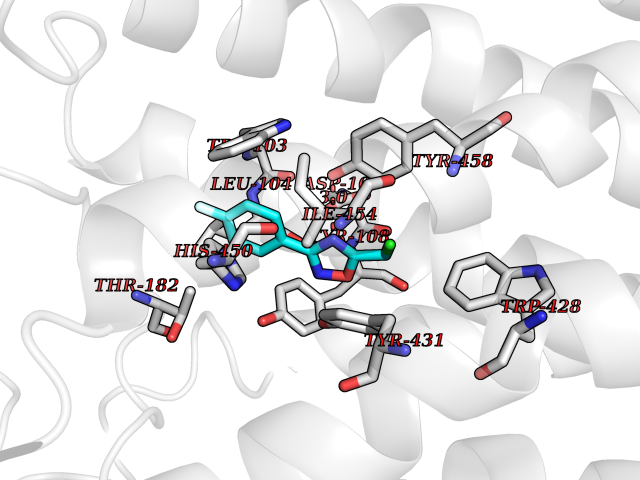 Pymol Map 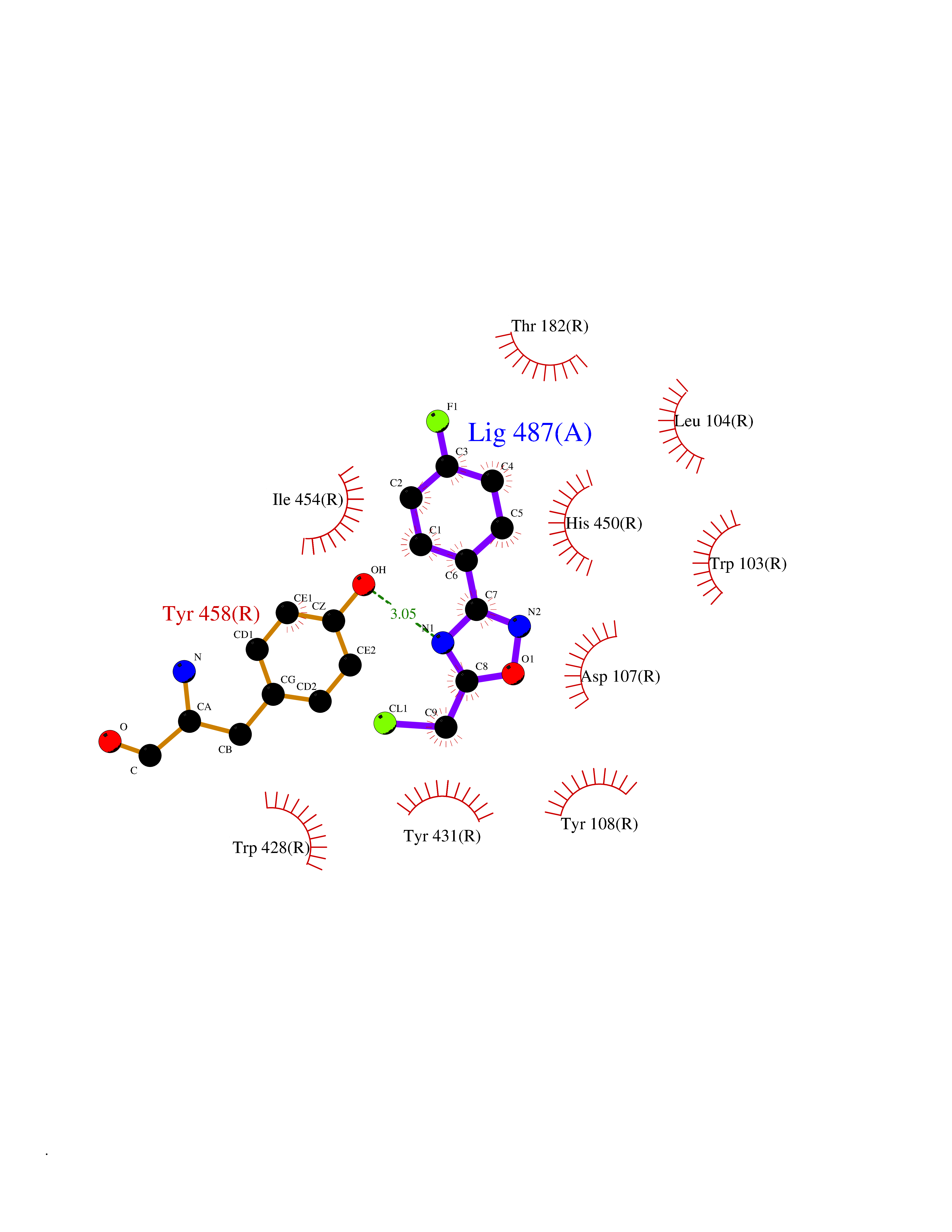 Ligplot Map 3D Binding mode Sequence MPLVVVLSTICLVTVGLNLLVLYAVRSERKLHTVGNLYIVSLSVADLIVGAVVMPMNILYLLMSKWSLGRPLCLFWLSMDYVASTASIFSVFILCIDRYRSVQQPLRYLKYRTKTRASATILGAWFLSFLWVIPILGWNHFMQQTSVRREDKCETDFYDVTWFKVMTAIINFYLPTLLMLWFYAKIYKAVRQHCLHMNRERKAAKQLGFIMAAFILCWIPYFIFFMVIAFCKNCCNEHLHMFTIWLGYINSTLNPLIYPLCNENFKKTFKRILHI Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Retinoic acid receptor alpha (RARA) | 3KMR | 5.77 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -7.87 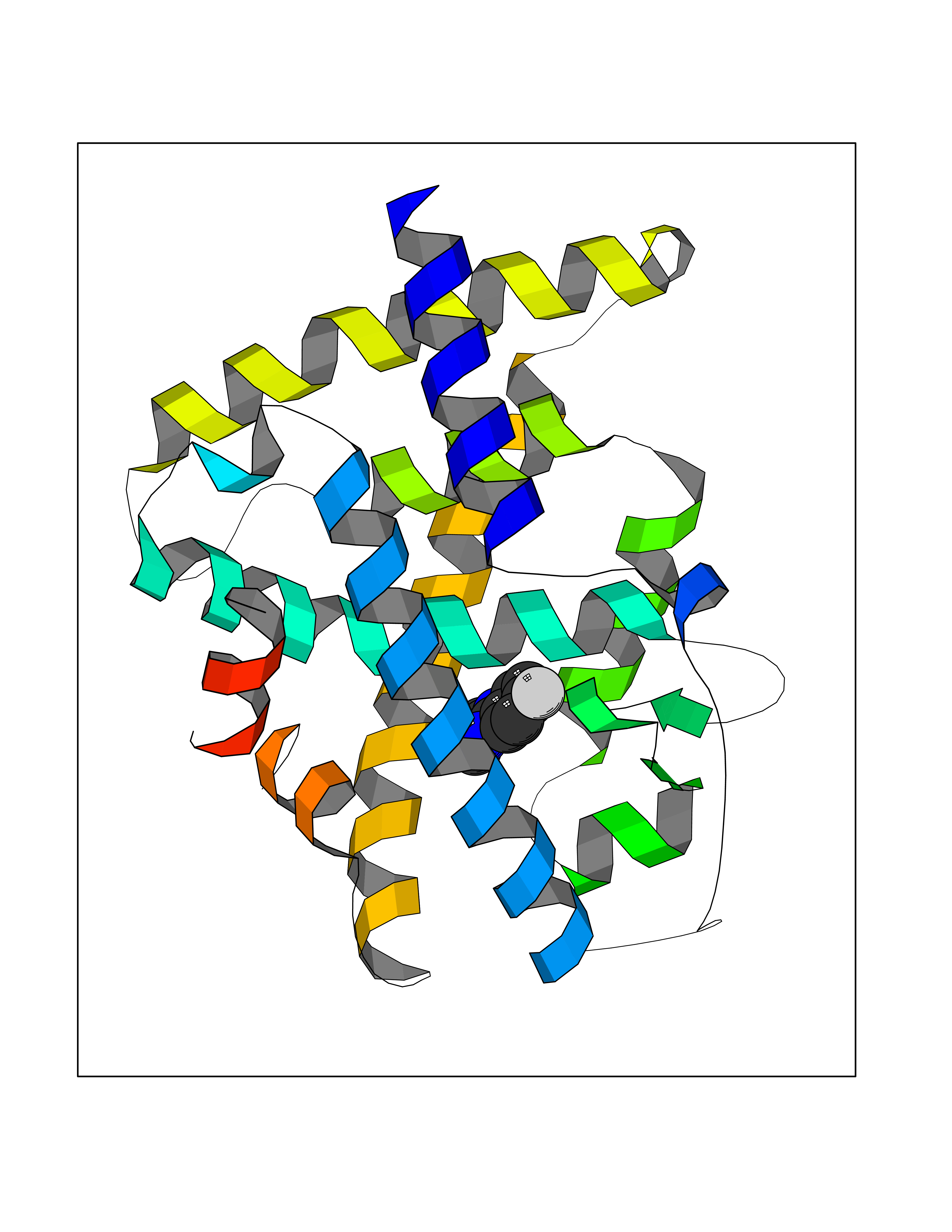 Molscript Map 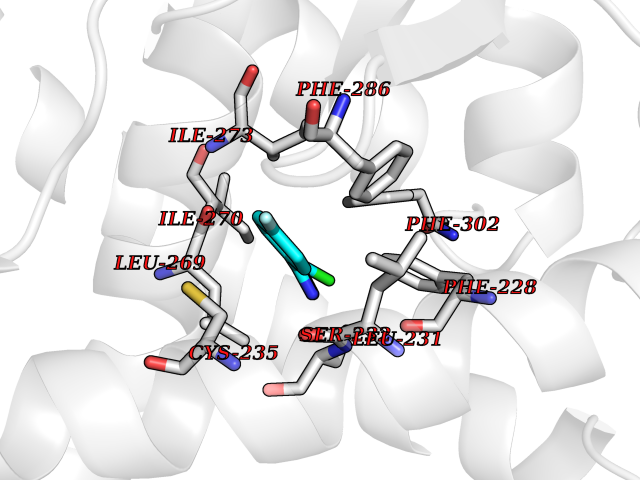 Pymol Map 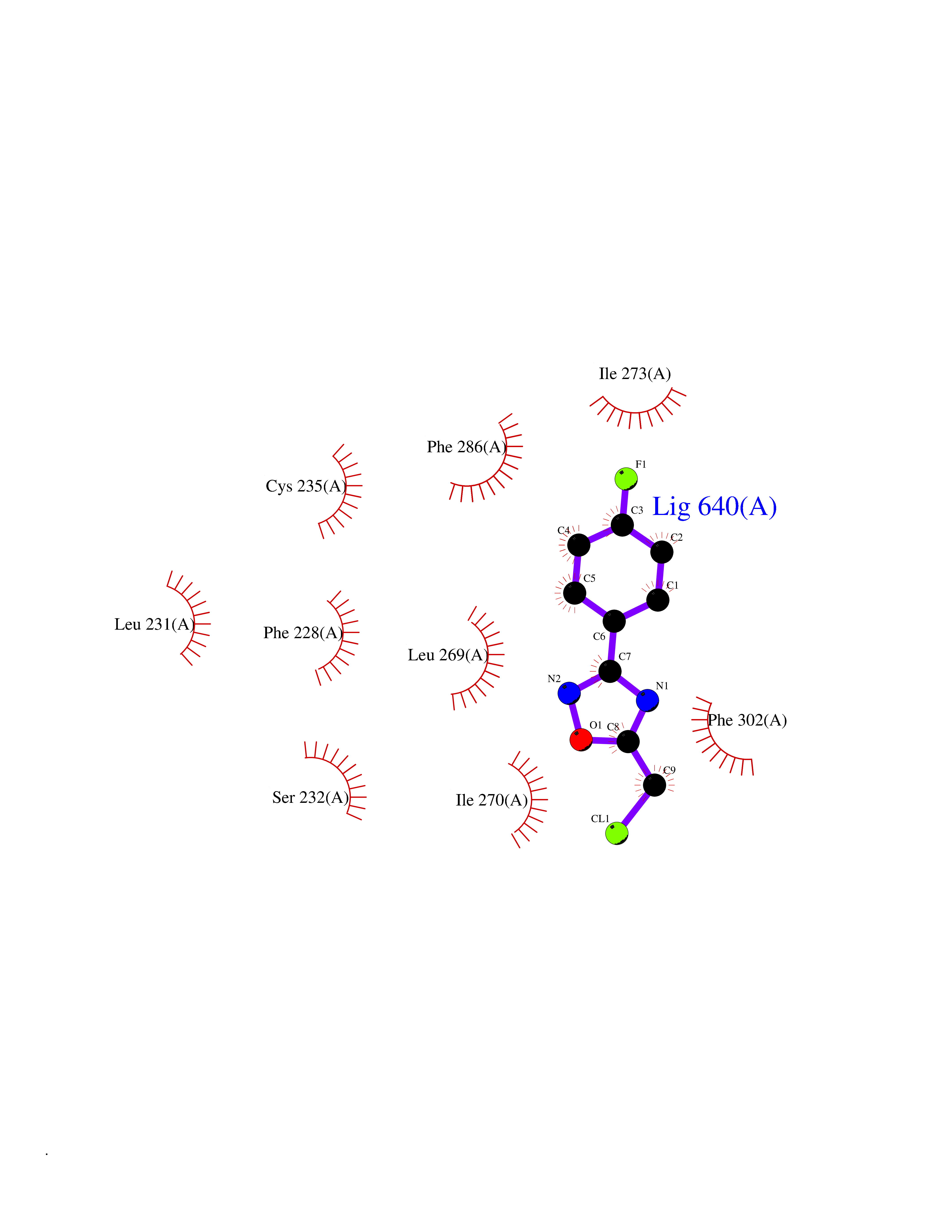 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Dopamine beta-hydroxylase | 4ZEL | 5.77 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.88 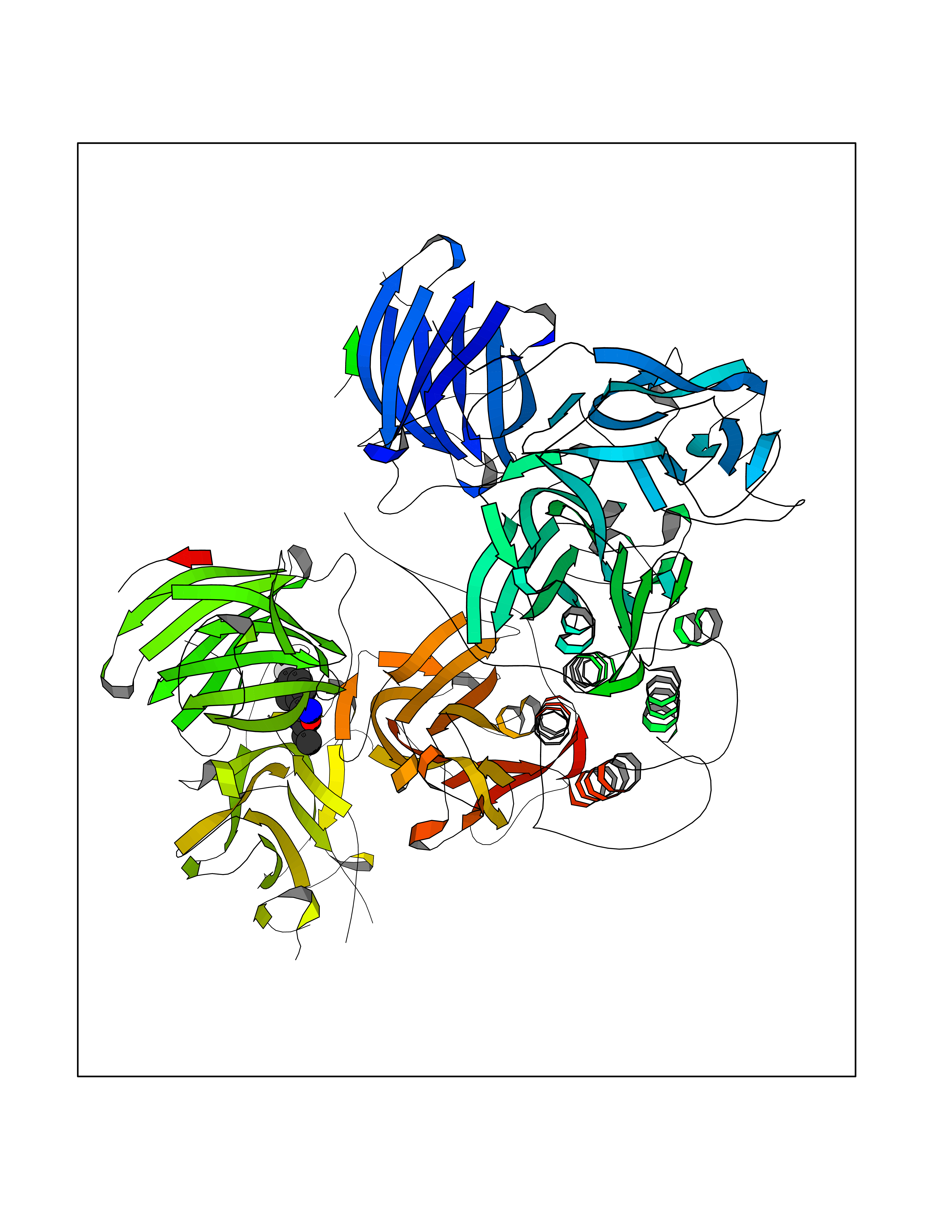 Molscript Map 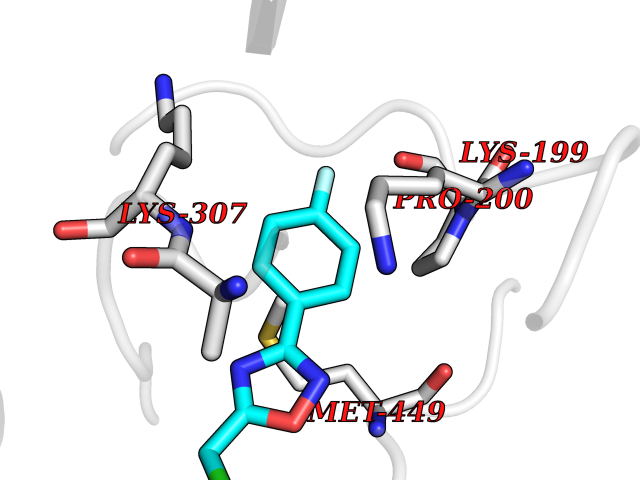 Pymol Map 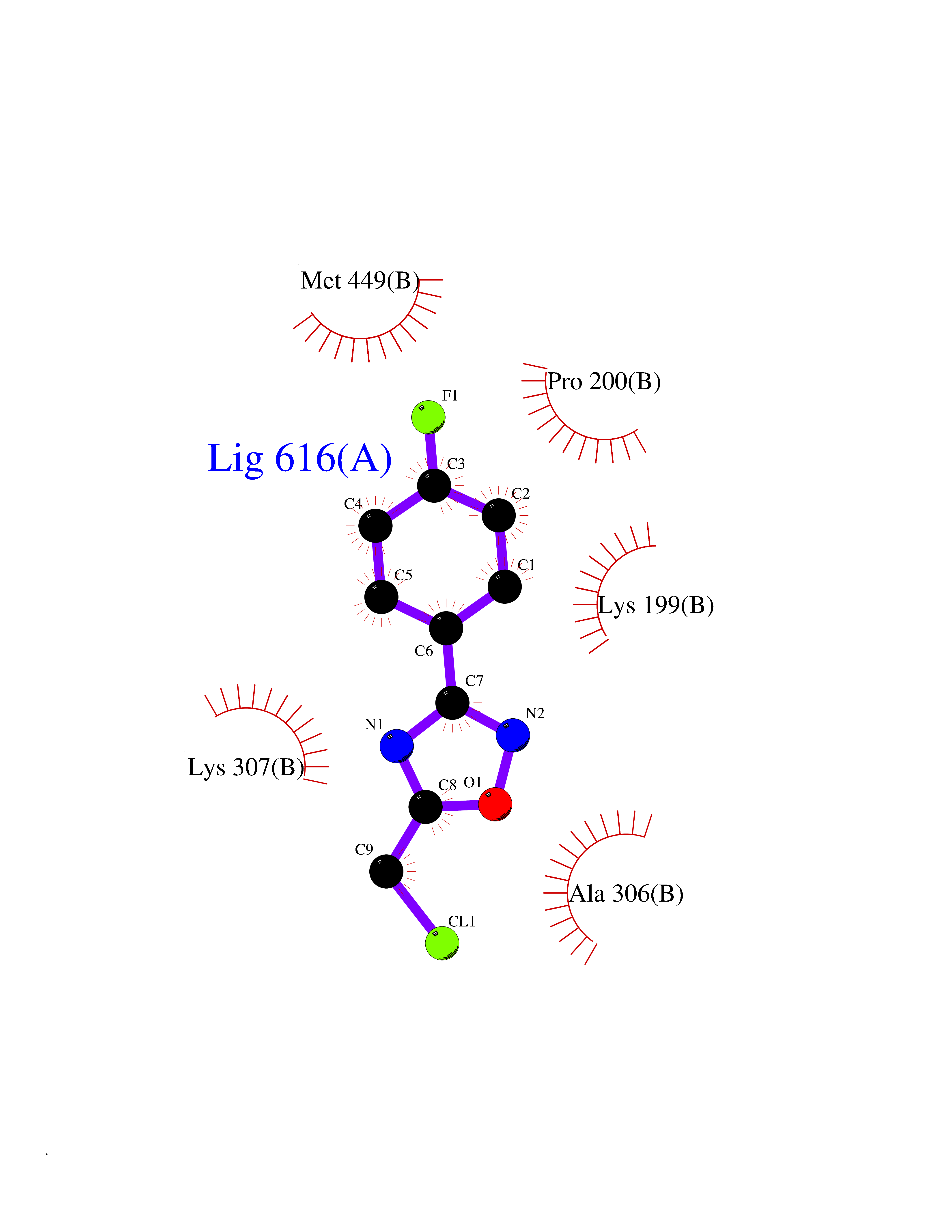 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | L-serine dehydratase/L-threonine deaminase | 1P5J | 5.77 | |
Target general information Gen name SDS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms SDH Protein family Serine/threonine dehydratase family Biochemical class Lyase Function L-serine ammonia-lyase activity.L-threonine ammonia-lyase activity.Protein homodimerization activity.Pyridoxal phosphate binding. Related diseases Immunodeficiency, common variable, 12, with autoimmunity (CVID12) [MIM:616576]: A primary immunodeficiency characterized by hypogammaglobulinemia and recurrent bacterial infections. About half of patients develop autoimmune features, including cytopenia, as well as generalized inflammation and lymphoproliferation manifest as lymphadenopathy or hepatosplenomegaly. {ECO:0000269|PubMed:26279205}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00114; DB00133 Interacts with Q8WTU0; O14964; Q96PF1 EC number 4.3.1.17; 4.3.1.19 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Gluconeogenesis; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33559.7 Length 319 Aromaticity 0.06 Instability index 34.65 Isoelectric point 7.2 Charge (pH=7) 0.42 2D Binding mode Binding energy (Kcal/mol) -7.87 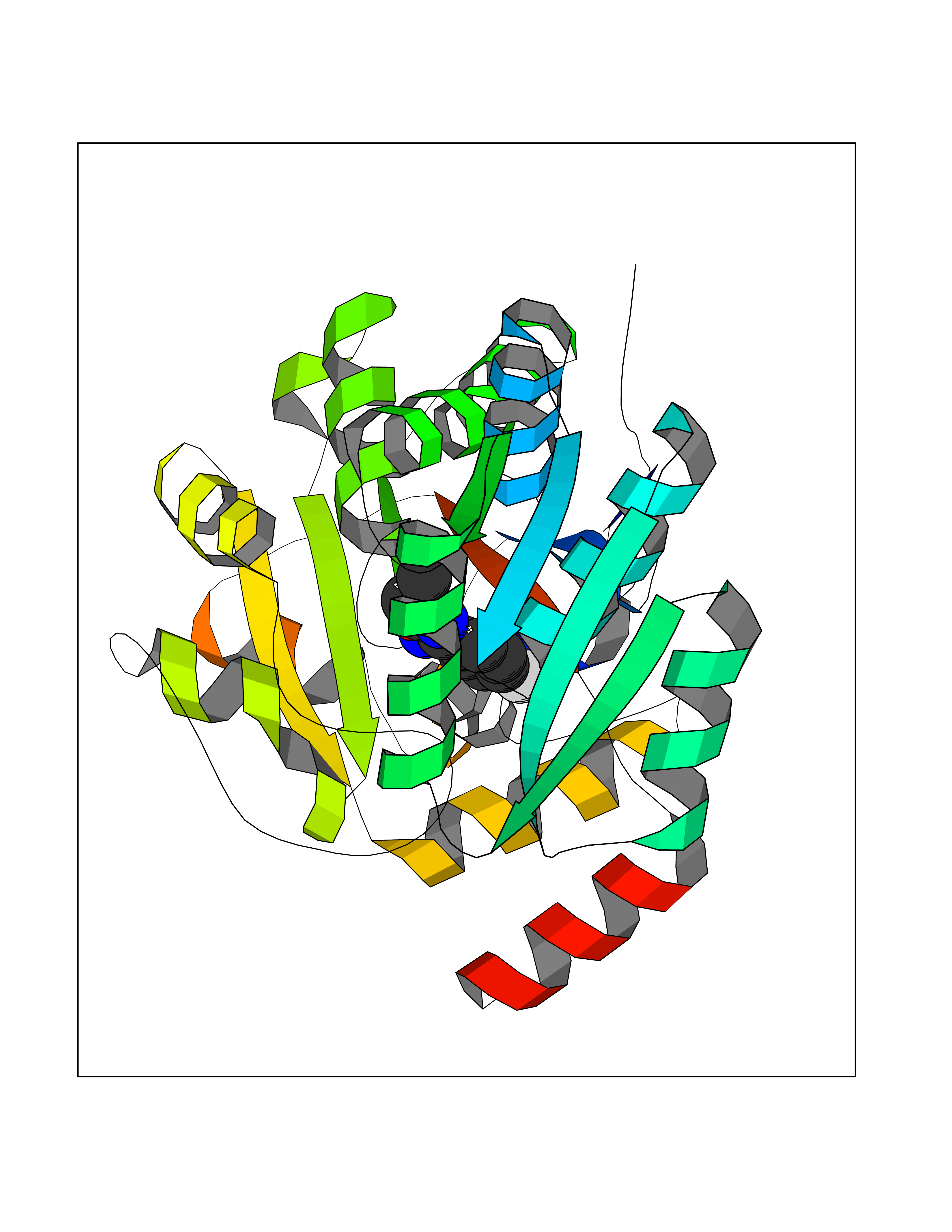 Molscript Map 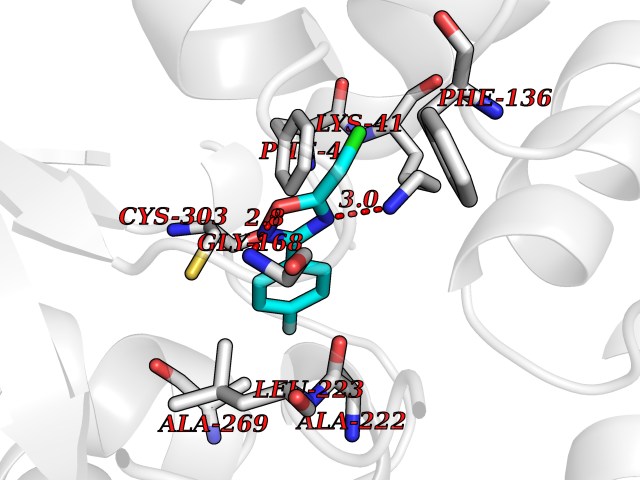 Pymol Map 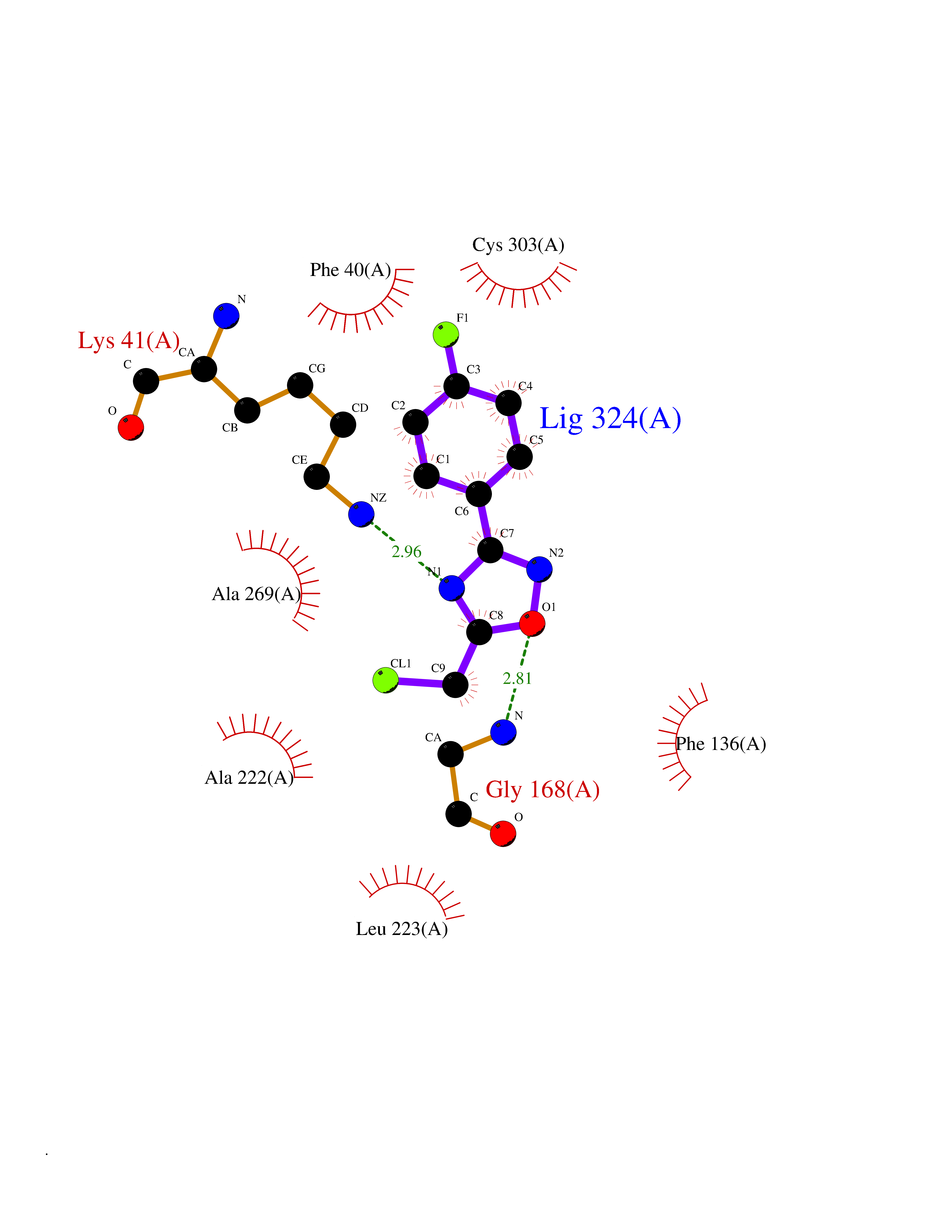 Ligplot Map 3D Binding mode Sequence GEPLHVKTPIRDSMALSKMAGTSVYLKMDSAQPSGSFKIRGIGHFCKRWAKQGCAHFVCSSAGNAGMAAAYAARQLGVPATIVVPGTTPALTIERLKNEGATCKVVGELLDEAFELAKALAKNNPGWVYIPPFDDPLIWEGHASIVKELKETLWEKPGAIALSVGGGGLLCGVVQGLQECGWGDVPVIAMETFGAHSFHAATTAGKLVSLPKITSVAKALGVKTVGSQALKLFQEHPIFSEVISDQEAVAAIEKFVDDEKILVEPACGAALAAVYSHVIQKLQLEGNLRTPLPSLVVIVCGGSNISLAQLRALKEQLGM Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Plasmepsin-2 | 2BJU | 5.77 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase A1 family Biochemical class Hydrolase Function Aspartic-type endopeptidase activity. Related diseases Short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) [MIM:610006]: Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features. {ECO:0000269|PubMed:10832746, ECO:0000269|PubMed:11013134, ECO:0000269|PubMed:16317551}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04378; DB04373; DB11638; DB01218; DB02505; DB03063 Interacts with NA EC number 3.4.23.39 Uniprot keywords 3D-structure; Aspartyl protease; Direct protein sequencing; Disulfide bond; Hydrolase; Membrane; Protease; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Vacuole; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 36923.5 Length 329 Aromaticity 0.13 Instability index 44.31 Isoelectric point 4.67 Charge (pH=7) -17.94 2D Binding mode Binding energy (Kcal/mol) -7.87  Molscript Map 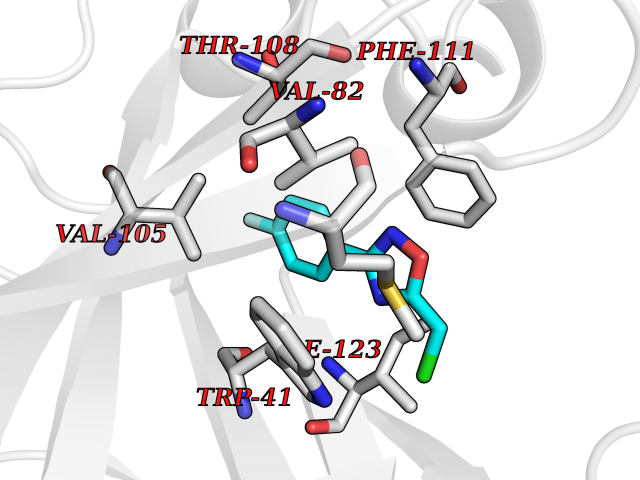 Pymol Map 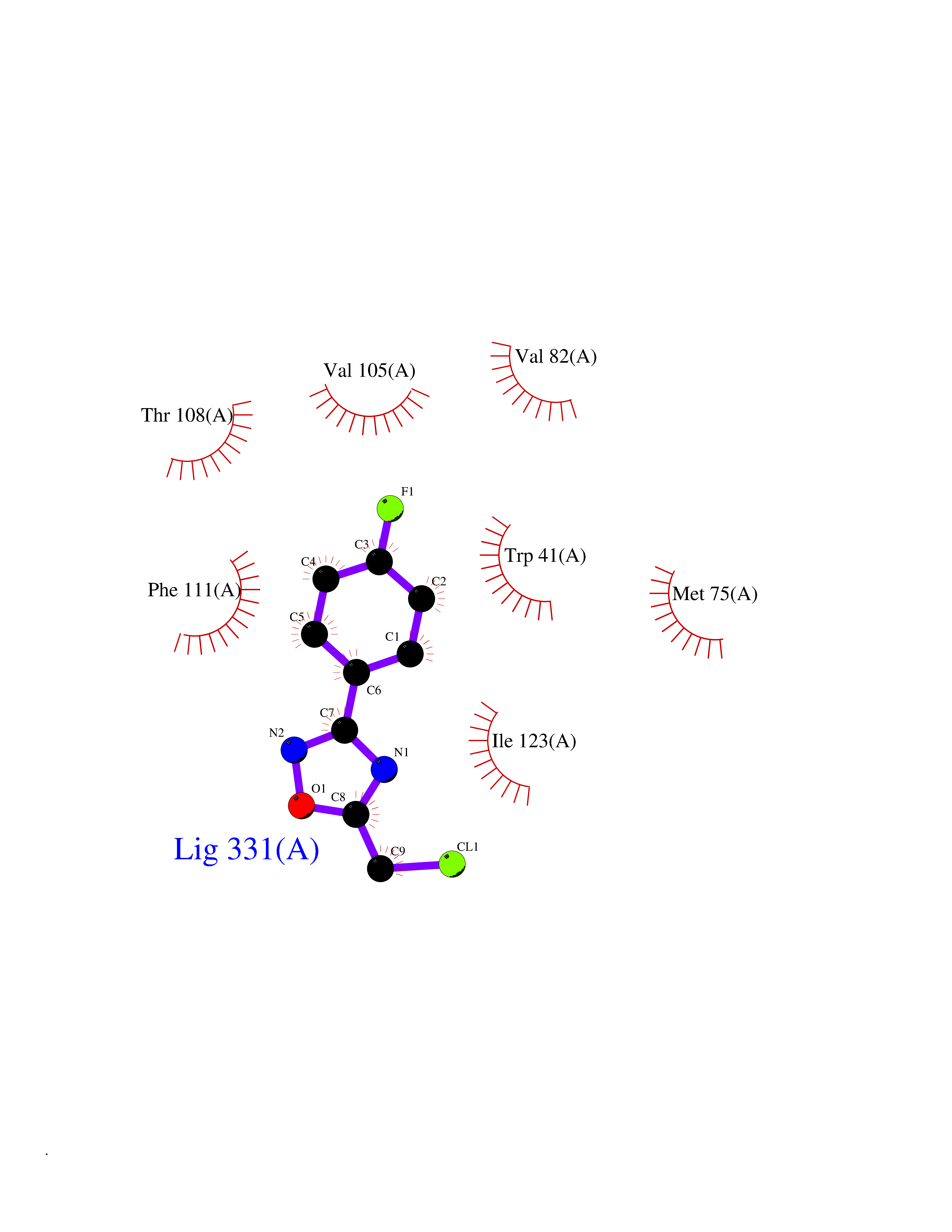 Ligplot Map 3D Binding mode Sequence SSNDNIELVDFQNIMFYGDAEVGDNQQPFTFILDTGSANLWVPSVKCTTAGCLTKHLYDSSKSRTYEKDGTKVEMNYVSGTVSGFFSKDLVTVGNLSLPYKFIEVIDTNGFEPTYTASTFDGILGLGWKDLSIGSVDPIVVELKNQNKIENALFTFYLPVHDKHTGFLTIGGIEERFYEGPLTYEKLNHDLYWQITLDAHVGNIMLEKANCIVDSGTSAITVPTDFLNKMLQNLDVIKVPFLPFYVTLCNNSKLPTFEFTSENGKYTLEPEYYLQHIEDVGPGLCMLNIIGLDFPVPTFILGDPFMRKYFTVFDYDNHSVGIALAKKNL Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Thiopurine S-methyltransferase | 2BZG | 5.77 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -7.87 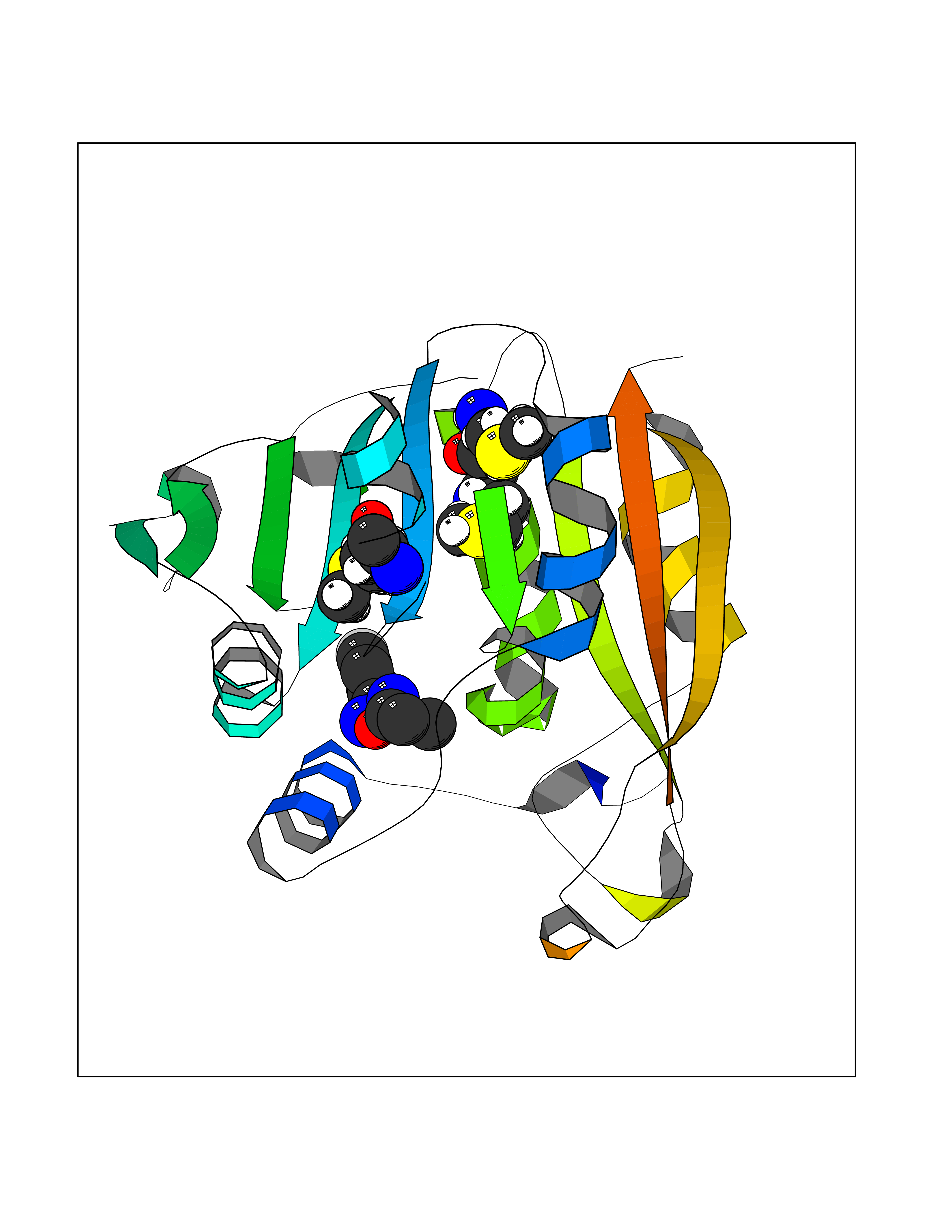 Molscript Map 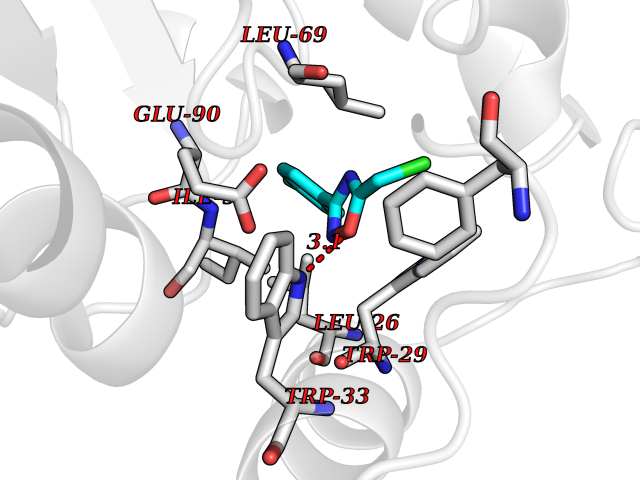 Pymol Map 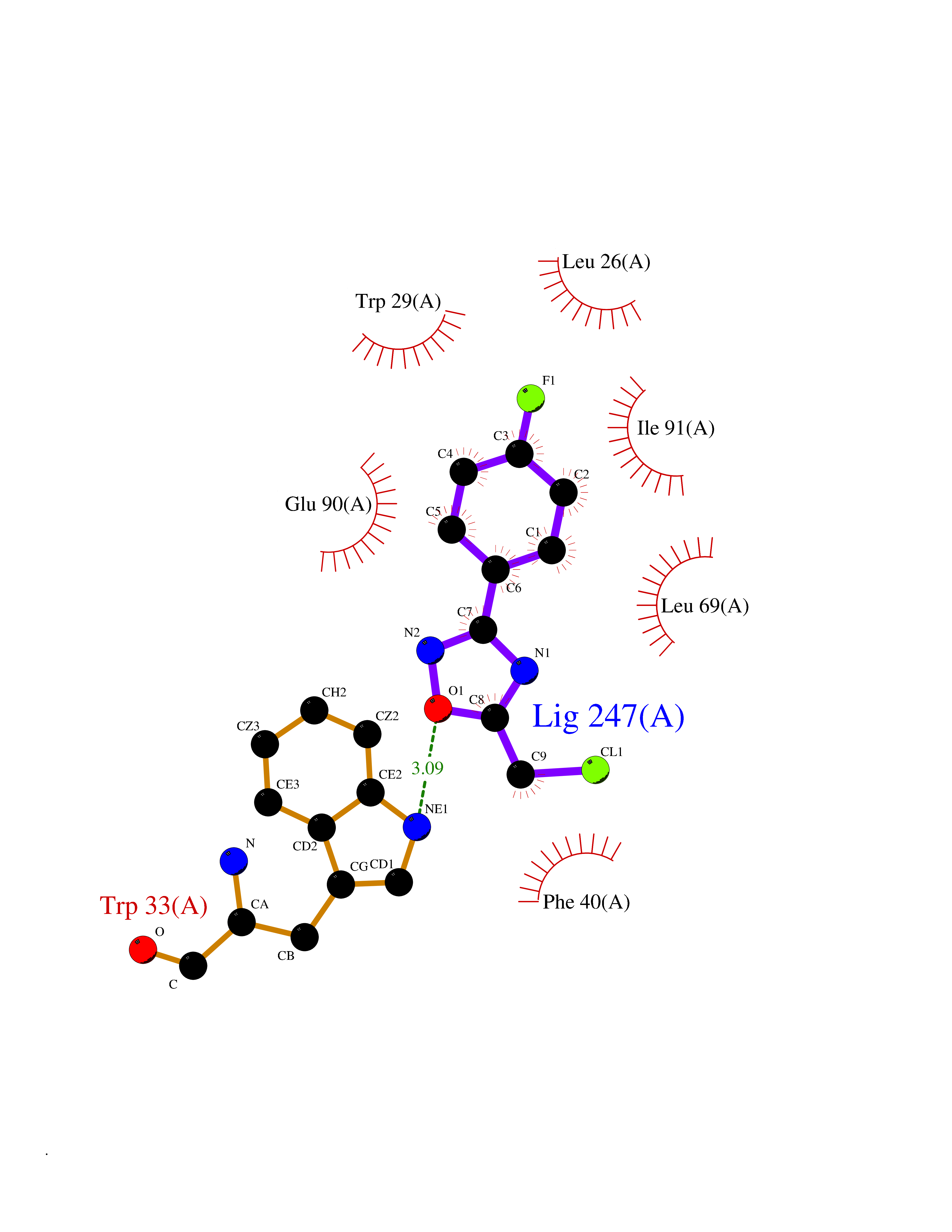 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 5.77 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -7.87 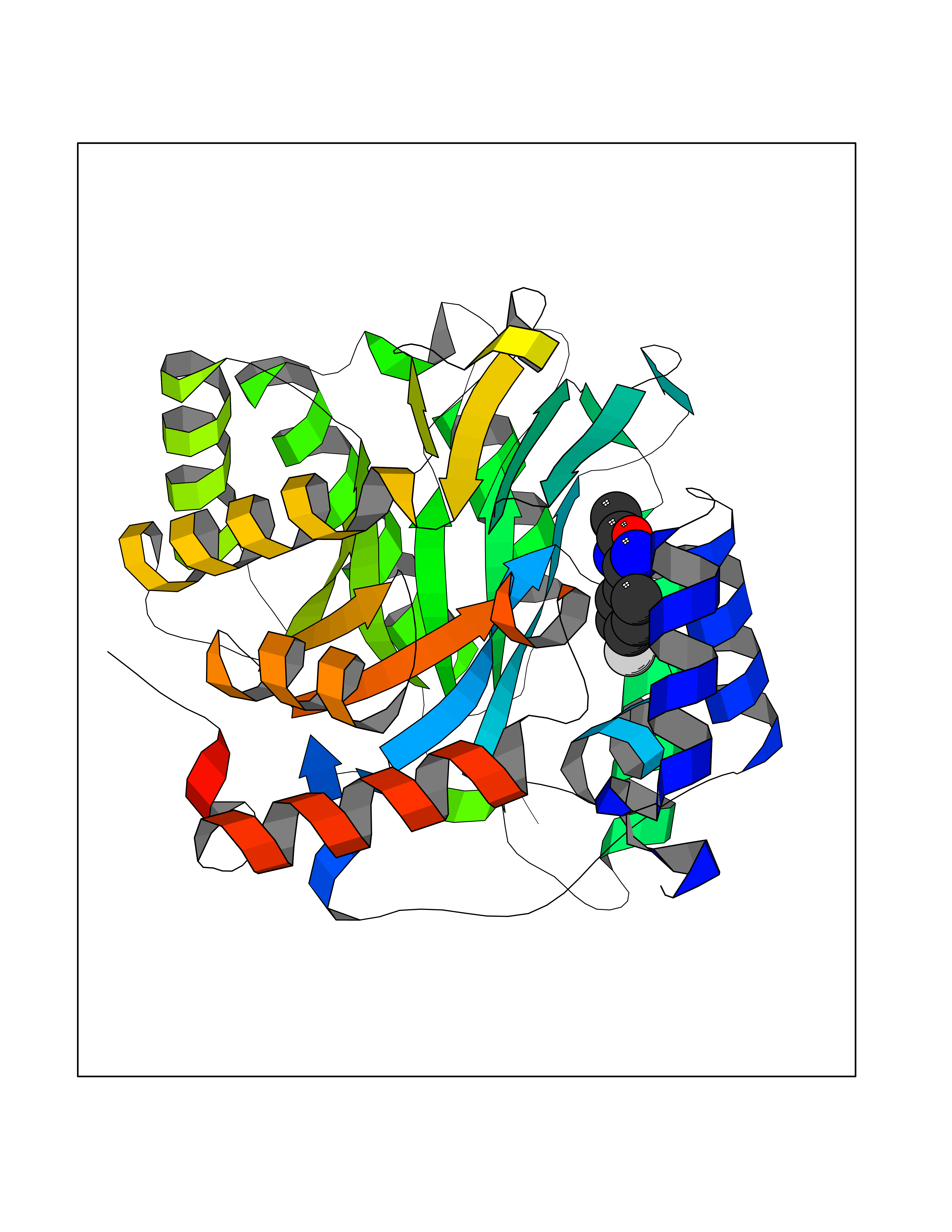 Molscript Map 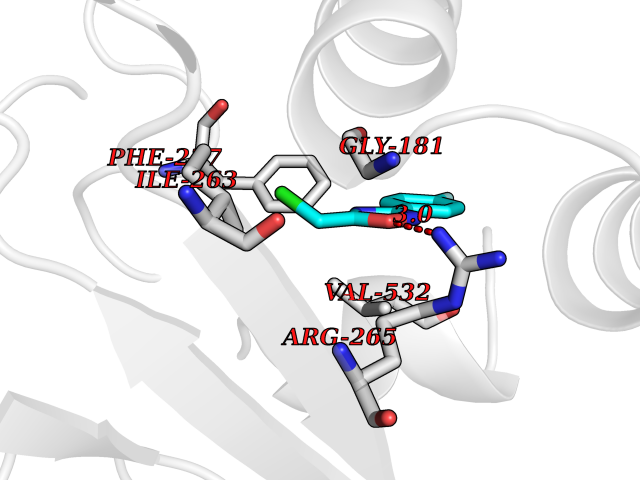 Pymol Map 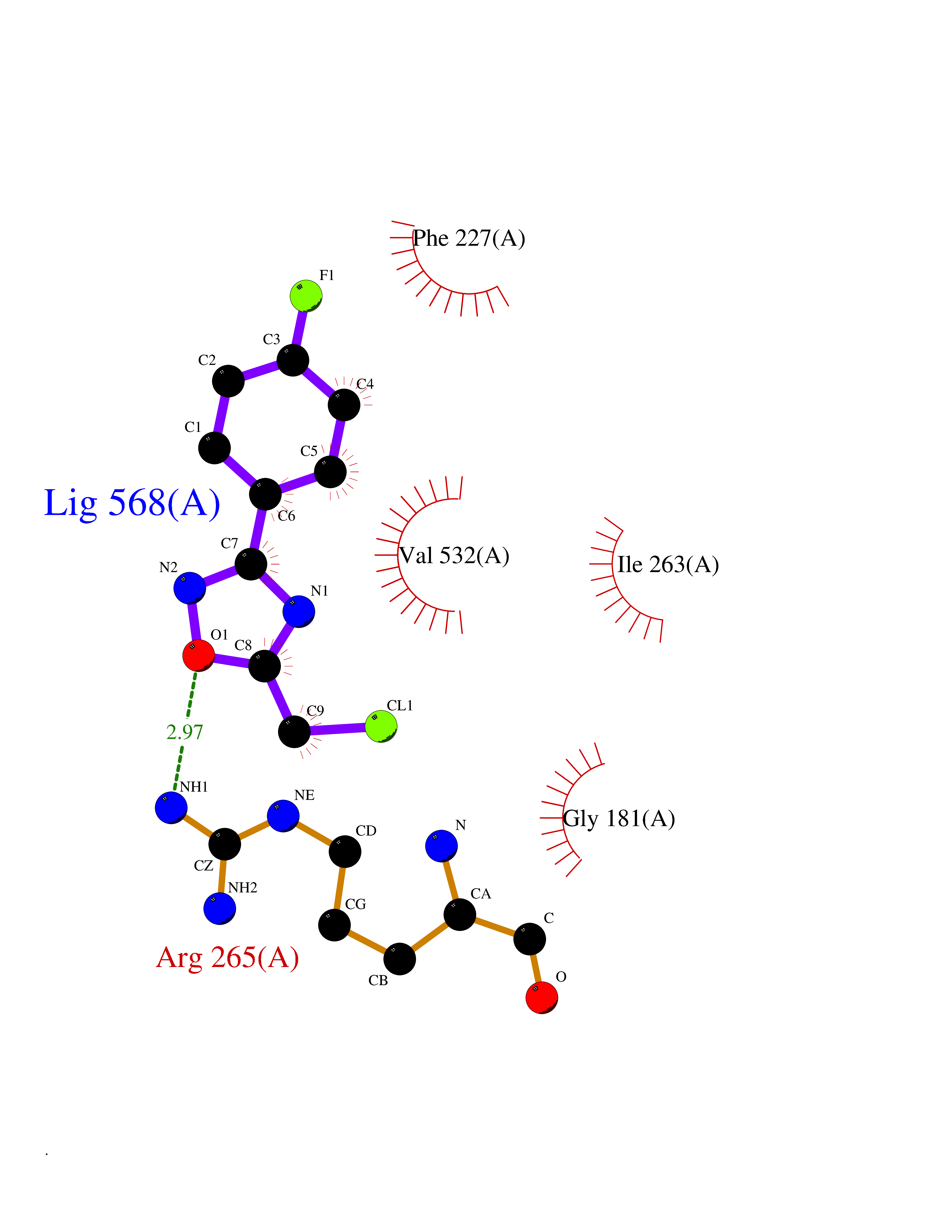 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Plasmodium DOXP reductoisomerase (Malaria DXR) | 3AU9 | 5.77 | |
Target general information Gen name Malaria DXR Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID Synonyms IspC; DXR; DXP reductoisomerase; DOXP reductoisomerase; 2-C-Methyl-d-erythritol 4-phosphate synthase; 1-deoxyxylulose-5-phosphate reductoisomerase Protein family DXR family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADP-dependent rearrangement and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol 4-phosphate (MEP). Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.1.267 Uniprot keywords 3D-structure; Apicoplast; Isoprene biosynthesis; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Plastid; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46644.4 Length 410 Aromaticity 0.09 Instability index 36.77 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -7.86 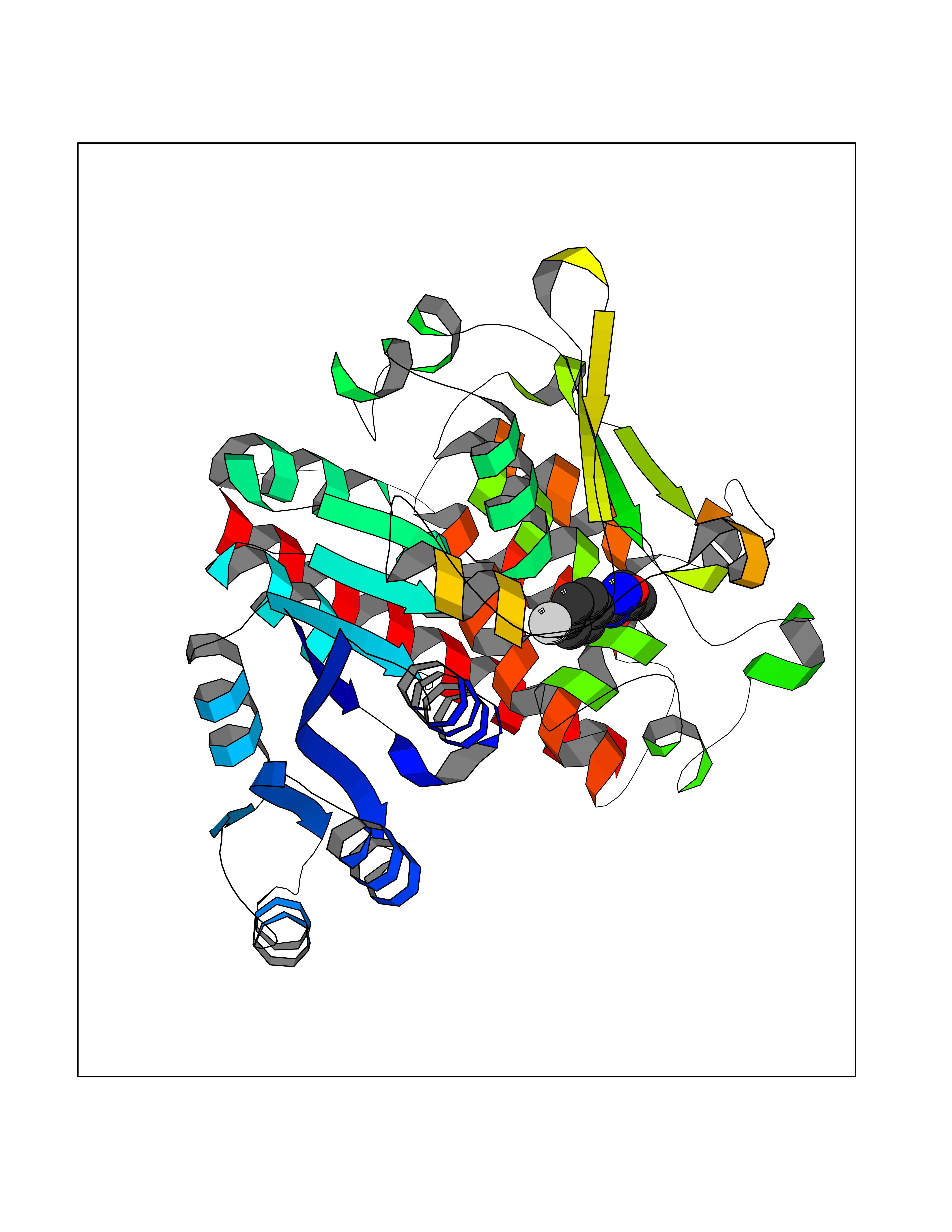 Molscript Map 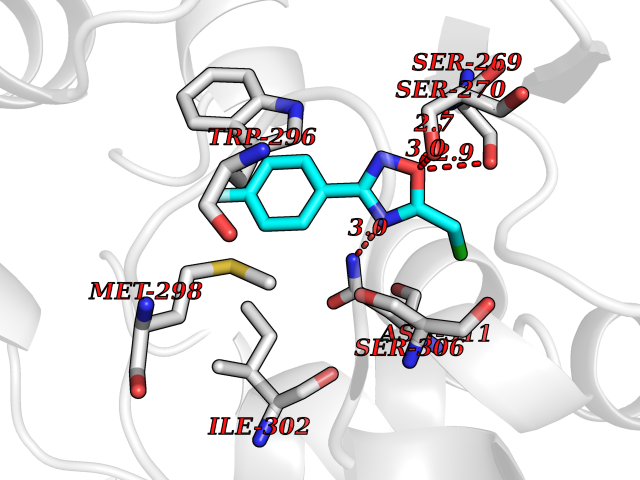 Pymol Map 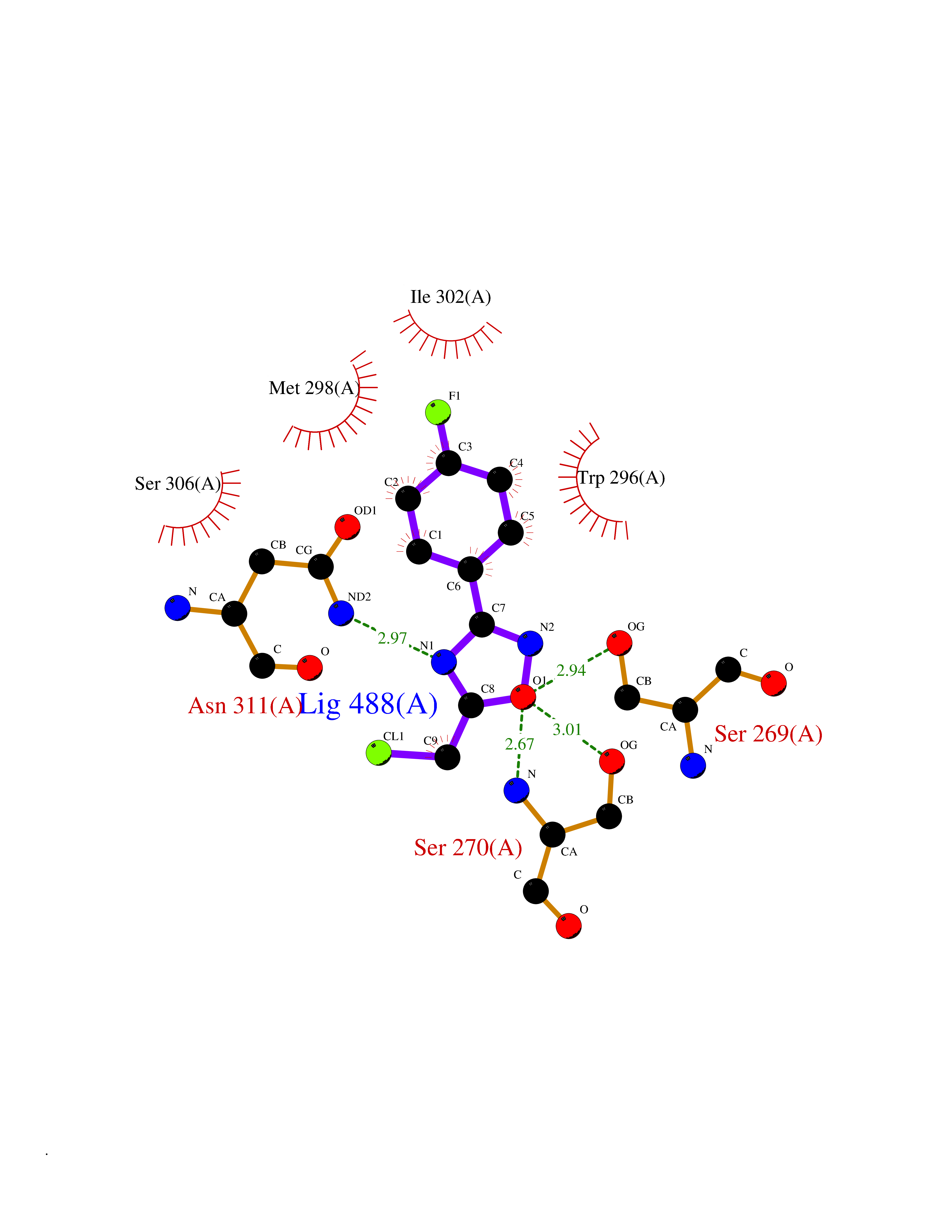 Ligplot Map 3D Binding mode Sequence PINVAIFGSTGSIGTNALNIIRECNKIENVFNVKALYVNKSVNELYEQAREFLPEYLCIHDKSVYEELKELVKNIKDYKPIILCGDEGMKEICSSNSIDKIVIGIDSFQGLYSTMYAIMNNKIVALANKESIVSAGFFLKKLLNIHKNAKIIPVDSEHSAIFQCLDNNKVLKTKCLQDNFSKINNINKIFLCSSGGPFQNLTMDELKNVTSENALKHPKWKMGKKITIDSATMMNKGLEVIETHFLFDVDYNDIEVIVHKECIIHSCVEFIDKSVISQMYYPDMQIPILYSLTWPDRIKTNLKPLDLAQVSTLTFHKPSLEHFPCIKLAYQAGIKGNFYPTVLNASNEIANNLFLNNKIKYFDISSIISQVLESFNSQKVSENSEDLMKQILQIHSWAKDKATDIYNKHN Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Wnt-7a protein (WNT7A) | 4UZQ | 5.77 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -7.87 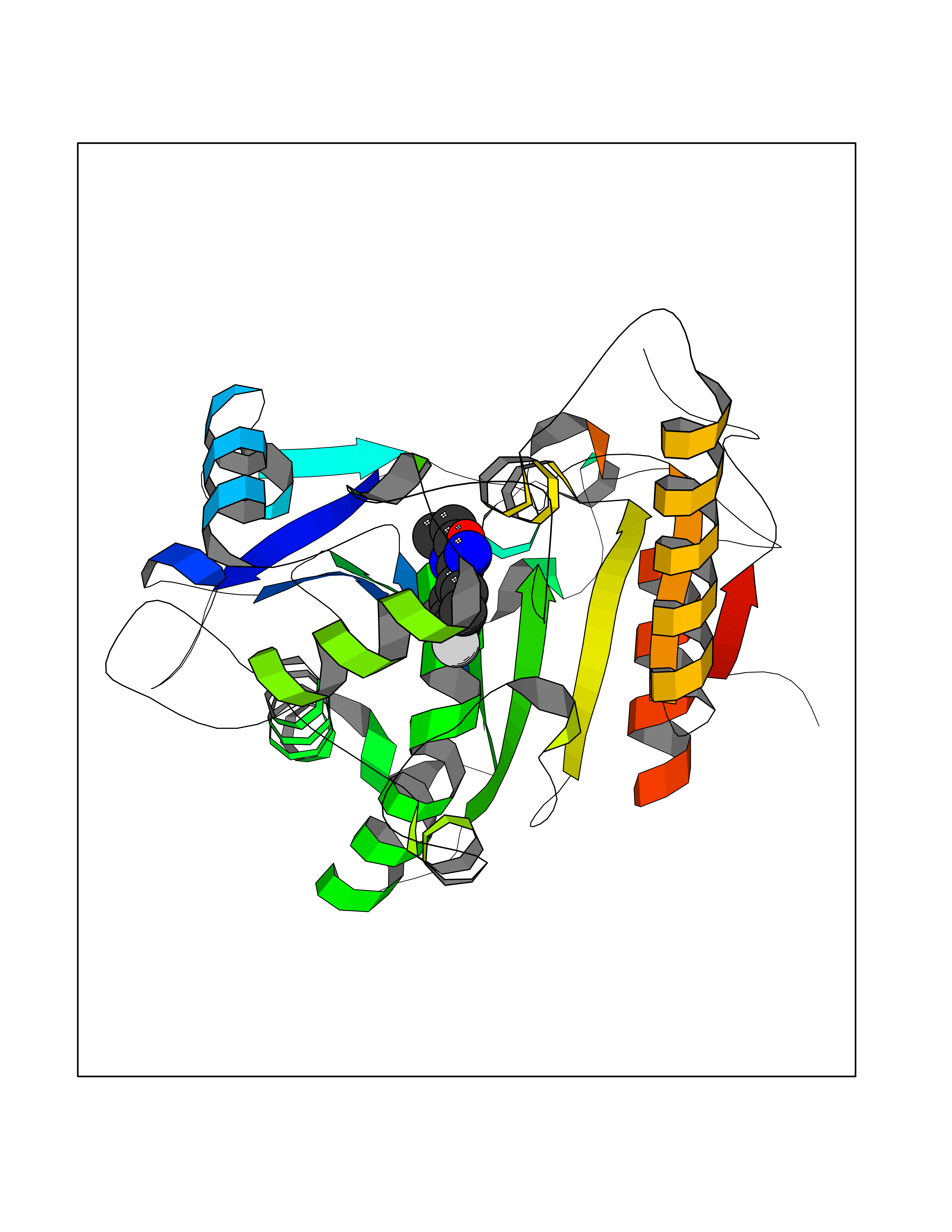 Molscript Map 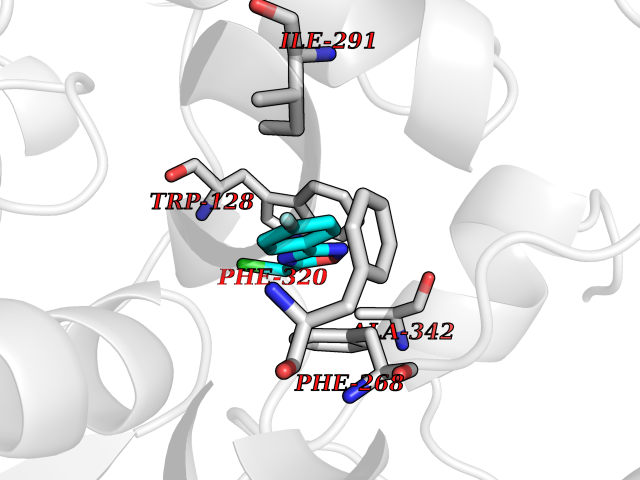 Pymol Map 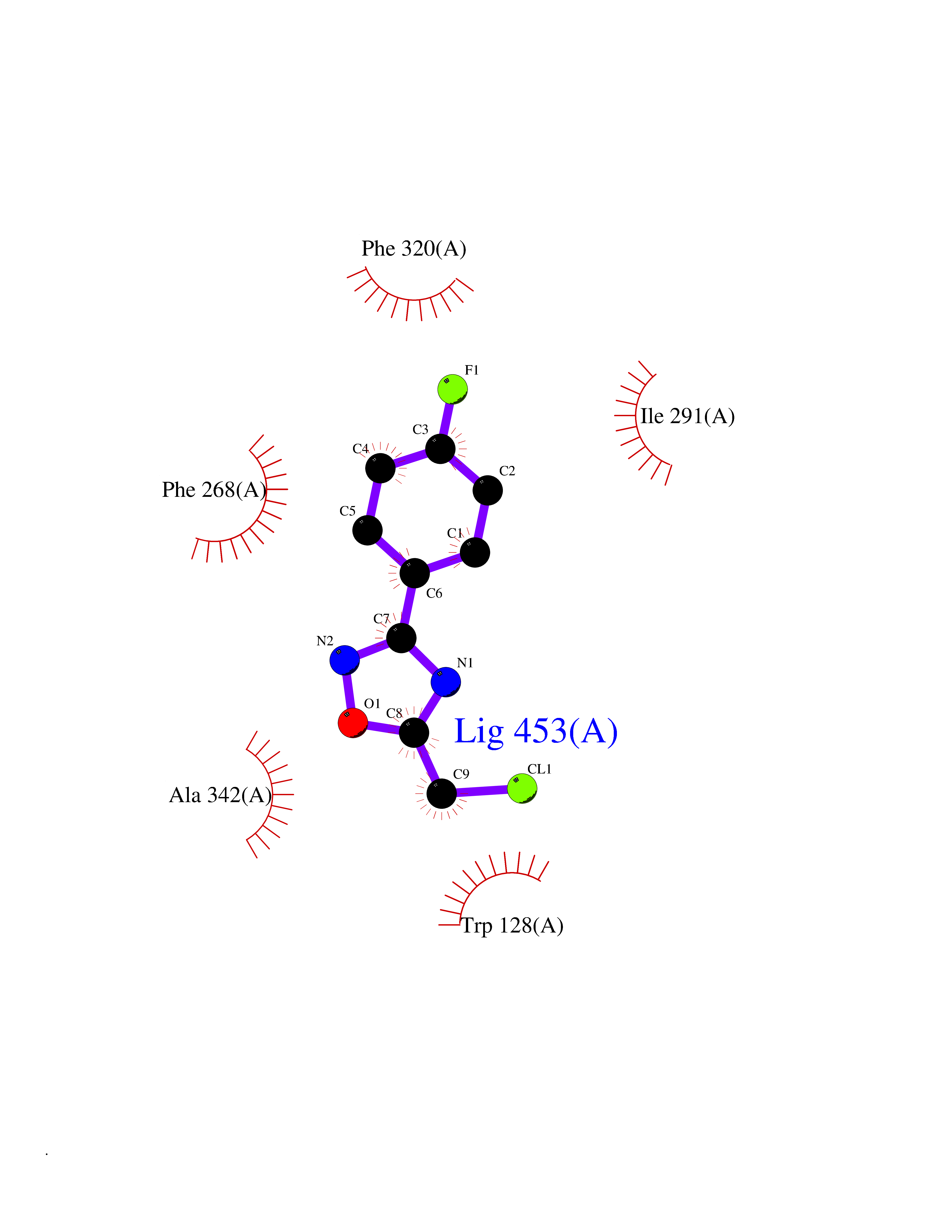 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Aldose reductase (AKR1B1) | 1US0 | 5.76 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -7.86  Molscript Map 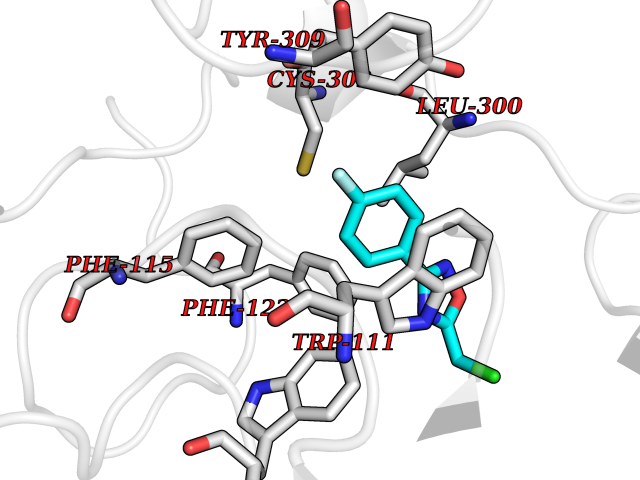 Pymol Map 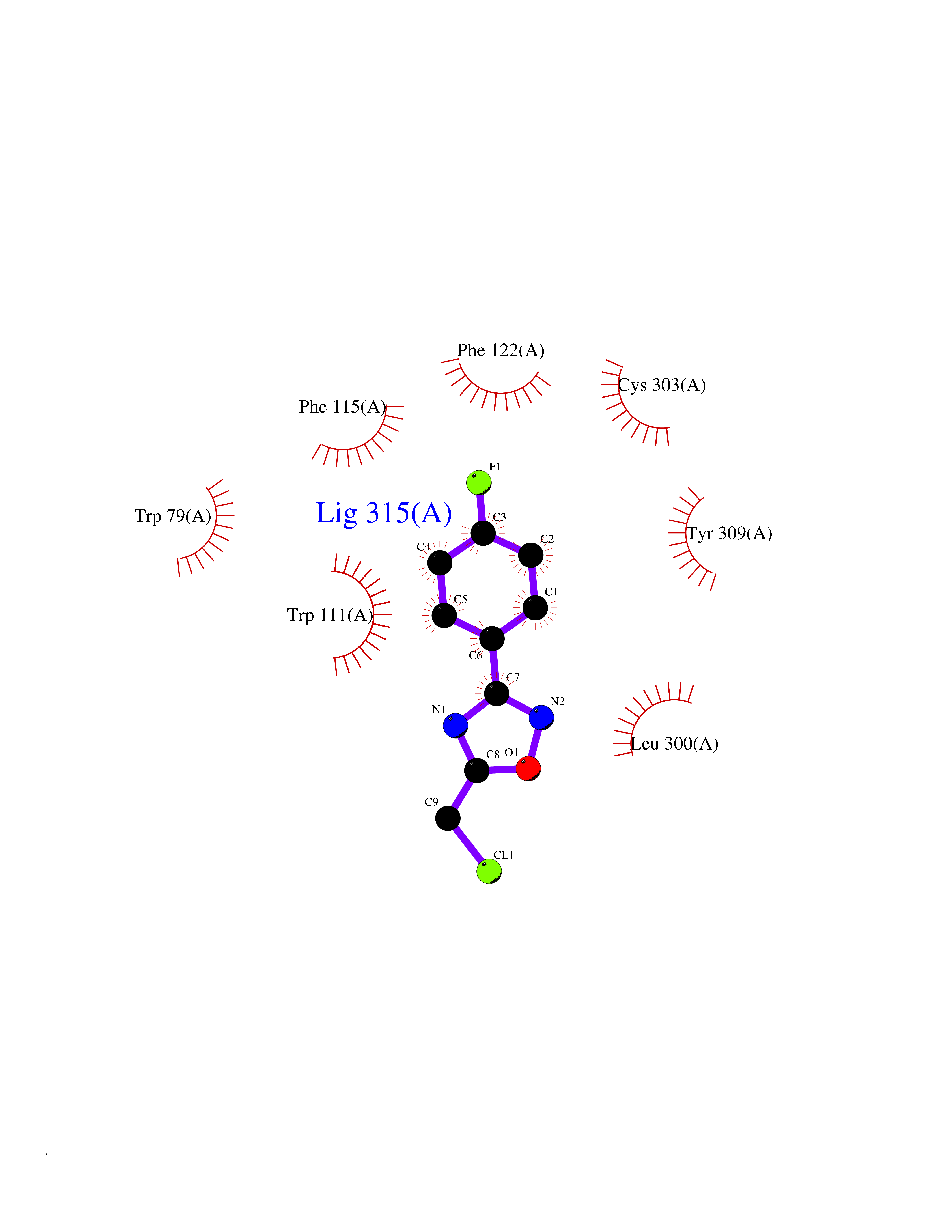 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Histamine H3 receptor (H3R) | 7F61 | 5.76 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -7.85 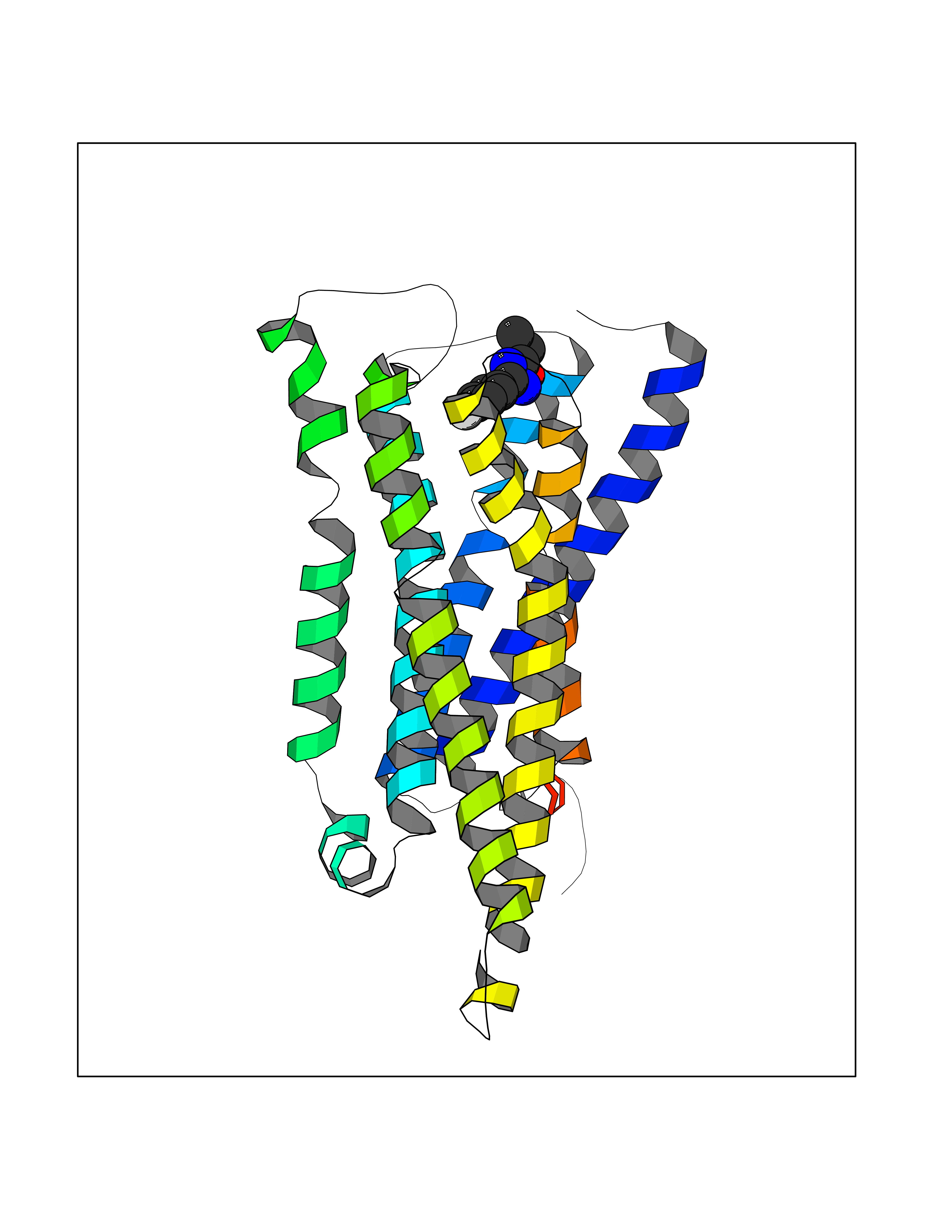 Molscript Map 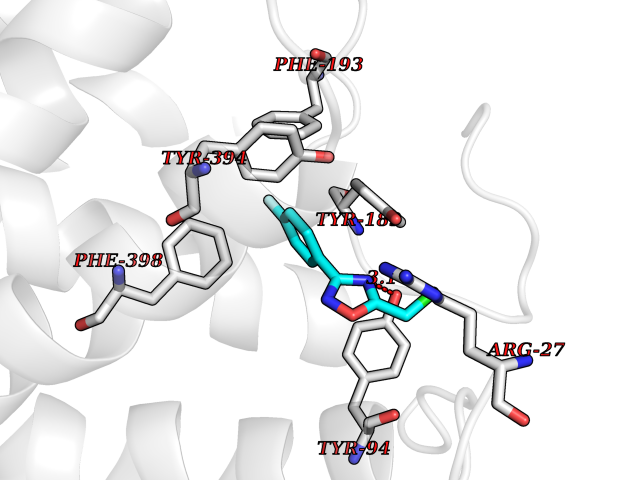 Pymol Map 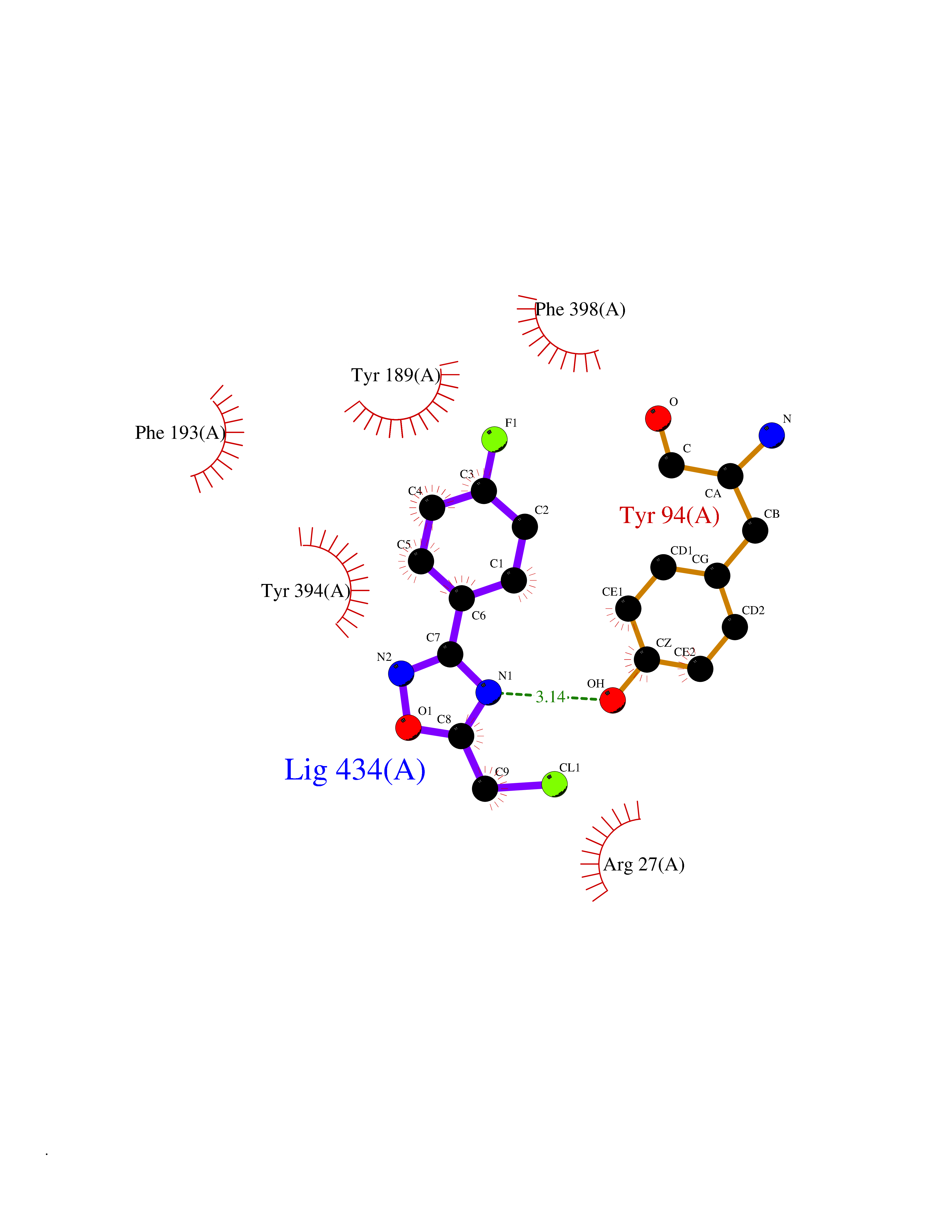 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||