Job Results:
Ligand
Structure
Job ID
176b63e19640f4b1f79a8fdfdb32fb77
Job name
NA
Time
2025-10-13 17:39:01
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Histone-lysine N-methyltransferase KMT5B (KMT5B) | 3S8P | 5.85 | |
Target general information Gen name KMT5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5B; Lysine-specific methyltransferase 5B; Suppressor of variegation 4-20 homolog 1; Su(var)4-20 homolog 1; Suv4-20h1; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5B is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Plays a role in myogenesis by regulating the expression of target genes, such as EID3. Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Intellectual developmental disorder, autosomal dominant 51 (MRD51) [MIM:617788]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:28191889, ECO:0000269|PubMed:29276005}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H2G4; Q61026 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Myogenesis; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 26230.2 Length 233 Aromaticity 0.11 Instability index 42.29 Isoelectric point 5.64 Charge (pH=7) -6.07 2D Binding mode Binding energy (Kcal/mol) -7.99  Molscript Map 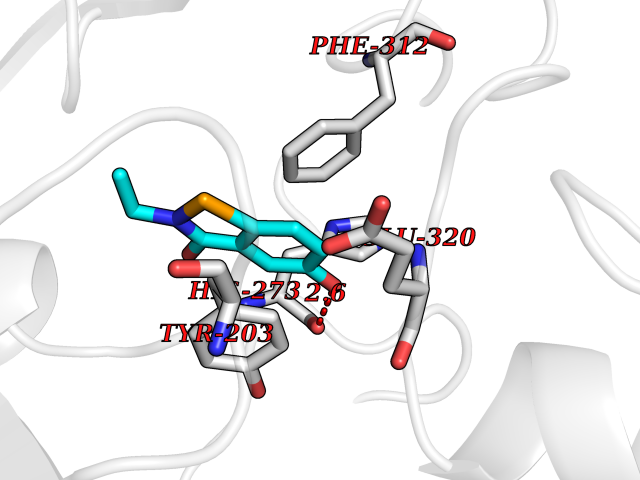 Pymol Map 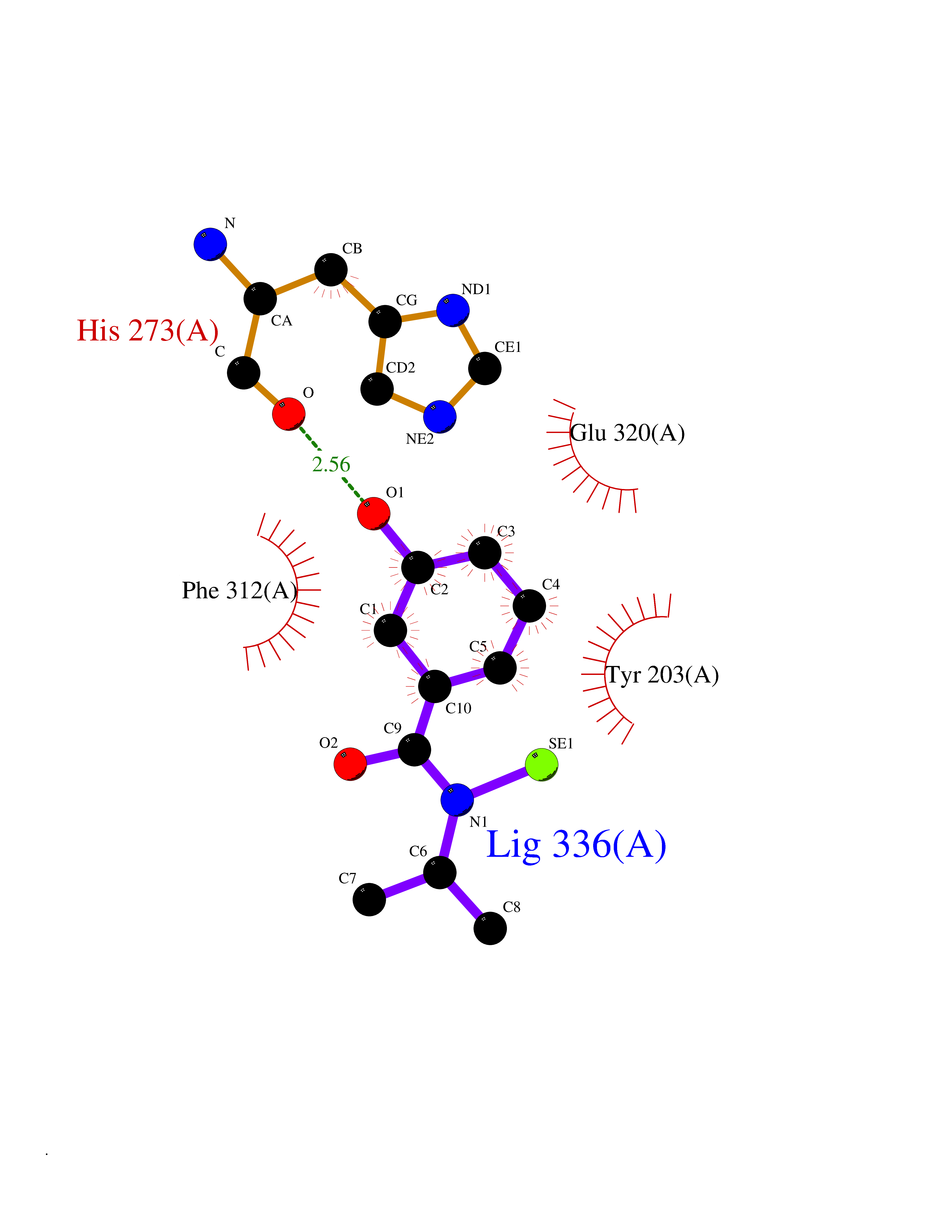 Ligplot Map 3D Binding mode Sequence XSAKELCENDDLATSLVLDPYLGFQTHKXNTRQEELKEVIERFKKDEHLEKAFKCLTSGEWARHYFLNKNKXQEKLFKEHVFIYLRXFATDSGFEILPCNRYSSEQNGAKIVATKEWKRNDKIELLVGCIAELSEIEENXLLRHGENDFSVXYSTRKNCAQLWLGPAAFINHDCRPNCKFVSTGRDTACVKALRDIEPGEEISCYYGDGFFGENNEFCECYTCERRGTGAFKS Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Macrophage migration inhibitory factor (MIF) | 3IJJ | 5.85 | |
Target general information Gen name MIF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phenylpyruvate tautomerase; MMIF; L-dopachrome tautomerase; L-dopachrome isomerase; Glycosylation-inhibiting factor; GLIF; GIF Protein family MIF family Biochemical class Intramolecular oxidoreductase Function Involved in the innate immune response to bacterial pathogens. The expression of MIF at sites of inflammation suggests a role as mediator in regulating the function of macrophages in host defense. Counteracts the anti-inflammatory activity of glucocorticoids. Has phenylpyruvate tautomerase and dopachrome tautomerase activity (in vitro), but the physiological substrate is not known. It is not clear whether the tautomerase activity has any physiological relevance, and whether it is important for cytokine activity. Pro-inflammatory cytokine. Related diseases Rheumatoid arthritis systemic juvenile (RASJ) [MIM:604302]: An inflammatory articular disorder with systemic onset beginning before the age of 16. It represents a subgroup of juvenile arthritis associated with severe extraarticular features and occasionally fatal complications. During active phases of the disorder, patients display a typical daily spiking fever, an evanescent macular rash, lymphadenopathy, hepatosplenomegaly, serositis, myalgia and arthritis. {ECO:0000269|PubMed:11508429}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01880; DB07888; DB08334; DB08335; DB08333; DB07718; DB08765; DB02728 Interacts with O43521-2; P00533; Q92743; P14174; Q96HA8 EC number EC 5.3.2.1 Uniprot keywords 3D-structure; Acetylation; Cytokine; Cytoplasm; Direct protein sequencing; Immunity; Inflammatory response; Innate immunity; Isomerase; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,C Molecular weight (Da) 24671.9 Length 228 Aromaticity 0.09 Instability index 31.45 Isoelectric point 8.37 Charge (pH=7) 2.26 2D Binding mode Binding energy (Kcal/mol) -7.98 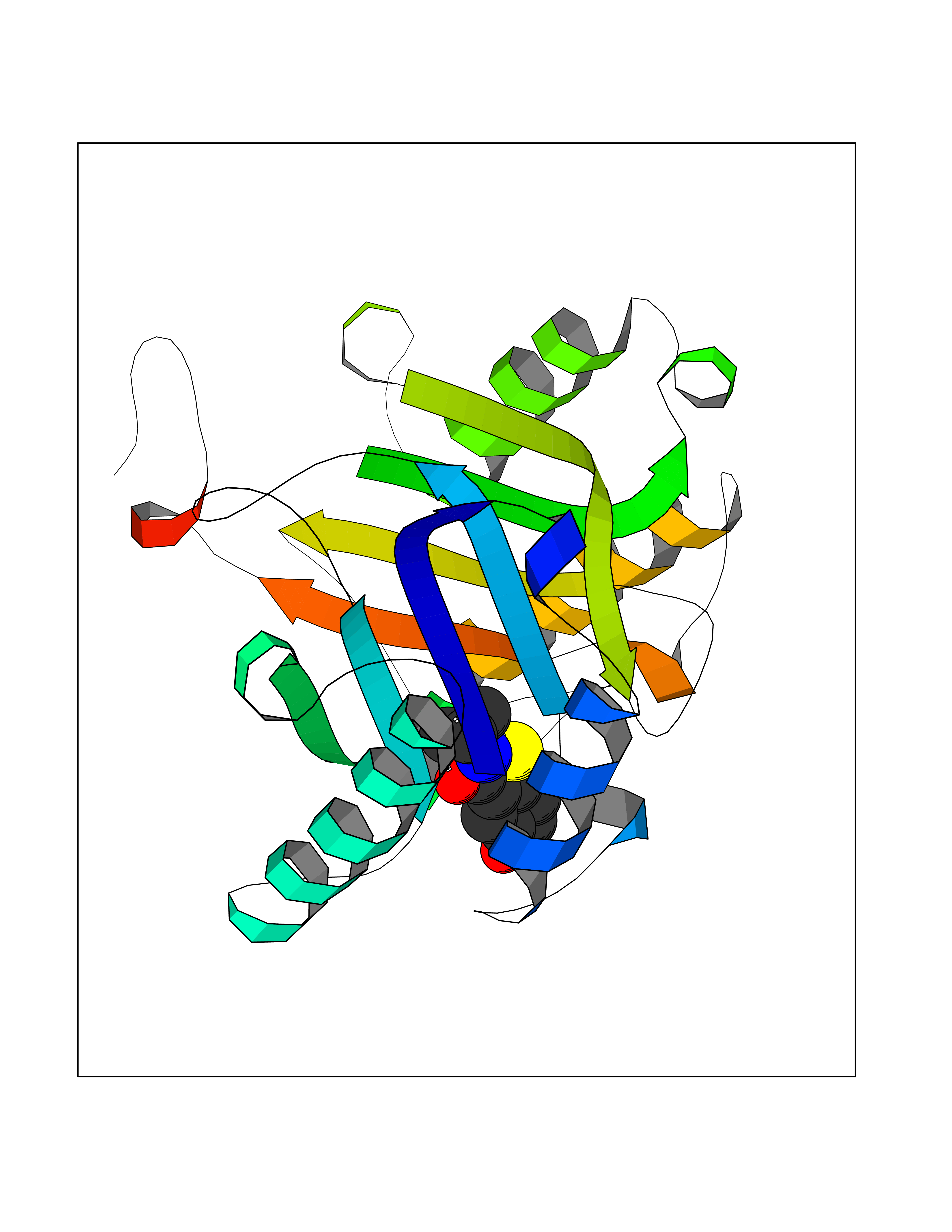 Molscript Map 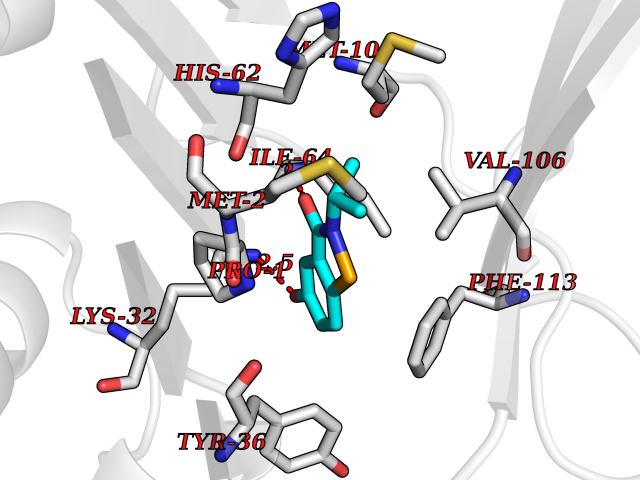 Pymol Map 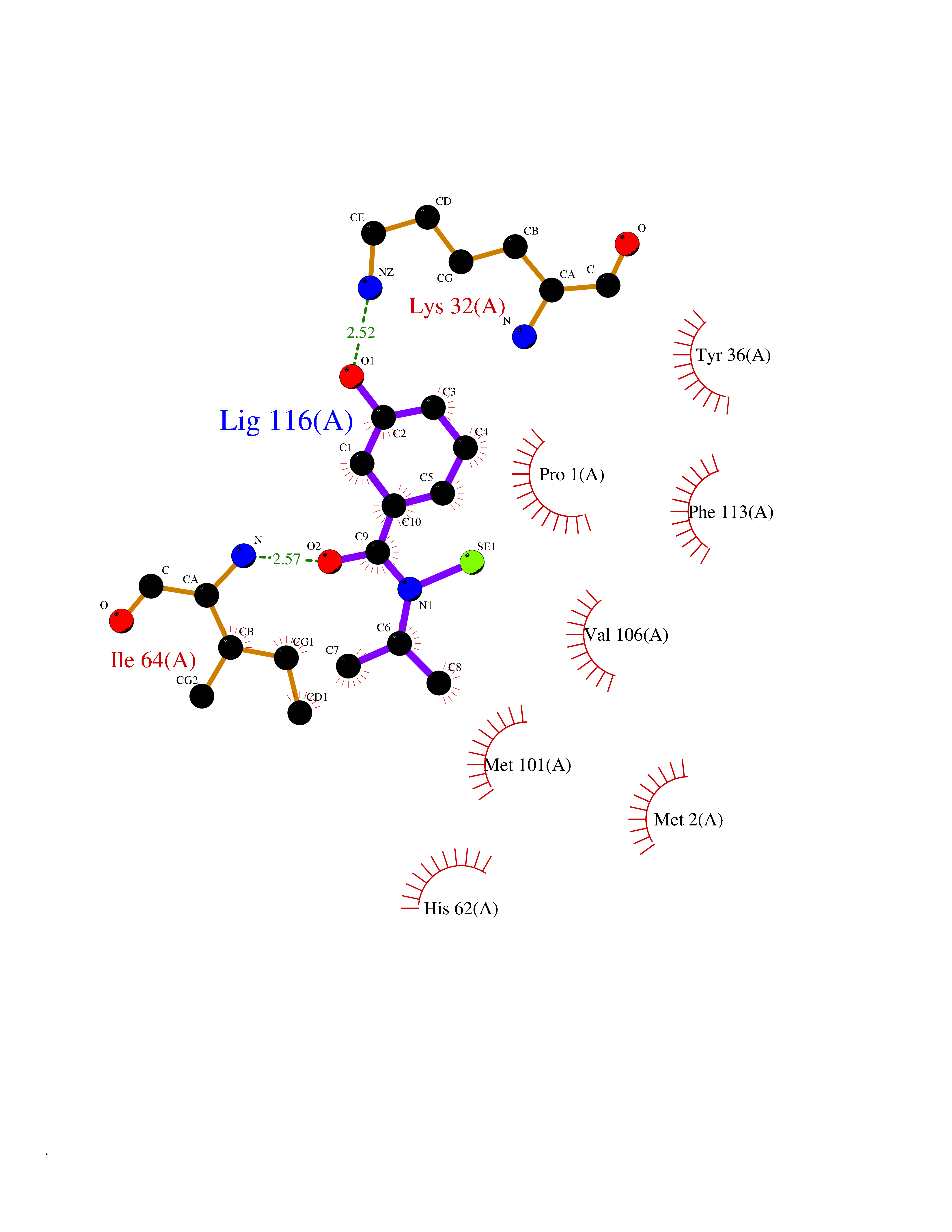 Ligplot Map 3D Binding mode Sequence PMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFAPMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFA Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Bacterial Threonine deaminase (Bact ilvA) | 1TDJ | 5.85 | |
Target general information Gen name Bact ilvA Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms ilvA; Putative threonine dehydratase Protein family Serine/threonine dehydratase family Biochemical class Carbon-nitrogen lyases Function Catalyzes the anaerobic formation of alpha-ketobutyrate and ammonia from threonine in a two-step reaction. The first step involved a dehydration of threonine and a production of enamine intermediates (aminocrotonate), which tautomerizes to its imine form(iminobutyrate). Both intermediates are unstable and short- lived. The second step is the nonenzymatic hydrolysis of the enamine/imine intermediates to form 2-ketobutyrate and free ammonia. In the low water environment of the cell, the second step is accelerated by RidA. Related diseases Familial male precocious puberty (FMPP) [MIM:176410]: In FMPP the receptor is constitutively activated. {ECO:0000269|PubMed:11134146, ECO:0000269|PubMed:11391350, ECO:0000269|PubMed:7629248, ECO:0000269|PubMed:7692306, ECO:0000269|PubMed:7714085, ECO:0000269|PubMed:7757065, ECO:0000269|PubMed:8281137, ECO:0000269|PubMed:8829636, ECO:0000269|PubMed:8929952, ECO:0000269|PubMed:9467560, ECO:0000269|PubMed:9661624}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Luteinizing hormone resistance (LHR) [MIM:238320]: An autosomal recessive disorder characterized by unresponsiveness to luteinizing hormone, defective sexual development in males, and defective follicular development and ovulation, amenorrhea and infertility in females. Two forms of the disorder have been defined in males. Type 1 is a severe form characterized by complete 46,XY male pseudohermaphroditism, low testosterone and high luteinizing hormone levels, total lack of responsiveness to luteinizing and chorionic gonadotropin hormones, lack of breast development, and absent development of secondary male sex characteristics. Type 2, a milder form, displays a broader range of phenotypic expression ranging from micropenis to severe hypospadias. {ECO:0000269|PubMed:12050206, ECO:0000269|PubMed:15372531, ECO:0000269|PubMed:15472221, ECO:0000269|PubMed:19551906, ECO:0000269|PubMed:7719343, ECO:0000269|PubMed:8559204, ECO:0000269|PubMed:9215288, ECO:0000269|PubMed:9514160, ECO:0000269|PubMed:9626144, ECO:0000269|PubMed:9626653}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 4.3.1.19 Uniprot keywords 3D-structure; Allosteric enzyme; Amino-acid biosynthesis; Branched-chain amino acid biosynthesis; Isoleucine biosynthesis; Lyase; Pyridoxal phosphate; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 53966.2 Length 494 Aromaticity 0.08 Instability index 41.16 Isoelectric point 5.88 Charge (pH=7) -8.91 2D Binding mode Binding energy (Kcal/mol) -7.98 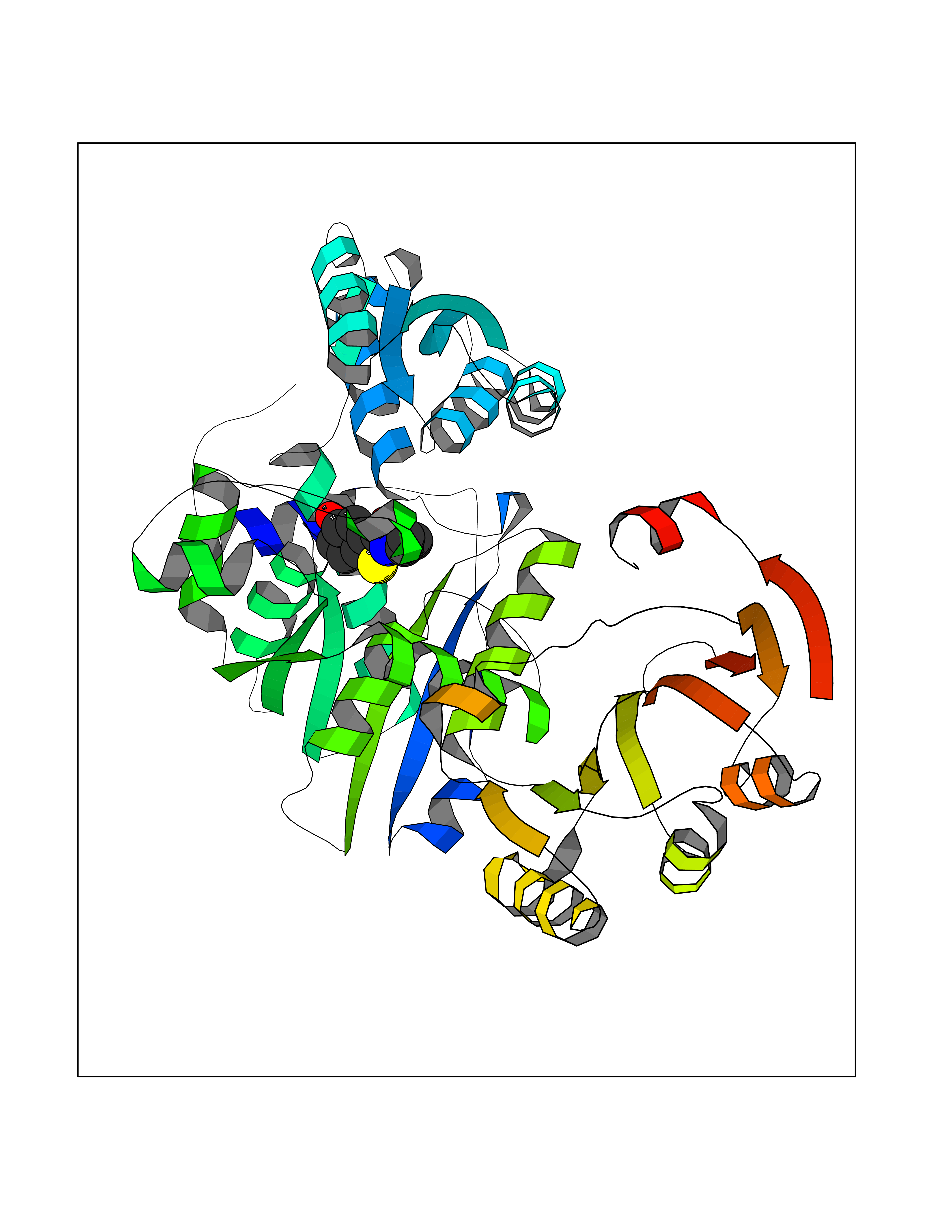 Molscript Map 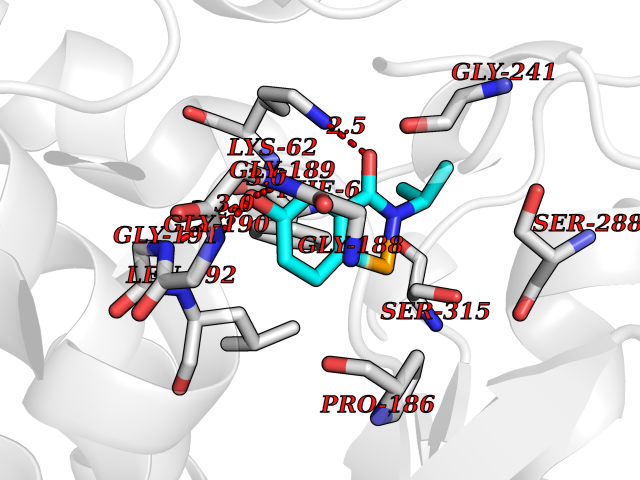 Pymol Map 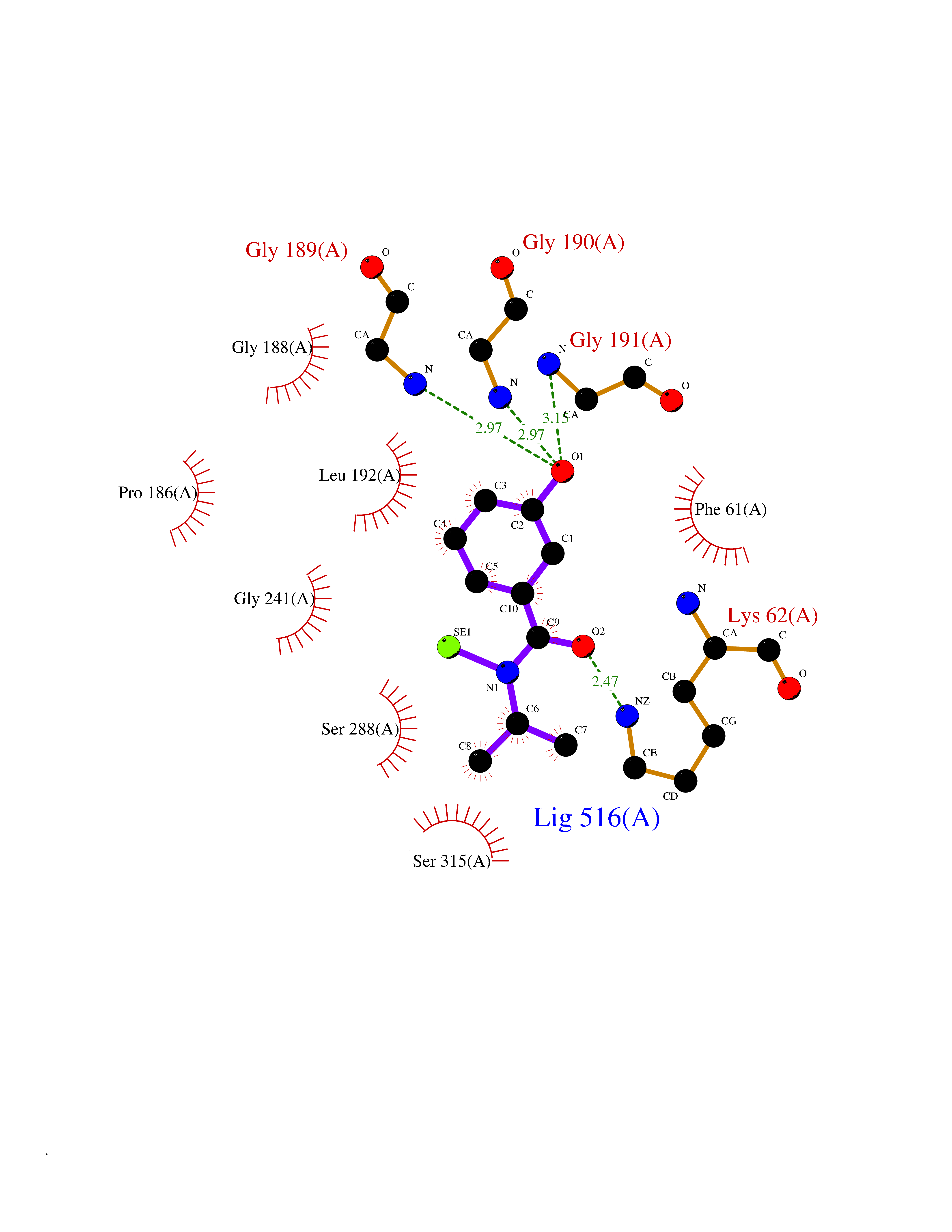 Ligplot Map 3D Binding mode Sequence QPLSGAPEGAEYLRAVLRAPVYEAAQVTPLQKMEKLSSRLDNVILVKREDRQPVHSFKLRGAYAMMAGLTEEQKAHGVITASAGNHAQGVAFSSARLGVKALIVMPTATADIKVDAVRGFGGEVLLHGANFDEAKAKAIELSQQQGFTWVPPFDHPMVIAGQGTLALELLQQDAHLDRVFVPVGGGGLAAGVAVLIKQLMPQIKVIAVEAEDSACLKAALDAGHPVDLPRVGLFAEGVAVKRIGDETFRLCQEYLDDIITVDSDAICAAMKDLFEDVRAVAEPSGALALAGMKKYIALHNIRGERLAHILSGANVNFHGLRYVSERCELGEQREALLAVTIPEEKGSFLKFCQLLGGRSVTEFNYRFADAKNACIFVGVRLSRGLEERKEILQMLNDGGYSVVDLSDDEMAKLHVRYMVGGRPSHPLQERLYSFEFPESPGALLRFLNTLGTYWNISLFHYRSHGTDYGRVLAAFEYDCHDETNNPAFRFFLAG Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Dopamine beta-hydroxylase | 4ZEL | 5.84 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.96  Molscript Map 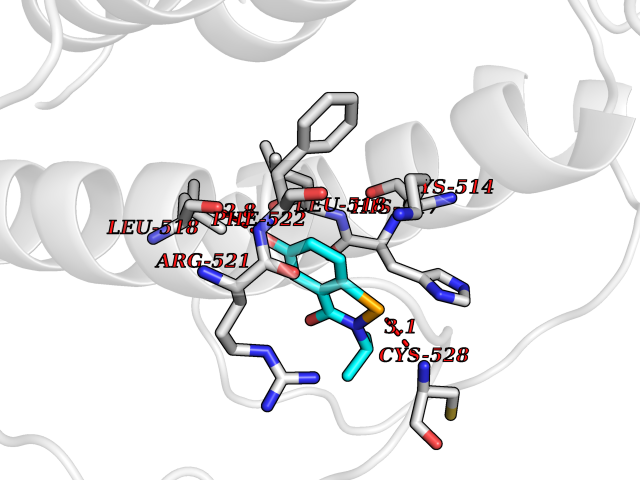 Pymol Map 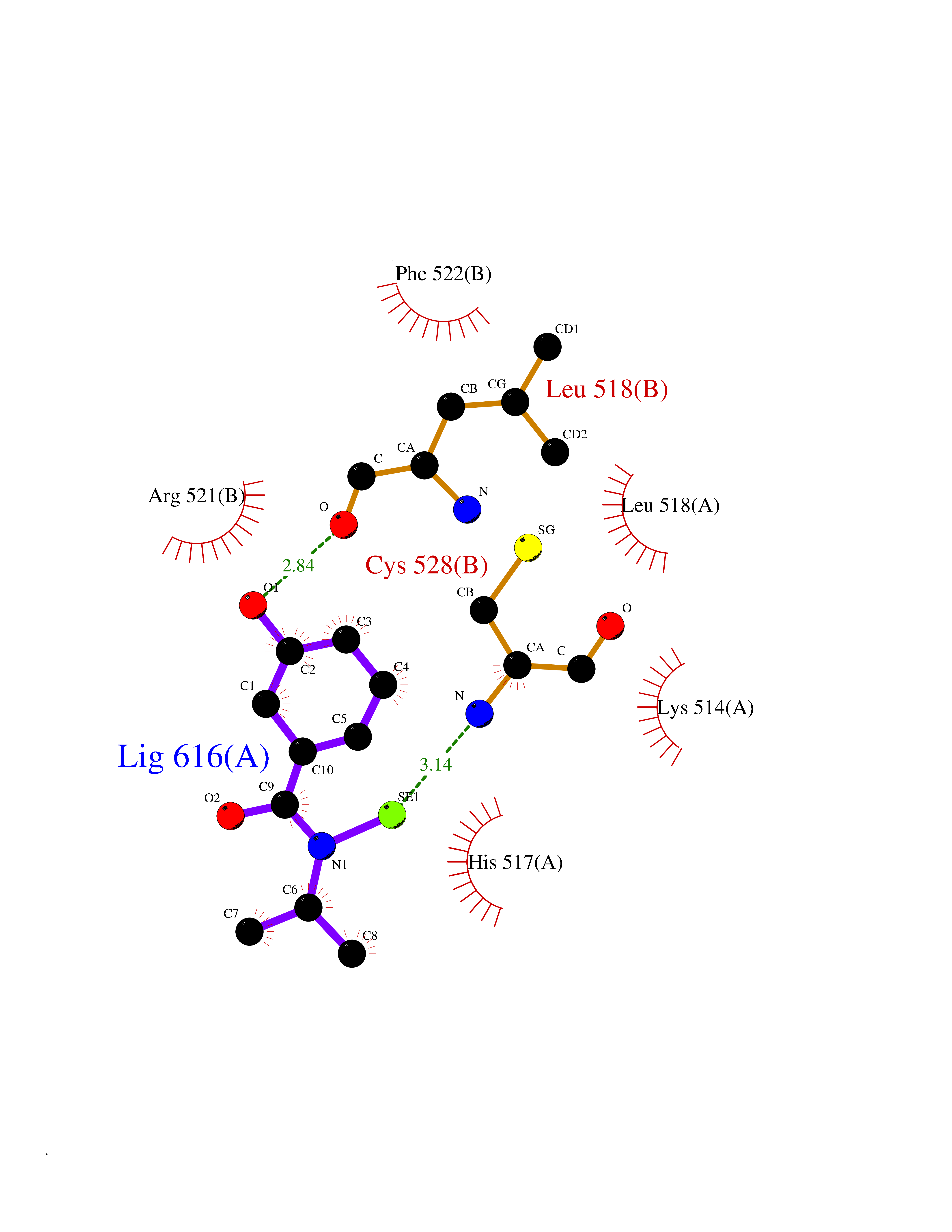 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Tyrosine aminotransferase | 3DYD | 5.84 | |
Target general information Gen name TAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Amino acid binding.L-phenylalanine:2-oxoglutarate aminotransferase activity.L-tyrosine:2-oxoglutarate aminotransferase activity.Pyridoxal phosphate binding. Related diseases Tyrosinemia 2 (TYRSN2) [MIM:276600]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, and oculocutaneous manifestations. Typical features include palmoplantar keratosis, painful corneal ulcers, and intellectual disability. {ECO:0000269|PubMed:1357662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142; DB00120; DB00114; DB00135 Interacts with P15104; P28799; P28799-2; P17735; Q05086; Q05086-3 EC number 2.6.1.5 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Intellectual disability; Palmoplantar keratoderma; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Tyrosine catabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42209.5 Length 380 Aromaticity 0.08 Instability index 51.79 Isoelectric point 5.29 Charge (pH=7) -10.66 2D Binding mode Binding energy (Kcal/mol) -7.96  Molscript Map 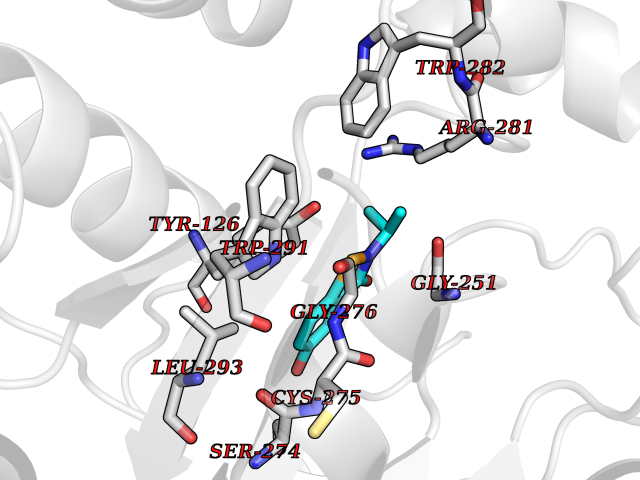 Pymol Map 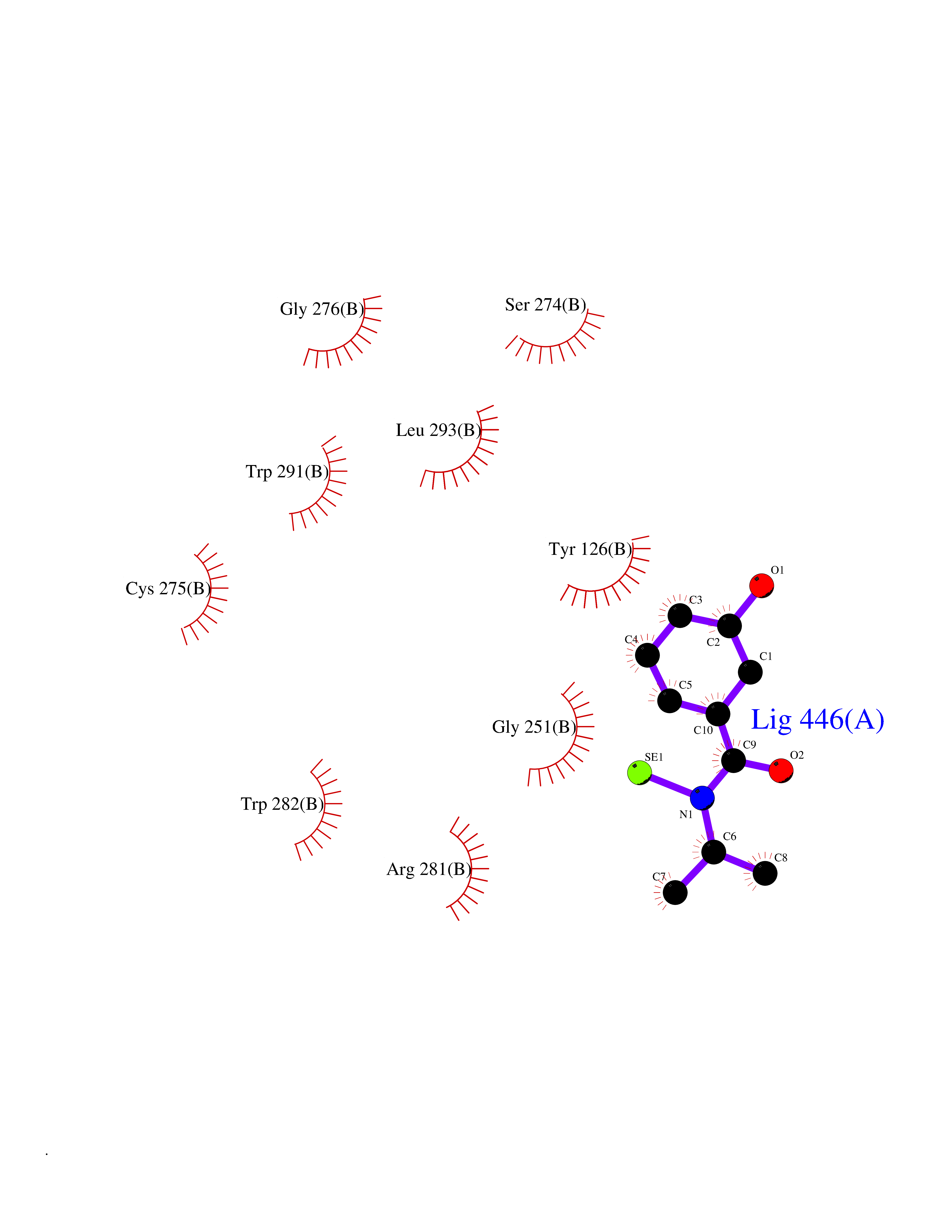 Ligplot Map 3D Binding mode Sequence VKPNPNKTMISLSIGDPTVFGNLPTDPEVTQAMKDALDSGKYNGYAPSIGFLSSREEIASYYHCPEAPLEAKDVILTSGCSQAIDLCLAVLANPGQNILVPRPGFSLYKTLAESMGIEVKLYNLLPEKSWEIDLKQLEYLIDEKTACLIVNNPSNPCGSVFSKRHLQKILAVAARQCVPILADEIYGDMVFSDCKYEPLATLSTDVPILSCGGLAKRWLVPGWRLGWILIHDRRDIFGNEIRDGLVKLSQRILGPCTIVQGALKSILCRTPGEFYHNTLSFLKSNADLCYGALAAIPGLRPVRPSGAMYLMVGIEMEHFPEFENDVEFTERLVAEQSVHCLPATCFEYPNFIRVVITVPEVMMLEACSRIQEFCEQHYHC Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Thiopurine S-methyltransferase | 2BZG | 5.84 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -7.97  Molscript Map 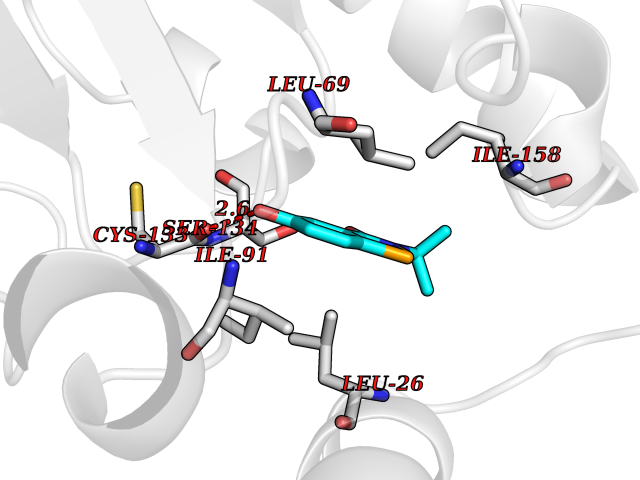 Pymol Map 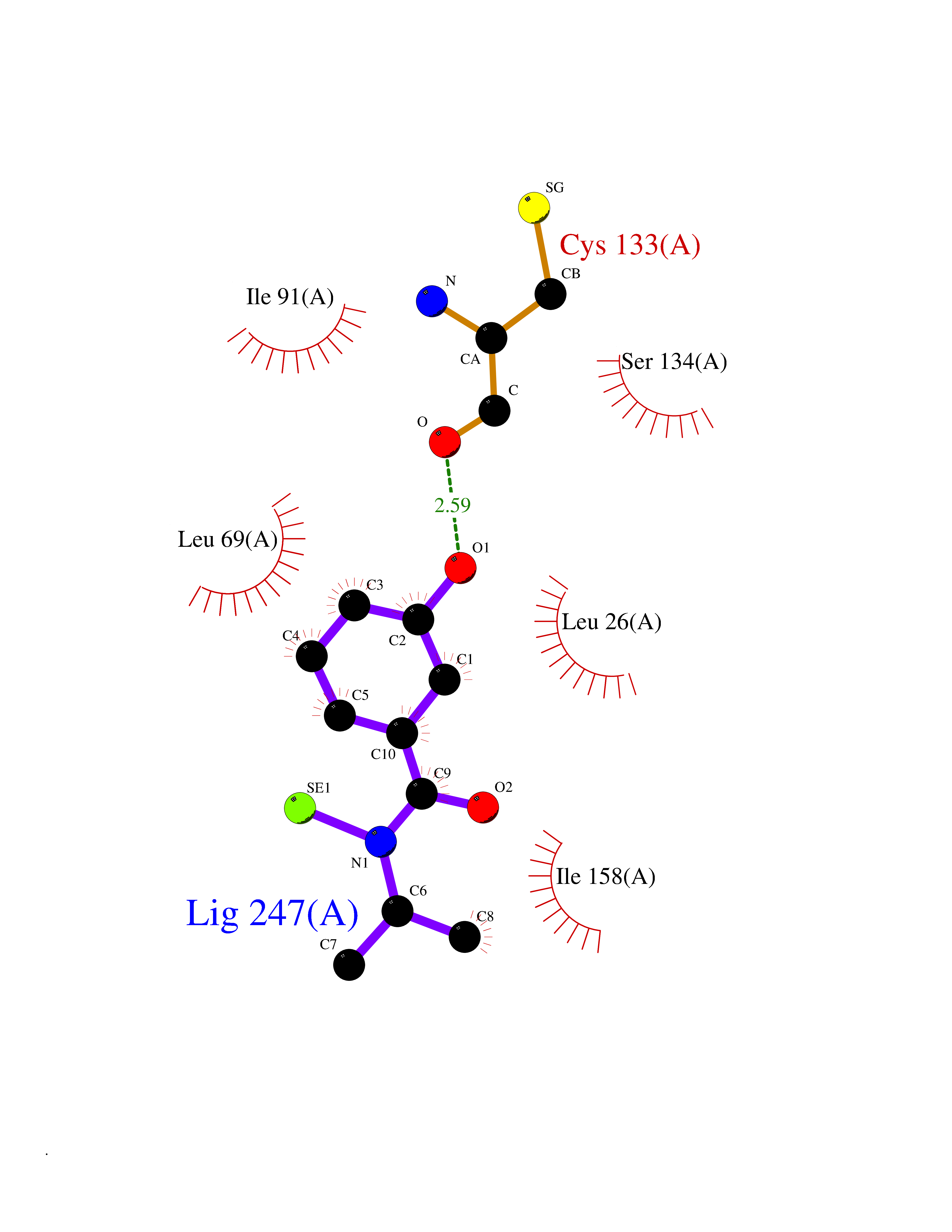 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Bacterial Botulinum toxin A (Bact botA) | 6XCF | 5.84 | |
Target general information Gen name Bact botA Organism Clostridium botulinum Uniprot ID TTD ID Synonyms botA Protein family Peptidase M27 family Biochemical class Peptidase Function Inhibits acetylcholine release. The botulinum toxin binds with high affinity to peripheral neuronal presynaptic membrane to the secretory vesicle protein SV2. It binds directly to the largest luminal loop of SV2A, SV2B and SV2C. It is then internalized by receptor-mediated endocytosis. The C-terminus of the heavy chain (H) is responsible for the adherence of the toxin to the cell surface while the N-terminus mediates transport of the light chain from the endocytic vesicle to the cytosol. After translocation, the light chain (L) hydrolyzes the 197-Gln-|-Arg- 198 bond in SNAP-25, thereby blocking neurotransmitter release. Inhibition of acetylcholine release results in flaccid paralysis, with frequent heart or respiratory failure. Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q02563; Q496J9; Q9Z2I6 EC number NA Uniprot keywords 3D-structure; Cell wall; Direct protein sequencing; Disulfide bond; Host cell membrane; Host cytoplasm; Host cytoplasmic vesicle; Host membrane; Host synapse; Hydrolase; Lipid-binding; Membrane; Metal-binding; Metalloprotease; Neurotoxin; Pharmaceutical; Protease; Secreted; Toxin; Transmembrane; Transmembrane helix; Ubl conjugation; Virulence; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 47759.6 Length 417 Aromaticity 0.13 Instability index 21.7 Isoelectric point 6.36 Charge (pH=7) -1.96 2D Binding mode Binding energy (Kcal/mol) -7.97  Molscript Map 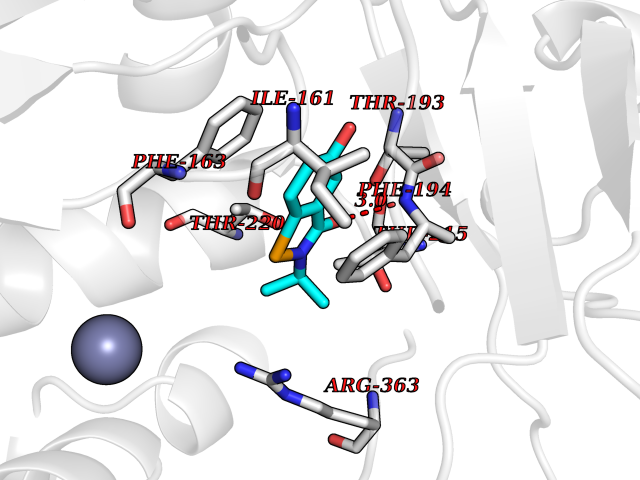 Pymol Map 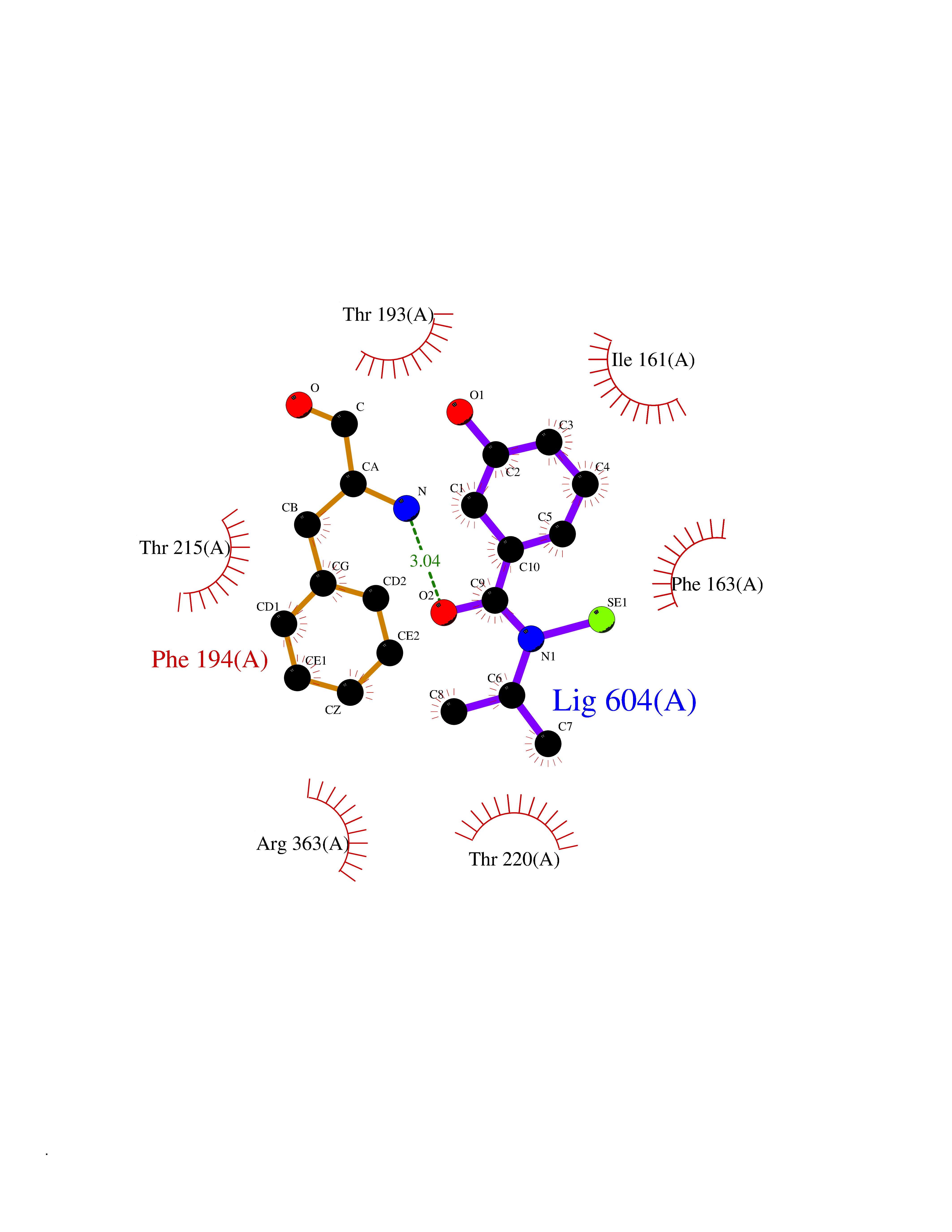 Ligplot Map 3D Binding mode Sequence MQFVNKQFNYKDPVNGVDIAYIKIPNVGQMQPVKAFKIHNKIWVIPERDTFTNPEEGDLNPPPPVSYYDSTYLSTDNEKDNYLKGVTKLFERIYSTDLGRMLLTSIVRGIPFWGGSTIDTELKVIDTNCINVIQPDGSYRSEELNLVIIGPSADIIQFECKSFGHEVLNLTRNGYGSTQYIRFSPDFTFGFEESLEVDTNPLLGAGKFATDPAVTLAHELIHAGHRLYGIAINPNRVFKVNTNAYYEMSGLEVSFEELRTFGGHDAKFIDSLQENEFRLYYYNKFKDIASTLNKAKSIVGTTASLQYMKNVFKEKYLLSEDTSGKFSVDKLKFDKLYKMLTEIYTEDNFVKFFKVLNRKTYLNFDKAVFKINIVPKVNYTIYDGFNLRNTNLAANFNGQNTEINNMNFTKLKNFTGL Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 5.84 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -7.97 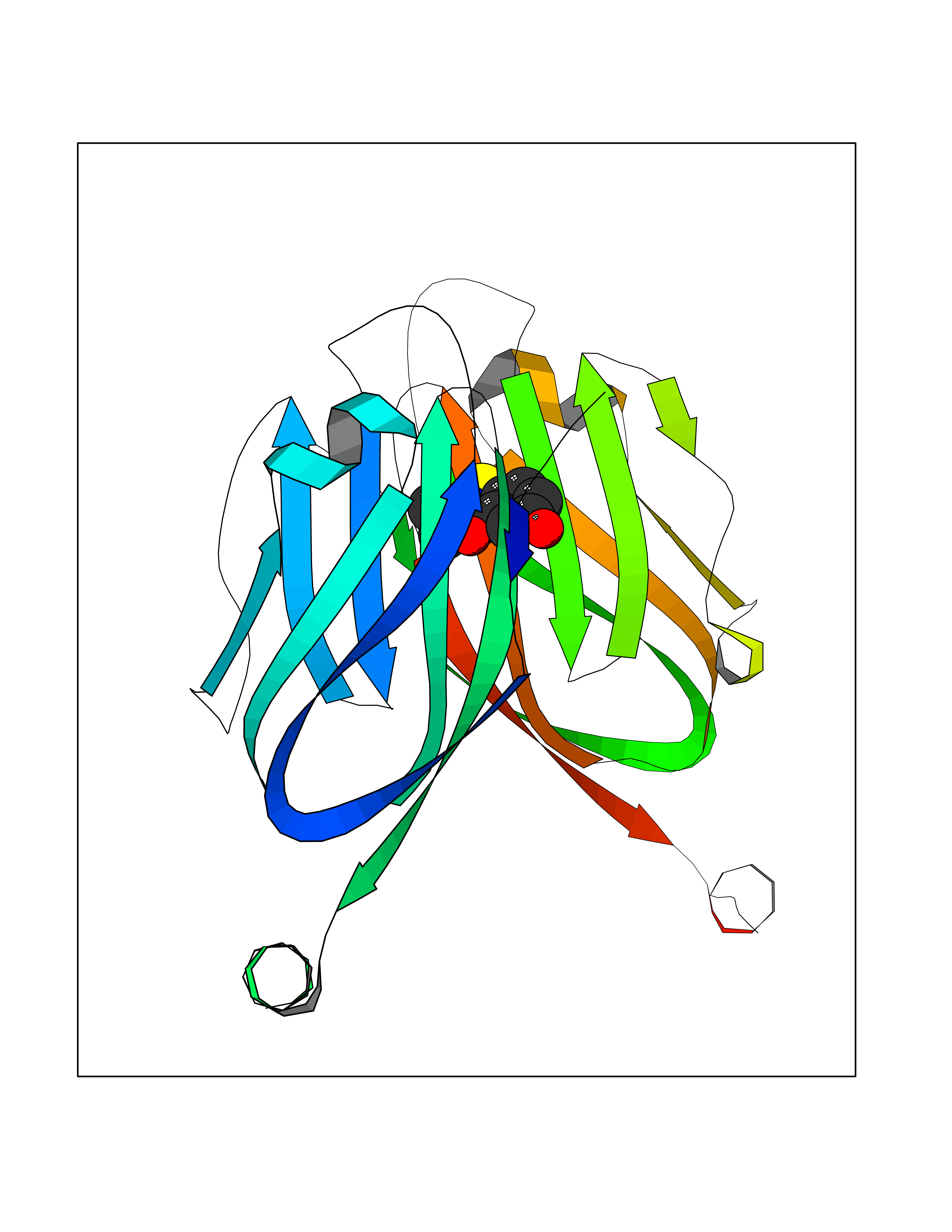 Molscript Map 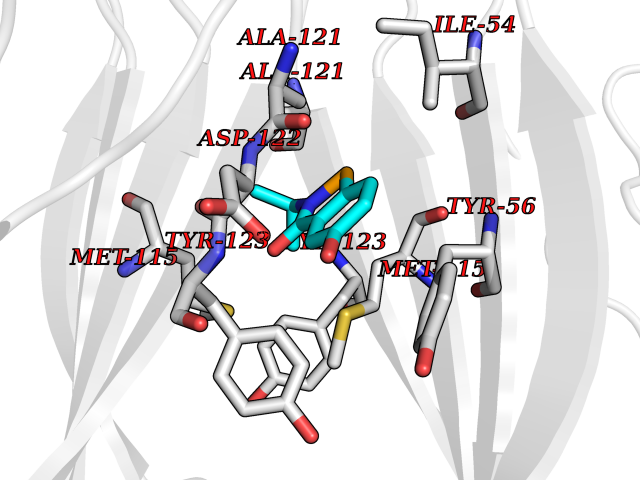 Pymol Map  Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 5.83 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -7.96  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 5.83 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -7.95 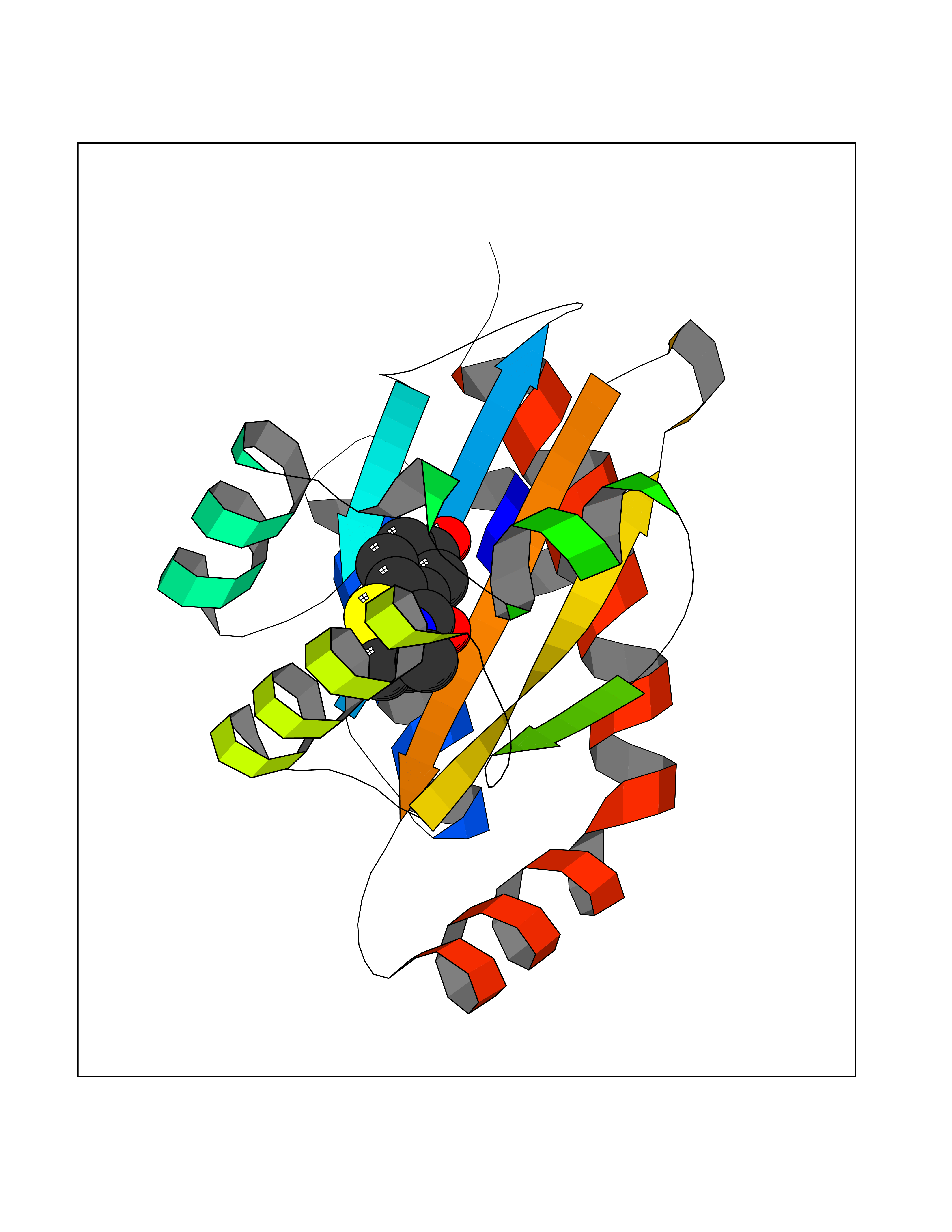 Molscript Map 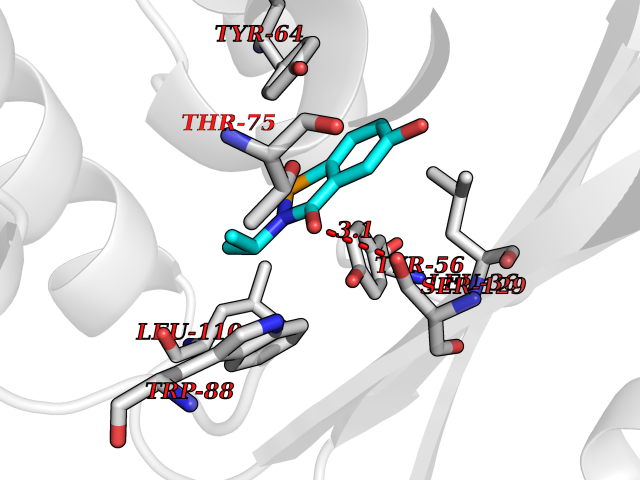 Pymol Map 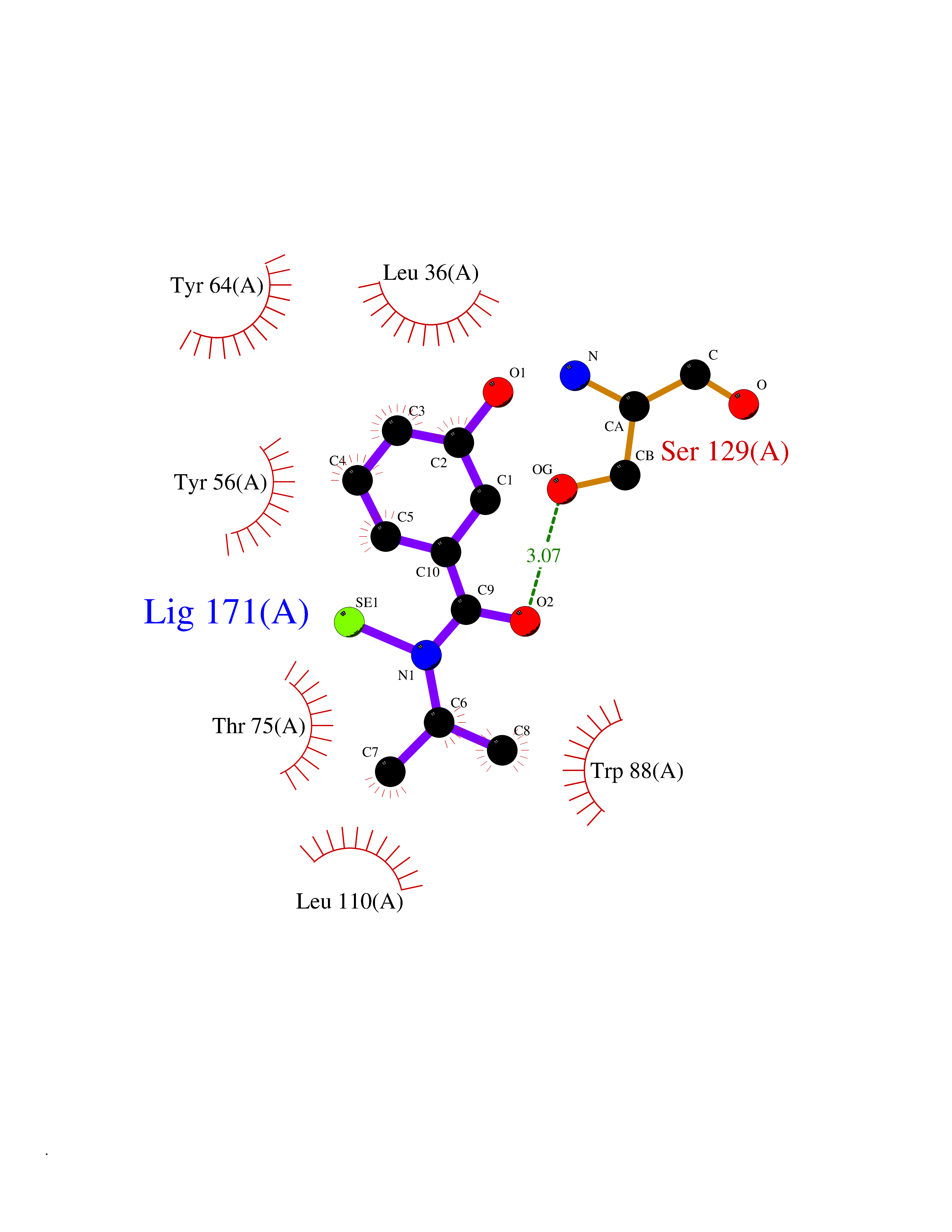 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Wnt-7a protein (WNT7A) | 4UZQ | 5.83 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -7.95 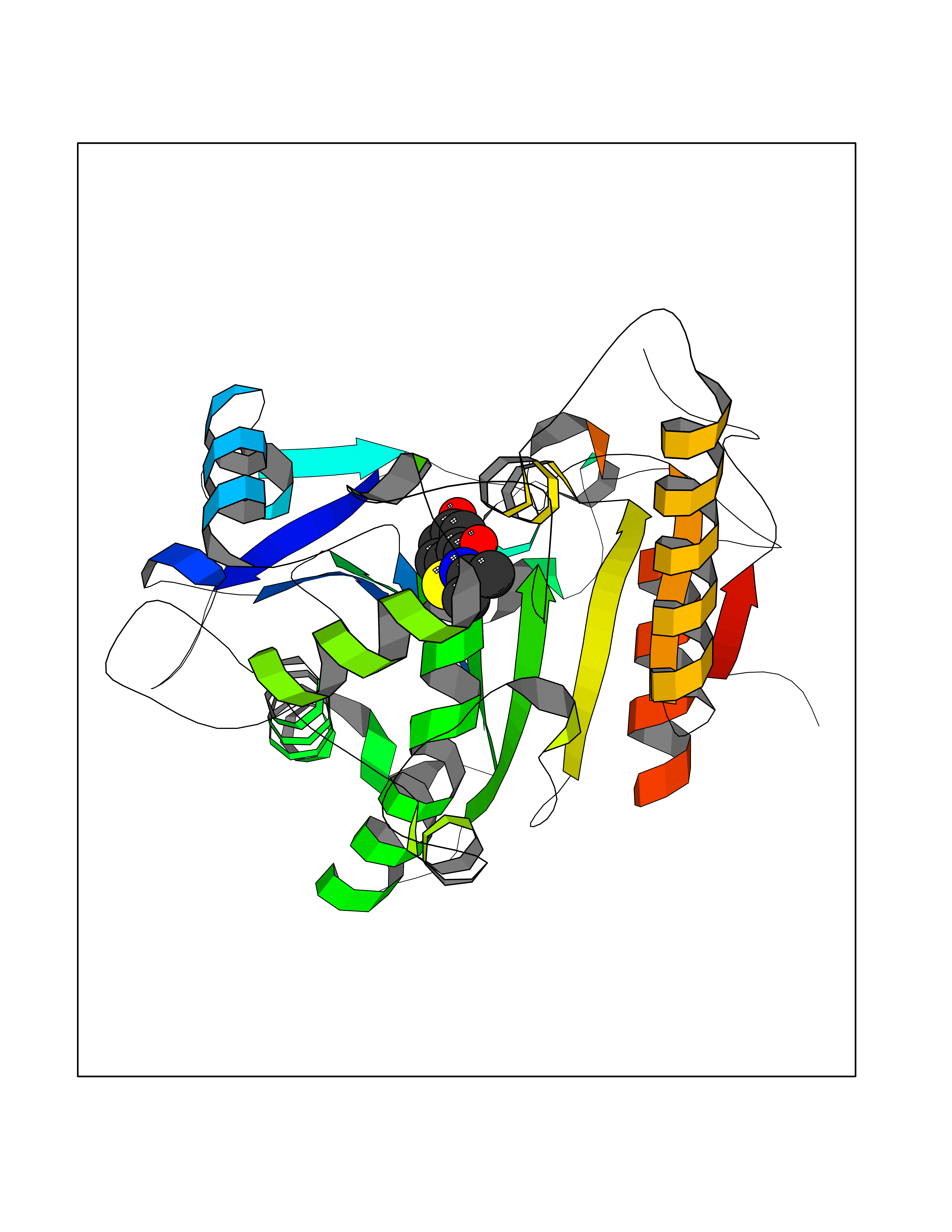 Molscript Map 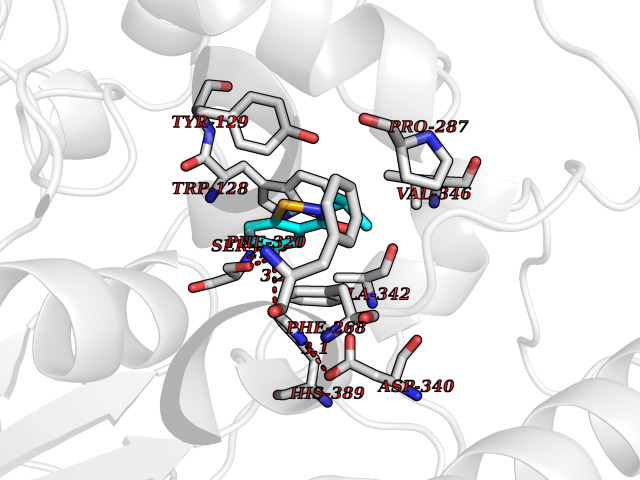 Pymol Map 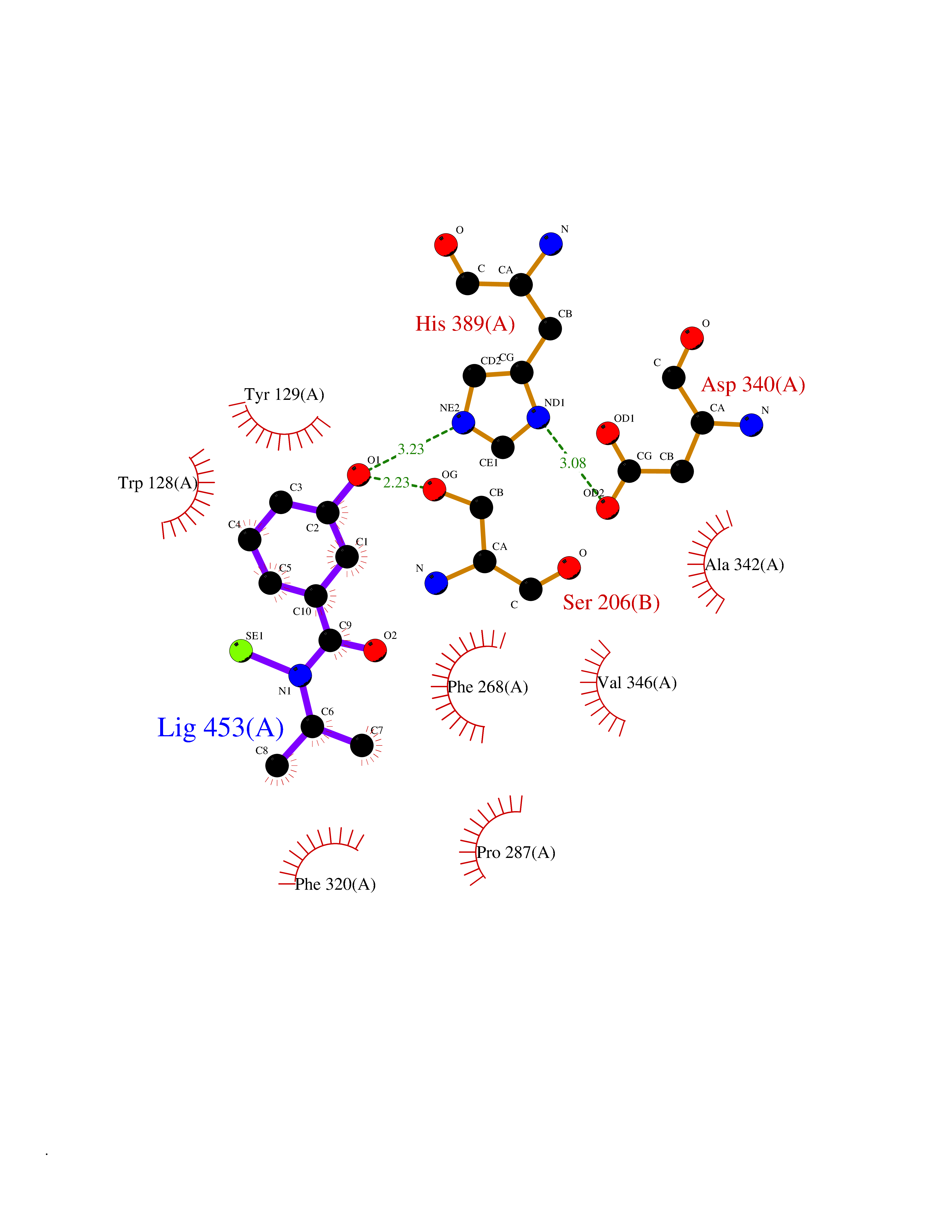 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase | 4WKC | 5.82 | |
Target general information Gen name mtnN Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms pfs;b0159;yadA;JW0155;mtn Protein family PNP/UDP phosphorylase family, MtnN subfamily Biochemical class hydrolase / hydrolase inhibitor Function Adenosylhomocysteine nucleosidase activity.Identical protein binding.Methylthioadenosine nucleosidase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02158; DB08606; DB02933; DB00173; DB02281 Interacts with P0AF12 EC number 3.2.2.9 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 24353.7 Length 232 Aromaticity 0.05 Instability index 22.1 Isoelectric point 5.09 Charge (pH=7) -9.9 2D Binding mode Binding energy (Kcal/mol) -7.93  Molscript Map 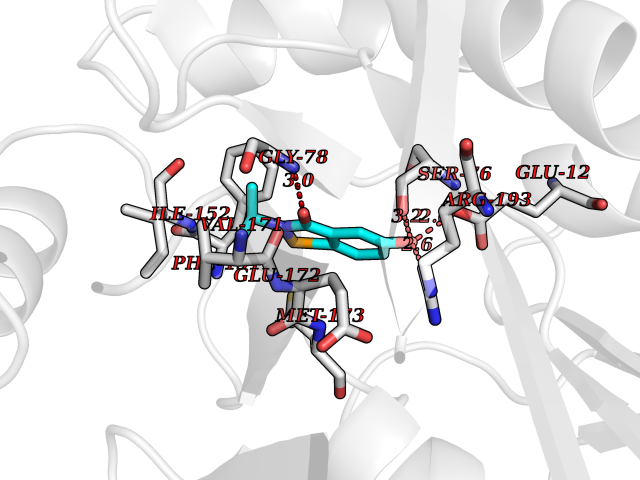 Pymol Map 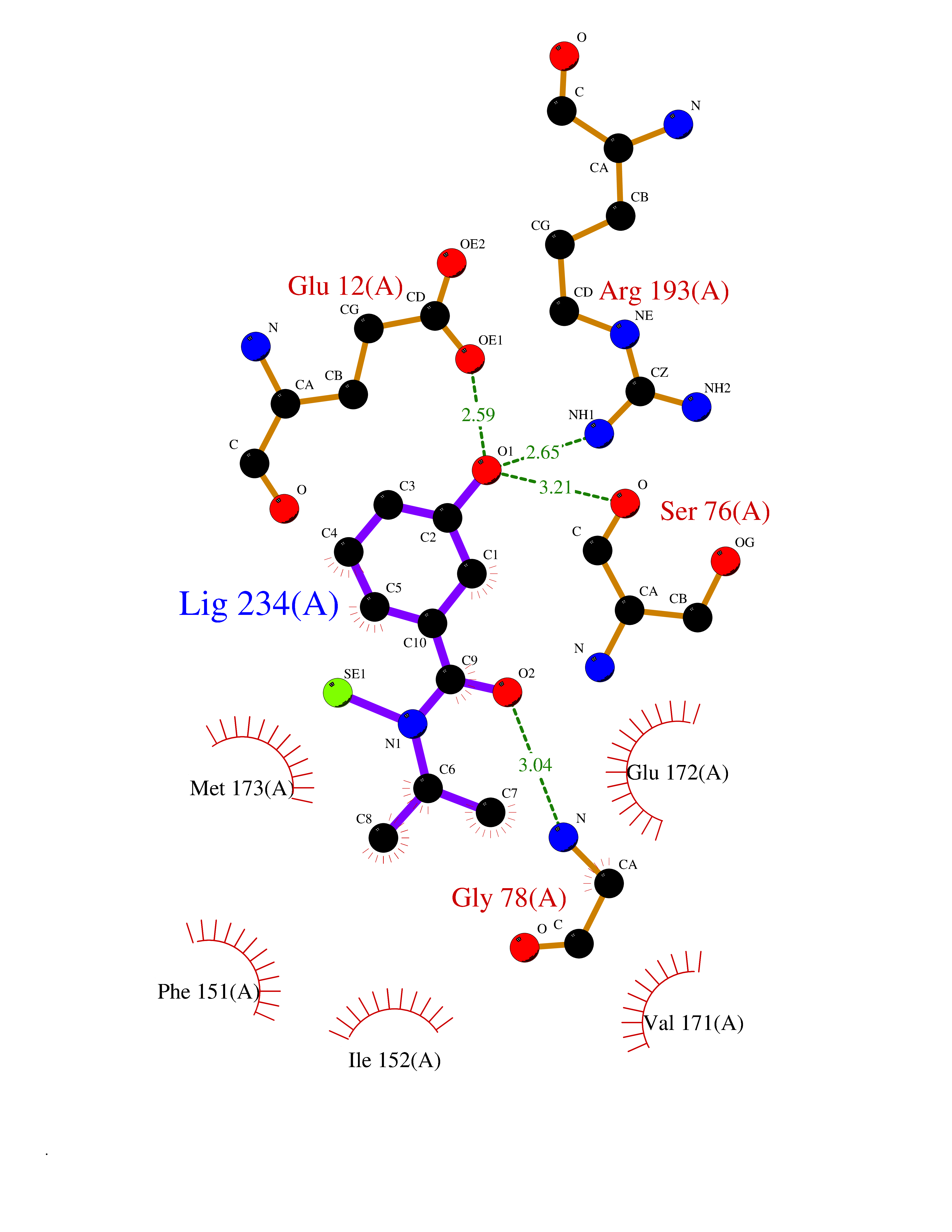 Ligplot Map 3D Binding mode Sequence MKIGIIGAMEEEVTLLRDKIENRQTISLGGCEIYTGQLNGTEVALLKSGIGKVAAALGATLLLEHCKPDVIINTGSAGGLAPTLKVGDIVVSDEARYHDADVTAFGYEYGQLPGCPAGFKADDKLIAAAEACIAELNLNAVRGLIVSGDAFINGSVGLAKIRHNFPQAIAVEMEATAIAHVCHNFNVPFVVVRAISDVADQQSHLSFDEFLAVAAKQSSLMVESLVQKLAHG Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 5.82 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -7.94 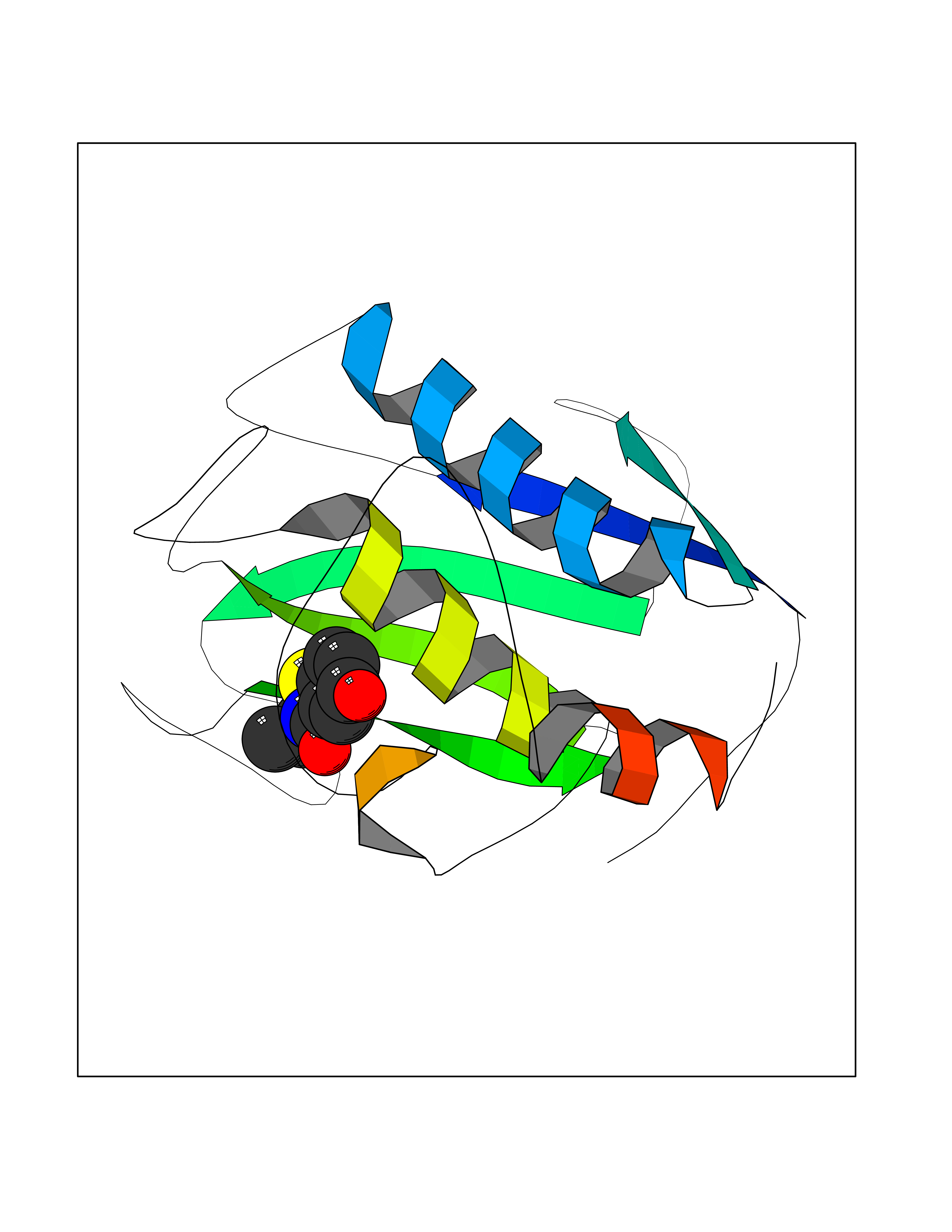 Molscript Map 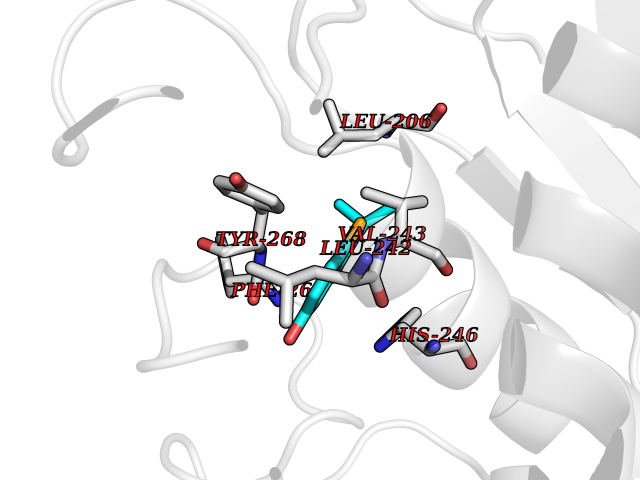 Pymol Map 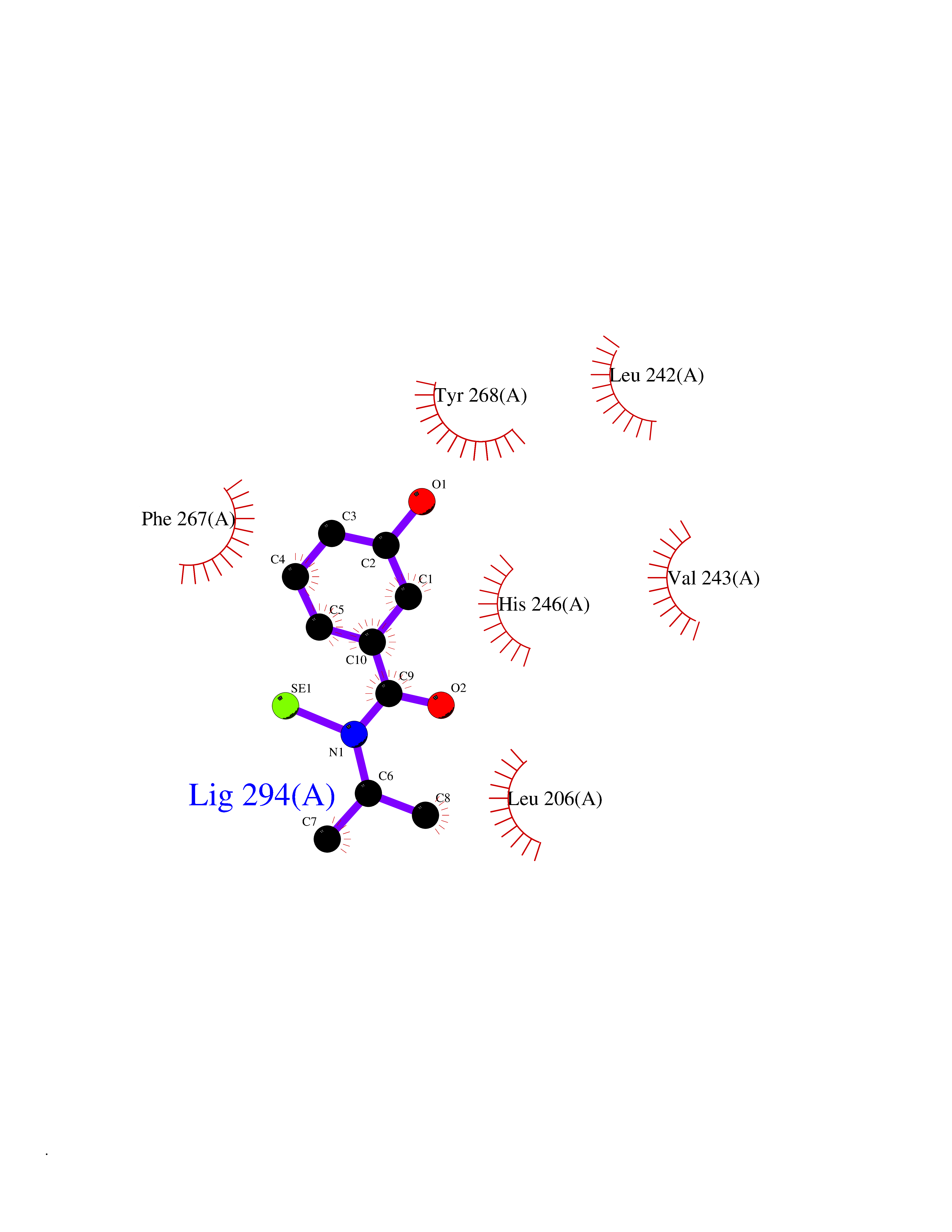 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Glutamate receptor ionotropic kainate 2 (GRIK2) | 5CMM | 5.82 | |
Target general information Gen name GRIK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate receptor ionotropic, kainate 2; Glutamate receptor 6; GluR6; GluR-6; GluK2; Excitatory amino acid receptor 4; EAA4 Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, GRIK2 subfamily Biochemical class Glutamate-gated ion channel Function L-glutamate acts as an excitatory neurotransmitter at many synapses in the central nervous system. Binding of the excitatory neurotransmitter L-glutamate induces a conformation change, leading to the opening of the cation channel, and thereby converts the chemical signal to an electrical impulse. The receptor then desensitizes rapidly and enters a transient inactive state, characterized by the presence of bound agonist. May be involved in the transmission of light information from the retina to the hypothalamus. Modulates cell surface expression of NETO2. Ionotropic glutamate receptor. Related diseases Intellectual developmental disorder, autosomal recessive 6 (MRT6) [MIM:611092]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT6 patients display mild to severe intellectual disability and psychomotor development delay in early childhood. Patients do not have neurologic problems, congenital malformations, or facial dysmorphism. Body height, weight, and head circumference are normal. {ECO:0000269|PubMed:17847003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with impaired language and ataxia and with or without seizures (NEDLAS) [MIM:619580]: An autosomal dominant disorder characterized by axial hypotonia and global developmental delay. Affected individuals show impaired intellectual development, delayed walking, poor speech, and behavioral abnormalities. Some patients have a more severe phenotype with early-onset seizures resembling epileptic encephalopathy, inability to walk or speak, and hypomyelination on brain imaging. {ECO:0000269|PubMed:28180184, ECO:0000269|PubMed:34375587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03425; DB01351; DB01352; DB01483; DB00237; DB00241; DB01353; DB01496; DB02852; DB00142; DB01354; DB01355; DB00463; DB00849; DB00312; DB01174; DB00794; DB02999; DB00418; DB00306; DB00599; DB00273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Isopeptide bond; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; RNA editing; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29150.1 Length 257 Aromaticity 0.1 Instability index 35.11 Isoelectric point 5.89 Charge (pH=7) -2.05 2D Binding mode Binding energy (Kcal/mol) -7.93  Molscript Map 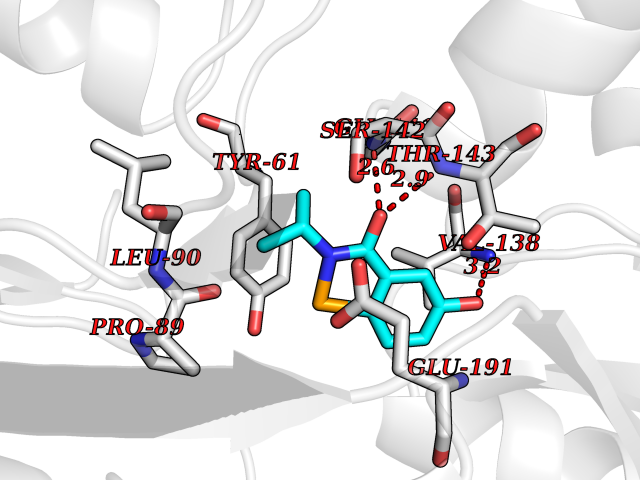 Pymol Map 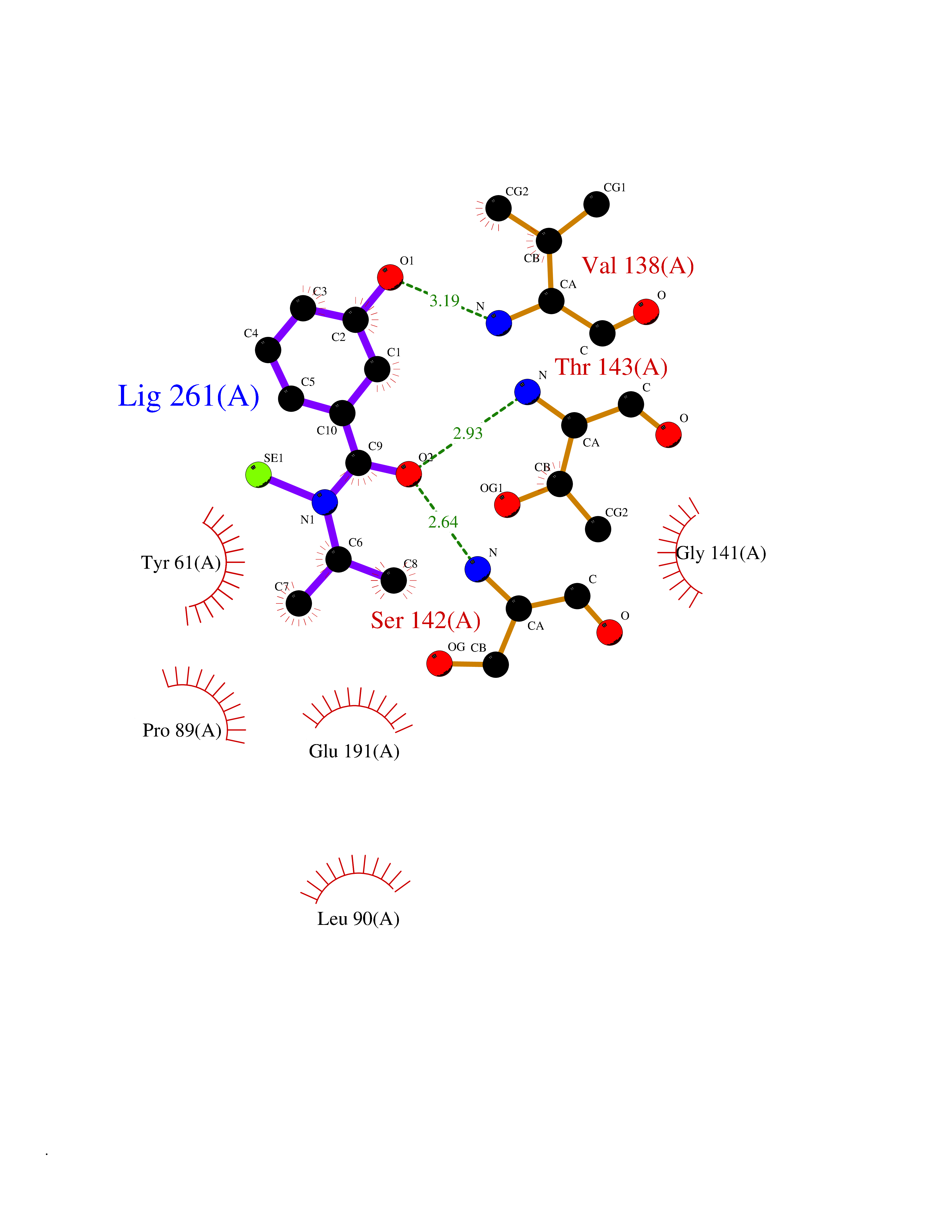 Ligplot Map 3D Binding mode Sequence GSNRSLIVTTILEEPYVLFKKSDKPLYGNDRFEGYCIDLLRELSTILGFTYEIRLVEDGKYGAQDDVNGQWNGMVRELIDHKADLAVAPLTITYVREKVIDFSKPFMTLGISILYRKGTPIDSADDLAKQTKIEYGAVEDGSTMTFFKKSKISTYDKMWAFMSSRRQSVLVKSSEEGIQRVLTSDYALLMESTTIEFVTQRNCNLTQIGGLIDSKGYGVGTPMGSPYRDKITIAILQLQEEGKLHMMKEKWWRGCPE Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Riboflavin kinase | 1NB0 | 5.82 | |
Target general information Gen name RFK Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Transferase Function ATP binding.Metal ion binding.Riboflavin kinase activity. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00140 Interacts with Q9NXG0-2; P19438; P19438-1 EC number 2.7.1.26 Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Flavoprotein; FMN; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 16749.9 Length 147 Aromaticity 0.12 Instability index 41.55 Isoelectric point 7.09 Charge (pH=7) 0.12 2D Binding mode Binding energy (Kcal/mol) -7.93  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RHLPYFCRGQVVRGFGRGSKQLGIPTANFPEQVVDNLPADISTGIYYGWASVGSGDVHKMVVSIGWNPYYKNTKKSMETHIMHTFKEDFYGEILNVAIVGYLRPEKNFDSLESLISAIQGDIEEAKKRLELPEYLKIKEDNFFQVSK Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 5.82 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -7.94  Molscript Map  Pymol Map 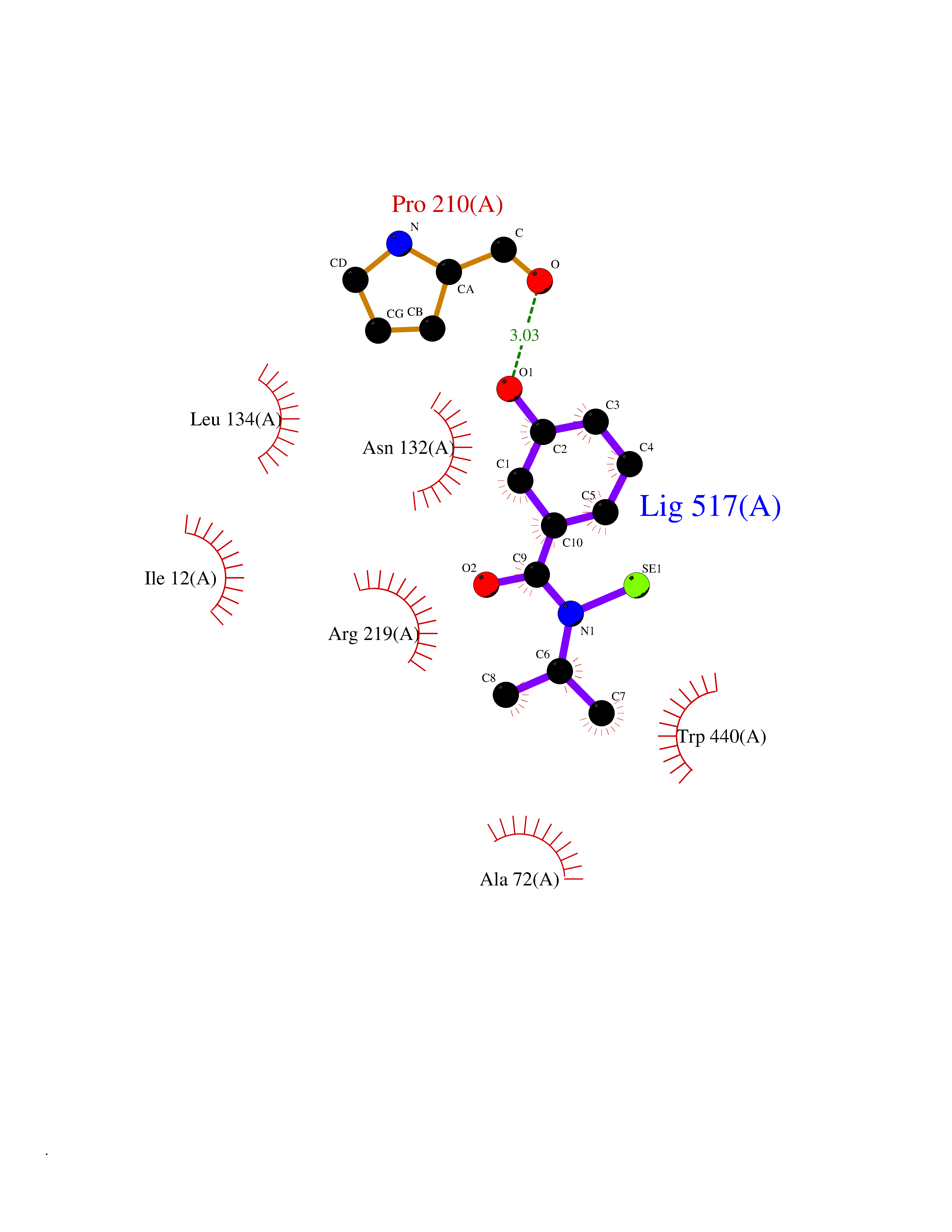 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 2CFI | 5.81 | |
Target general information Gen name ALDH1L1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FTHFD Protein family GART family; Aldehyde dehydrogenase family, ALDH1L subfamily Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Catalytic activity.Formyltetrahydrofolate dehydrogenase activity.Hydroxymethyl-, formyl- and related transferase activity. Related diseases Developmental and epileptic encephalopathy 39 with leukodystrophy (DEE39) [MIM:612949]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE39 is characterized by global hypomyelination of the central nervous system, with the gray matter appearing relatively unaffected. Inheritance is autosomal recessive. {ECO:0000269|PubMed:19641205, ECO:0000269|PubMed:24515575}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00116 Interacts with Q3SY69; Q92624 EC number 1.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; One-carbon metabolism; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33869.5 Length 308 Aromaticity 0.08 Instability index 30.42 Isoelectric point 6.09 Charge (pH=7) -3.83 2D Binding mode Binding energy (Kcal/mol) -7.93  Molscript Map 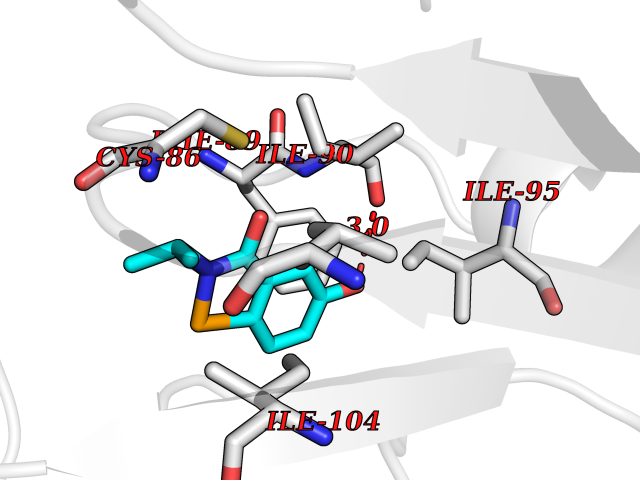 Pymol Map 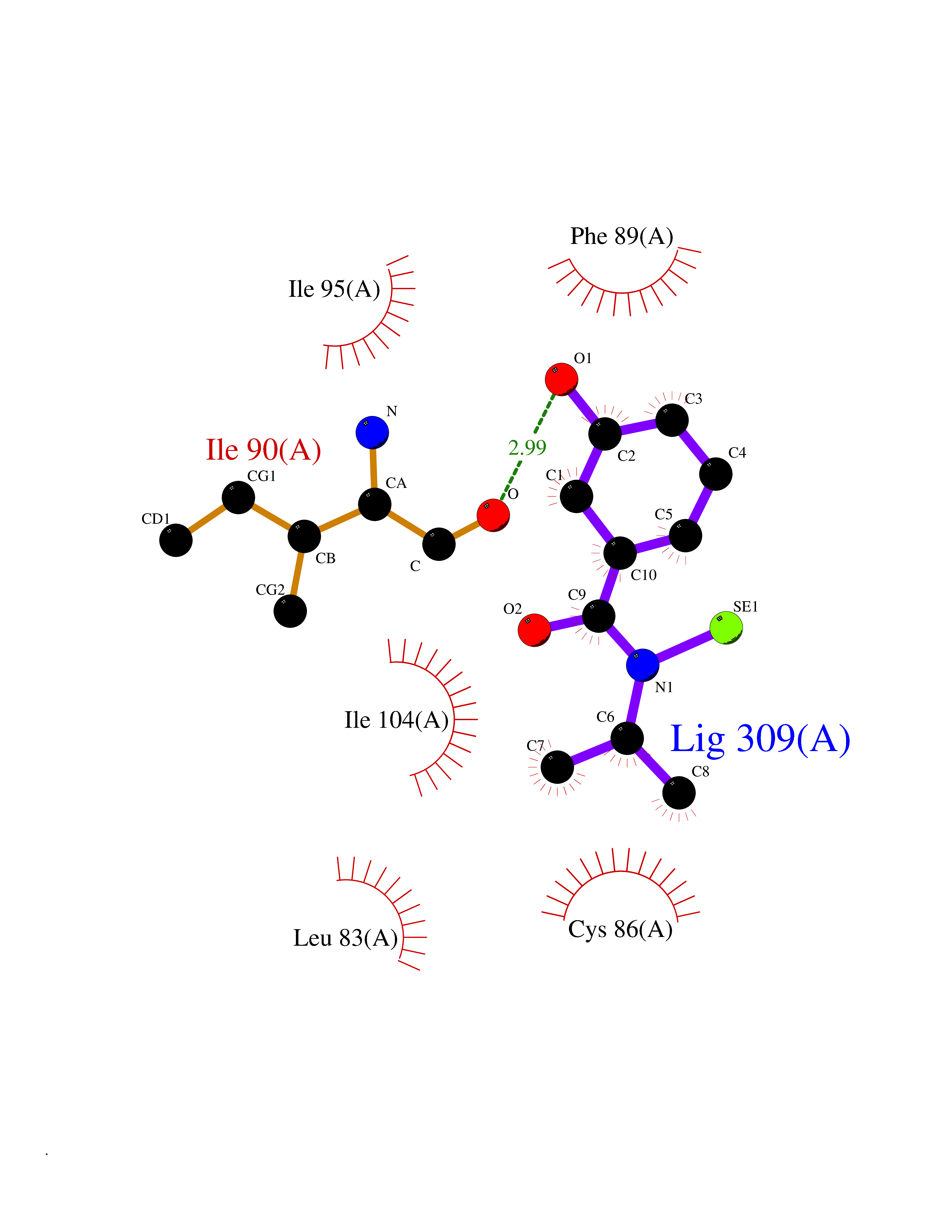 Ligplot Map 3D Binding mode Sequence SMKIAVIGQSLFGQEVYCHLRKEGHEVVGVFTVPDKDGKADPLGLEAEKDGVPVFKYSRWRAKGQALPDVVAKYQALGAELNVLPFCSQFIPMEIISAPRHGSIIYHPSLLPRHRGASAINWTLIHGDKKGGFSIFWADDGLDTGDLLLQKECEVLPDDTVSTLYNRFLFPEGIKGMVQAVRLIAEGKAPRLPQPEEGATYEGIQKKETAKINWDQPAEAIHNWIRGNDKVPGAWTEACEQKLTFFNSTLNTSGLVPEGDALPIPGAHRPGVVTKAGLILFGNDDKMLLVKNIQLEDGKMILASNFFK Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 5.81 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -7.92 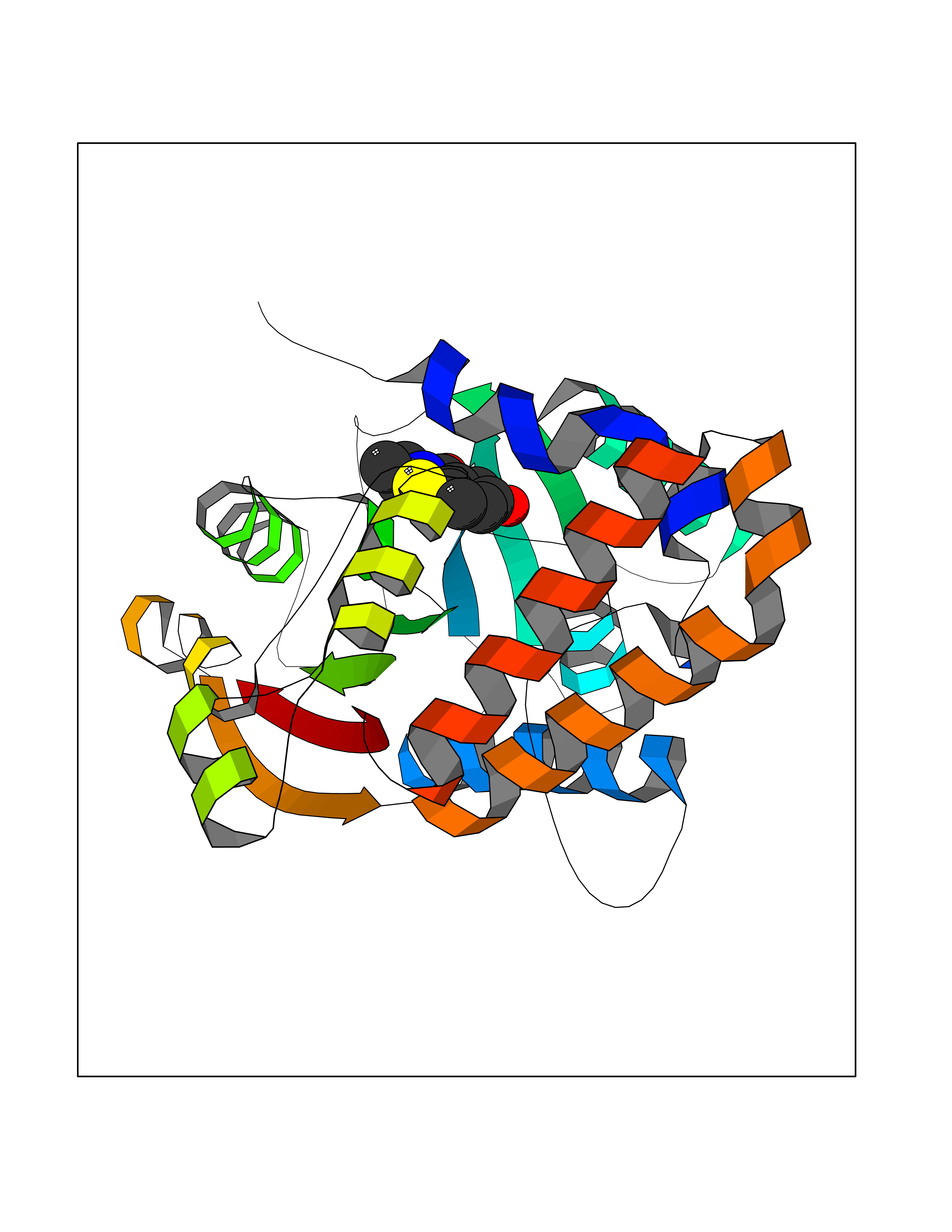 Molscript Map 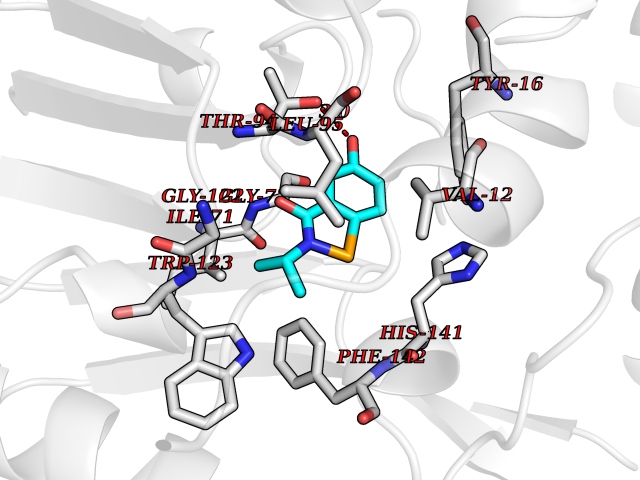 Pymol Map 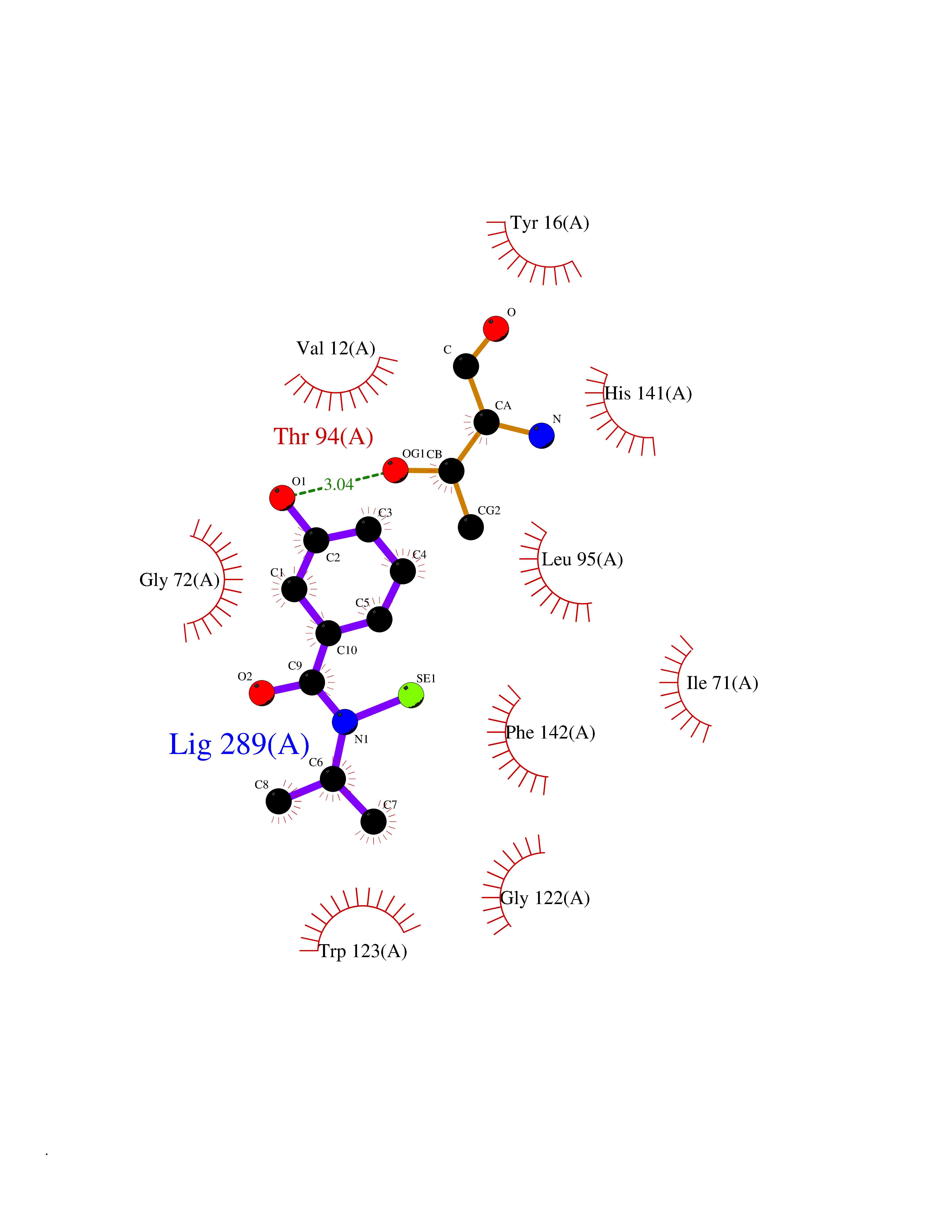 Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | p53-binding protein Mdm4 (MDM4) | 6Q9Y | 5.81 | |
Target general information Gen name MDM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Mdmx; Mdm2-like p53-binding protein; Double minute 4 protein Protein family MDM2/MDM4 family Biochemical class MDM2/MDM4 family Function Inhibits p53/TP53- and TP73/p73-mediated cell cycle arrest and apoptosis by binding its transcriptional activation domain. Inhibits degradation of MDM2. Can reverse MDM2-targeted degradation of TP53 while maintaining suppression of TP53 transactivation and apoptotic functions. Related diseases Bone marrow failure syndrome 6 (BMFS6) [MIM:618849]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS6 is an autosomal dominant form characterized by intermittent neutropenia, lymphopenia, or anemia associated with hypocellular bone marrow, and increased susceptibility to cancer. {ECO:0000269|PubMed:32300648}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NX04; P10415; Q7Z479; O95971; P48729; Q00987; Q13064; P41227; P06400; Q9Y4L5; P23297; P29034; P33763; P04271; P31947; P04637; P62837; Q93009; O14972; P61964; P62258; P61981; P63104; Q9BRR0; A0A0S2Z6X0; Q3YBA8; P03255-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19722 Length 173 Aromaticity 0.08 Instability index 50.78 Isoelectric point 8.48 Charge (pH=7) 2.27 2D Binding mode Binding energy (Kcal/mol) -7.92 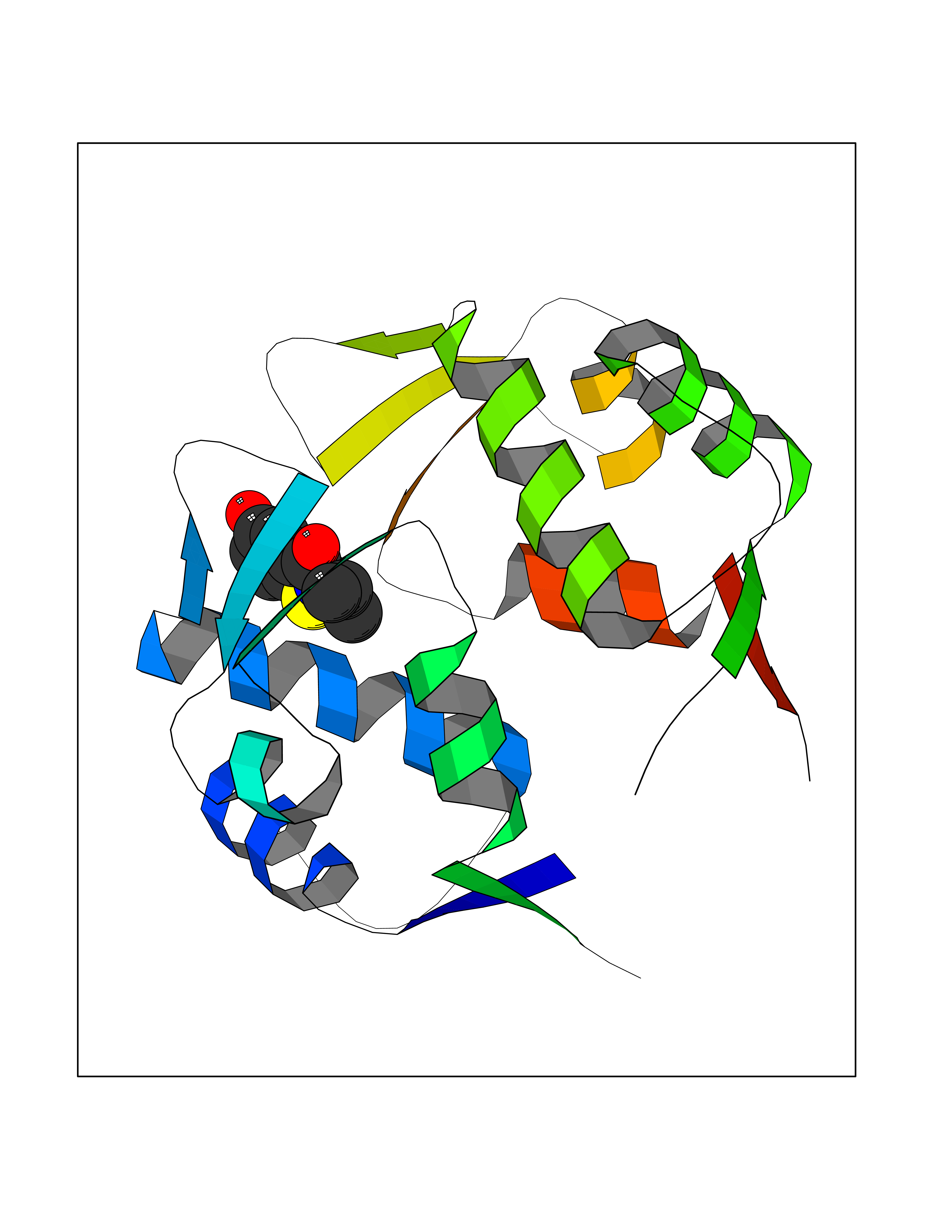 Molscript Map 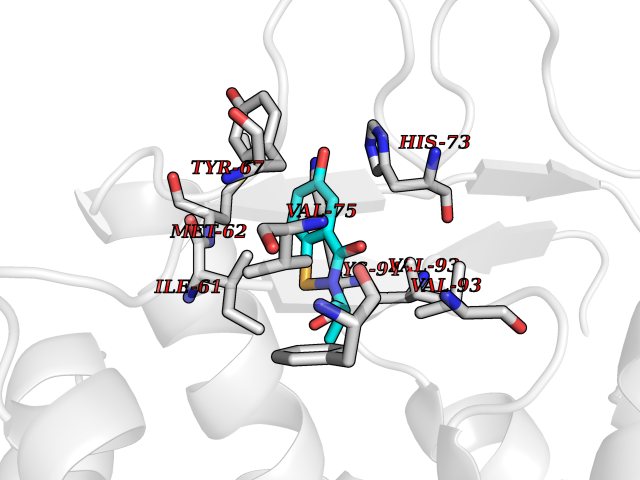 Pymol Map 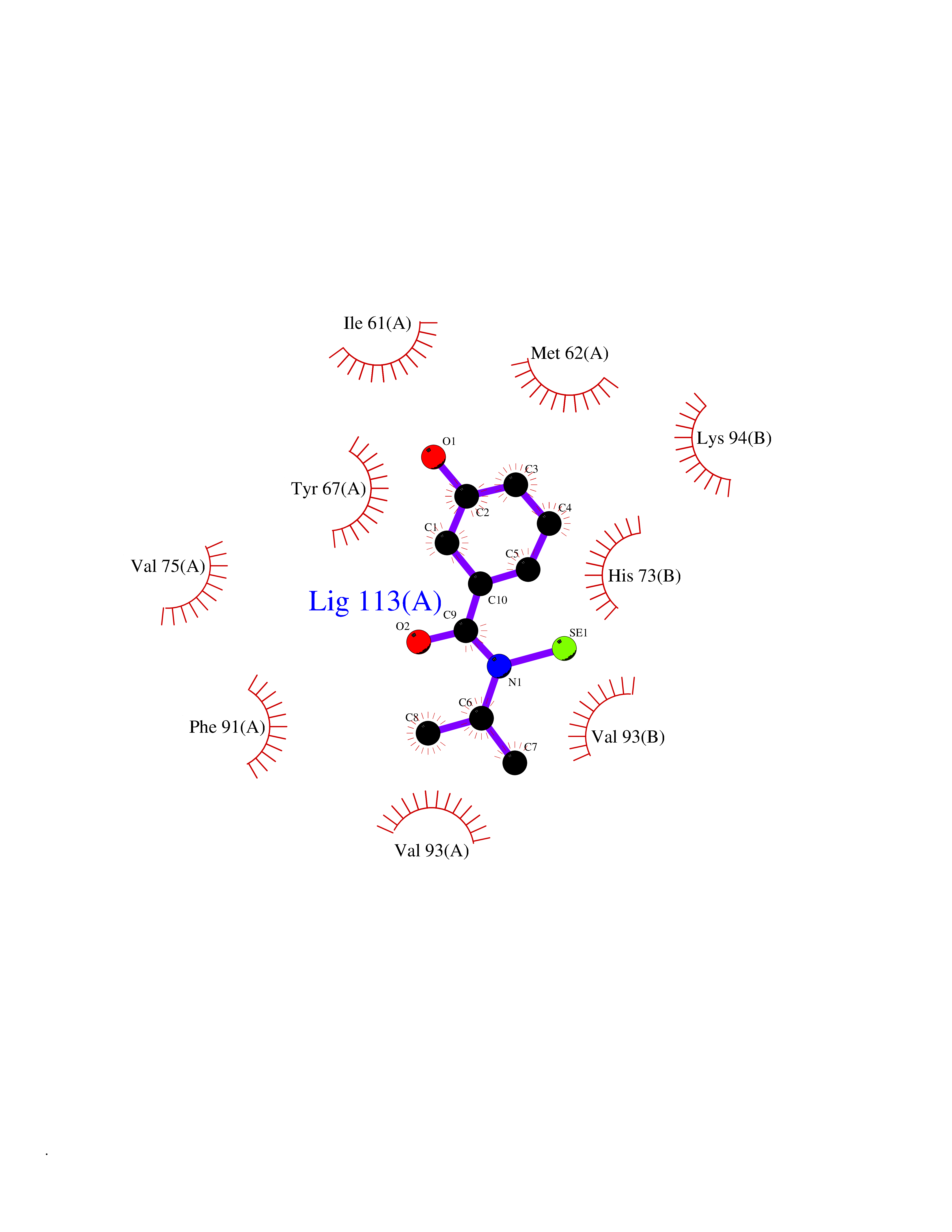 Ligplot Map 3D Binding mode Sequence QVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLAQINQVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLA Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Folate receptor alpha (FOLR1) | 4LRH | 5.81 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -7.93 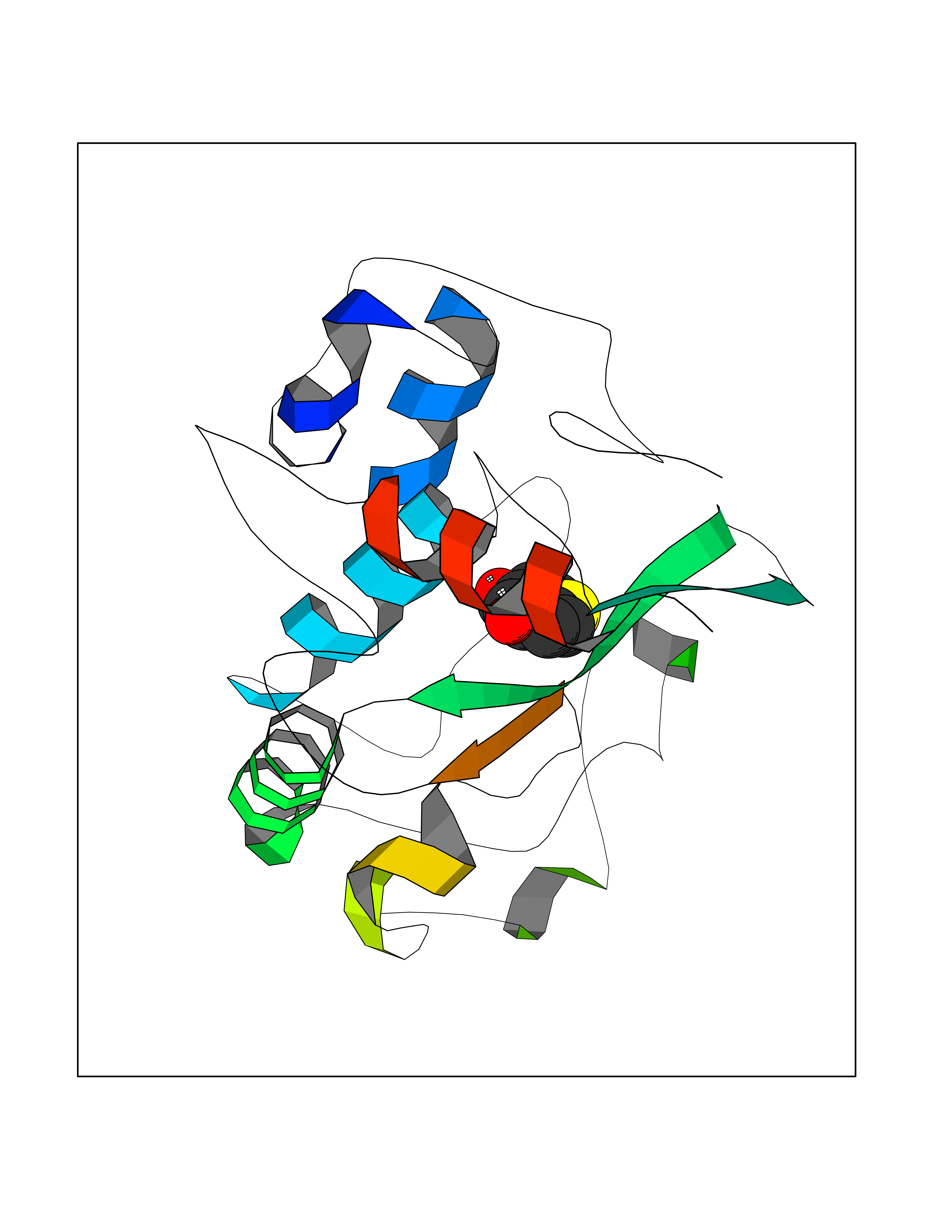 Molscript Map 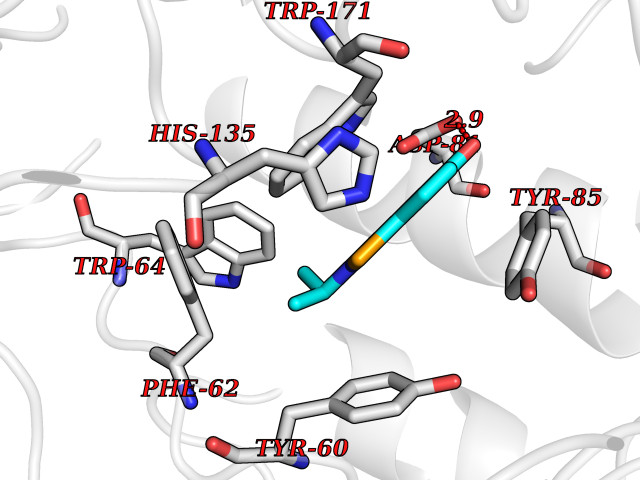 Pymol Map 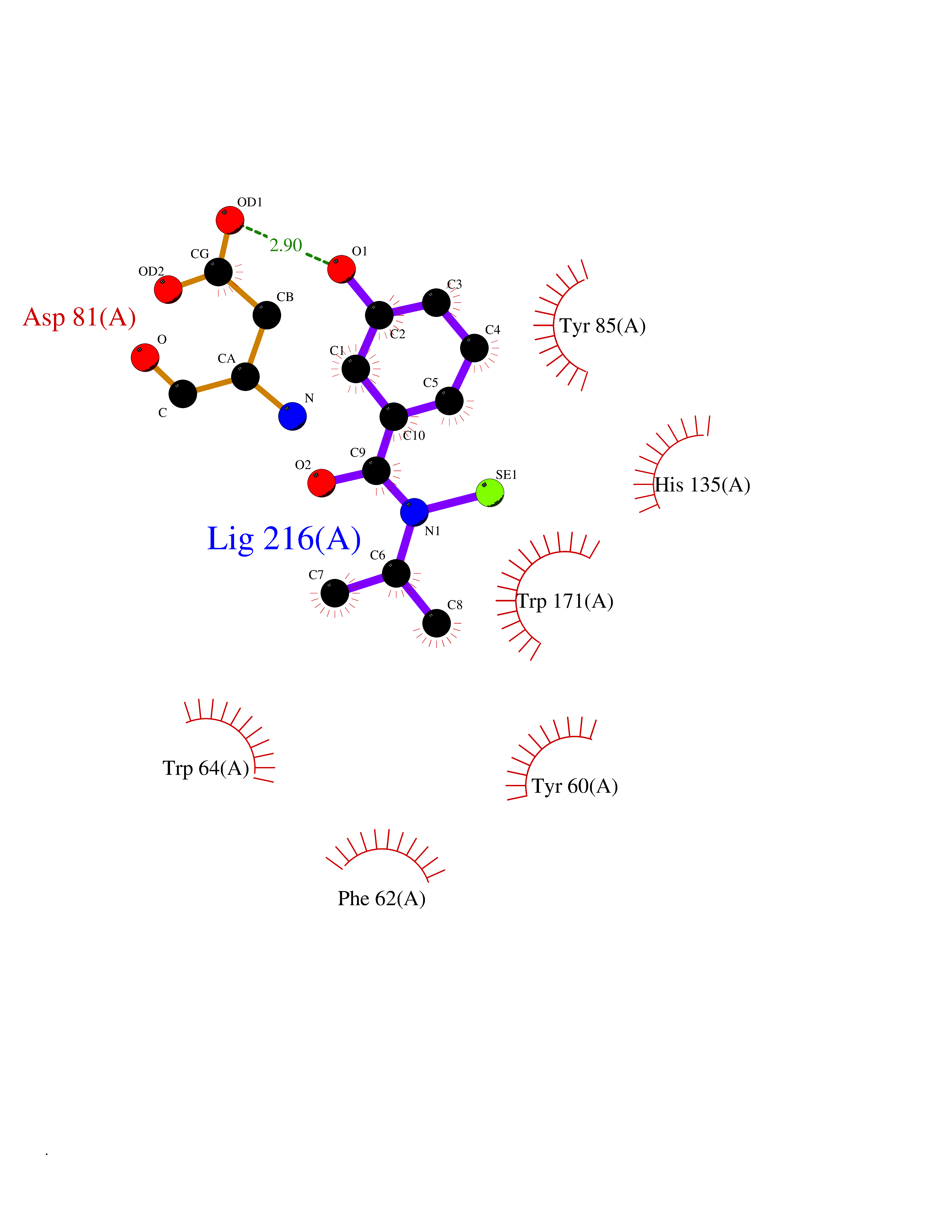 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||