Job Results:
Ligand
Structure
Job ID
8fa392a7f47e3bb9cb55af0501d4ab6f
Job name
NA
Time
2025-11-13 18:42:40
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Plasma retinol-binding protein (RBP4) | 5NU7 | 7.70 | |
Target general information Gen name RBP4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinol-binding protein 4; RBP4; RBP; Plasma retinol-binding protein(1-176); PRBP Protein family Calycin superfamily, Lipocalin family Biochemical class Calycin family Function Delivers retinol from the liver stores to the peripheral tissues. In plasma, the RBP-retinol complex interacts with transthyretin, this prevents its loss by filtration through the kidney glomeruli. Related diseases Retinal dystrophy, iris coloboma, and comedogenic acne syndrome (RDCCAS) [MIM:615147]: A disease characterized by retinal degeneration, ocular colobomas involving both the anterior and posterior segment, impaired night vision and loss of visual acuity. Additional characteristic features include developmental abnormalities and severe acne. {ECO:0000269|PubMed:10232633, ECO:0000269|PubMed:23189188, ECO:0000269|PubMed:9888420}. The disease is caused by variants affecting the gene represented in this entry. Loss of functional RBP4 protein results in serum retinol deficiency. Lack of normal levels of retinol impairs the visual cycle leading to night blindness at early stages; prolonged deficiency may lead to retinal degeneration. Additionally, retinol deficiency may result in dry skin, increased susceptibility to infection and acne (PubMed:23189188). {ECO:0000269|PubMed:23189188}.; DISEASE: Microphthalmia/Coloboma 10 (MCOPCB10) [MIM:616428]: A disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues. Ocular abnormalities like opacities of the cornea and lens, scaring of the retina and choroid, and other abnormalities may also be present. Ocular colobomas are a set of malformations resulting from abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). {ECO:0000269|PubMed:25910211}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06985; DB06755; DB05076; DB03917; DB00755; DB00162 Interacts with Q9UBX0; P02766; O55245 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Methylation; Microphthalmia; Proteomics identification; Reference proteome; Retinol-binding; Secreted; Sensory transduction; Signal; Transport; Vision; Vitamin A Protein physicochemical properties Chain ID A Molecular weight (Da) 20030.2 Length 174 Aromaticity 0.13 Instability index 28.54 Isoelectric point 5.24 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -10.51  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTEDPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQEELCLARQYRLIVHNGYC Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Diamine oxidase (AOC1) | 3HII | 7.70 | |
Target general information Gen name AOC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kidney amine oxidase; KAO; Histaminase; Amiloride-binding protein; AOC1; ABP Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the degradation of compounds such as putrescine, histamine, spermine, and spermidine, substances involved in allergic and immune responses, cell proliferation, tissue differentiation, tumor formation, and possibly apoptosis. Placental DAO is thought to play a role in the regulation of the female reproductive function. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00594; DB01373; DB09130; DB03608; DB05383 Interacts with Q15038; O75593; Q8IUC2; Q96HA8; Q7Z3K3; Q6ZRY4; Q01085-2; O43711; Q96K80 EC number EC 1.4.3.22 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Heparin-binding; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal; TPQ Protein physicochemical properties Chain ID A,B Molecular weight (Da) 162607 Length 1425 Aromaticity 0.13 Instability index 45.72 Isoelectric point 6.76 Charge (pH=7) -4.07 2D Binding mode Binding energy (Kcal/mol) -10.5  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PRKAGVFSDLSNQELKAVHSFLWSKKELRLQPSSTTTMAKNTVFLIEMLLPKKYHVLRFLDKGERHPVREARAVIFFGDQEHPNVTEFAVGPLPGPCYMRALSPRPGYQSSWASRPISTAEYALLYHTLQEATKPLHQFFLNTTGFSFQDCHDRCLAFTDVAPRGVASGQRRSWLIIQRYVEGYFLHPTGLELLVDHGSTDAGHWAVEQVWYNGKFYGSPEELARKYADGEVDVVVLEDPLEPPLFSSHKPRGDFPSPIHVSGPRLVQPHGPRFRLEGNAVLYGGWSFAFRLRSSSGLQVLNVHFGGERIAYEVSVQEAVALYGGHTPAGMQTKYLDVGWGLGSVTHELAPGIDCPETATFLDTFHYYDADDPVHYPRALCLFEMPTGVPLRRHFNSNFKGGFNFYAGLKGQVLVLRTTSTVYNXDYIWDFIFYPNGVMEAKMHATGYVHATFYTPEGLRHGTRLHTHLIGNIHTHLVHYRVDLDVAGTKNSFQTLQMKLENITNPWSPRHRVVQPTLEQTQYSWERQAAFRFKRKLPKYLLFTSPQENPWGHKRSYRLQIHSMADQVLPPGWQEEQAITWARYPLAVTKYRESELCSSSIYHQNDPWDPPVVFEQFLHNNENIENEDLVAWVTVGFLHIPHSEDIPNTATPGNSVGFLLRPFNFFPEDPSLASRDTVIVWPRDNGPNYVQRWIPEDRDCSMPPPFSYNGTYRPVRKAGVFSDLSNQELKAVHSFLWSKKELRLQPSSTTTMAKNTVFLIEMLLPKKYHVLRFLDKGERHPVREARAVIFFGDQEHPNVTEFAVGPLPGPCYMRALSPRPGYQSSWASRPISTAEYALLYHTLQEATKPLHQFFLNTTGFSFQDCHDRCLAFTDVAPRGVASGQRRSWLIIQRYVEGYFLHPTGLELLVDHGSTDAGHWAVEQVWYNGKFYGSPEELARKYADGEVDVVVLEPPLFSSHKPRGDFPSPIHVSGPRLVQPHGPRFRLEGNAVLYGGWSFAFRLRSSSGLQVLNVHFGGERIAYEVSVQEAVALYGGHTPAGMQTKYLDVGWGLGSVTHELAPGIDCPETATFLDTFHYYDADDPVHYPRALCLFEMPTGVPLRRHFNSNFKGGFNFYAGLKGQVLVLRTTSTVYNXDYIWDFIFYPNGVMEAKMHATGYVHATFYTPEGLRHGTRLHTHLIGNIHTHLVHYRVDLDVAGTKNSFQTLQMKLENITNPWSPRHRVVQPTLEQTQYSWERQAAFRFKRKLPKYLLFTSPQENPWGHKRSYRLQIHSMADQVLPPGWQEEQAITWARYPLAVTKYRESELCSSSIYHQNDPWDPPVVFEQFLHNNENIENEDLVAWVTVGFLHIPHSEDIPNTATPGNSVGFLLRPFNFFPEDPSLASRDTVIVWPRDNGPNYVQRWIPEDRDCSMPPPFSYNGTYRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Vitamin D3 receptor (VDR) | 3B0T | 7.68 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -10.48  Molscript Map  Pymol Map 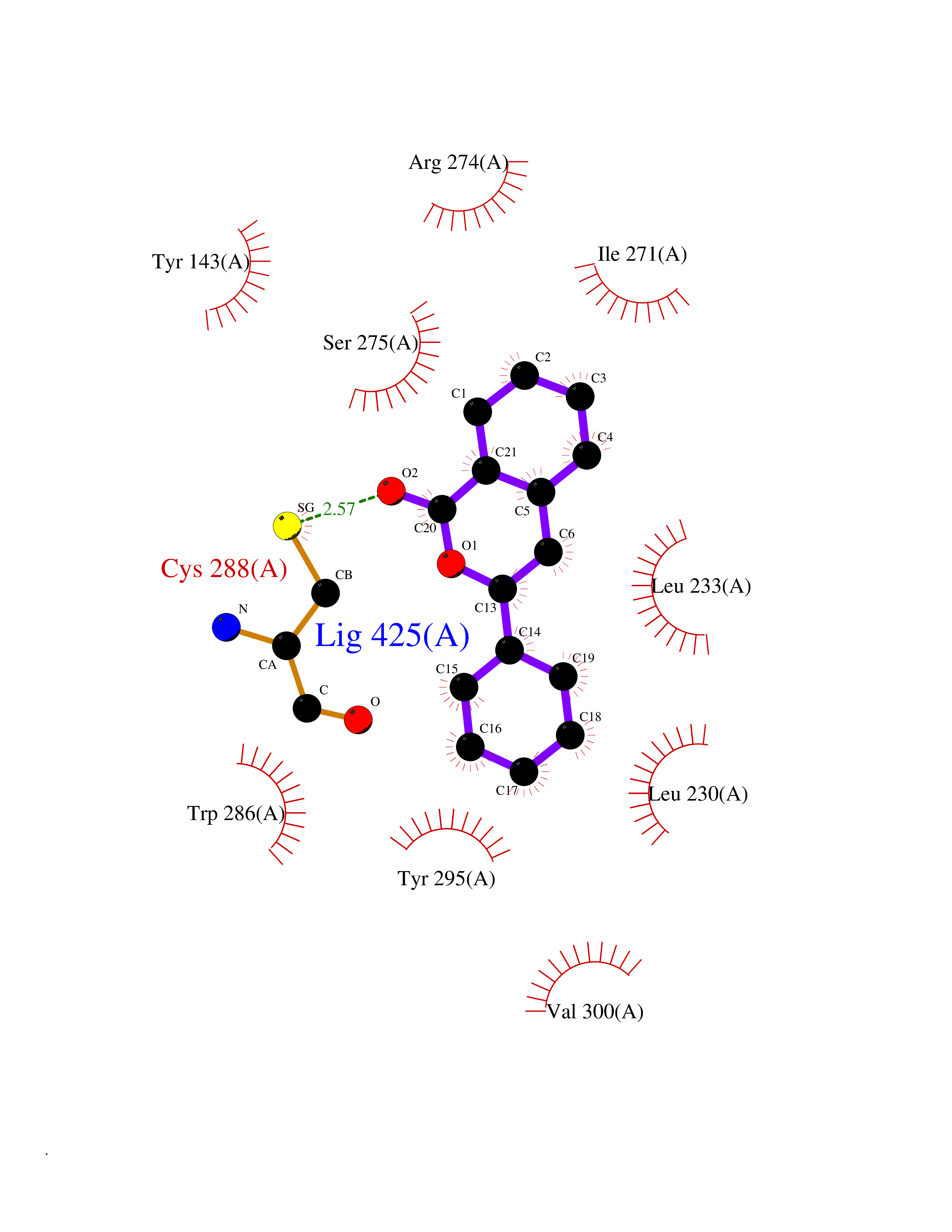 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Thyroid hormone receptor alpha (THRA) | 3ILZ | 7.68 | |
Target general information Gen name THRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms V-erbA-related protein 7; THRA2; THRA1; Nuclear receptor subfamily 1 group A member 1; NR1A1; ERBA1; EAR7; EAR-7; C-erbA-alpha; C-erbA-1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Isoform Alpha-1: Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Hypothyroidism, congenital, non-goitrous, 6 (CHNG6) [MIM:614450]: A disease characterized by growth retardation, developmental retardation, skeletal dysplasia, borderline low thyroxine levels and high triiodothyronine levels. There is differential sensitivity to thyroid hormone action, with retention of hormone responsiveness in the hypothalamic pituitary axis and liver but skeletal, gastrointestinal, and myocardial resistance. {ECO:0000269|PubMed:22168587, ECO:0000269|PubMed:24969835, ECO:0000269|PubMed:25670821, ECO:0000269|PubMed:26037512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01118; DB00509; DB04855; DB05035; DB03176; DB00451; DB00279; DB01583; DB05235; DB09100 Interacts with Q9Y2J4; Q9Y2J4-4; O95971; Q8TAP6; Q96JM7; Q15648; Q6FHY5; P31321; Q96A49; O75410-7; Q9JLI4 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Congenital hypothyroidism; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29910.1 Length 267 Aromaticity 0.07 Instability index 52.75 Isoelectric point 5.31 Charge (pH=7) -11.32 2D Binding mode Binding energy (Kcal/mol) -10.48 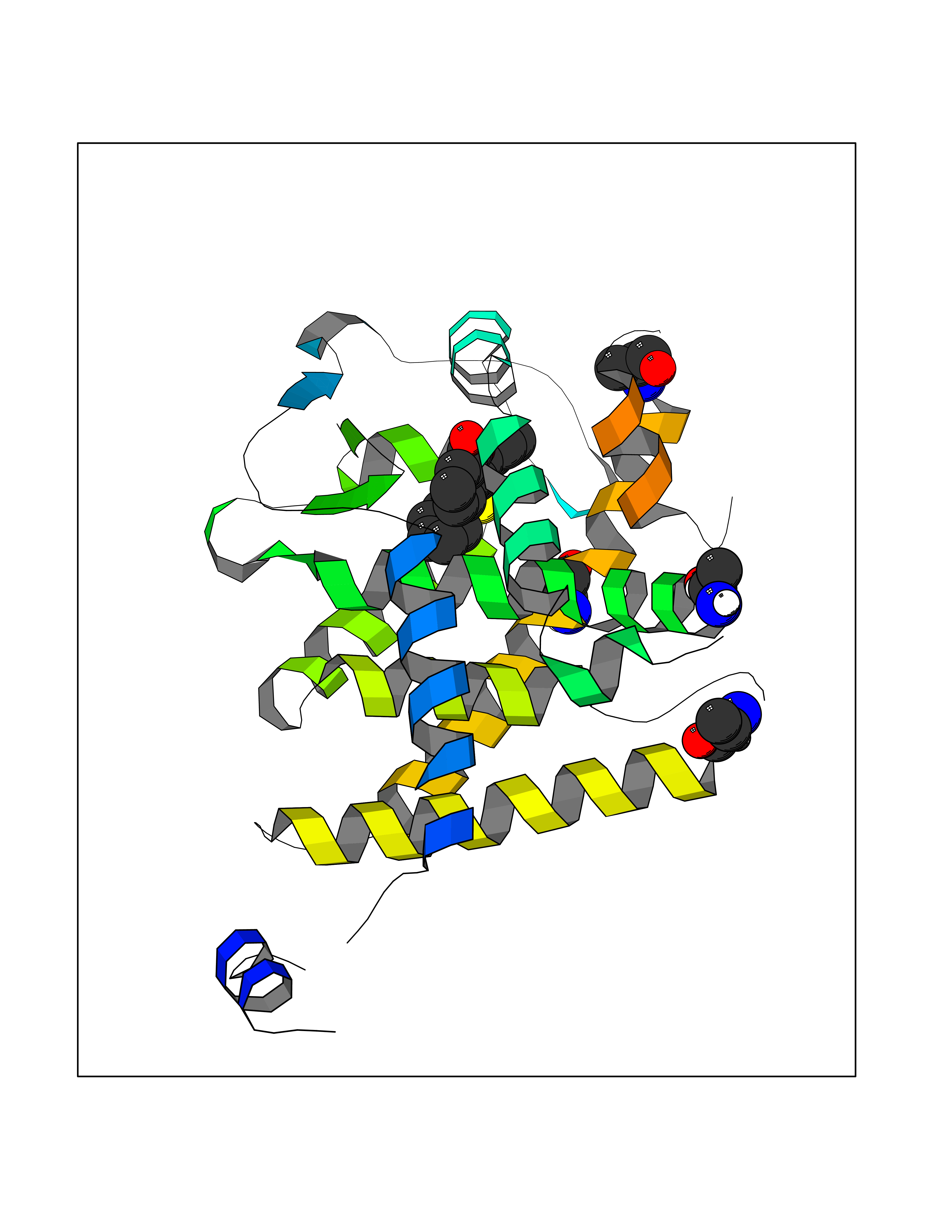 Molscript Map 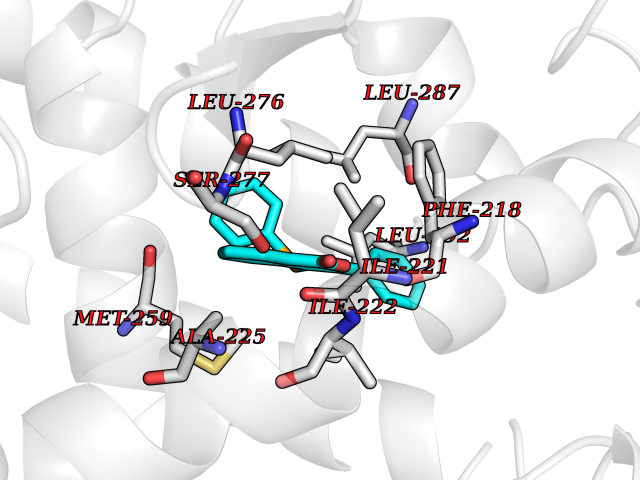 Pymol Map 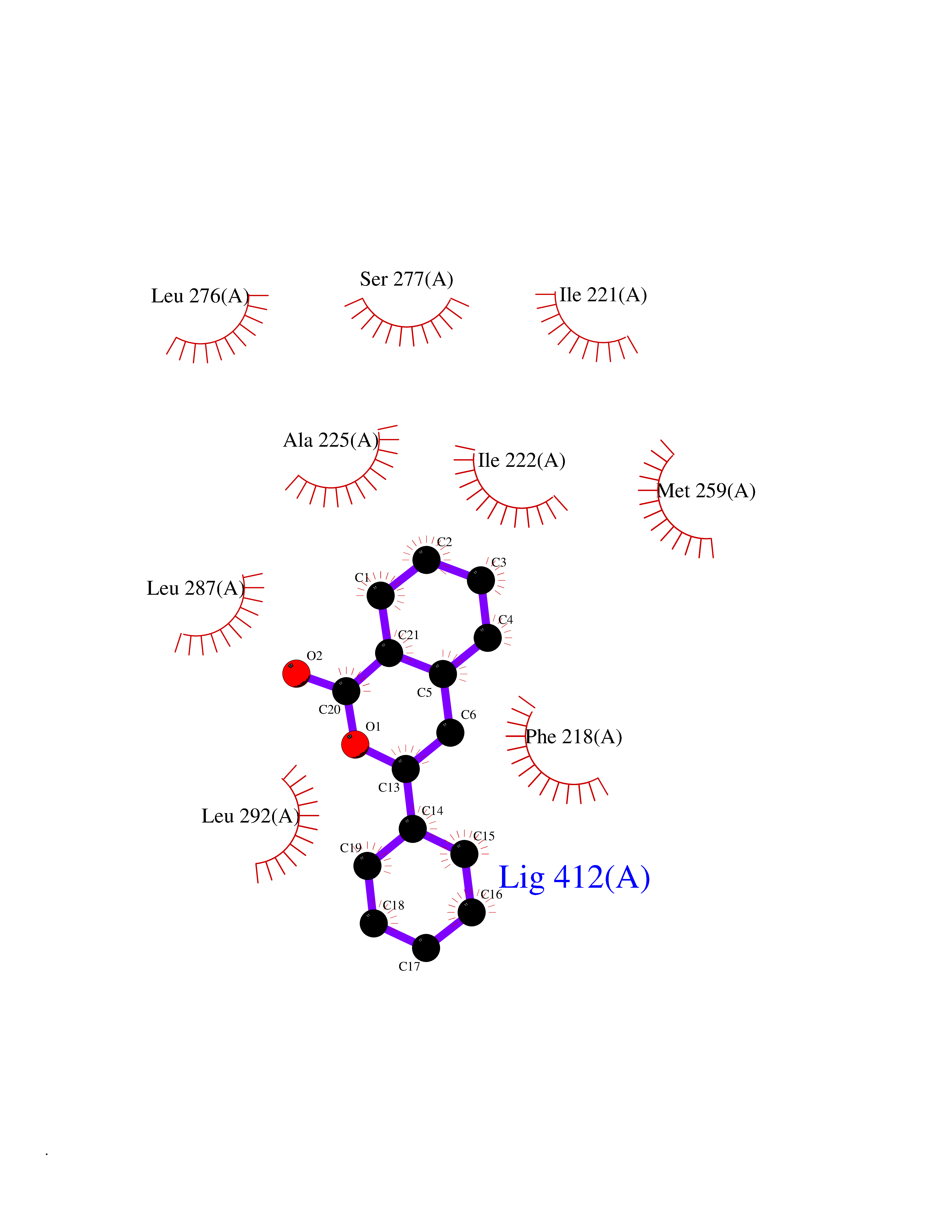 Ligplot Map 3D Binding mode Sequence GSHMEEMIRSLQQRPEPTPEEWDLIHIATEAHRSTNAQGSHWKQRRKFLPDDIGQSPIVSMPDGDKVDLEAFSEFTKIITPAITRVVDFAKKLPMFSELPXEDQIILLKGCCMEIMSLRAAVRYDPESDTLTLSGEMAVKREQLKNGGLGVVSDAIFELGKSLSAFNLDDTEVALLQAVLLMSTDRSGLLXVDKIEKSQEAYLLAFEHYVNHRKHNIPHFWPKLLMKVTDLRMIGAXHASRFLHMKVEXPTELFPPLFLEVFEDQEV Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Dimethylglycine oxidase | 1PJ5 | 7.67 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -10.46 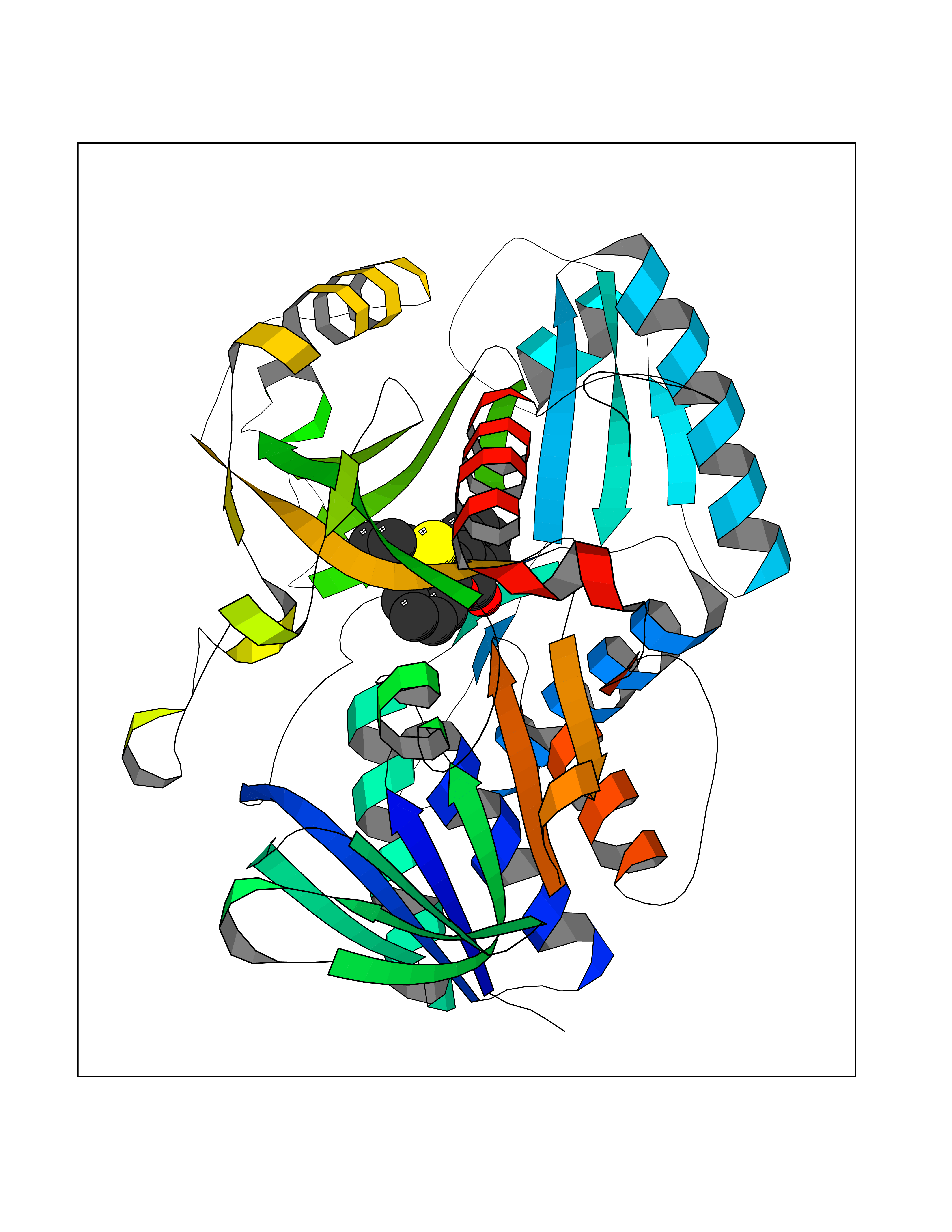 Molscript Map 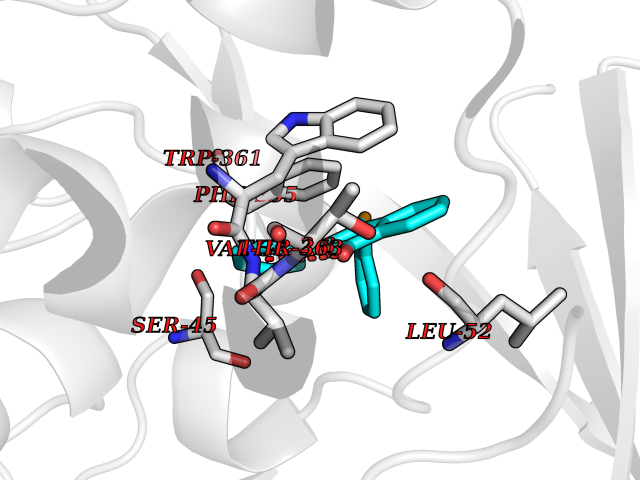 Pymol Map 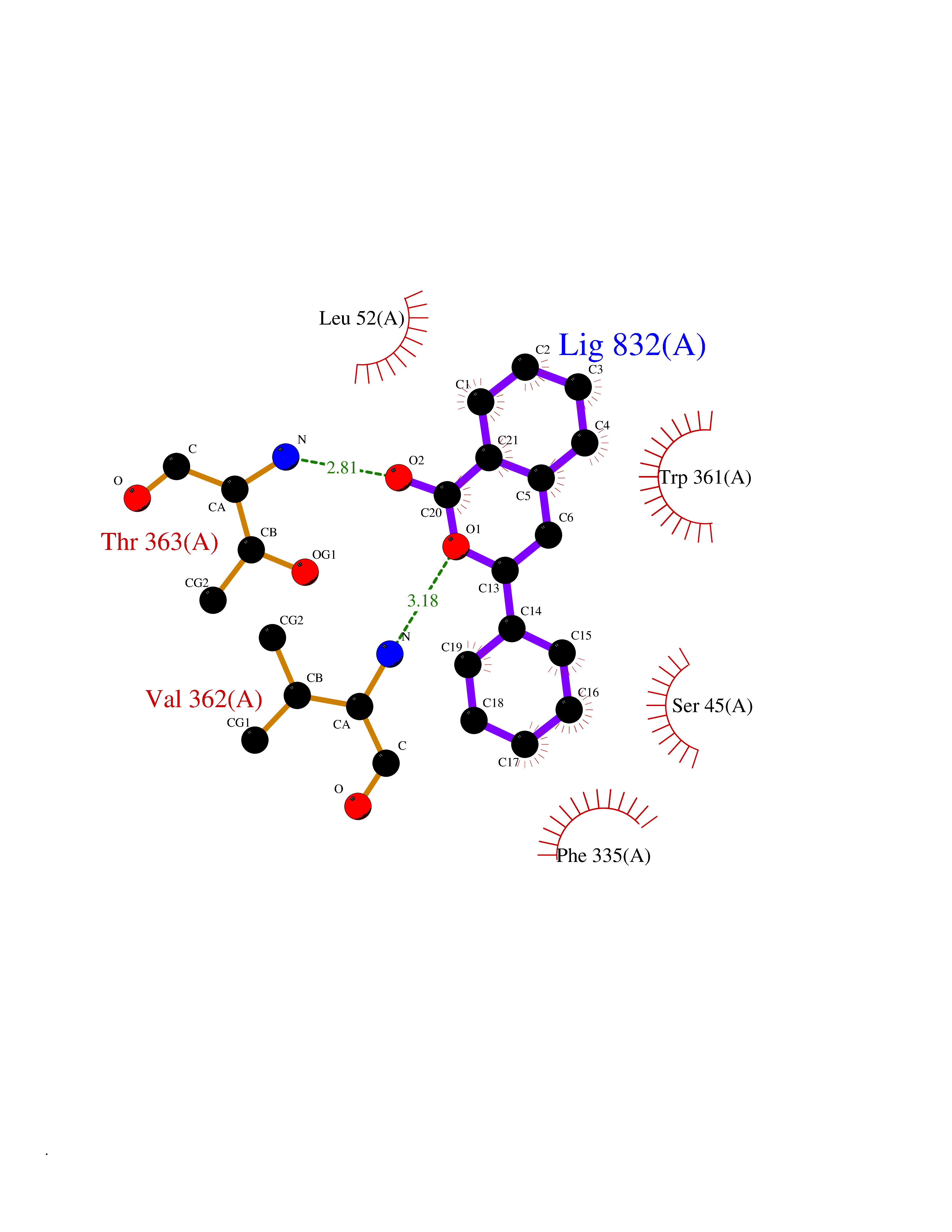 Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Retinoic acid receptor gamma (RARG) | 1FCY | 7.66 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -10.45 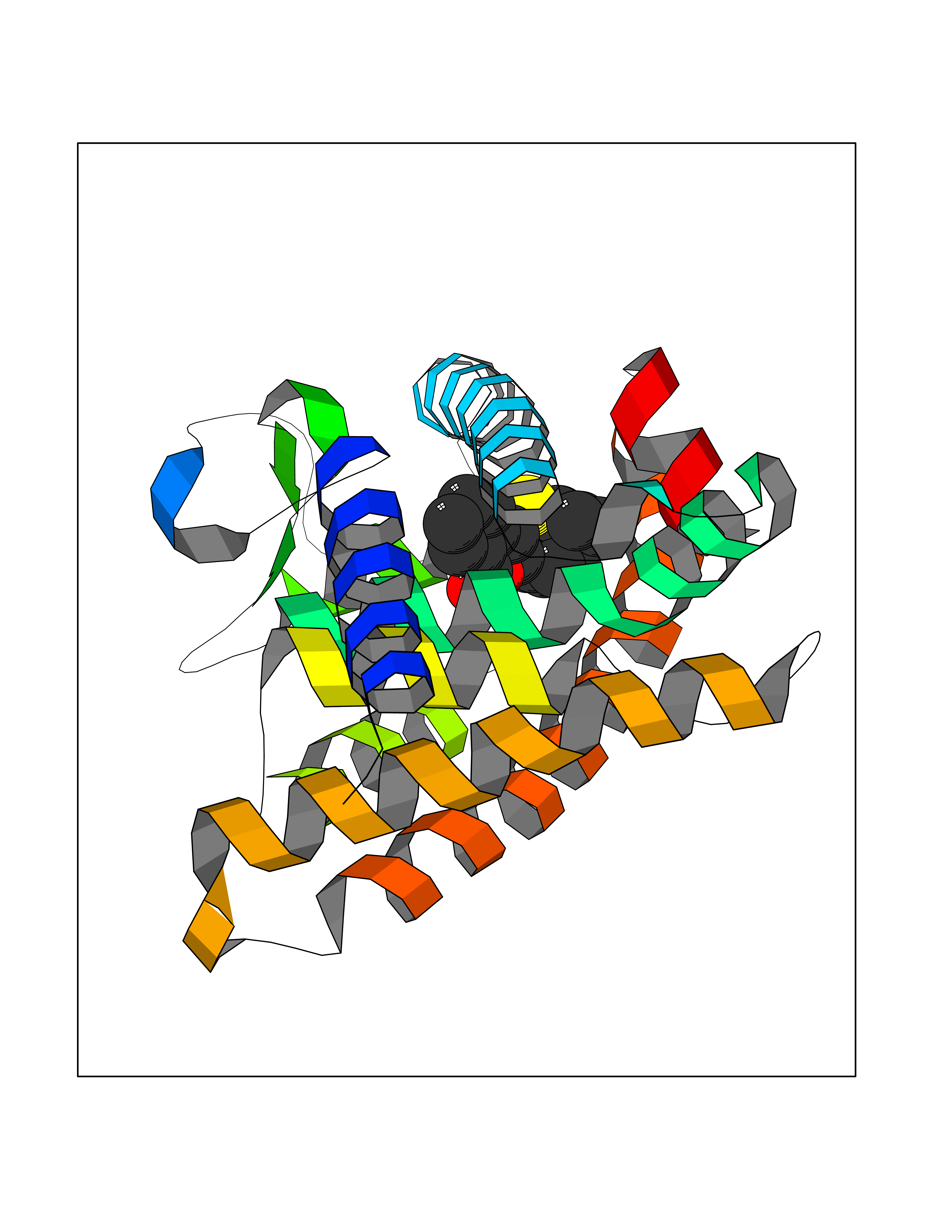 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Cytochrome P450 1A2 | 2HI4 | 7.66 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -10.45  Molscript Map  Pymol Map 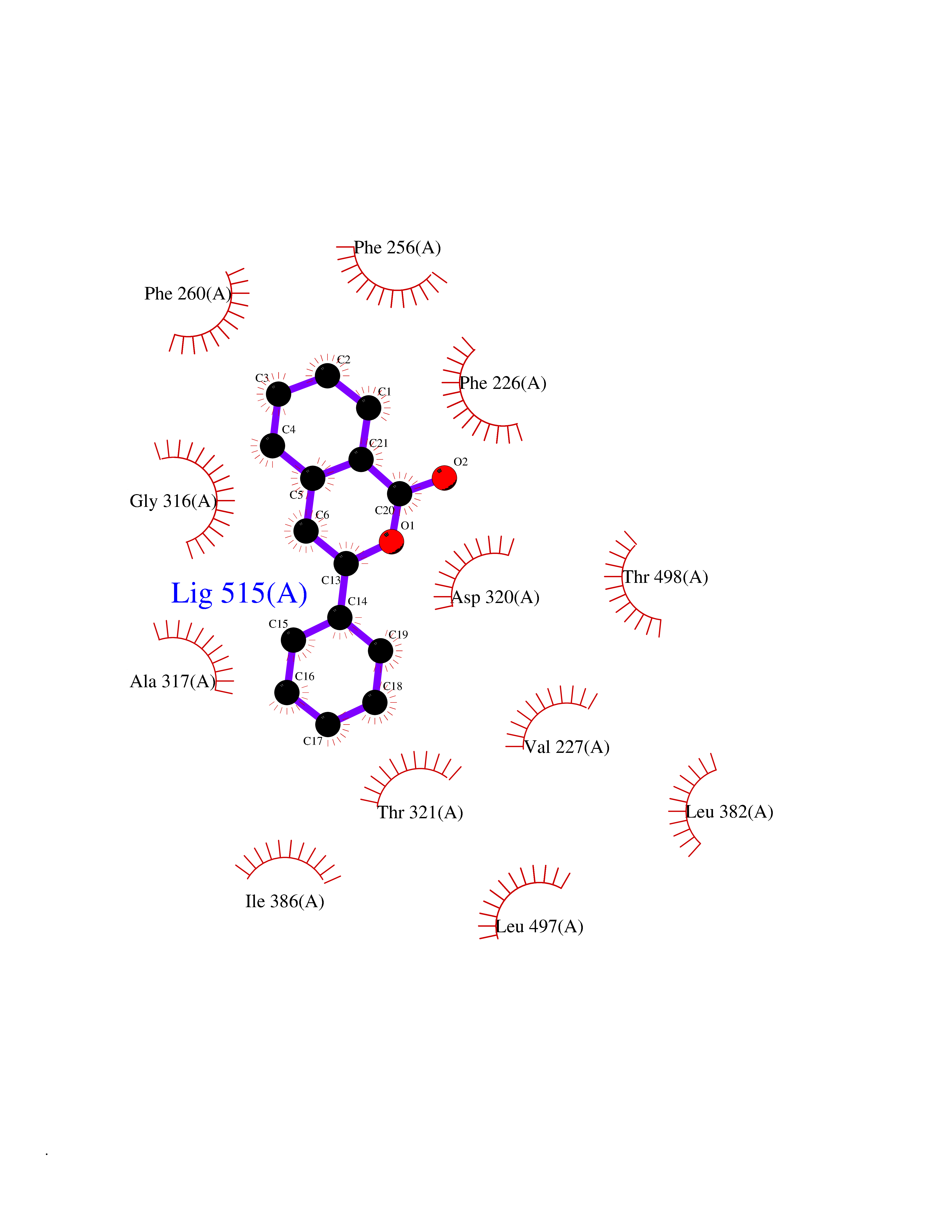 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Muscarinic acetylcholine receptor M4 (CHRM4) | 5DSG | 7.65 | |
Target general information Gen name CHRM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms M4 receptor; CHRM4 Protein family G-protein coupled receptor 1 family, Muscarinic acetylcholine receptor subfamily, CHRM4 sub-subfamily Biochemical class GPCR rhodopsin Function The muscarinic acetylcholine receptor mediates various cellular responses, including inhibition of adenylate cyclase, breakdown of phosphoinositides and modulation of potassium channels through the action of G proteins. Primary transducing effect is inhibition of adenylate cyclase. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03128; DB08897; DB05752; DB00321; DB00543; DB01238; DB14185; DB00572; DB00767; DB01019; DB00835; DB00411; DB01239; DB00568; DB00363; DB00496; DB01151; DB09167; DB01142; DB00366; DB09194; DB06702; DB00986; DB06787; DB11181; DB00725; DB00424; DB00458; DB01625; DB01221; DB00408; DB00934; DB00454; DB06709; DB00940; DB01403; DB00340; DB00622; DB00540; DB00334; DB00715; DB01085; DB00387; DB01069; DB00777; DB12278; DB01224; DB11855; DB13581; DB00747; DB01591; DB00342; DB11235; DB01409; DB01036; DB00376; DB09089; DB00726; DB00809; DB09076; DB09185; DB00246 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32545.4 Length 287 Aromaticity 0.14 Instability index 30.63 Isoelectric point 9.34 Charge (pH=7) 11.91 2D Binding mode Binding energy (Kcal/mol) -10.44 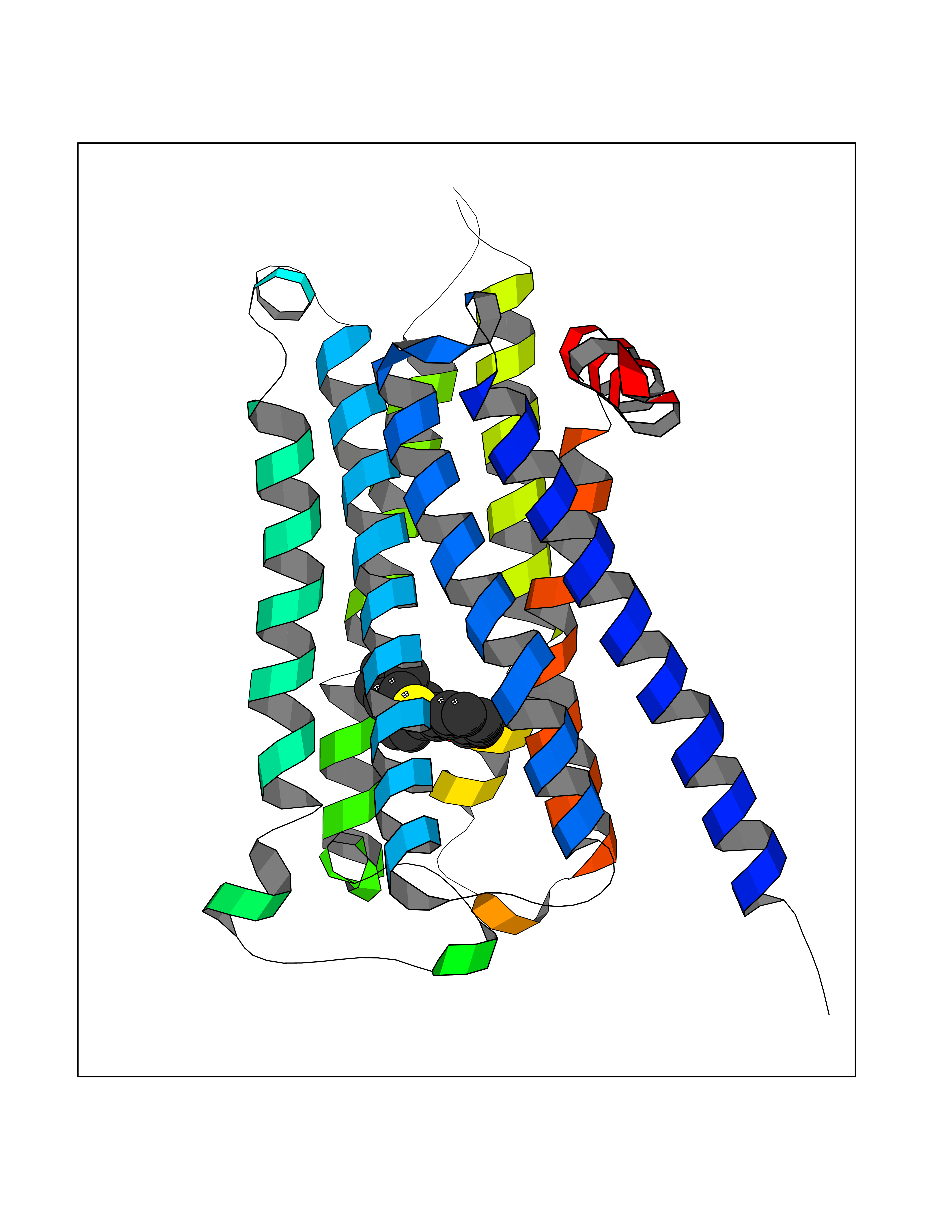 Molscript Map 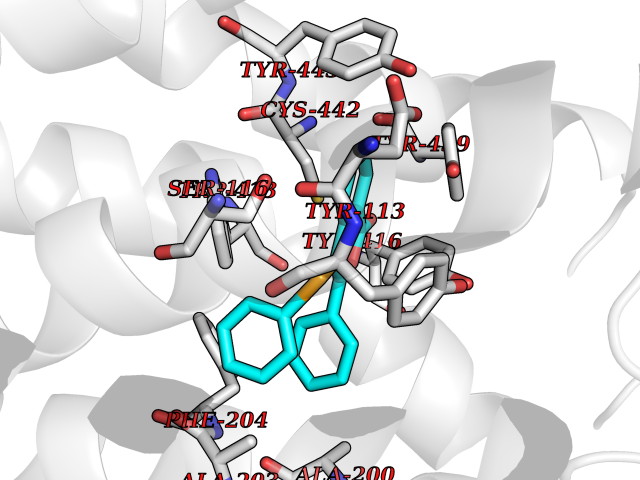 Pymol Map 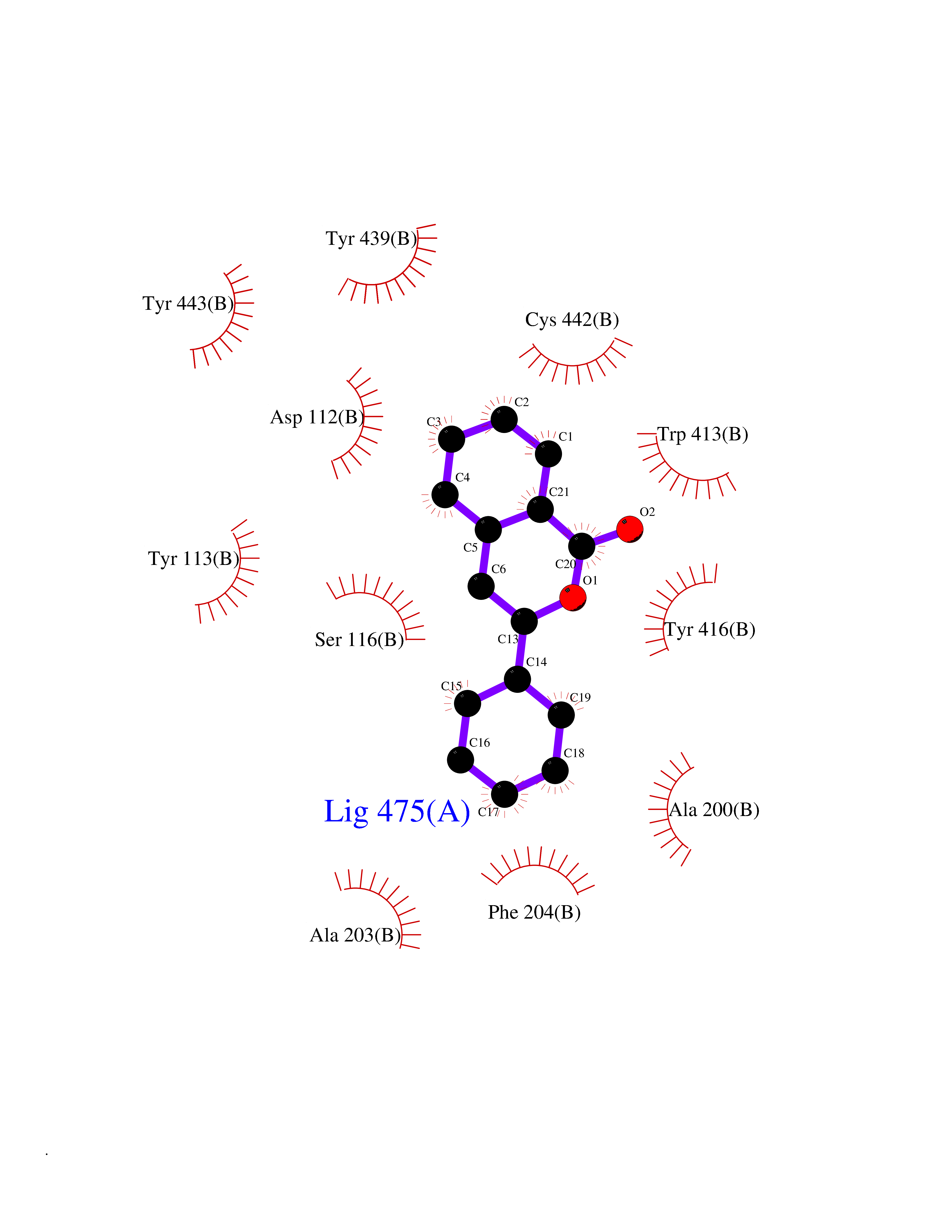 Ligplot Map 3D Binding mode Sequence HNRYETVEMVFIATVTGSLSLVTVVGNILVMLSIKVNRQLQTVNNYFLFSLACADLIIGAFSMNLYTVYIIKGYWPLGAVVCDLWLALDYVVSNASVMNLLIISFDRYFCVTKPLTYPARRTTKMAGLMIAAAWVLSFVLWAPAILFWQFVVGKRTVPDNQCFIQFLSNPAVTFGTAIAAFYLPVVIMTVLYIHISLASRSRVQMAARERKVTRTIFAILLAFILTWTPYNVMVLVNTFCQSCIPDTVWSIGYWLCYVNSTINPACYALCNATFKKTFRHLLLCQYR Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | cAMP-dependent protein kinase A type I (PRKAR1A) | 5KJZ | 7.65 | |
Target general information Gen name PRKAR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-dependent protein kinase type I-alpha regulatory subunit; Tissue-specific extinguisher 1; TSE1; PRKAR1; PKR1 Protein family CAMP-dependent kinase regulatory chain family Biochemical class Kinase Function Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. Related diseases Carney complex 1 (CNC1) [MIM:160980]: CNC is a multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and psammomatous melanotic schwannomas. {ECO:0000269|PubMed:15371594, ECO:0000269|PubMed:18241045, ECO:0000269|PubMed:22785148, ECO:0000269|PubMed:23323113, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Intracardiac myxoma (INTMYX) [MIM:255960]: Inheritance is autosomal recessive. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Primary pigmented nodular adrenocortical disease 1 (PPNAD1) [MIM:610489]: A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes. PPNAD1 is most often diagnosed in patients with Carney complex, a multiple neoplasia syndrome. However it can also be observed in patients without other manifestations. {ECO:0000269|PubMed:12213893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acrodysostosis 1, with or without hormone resistance (ACRDYS1) [MIM:101800]: A form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin. However, not all patients show endocrine abnormalities. {ECO:0000269|PubMed:21651393, ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:22723333, ECO:0000269|PubMed:23043190, ECO:0000269|PubMed:23425300, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01790; DB02527; DB02315; DB05798 Interacts with Q9GZX7; P24588; O43687-2; Q9BSF0; Q9H6J7-2; Q86Y01; P0C7A2-2; Q9H0R8; Q9H8W4; P17612; P31321; P51817; P35250; Q86UC2; Q01105; Q8N0X7; O96006; P03259-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; cAMP; cAMP-binding; Cell membrane; Cushing syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 17007.4 Length 149 Aromaticity 0.08 Instability index 49.36 Isoelectric point 6.36 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -10.44  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SILMGSTLRKRKMYEEFLSKVSILESLDKWERLTVADALEPVQFEDGQKIVVQGEPGDEFFIILEGSAAVLQRRSENEEFVEVRRLGPSDYFGEIALLMNRPRTATVVARGPLKCVKLDRPRFERVLGPCSDILKRNIQQYNSFVSLSV Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Retinoic acid receptor RXR-beta (RXRB) | 5HJP | 7.64 | |
Target general information Gen name RXRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor beta; Nuclear receptor subfamily 2 group B member 2; NR2B2 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE). Receptor for retinoic acid. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB00459; DB00210; DB00523; DB00307; DB01393; DB03756; DB00926; DB01941; DB07929; DB02746; DB00412; DB00799; DB07080; DB00755 Interacts with Q00975; Q9HB07; F1D8P7; Q13133; Q13133-3; Q96RI1-1; P04150; Q9NRD5; P37231; P10276; P10276-2; P10826-2; P13631; Q6IQ16; Q13137; Q96B26; Q08379; Q6A162; Q9UJV3-2; Q13133-3; Q96RI1-1; O43586; P10276; P10826-2; Q8IUQ4-2; O75528; Q12800; Q9UBB9; Q05BL1; P14373; O94972; Q96S82 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 28845.8 Length 251 Aromaticity 0.08 Instability index 54.86 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -10.42  Molscript Map 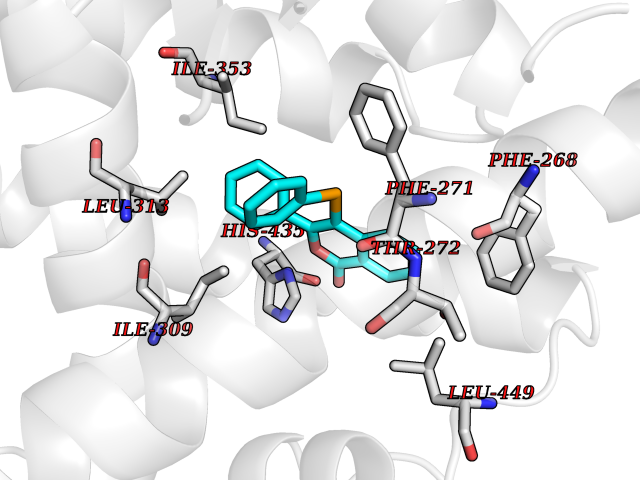 Pymol Map 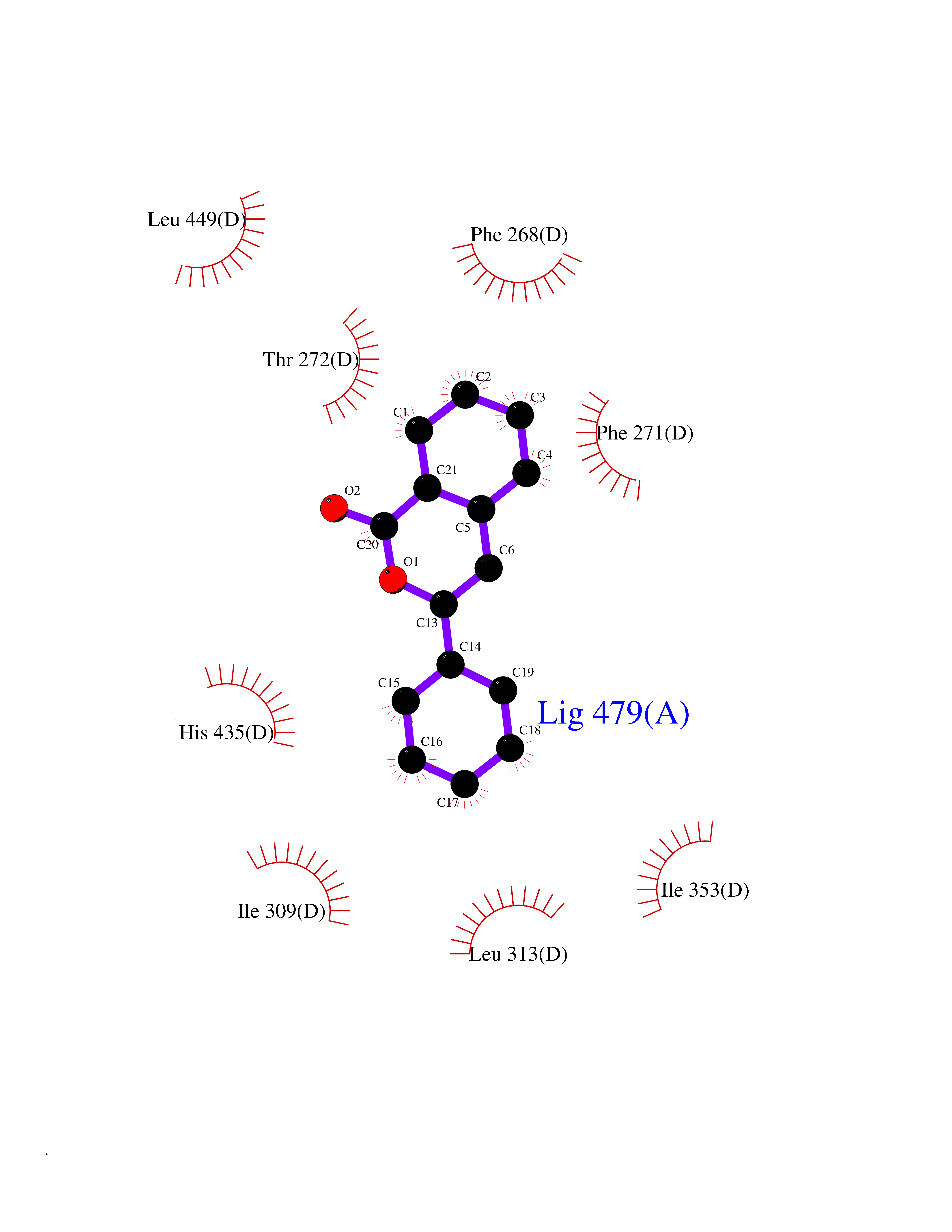 Ligplot Map 3D Binding mode Sequence QLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPSASQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDVHEGSGSGSHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Histamine N-methyltransferase (HNMT) | 2AOT | 7.64 | |
Target general information Gen name HNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine-N-methyltransferase; HNMT; HMT Protein family Class I-like SAM-binding methyltransferase superfamily, HNMT family Biochemical class Methyltransferase Function Inactivates histamine by N-methylation. Plays an important role in degrading histamine and in regulating the airway response to histamine. Related diseases Intellectual developmental disorder, autosomal recessive 51 (MRT51) [MIM:616739]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26206890}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) DB00613; DB13875; DB05381; DB04655; DB01103; DB01752; DB07106 Interacts with NA EC number EC 2.1.1.8 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32712 Length 288 Aromaticity 0.1 Instability index 36.38 Isoelectric point 5.18 Charge (pH=7) -9.97 2D Binding mode Binding energy (Kcal/mol) -10.43 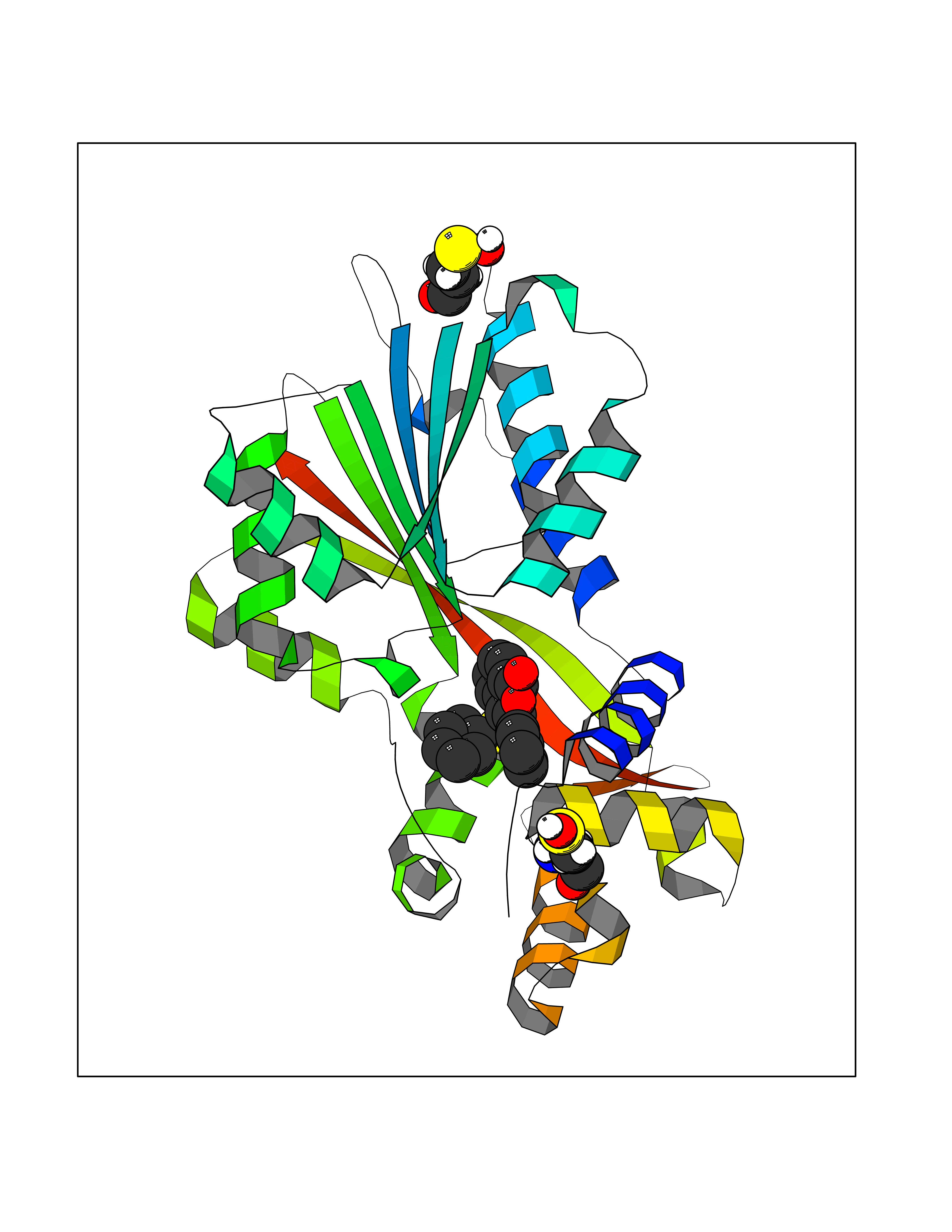 Molscript Map 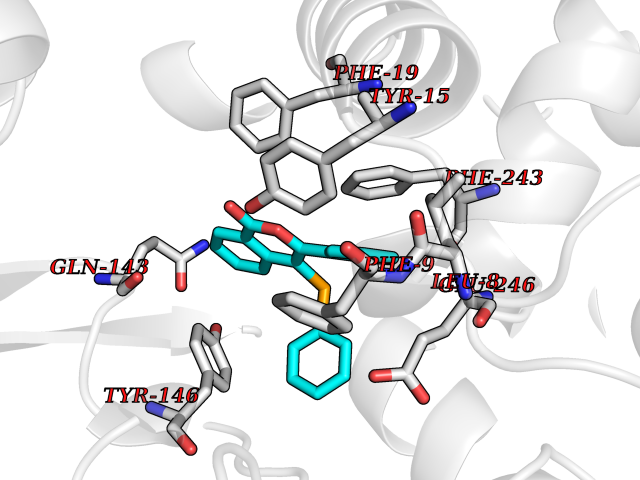 Pymol Map 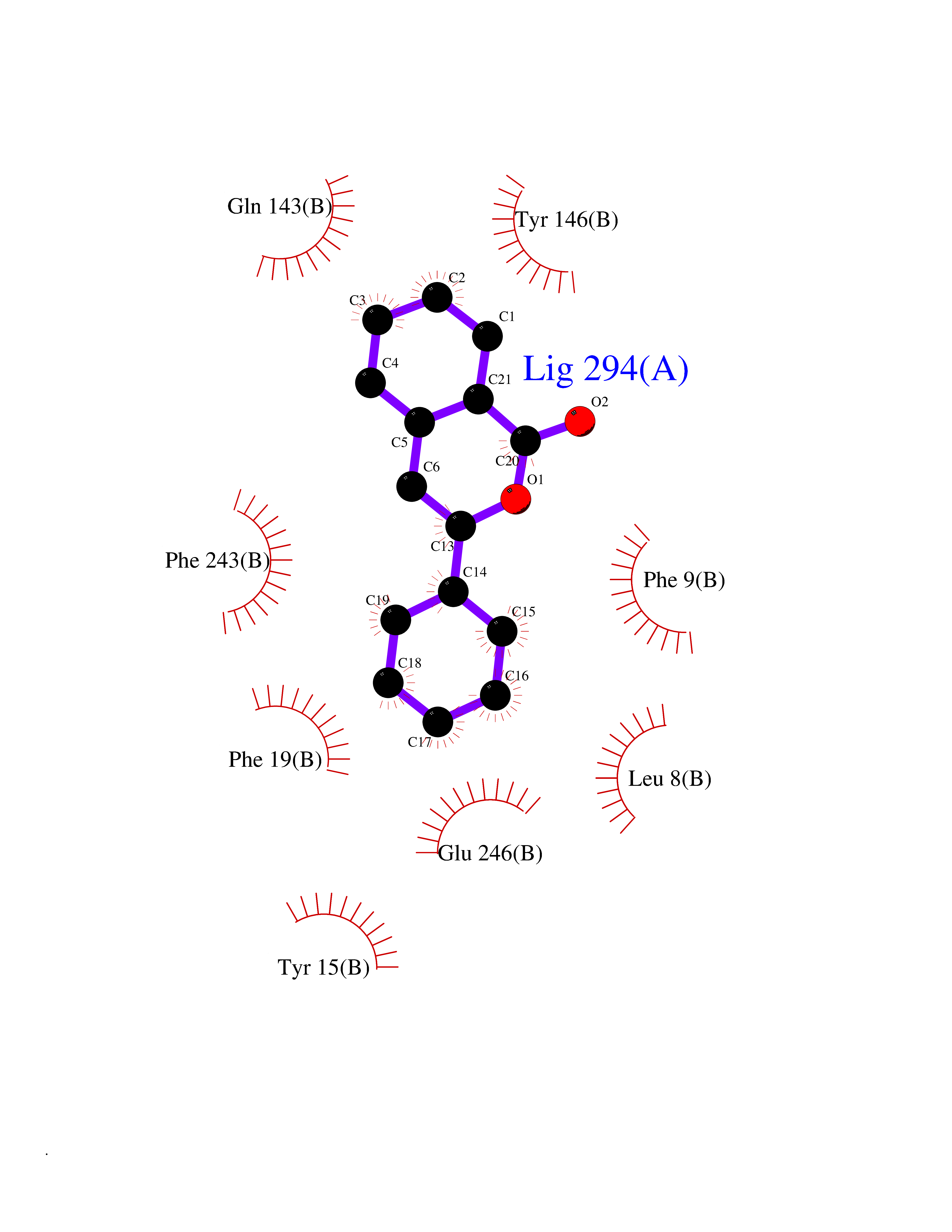 Ligplot Map 3D Binding mode Sequence MRSLFSDHGKYVESFRRFLNHSTEHQCMQEFMDKKLPGIIGRIGDTKSEIKILSIGGGAGEIDLQILSKVQAQYPGVXINNEVVEPSAEQIAKYKELVAKTSNLENVKFAWHKETSSEYQSRMLEKKELQKWDFIHMIQMLYYVKDIPATLKFFHSLLGTNAKMLIIVVSGSSGWDKLWKKYGSRFPQDDLCQYITSDDLTQMLDNLGLKYECYDLLSTMDISDCFIDGNENGDLLWDFLTETXNFNATAPPDLRAELGKDLQEPEFSAKKEGKVLFNNTLSFIVIEA Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Pyruvate synthase | 2C42 | 7.64 | |
Target general information Gen name por Organism Desulfocurvibacter africanus (Desulfovibrio africanus) Uniprot ID TTD ID NA Synonyms NA Protein family Pyruvate:ferredoxin/flavodoxin oxidoreductase family Biochemical class Oxidoreductase Function 4 iron, 4 sulfur cluster binding.Iron ion binding.Pyruvate synthase activity.Thiamine pyrophosphate binding. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02410; DB01987; DB00507 Interacts with NA EC number 1.2.7.1 Uniprot keywords 3D-structure; 4Fe-4S; Calcium; Cytoplasm; Direct protein sequencing; Disulfide bond; Electron transport; Iron; Iron-sulfur; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Thiamine pyrophosphate; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 115569 Length 1065 Aromaticity 0.09 Instability index 31.51 Isoelectric point 6.32 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -10.42  Molscript Map 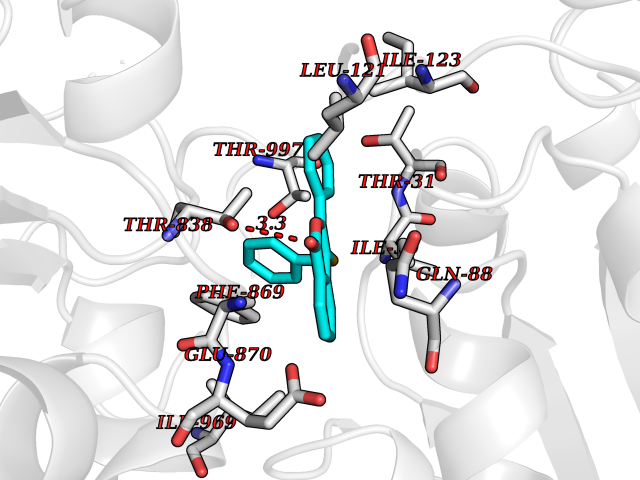 Pymol Map 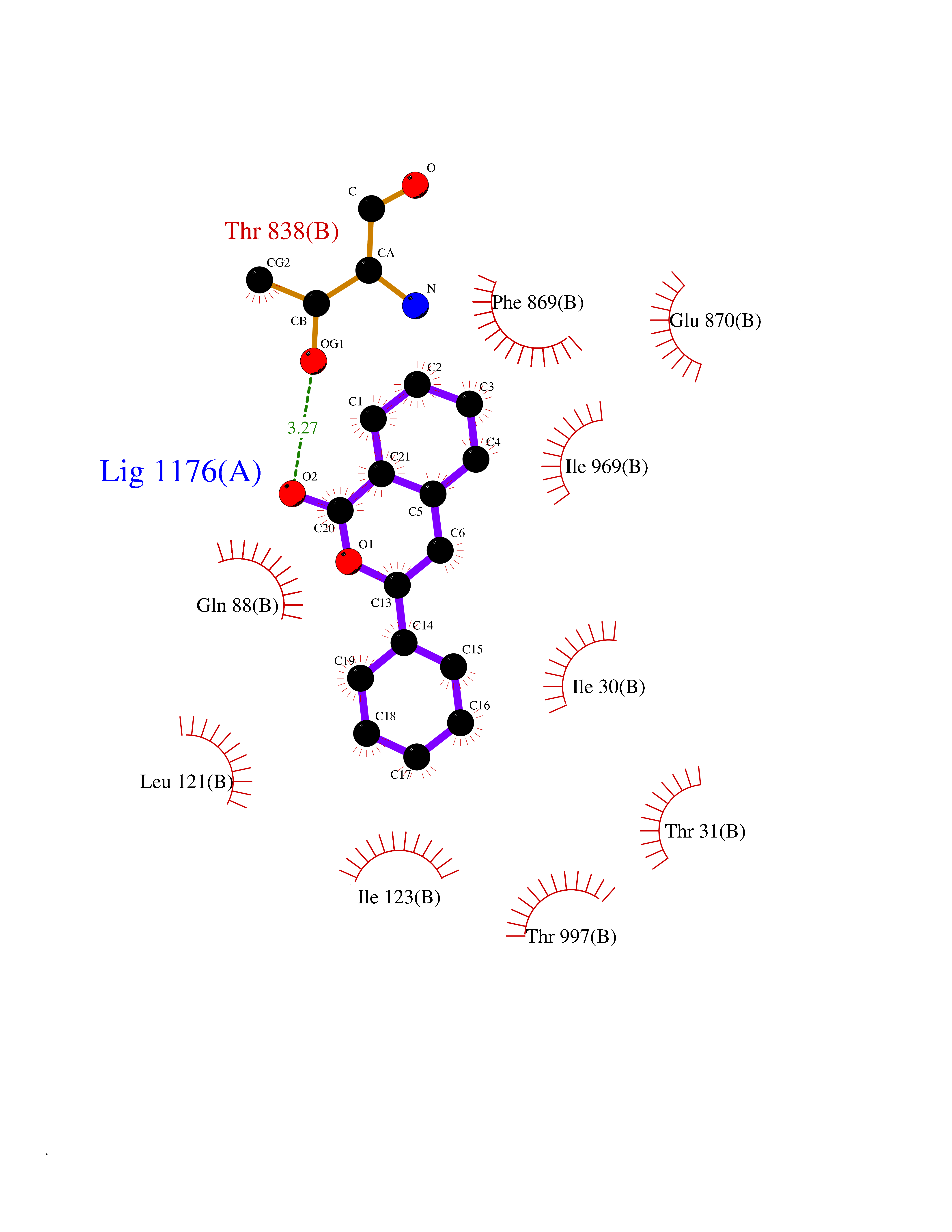 Ligplot Map 3D Binding mode Sequence GKKMMTTDGNTATAHVAYAMSEVAAIYPITPSSTMGEEADDWAAQGRKNIFGQTLTIREMQSEAGAAGAVHGALAAGALTTTFTASQGLLLMIPNMYKISGELLPGVFHVTARAIAAHALSIFGDHQDIYAARQTGFAMLASSSVQEAHDMALVAHLAAIESNVPFMHFFDGFRTSHEIQKIEVLDYADMASLVNQKALAEFRAKSPGIVAEYMQKVASLTGRSYKLFDYVGAPDAERVIVSMGSSCETIEEVINHLAAKGEKIGLIKVRLYRPFVSEAFFAALPASAKVITVLDRTKEPGAPGDPLYLDVCSAFVERGEAMPKILAGRYGLGSKEFSPAMVKSVYDNMSGAKKNHFTVGIEDDVTGTSLPVDNAFADTTPKGTIQCQFWGLGADGTVGANKQAIKIIGDNTDLFAQGYFSYDSKKSGGITISHLRFGEKPIQSTYLVNRADYVACHNPAYVGIYDILEGIKDGGTFVLNSPWSSLEDMDKHLPSGIKRTIANKKLKFYNIDAVKIATDVGLGGRINMIMQTAFFKLAGVLPFEKAVDLLKKSIHKAYGKKGEKIVKMNTDAVDQAVTSLQEFKYPDSWKDAPAETKAEPMTNEFFKNVVKPILTQQGDKLPVSAFEADGRFPLGTSQFEKRGVAINVPQWVPENCIQCNQCAFVCPHSAILPVLAKEEELVGAPANFTALEAKGKELKGYKFRIQINTLDCMGCGNCADICPPKEKALVMQPLDTQRDAQVPNLEYAARIPVKSEVLPRDSLKGSQFQEPLMEFSGACSGCGETPYVRVITQLFGERMFIANATGCSSIWGASAPSMPYKTNRLGQGPAWGNSLFEDAAEYGFGMSVWIFGGDGWAYDIGYGGLDHVLASGEDVNVFVMDTEVYSNTGGQSSKATPTGAVAKFAAAGKRTGKKDLARMVMTYGYVYVATVSMGYSKQQFLKVLKEAESFPGPSLVIAYATCINQGLRKGMGKSQDVMNTAVKSGYWPLFRYDPRLAAQGKNPFQLDSKAPDGSVEEFLMAQNRFAVLDRSFPEDAKRLRAQVAHELDVRFKELEHMAATNIFES Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | PRKR-like endoplasmic reticulum kinase (PERK) | 4G31 | 7.64 | |
Target general information Gen name EIF2AK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PEK Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class Kinase Function Converts phosphorylated eIF-2-alpha/EIF2S1 either in a global protein synthesis inhibitor, leading to a reduced overall utilization of amino acids, or to a translation initiation activator of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Serves as a critical effector of unfolded protein response (UPR)-induced G1 growth arrest due to the loss of cyclin-D1 (CCND1). Involved in control of mitochondrial morphology and function. Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' during the unfolded protein response (UPR) and in response to low amino acid availability. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NZJ5; P11021 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ADP-ribosylation; ATP-binding; Diabetes mellitus; Disease variant; Endoplasmic reticulum; Glycoprotein; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Signal; Stress response; Transferase; Translation regulation; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Unfolded protein response Protein physicochemical properties Chain ID A Molecular weight (Da) 29033.5 Length 248 Aromaticity 0.1 Instability index 43.71 Isoelectric point 7.75 Charge (pH=7) 1.27 2D Binding mode Binding energy (Kcal/mol) -10.42  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GRYLTDFEPIQCLGRGGVVFEAKNKVDDCNYAIKRIRLPNRELAREKVMREVKALAKLEHPGIVRYFNAWLEKNKVYLYIQMQLCRKENLKDWMNGRCTIEERERSVCLHIFLQIAEAVEFLHSKGLMHRDLKPSNIFFTMDDVVKVGDFGLVGTKLYMSPEQIHGNSYSHKVDIFSLGLILFELLYPFSTQMERVRTLTDVRNLKFPPLFTQKYPCEYVMVQDMLSPSPMERPEAINIIENAVFEDL Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Tankyrase-2 (TNKS-2) | 3U9H | 7.64 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -10.42  Molscript Map 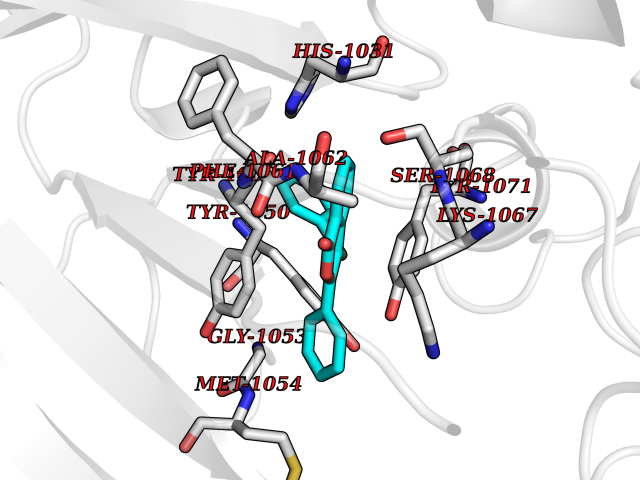 Pymol Map 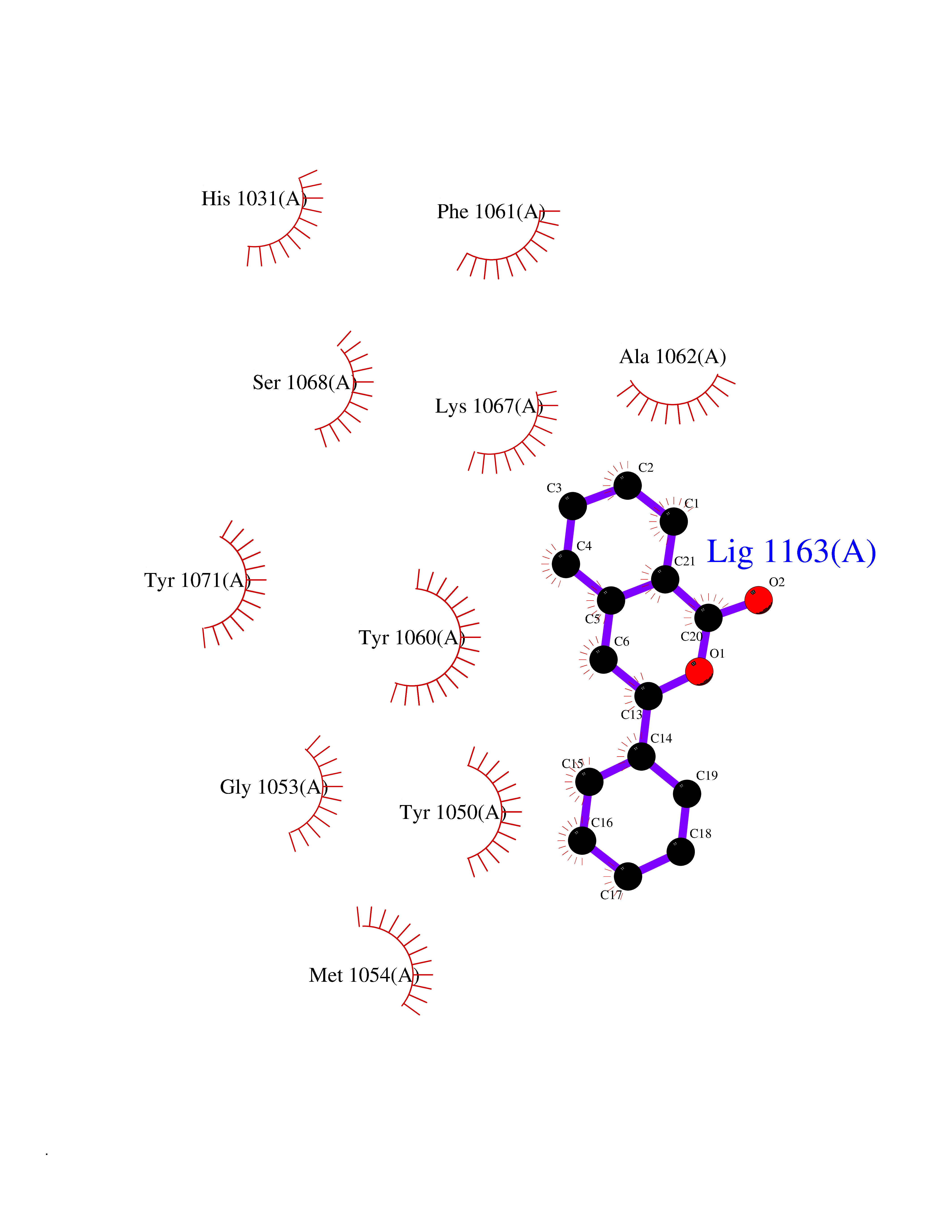 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Wnt-7a protein (WNT7A) | 4UZQ | 7.64 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -10.41 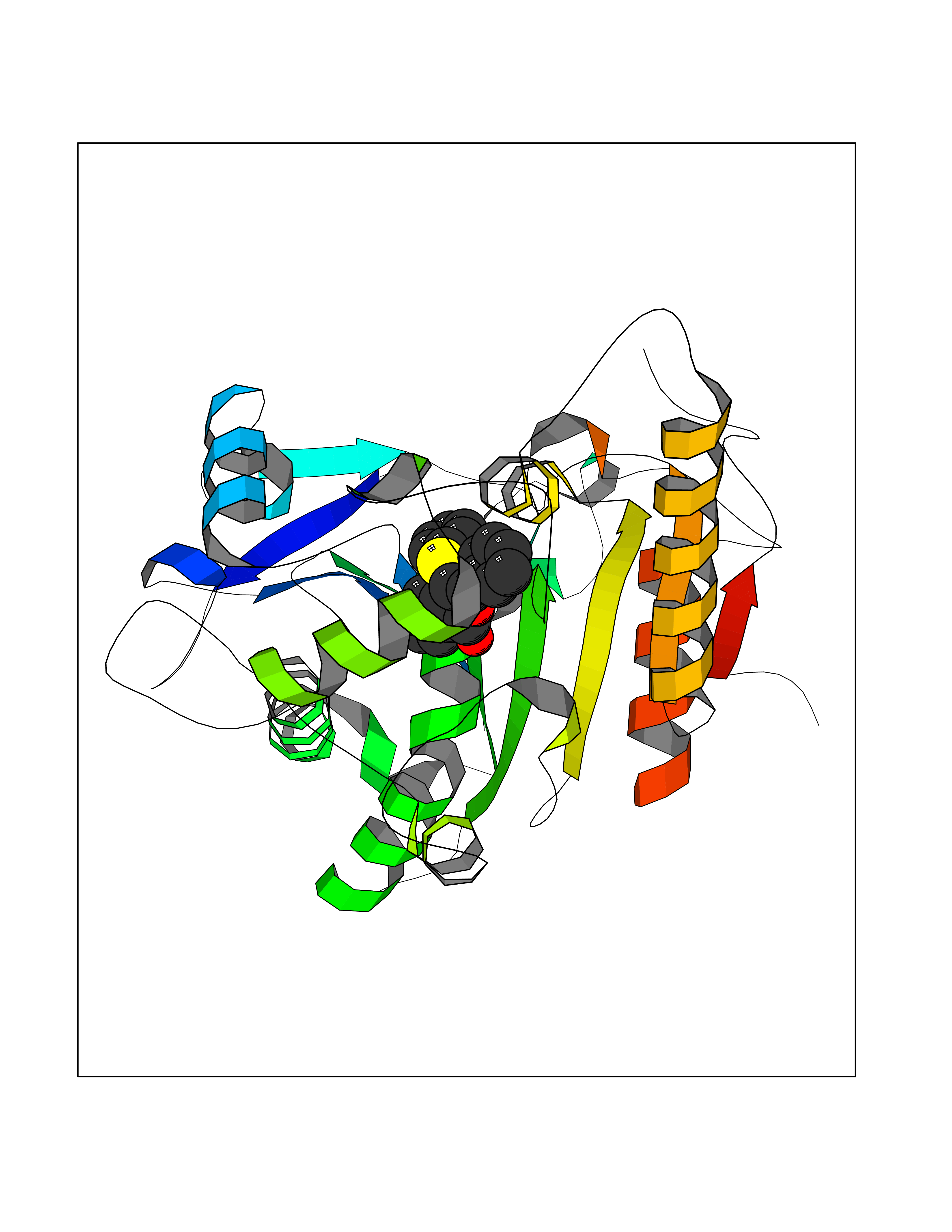 Molscript Map 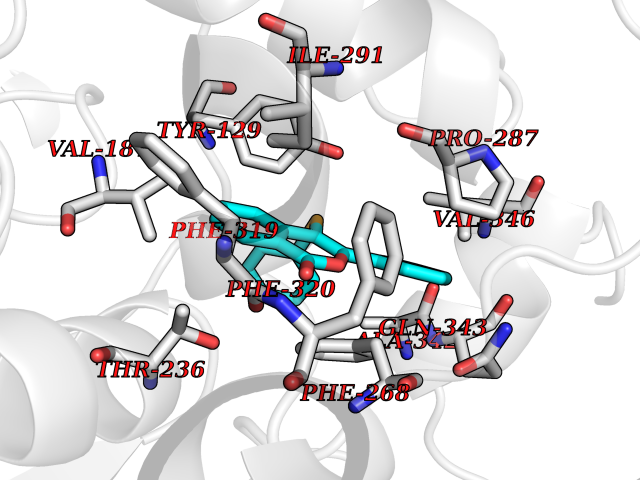 Pymol Map 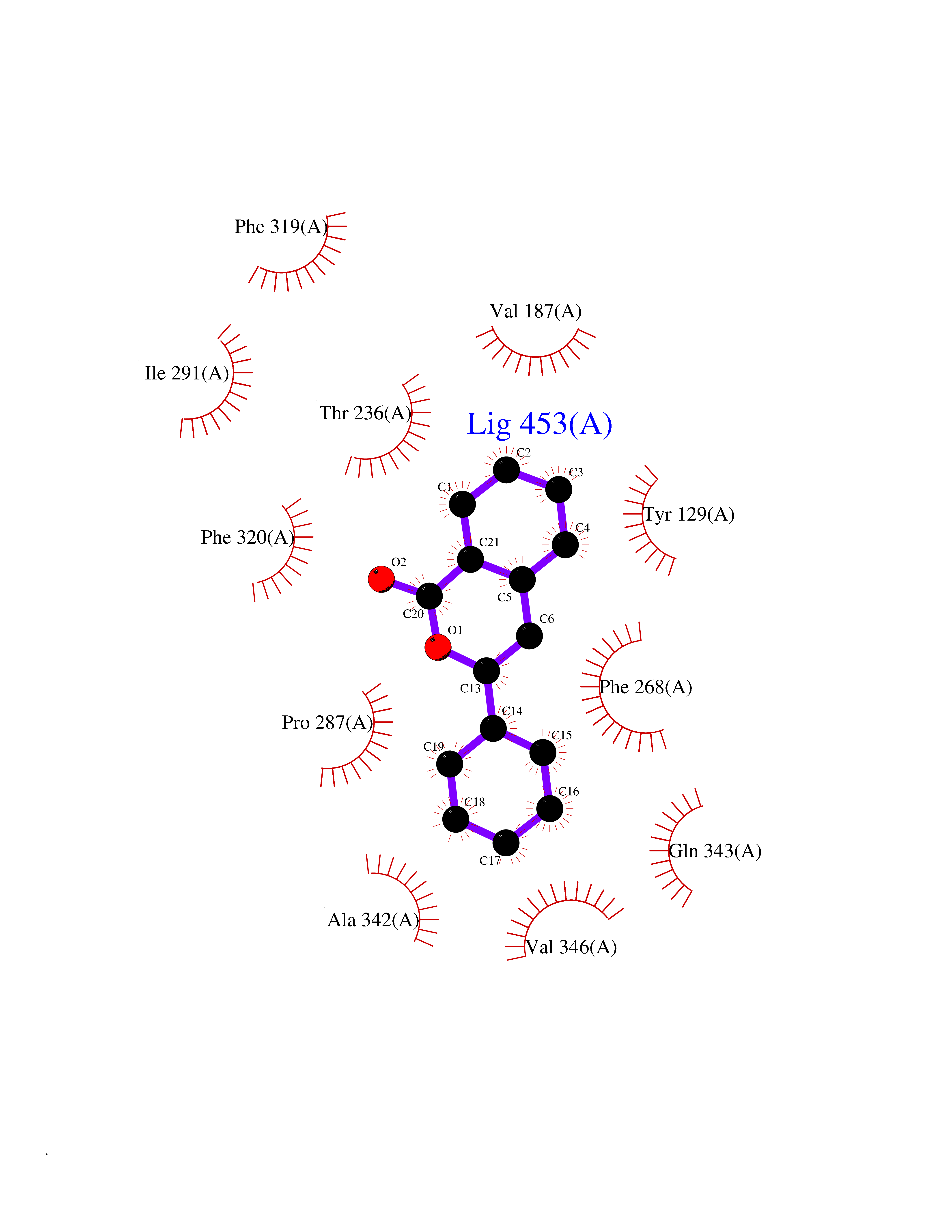 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Histamine N-methyltransferase (HNMT) | 2AOT | 7.63 | |
Target general information Gen name HNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine-N-methyltransferase; HNMT; HMT Protein family Class I-like SAM-binding methyltransferase superfamily, HNMT family Biochemical class Methyltransferase Function Inactivates histamine by N-methylation. Plays an important role in degrading histamine and in regulating the airway response to histamine. Related diseases Intellectual developmental disorder, autosomal recessive 51 (MRT51) [MIM:616739]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26206890}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) DB00613; DB13875; DB05381; DB04655; DB01103; DB01752; DB07106 Interacts with NA EC number EC 2.1.1.8 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32712 Length 288 Aromaticity 0.1 Instability index 36.38 Isoelectric point 5.18 Charge (pH=7) -9.97 2D Binding mode Binding energy (Kcal/mol) -10.41  Molscript Map 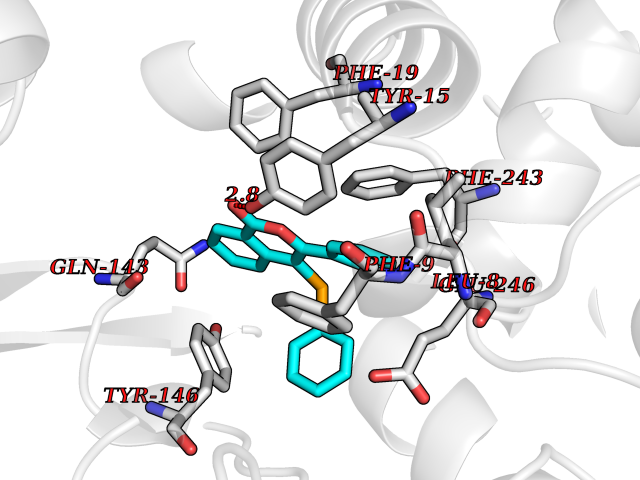 Pymol Map 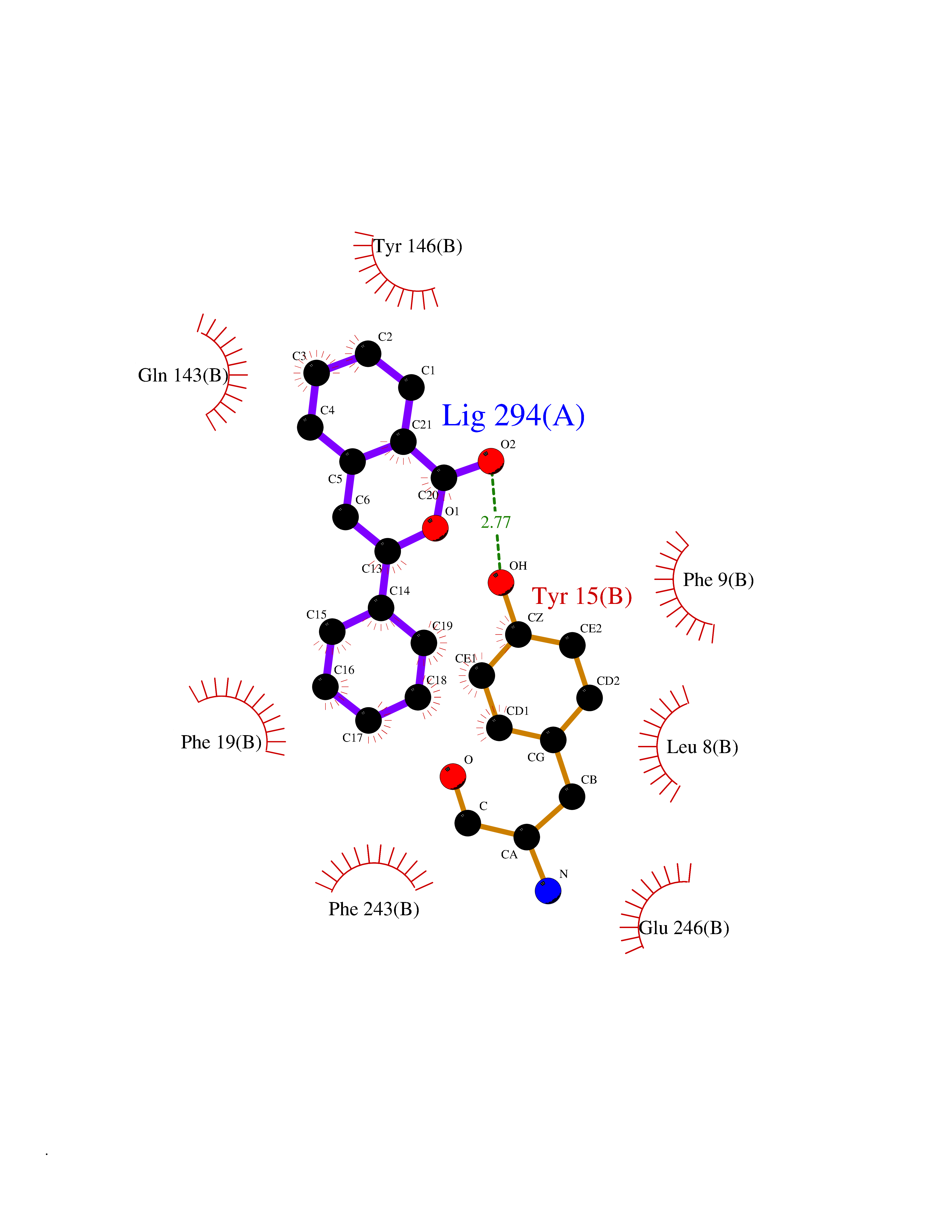 Ligplot Map 3D Binding mode Sequence MRSLFSDHGKYVESFRRFLNHSTEHQCMQEFMDKKLPGIIGRIGDTKSEIKILSIGGGAGEIDLQILSKVQAQYPGVXINNEVVEPSAEQIAKYKELVAKTSNLENVKFAWHKETSSEYQSRMLEKKELQKWDFIHMIQMLYYVKDIPATLKFFHSLLGTNAKMLIIVVSGSSGWDKLWKKYGSRFPQDDLCQYITSDDLTQMLDNLGLKYECYDLLSTMDISDCFIDGNENGDLLWDFLTETXNFNATAPPDLRAELGKDLQEPEFSAKKEGKVLFNNTLSFIVIEA Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Melatonin receptor type 1B (MTNR1B) | 6ME9 | 7.63 | |
Target general information Gen name MTNR1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1b receptor; Mel1b melatonin receptor; Mel-1B-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071; DB15133 Interacts with P28335; P48039; O76081; Q14669 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50184.9 Length 448 Aromaticity 0.11 Instability index 37.2 Isoelectric point 5.72 Charge (pH=7) -5.68 2D Binding mode Binding energy (Kcal/mol) -10.41 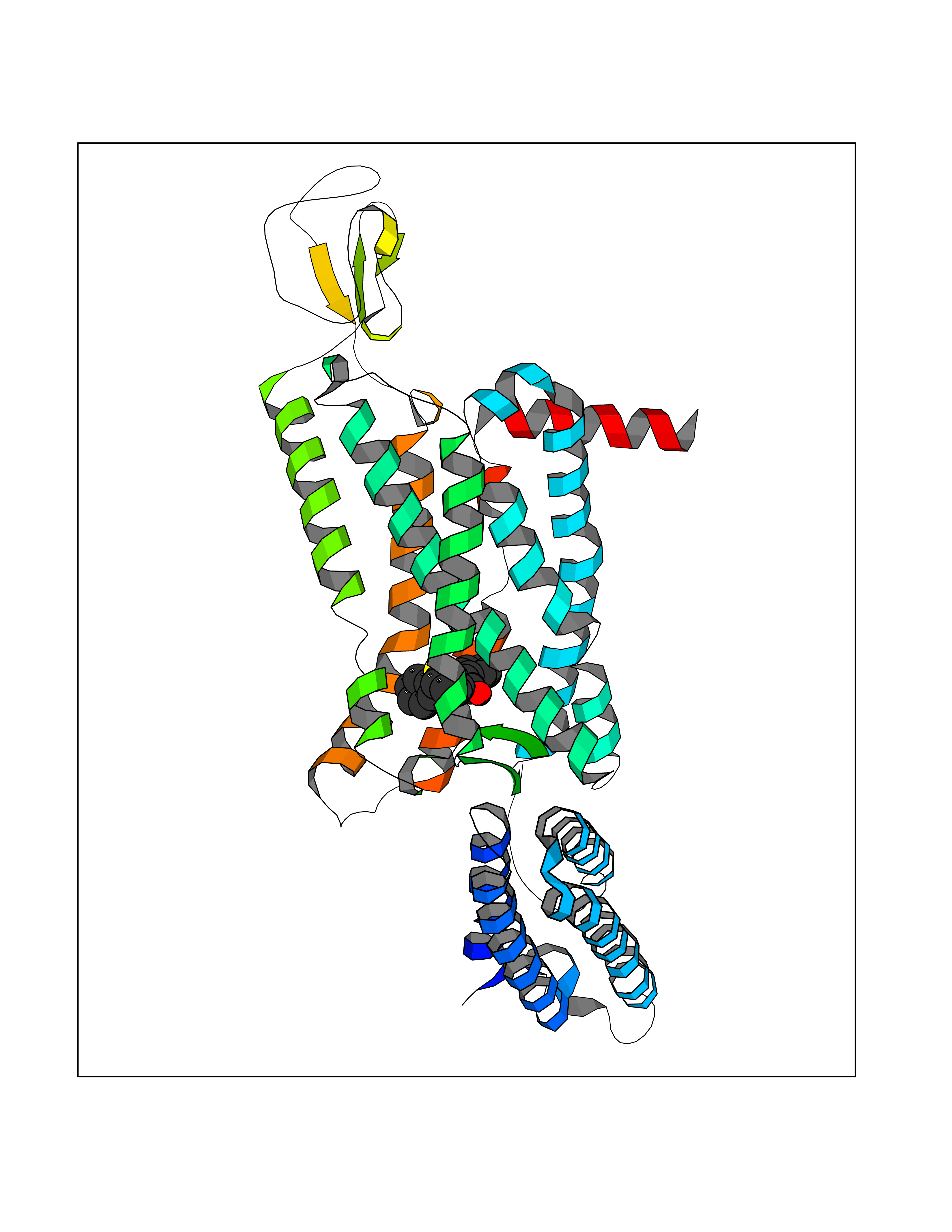 Molscript Map 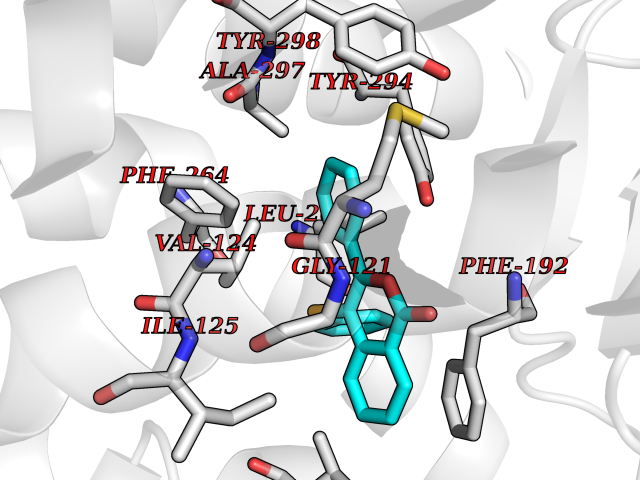 Pymol Map 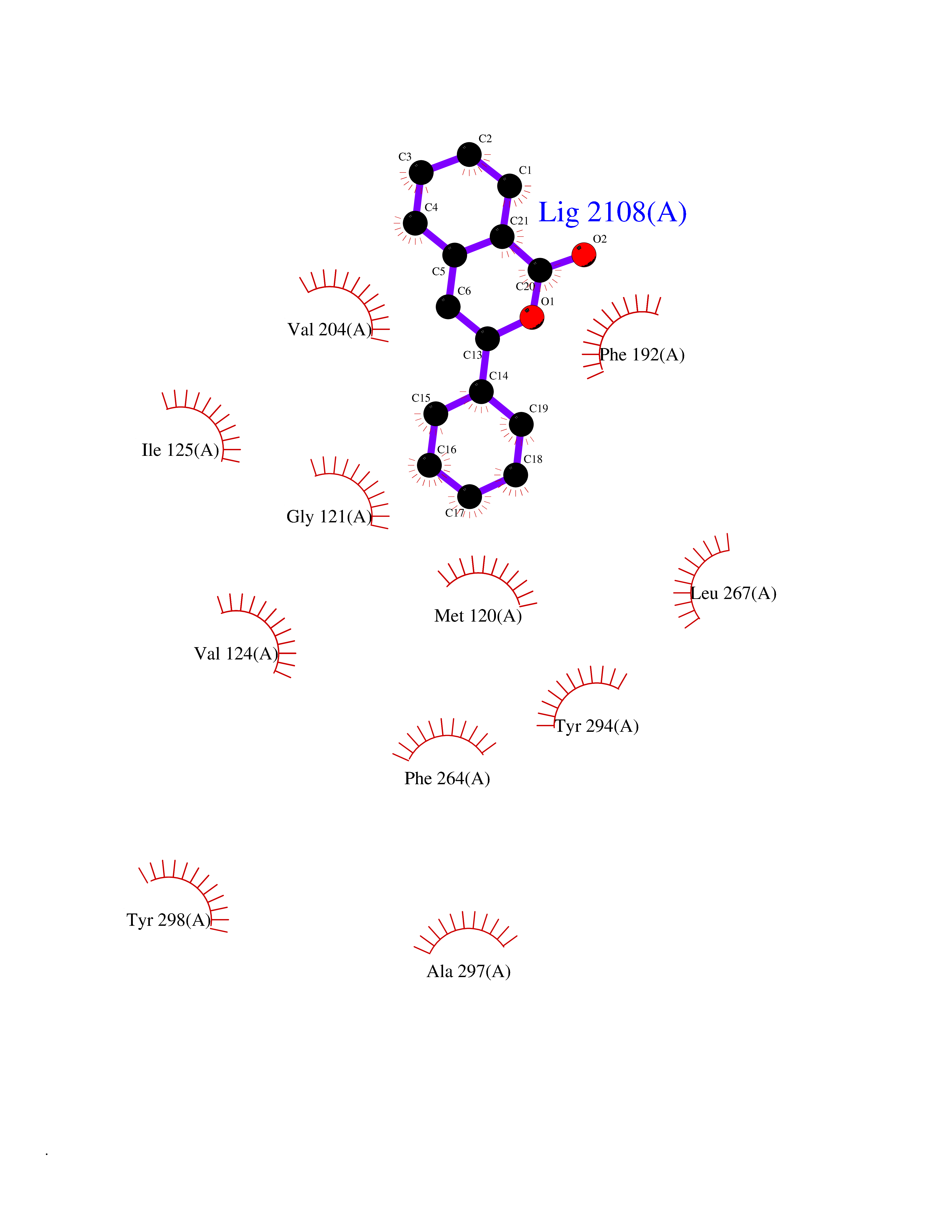 Ligplot Map 3D Binding mode Sequence ADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLGDGARPSWVAPALSAVLIVTTAVDVVGNLLVILSVLRNRKLRNAGNLFLVSLALANLVVAFYPYPLILVAIFYDGWAFGEEHCKASAFVMGLSVIGSVWNITAIAIDRYLYICHSMAYHRIYRRWHTPLHICLIWLLTVVALLPNFFVGSLEYDPRIYSCTFIQTASTQYTAAVVVIHFLLPIAVVSFCYLRIWVLVLQARMKKYTCTVCGYIYNPEDGDPDNGVNPGTDFKDIPDDWVCPLCGVGKDQFEEVECLKPSDLRSFLTMFVVFVIFAICFAPLNCIGLAVAINPQEMAPQIPEGLFVTSYLLAYFNSCLNPIVYGLLDQNFRREYKRILLALWN Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Multidrug efflux pump subunit AcrB | 5ENQ | 7.62 | |
Target general information Gen name acrB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0462;acrE;JW0451 Protein family Resistance-nodulation-cell division (RND) (TC 2.A.6) family Biochemical class Transport protein Function Drug:proton antiporter activity.Drug transmembrane transporter activity.Identical protein binding. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03619; DB04209; DB03825 Interacts with P31224; P0AAW9; P0ADZ7 EC number NA Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Membrane; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 60242.4 Length 553 Aromaticity 0.08 Instability index 29.1 Isoelectric point 4.76 Charge (pH=7) -18.86 2D Binding mode Binding energy (Kcal/mol) -10.4 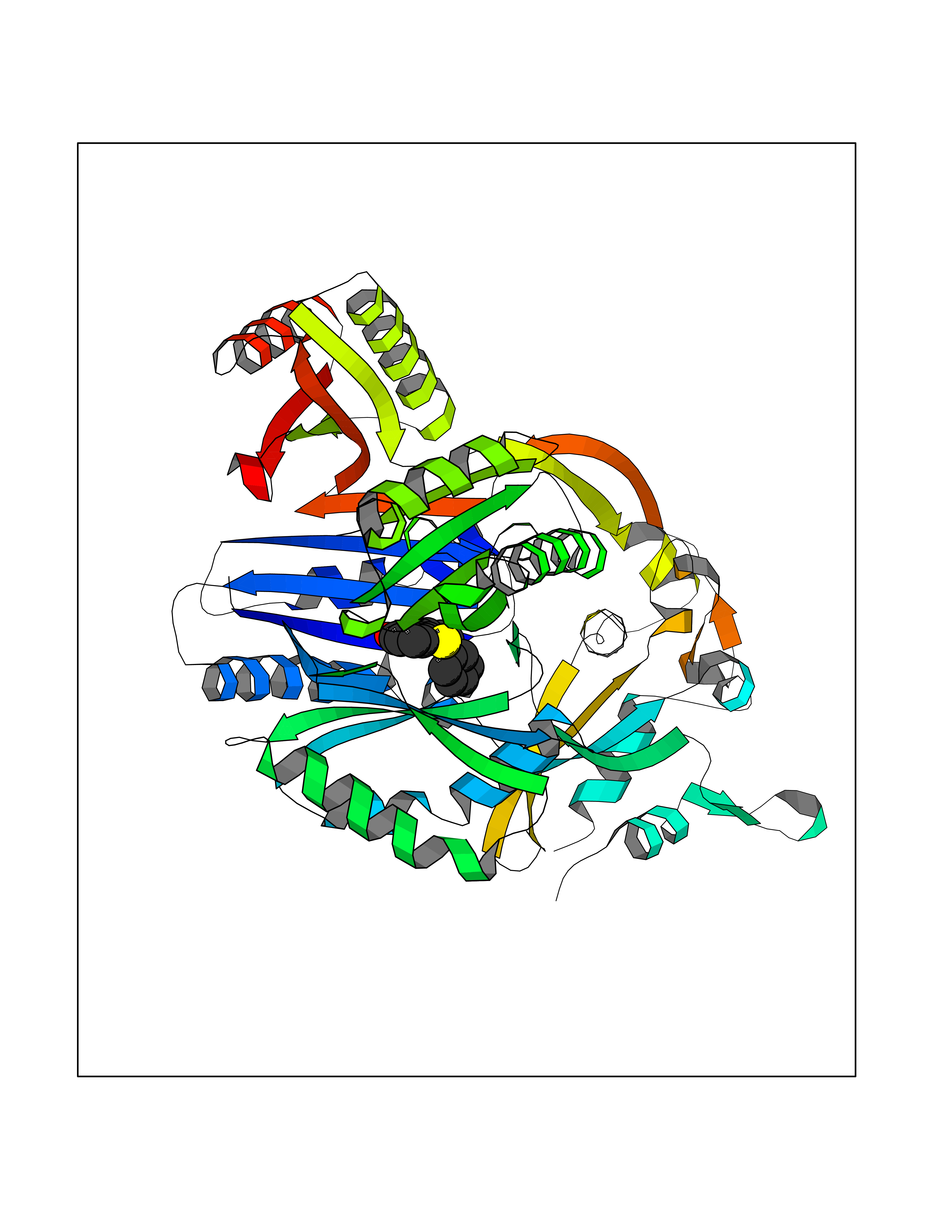 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence APPAVTISASYPGADAKTVQDTVTQVIEQNMNGIDNLMYMSSNSDSTGTVQITLTFESGTDADIAQVQVQNKLQLAMPLLPQEVQQQGVSVEKSSSSFLMVVGVINTDGTMTQEDISDYVAANMKDAISRTSGVGDVQLFGSQYAMRIWMNPNELNKFQLTPVDVITAIKAQNATRLTSTEEFGKILLKVNQDGSRVLLRDVAKIELGGENYDIIAEFNGQPASGLGIKLATGANALDTAAAIRAELAKMEPFFPSGLKIVYPYDTGVFMTMVQLPAGATQERTQKVLNEVTHYYLTKEKNNVESVFAVNGFGFAGRGQNTGIAFVSLKDWADRPGEENKVEAITMRATRAFSQIKDAMVFAFNLATGFDFELIDQAGLGHEKLTQARNQLLAEAAKHPDMLTSVRPNGLEDTPQFKIDIDQEKAQALGVSINDINTTLGAAWGGSYVNDFIDRGRVKKVYVMSEAKYRMLPDDIGDWYVRAADGQMVPFSAFSSSRWEYGSPRLERYNGLPSMEILGQAAPGKSTGEAMELMEQLASKLPTGVGYDWTGMSY Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Androgen receptor (AR) | 2AM9 | 7.62 | |
Target general information Gen name AR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Testosterone receptor; Nuclear receptor subfamily 3 group C member 4; NR3C4; Dihydrotestosterone receptor; DHTR Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Transcription factor activity is modulated by bound coactivator and corepressor proteins like ZBTB7A that recruits NCOR1 and NCOR2 to the androgen response elements/ARE on target genes, negatively regulating androgen receptor signaling and androgen-induced cell proliferation. Transcription activation is also down-regulated by NR0B2. Activated, but not phosphorylated, by HIPK3 and ZIPK/DAPK3. Steroid hormone receptors are ligand-activated transcription factors that regulate eukaryotic gene expression and affect cellular proliferation and differentiation in target tissues. Related diseases Androgen insensitivity syndrome (AIS) [MIM:300068]: An X-linked recessive form of pseudohermaphroditism due end-organ resistance to androgen. Affected males have female external genitalia, female breast development, blind vagina, absent uterus and female adnexa, and abdominal or inguinal testes, despite a normal 46,XY karyotype. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10221770, ECO:0000269|PubMed:10404311, ECO:0000269|PubMed:10458483, ECO:0000269|PubMed:10571951, ECO:0000269|PubMed:10590024, ECO:0000269|PubMed:10690872, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:11744994, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1426313, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:1464650, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:1480178, ECO:0000269|PubMed:1487249, ECO:0000269|PubMed:1569163, ECO:0000269|PubMed:1609793, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:16595706, ECO:0000269|PubMed:1775137, ECO:0000269|PubMed:1999491, ECO:0000269|PubMed:2082179, ECO:0000269|PubMed:2594783, ECO:0000269|PubMed:7537149, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7633398, ECO:0000269|PubMed:7641413, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7962294, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:7981689, ECO:0000269|PubMed:7993455, ECO:0000269|PubMed:8040309, ECO:0000269|PubMed:8096390, ECO:0000269|PubMed:8103398, ECO:0000269|PubMed:8162033, ECO:0000269|PubMed:8224266, ECO:0000269|PubMed:8281140, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8339746, ECO:0000269|PubMed:8413310, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8626869, ECO:0000269|PubMed:8647313, ECO:0000269|PubMed:8683794, ECO:0000269|PubMed:8723113, ECO:0000269|PubMed:8768864, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8830623, ECO:0000269|PubMed:8918984, ECO:0000269|PubMed:8990010, ECO:0000269|PubMed:9001799, ECO:0000269|PubMed:9007482, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9106550, ECO:0000269|PubMed:9160185, ECO:0000269|PubMed:9252933, ECO:0000269|PubMed:9255042, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9328206, ECO:0000269|PubMed:9544375, ECO:0000269|PubMed:9554754, ECO:0000269|PubMed:9610419, ECO:0000269|PubMed:9627582, ECO:0000269|PubMed:9698822, ECO:0000269|PubMed:9851768, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.116, ECO:0000269|Ref.182}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal and bulbar muscular atrophy X-linked 1 (SMAX1) [MIM:313200]: An X-linked recessive form of spinal muscular atrophy. Spinal muscular atrophy refers to a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAX1 occurs only in men. Age at onset is usually in the third to fifth decade of life, but earlier involvement has been reported. It is characterized by slowly progressive limb and bulbar muscle weakness with fasciculations, muscle atrophy, and gynecomastia. The disorder is clinically similar to classic forms of autosomal spinal muscular atrophy. {ECO:0000269|PubMed:15851746}. The disease is caused by variants affecting the gene represented in this entry. Caused by trinucleotide CAG repeat expansion. In SMAX1 patients the number of Gln ranges from 38 to 62. Longer expansions result in earlier onset and more severe clinical manifestations of the disease.; DISEASE: Prostate cancer, hereditary, X-linked 3 (HPCX3) [MIM:301120]: A condition associated with familial predisposition to cancer of the prostate. Most prostate cancers are adenocarcinomas that develop in the acini of the prostatic ducts. Other rare histopathologic types of prostate cancer that occur in approximately 5% of patients include small cell carcinoma, mucinous carcinoma, prostatic ductal carcinoma, transitional cell carcinoma, squamous cell carcinoma, basal cell carcinoma, adenoid cystic carcinoma (basaloid), signet-ring cell carcinoma and neuroendocrine carcinoma. {ECO:0000269|PubMed:8530589}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Defects in AR may play a role in metastatic prostate cancer. The mutated receptor stimulates prostate growth and metastases development despite of androgen ablation. This treatment can reduce primary and metastatic lesions probably by inducing apoptosis of tumor cells when they express the wild-type receptor. {ECO:0000269|PubMed:10363963, ECO:0000269|PubMed:10569618, ECO:0000269|PubMed:1562539, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:17311914, ECO:0000269|PubMed:2260966, ECO:0000269|PubMed:25091737, ECO:0000269|PubMed:8187068, ECO:0000269|PubMed:8274409, ECO:0000269|PubMed:8827083}.; DISEASE: Androgen insensitivity, partial (PAIS) [MIM:312300]: A disorder that is characterized by hypospadias, hypogonadism, gynecomastia, genital ambiguity, normal XY karyotype, and a pedigree pattern consistent with X-linked recessive inheritance. Some patients present azoospermia or severe oligospermia without other clinical manifestations. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10470409, ECO:0000269|PubMed:10502786, ECO:0000269|PubMed:10543676, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:1303262, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1424203, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:2010552, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7649358, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7909256, ECO:0000269|PubMed:7910529, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:8033918, ECO:0000269|PubMed:8097257, ECO:0000269|PubMed:8126121, ECO:0000269|PubMed:8205256, ECO:0000269|PubMed:8281139, ECO:0000269|PubMed:8325932, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8550758, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8823308, ECO:0000269|PubMed:8824883, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9196614, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9329414, ECO:0000269|PubMed:9543136, ECO:0000269|PubMed:9607727, ECO:0000269|PubMed:9768671, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.124}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypospadias 1, X-linked (HYSP1) [MIM:300633]: A common malformation in which the urethra opens on the ventral side of the penis, due to developmental arrest of urethral fusion. The opening can be located glandular, penile, or even more posterior in the scrotum or perineum. Hypospadias is a feature of several syndromic disorders, including the androgen insensitivity syndrome and Opitz syndrome. {ECO:0000269|PubMed:8097257}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07422; DB07039; DB04709; DB07717; DB07454; DB02932; DB08035; DB01481; DB08088; DB08461; DB08087; DB07421; DB01063; DB07423; DB11901; DB01128; DB07286; DB01541; DB14639; DB01564; DB12499; DB04839; DB01406; DB12941; DB09123; DB00255; DB06133; DB01395; DB00858; DB15488; DB11219; DB08899; DB13155; DB00655; DB09086; DB02266; DB01185; DB00623; DB00499; DB11619; DB11064; DB01026; DB15647; DB00367; DB08089; DB05234; DB13934; DB11425; DB06710; DB02998; DB11429; DB00648; DB08804; DB00984; DB00665; DB06713; DB00717; DB09371; DB00957; DB09389; DB00621; DB01428; DB06412; DB01608; DB11447; DB01708; DB00396; DB07419; DB07769; DB14583; DB00421; DB02901; DB13951; DB06718; DB00675; DB00624; DB13943; DB13944; DB01420; DB13946; DB06870; DB08604; DB08867 Interacts with P00519; Q9UBL3; P51451; Q8WV28; O60885-1; P78543; Q14790; P24385; Q92793; O14595; P35222; Q9UER7; P20711; P11308; P07332; P09769; Q02790; P55317; O75593; Q14451; P06396; P56524; Q16665; Q16666; O15357; Q15652; O95251; Q9BY66; Q9BY66-3; Q03164; O14686; P06239; P07948; P20794; P42679; Q00987; Q15596; Q14686; O96028; Q99497; P27986; O00459; Q92569; P19174; P16885; Q06830; P78527; Q06124; P20936; Q9UBS8; Q9Y252; O14796; Q9NP31; P29353; Q6S5L8; Q5VZ18; Q15797; O14544; P12931; Q9ULZ2; P63165; Q9HBL0; P07947; Q9R1E0; Q06986 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Isopeptide bond; Lipid-binding; Lipoprotein; Metal-binding; Neurodegeneration; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Pseudohermaphroditism; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Triplet repeat expansion; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29137.9 Length 250 Aromaticity 0.11 Instability index 42.11 Isoelectric point 8.94 Charge (pH=7) 5.43 2D Binding mode Binding energy (Kcal/mol) -10.39  Molscript Map  Pymol Map 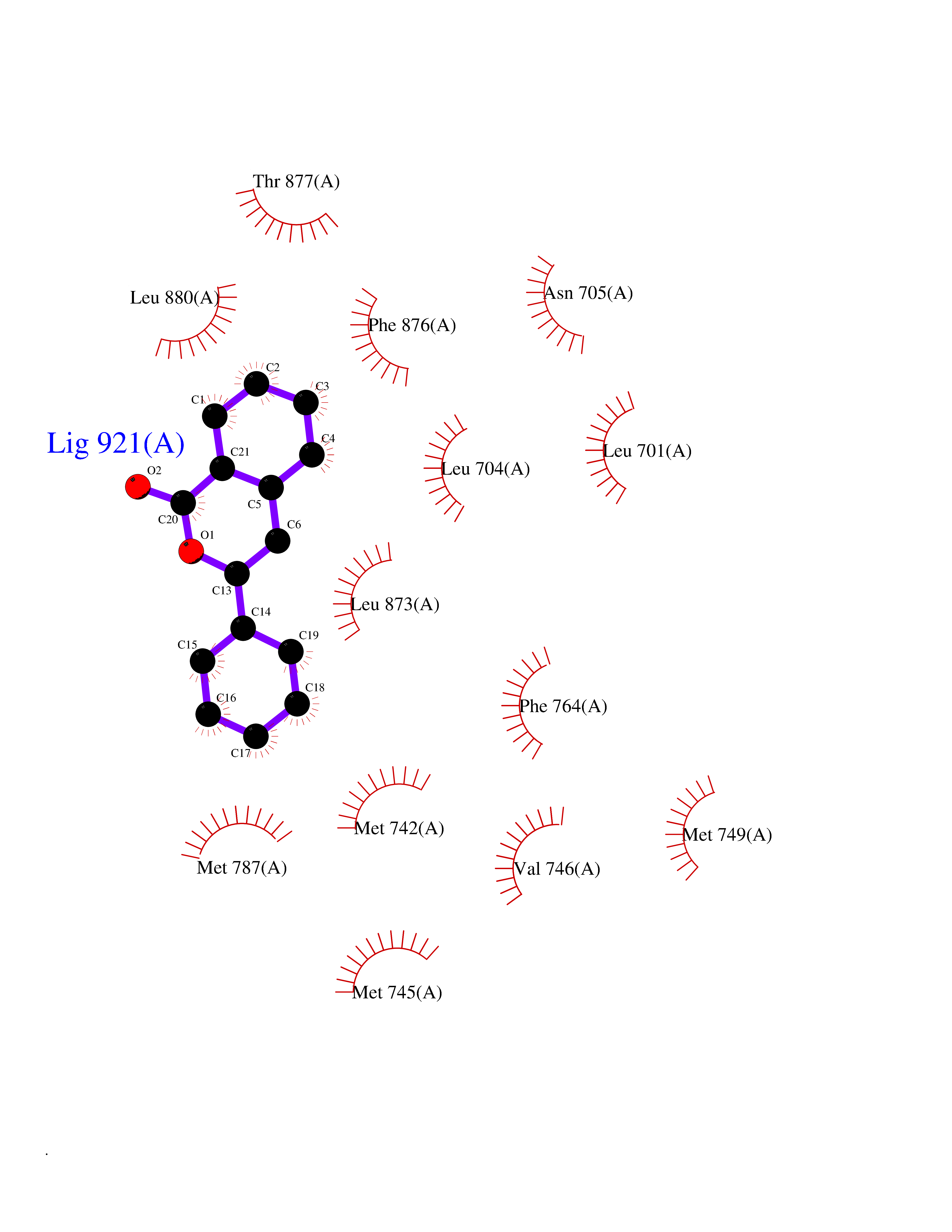 Ligplot Map 3D Binding mode Sequence QPIFLNVLEAIEPGVVCAGHDNNQPDSFAALLSSLNELGERQLVHVVKWAKALPGFRNLHVDDQMAVIQYSWMGLMVFAMGWRSFTNVNSRMLYFAPDLVFNEYRMHKSRMYSQCVRMRHLSQEFGWLQITPQEFLCMKALLLFSIIPVDGLKNQKFFDELRMNYIKELDRIIACKRKNPTSCSRRFYQLTKLLDSVQPIARELHQFTFDLLIKSHMVSVDFPEMMAEIISVQVPKILSGKVKPIYFHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Sphingosine-1-phosphate receptor 5 (S1PR5) | 7EW1 | 7.62 | |
Target general information Gen name S1PR5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-8; S1PR5; S1P5; S1P receptor Edg-8; S1P receptor 5; Endothelial differentiation G-protein-coupled receptor 8 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the lysosphingolipid sphingosine 1- phosphate (S1P). S1P is a bioactive lysophospholipid that elicits diverse physiological effect on most types of cells and tissues. Is coupled to both the G(i/0)alpha and G(12) subclass of heteromeric G-proteins. May play a regulatory role in the transformation of radial glial cells into astrocytes and may affect proliferative activity of these cells. Related diseases Hereditary non-polyposis colorectal cancer 6 (HNPCC6) [MIM:614331]: An autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. HNPCC is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, HNPCC is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical HNPCC is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected HNPCC' or 'incomplete HNPCC' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:9590282}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Esophageal cancer (ESCR) [MIM:133239]: A malignancy of the esophagus. The most common types are esophageal squamous cell carcinoma and adenocarcinoma. Cancer of the esophagus remains a devastating disease because it is usually not detected until it has progressed to an advanced incurable stage. {ECO:0000269|PubMed:10789724}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Loeys-Dietz syndrome 2 (LDS2) [MIM:610168]: An aortic aneurysm syndrome with widespread systemic involvement, characterized by arterial tortuosity and aneurysms, hypertelorism, and bifid uvula or cleft palate. Physical findings include prominent joint laxity, easy bruising, wide and atrophic scars, velvety and translucent skin with easily visible veins, spontaneous rupture of the spleen or bowel, and catastrophic complications of pregnancy, including rupture of the gravid uterus and the arteries, either during pregnancy or in the immediate postpartum period. Some patients have craniosynostosis, exotropy, micrognathia and retrognathia, structural brain abnormalities, and intellectual deficit. {ECO:0000269|PubMed:15235604, ECO:0000269|PubMed:15731757, ECO:0000269|PubMed:16027248, ECO:0000269|PubMed:16251899, ECO:0000269|PubMed:19533785, ECO:0000269|PubMed:19883511, ECO:0000269|PubMed:20101701, ECO:0000269|PubMed:20358619, ECO:0000269|PubMed:21949523, ECO:0000269|PubMed:22113417}. The disease is caused by variants affecting the gene represented in this entry. TGFBR2 mutations Cys-460 and His-460 have been reported to be associated with thoracic aortic aneurysms and dissection (TAAD). This phenotype, also known as thoracic aortic aneurysms type 3 (AAT3), is distinguised from LDS2 by having aneurysms restricted to thoracic aorta. As individuals carrying these mutations also exhibit descending aortic disease and aneurysms of other arteries (PubMed:16027248), they have been considered as LDS2 by the OMIM resource. {ECO:0000269|PubMed:16027248}. Drugs (DrugBank ID) DB14766; DB08868; DB12612; DB12371 Interacts with P48165; Q8WWF3 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 28613.3 Length 269 Aromaticity 0.08 Instability index 43.53 Isoelectric point 9.76 Charge (pH=7) 13.81 2D Binding mode Binding energy (Kcal/mol) -10.39 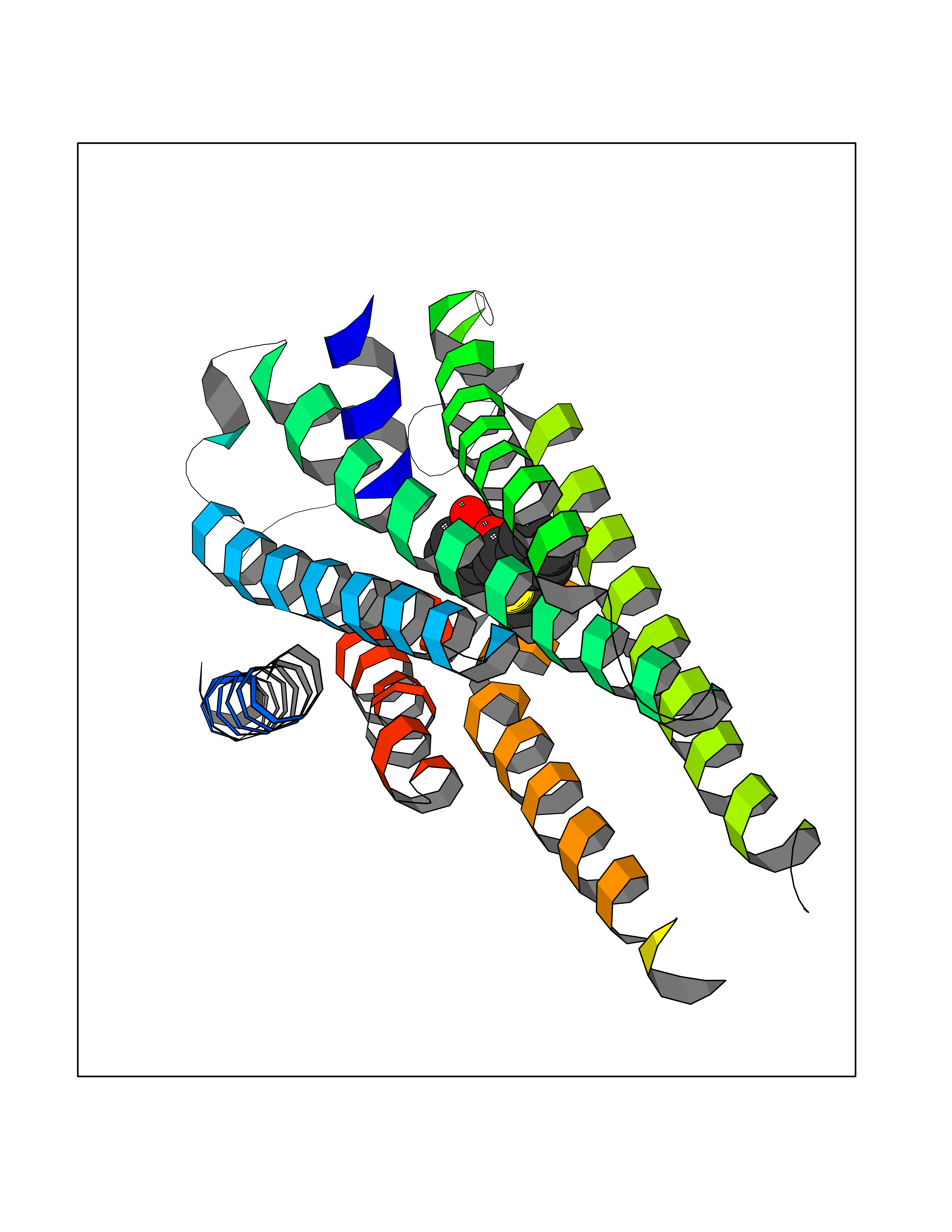 Molscript Map 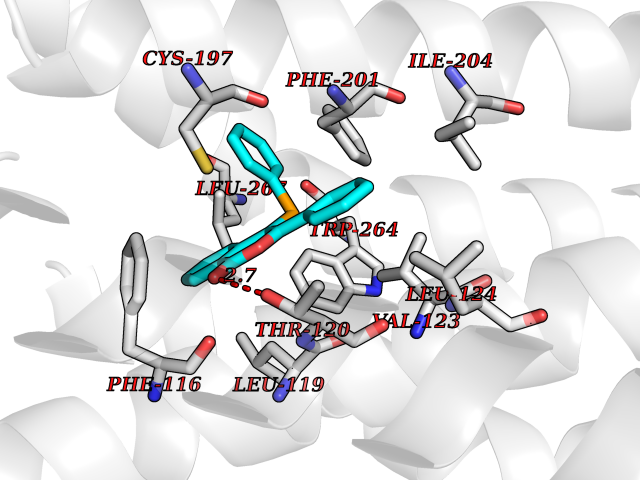 Pymol Map 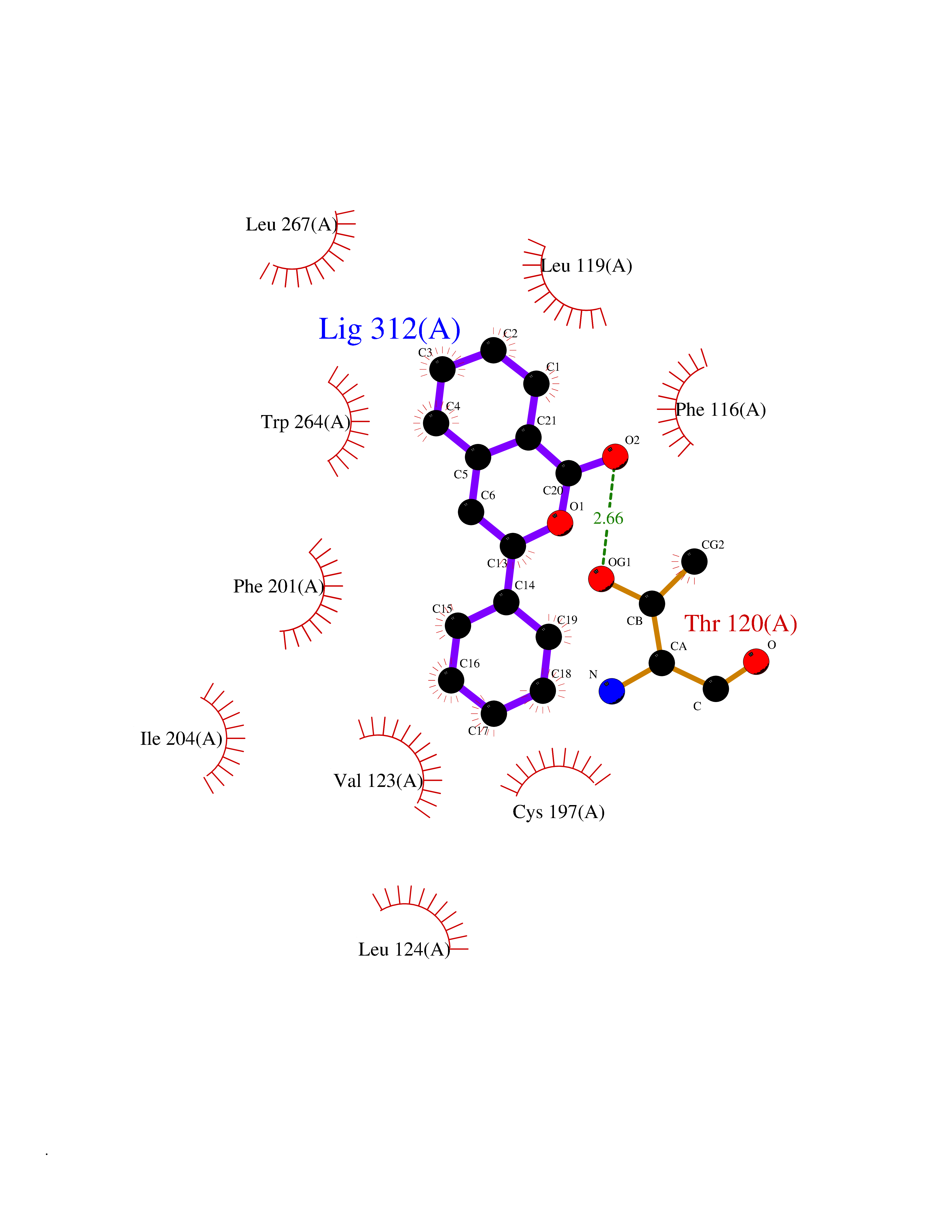 Ligplot Map 3D Binding mode Sequence VIVLHYNYTGKLRGRADAVVCLAVCAFIVLENLAVLLVLGRHAPMFLLLGSLTLSDLLAGAAYAANILLSGPLTLKLSPALWFAREGGVFVALTASVLSLLAIALERSLTMARRGPAPVSSRGRTLAMAAAAWGVSLLLGLLPALGWNCLGRLDACSTVLPLYAKAYVLFCVLAFVGILAAICALYARIYCQVRANARRLRKPRSLALLRTLSVVLLAFVACWGPLFLLLLLDVACPARTCPVLLQADPFLGLAMANSLLNPIIYTLTN Hydrogen bonds contact Hydrophobic contact | ||||