Job Results:
Ligand
Structure
Job ID
b9ed182cddc54993fbd50fd04d8e2354
Job name
NA
Time
2025-09-26 08:49:45
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Aldo-keto reductase family 1 member C3 | 1S1P | 7.87 | |
Target general information Gen name AKR1C3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PGFS;DDH1;HSD17B5;KIAA0119 Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 15-hydroxyprostaglandin-D dehydrogenase (NADP+) activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase activity.Delta4-3-oxosteroid 5beta-reductase activity.Dihydrotestosterone 17-beta-dehydrogenase activity.Geranylgeranyl reductase activity.Indanol dehydrogenase activity.Ketoreductase activity.Ketosteroid monooxygenase activity.NADP-retinol dehydrogenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Prostaglandin D2 11-ketoreductase activity.Prostaglandin-F synthase activity.Prostaglandin H2 endoperoxidase reductase activity.Retinal dehydrogenase activity.Retinol dehydrogenase activity.Testosterone 17-beta-dehydrogenase (NADP+) activity.Testosterone dehydrogenase (NAD+) activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Neurodevelopmental disorder with language impairment and behavioral abnormalities (NEDLIB) [MIM:618917]: A neurodevelopmental disorder characterized by global developmental delay, impaired intellectual development, poor or absent speech, and behavioral abnormalities, such as autism spectrum disorder, repetitive behaviors, and hyperactivity. Some patients develop seizures and manifest developmental regression. {ECO:0000269|PubMed:31300657}. The disease is caused by variants affecting the gene represented in this entry. The genetic variation producing the missense variant p.Q607E, associated with NEDLIB, is predicted to deeply affect RNA editing. In a physiological context, the adenosine (A) residue of the original glutamine (Q) codon CAG is post-transcriptionaly edited to inosine (I) by ADAR2, leading to a codon recognized by the ribosome as arginine (R). The glutamate (E) codon GAG, resulting from the genetic variation, is predicted to be edited 90% less than the normal CAG codon. If edited, the codon GIG would be translated as p.Q607G. {ECO:0000305|PubMed:31300657}. Drugs (DrugBank ID) DB07700; DB01561; DB01536; DB00997; DB01039; DB02266; DB13751; DB00328; DB06077; DB00959; DB00157; DB03461; DB09074; DB00776; DB02056; DB01698; DB02901 Interacts with P17516 EC number 1.1.1.-; 1.1.1.188; 1.1.1.210; 1.1.1.239; 1.1.1.357; 1.1.1.53; 1.1.1.62; 1.1.1.64 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid metabolism; NAD; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35846.8 Length 315 Aromaticity 0.09 Instability index 47.59 Isoelectric point 8.54 Charge (pH=7) 4.51 2D Binding mode Binding energy (Kcal/mol) -10.73  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence QCVKLNDGHFMPVLGFGTYAPPEVPRSKALEVTKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWSTFHRPELVRPALENSLKKAQLDYVDLYLIHSPMSLKPGEELSPTDENGKVIFDIVDLCTTWEAMEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNRSKLLDFCKSKDIVLVAYSALGSQRDKRWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTAEDMKAIDGLDRNLHYFNSDSFASHPNYPYS Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Monoamine oxidase type B (MAO-B) | 2V5Z | 7.86 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -10.72  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Cannabinoid receptor 1 (CB1) | 5U09 | 7.84 | |
Target general information Gen name CNR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cannabinoid CB1 receptor; CNR; CB-R; CANN6 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Mediates many cannabinoid-induced effects, acting, among others, on food intake, memory loss, gastrointestinal motility, catalepsy, ambulatory activity, anxiety, chronic pain. Signaling typically involves reduction in cyclic AMP. In the hypothalamus, may have a dual effect on mitochondrial respiration depending upon the agonist dose and possibly upon the cell type. Increases respiration at low doses, while decreases respiration at high doses. At high doses, CNR1 signal transduction involves G-protein alpha-i protein activation and subsequent inhibition of mitochondrial soluble adenylate cyclase, decrease in cyclic AMP concentration, inhibition of protein kinase A (PKA)-dependent phosphorylation of specific subunits of the mitochondrial electron transport system, including NDUFS2. In the hypothalamus, inhibits leptin-induced reactive oxygen species (ROS) formation and mediates cannabinoid-induced increase in SREBF1 and FASN gene expression. In response to cannabinoids, drives the release of orexigenic beta-endorphin, but not that of melanocyte-stimulating hormone alpha/alpha-MSH, from hypothalamic POMC neurons, hence promoting food intake. In the hippocampus, regulates cellular respiration and energy production in response to cannabinoids. Involved in cannabinoid-dependent depolarization-induced suppression of inhibition (DSI), a process in which depolarization of CA1 postsynaptic pyramidal neurons mobilizes eCBs, which retrogradely activate presynaptic CB1 receptors, transiently decreasing GABAergic inhibitory neurotransmission. Also reduces excitatory synaptic transmission. In superior cervical ganglions and cerebral vascular smooth muscle cells, inhibits voltage-gated Ca(2+) channels in a constitutive, as well as agonist-dependent manner. In cerebral vascular smooth muscle cells, cannabinoid-induced inhibition of voltage-gated Ca(2+) channels leads to vasodilation and decreased vascular tone. Induces leptin production in adipocytes and reduces LRP2-mediated leptin clearance in the kidney, hence participating in hyperleptinemia. In adipose tissue, CNR1 signaling leads to increased expression of SREBF1, ACACA and FASN genes. In the liver, activation by endocannabinoids leads to increased de novo lipogenesis and reduced fatty acid catabolism, associated with increased expression of SREBF1/SREBP-1, GCK, ACACA, ACACB and FASN genes. May also affect de novo cholesterol synthesis and HDL-cholesteryl ether uptake. Peripherally modulates energy metabolism. In high carbohydrate diet-induced obesity, may decrease the expression of mitochondrial dihydrolipoyl dehydrogenase/DLD in striated muscles, as well as that of selected glucose/ pyruvate metabolic enzymes, hence affecting energy expenditure through mitochondrial metabolism. In response to cannabinoid anandamide, elicits a proinflammatory response in macrophages, which involves NLRP3 inflammasome activation and IL1B and IL18 secretion. In macrophages infiltrating pancreatic islets, this process may participate in the progression of type-2 diabetes and associated loss of pancreatic beta-cells. G-protein coupled receptor for endogenous cannabinoids (eCBs), including N-arachidonoylethanolamide (also called anandamide or AEA) and 2-arachidonoylglycerol (2-AG), as well as phytocannabinoids, such as delta(9)-tetrahydrocannabinol (THC). Related diseases Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:18177726}. The protein represented in this entry may be involved in disease pathogenesis. May contribute to the development of diet-induced obesity and several obesity-associated features, such as dyslipidemia and liver steatosis, regulating peripheral lipogenesis, energy expenditure and feeding behavior. CNR1 inverse agonists have been shown to reduce body weight and improve metabolic abnormalities in obese subjects, although adverse neuropsychiatric effects, including anxiety, irritability, and depressed mood, halted their therapeutic development (PubMed:18177726). In obese mice, peripherally restricted CNR1 inverse agonists have been shown to normalize metabolic abnormalities, including insulin resistance and fatty liver, and to reverse leptin resistance. {ECO:0000269|PubMed:18177726}.; DISEASE: Dysfunction of the endogenous cannabinoid system including CNR1 has been implicated in the pathogenesis of a number of central nervous system disorders, including Huntington disease, Parkinson disease, and Alzheimer disease (PubMed:32549916). In post-mortem brains from Huntington disease patients, a progressive CNR1 loss has been observed in the caudate nucleus, putamen, and substantia nigra pars reticulata, and altered expression and abnormal endocannabinoid levels precede motor symptoms in a disease mouse model (PubMed:10828533, PubMed:19524019, PubMed:8255419). In Parkinson disease, low CNR1 expression in mid-superior frontal gyrus and mid-cingulate cortex has been associated with poor mind, poor executive functioning and poor episode memory, while patients with more severe visuospatial dysfunction showed decreased receptor availability in the precuneus, mid-cingulate, supplementary motor cortex, inferior orbitofrontal gyrus and thalamus (PubMed:31342135). In an animal model for Alzheimer disease, CNR1 heterozygous deletion has been associated with decreased levels of postsynaptic density protein 95 (DLG4/PSD95) and accelerated memory impairment, suggesting synaptic dysfunction and a crucial role for CNR1 in the progression of disease symptoms (PubMed:10828533, PubMed:19524019, PubMed:30096288, PubMed:31342135, PubMed:8255419). {ECO:0000269|PubMed:10828533, ECO:0000269|PubMed:19524019, ECO:0000269|PubMed:30096288, ECO:0000269|PubMed:31342135, ECO:0000269|PubMed:32549916, ECO:0000269|PubMed:8255419}. Drugs (DrugBank ID) DB05750; DB09061; DB00470; DB14009; DB00486; DB14011; DB11745; DB09288; DB02955; DB06155; DB05077; DB11755; DB05201 Interacts with P29274; P21554 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurodegeneration; Obesity; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32070.3 Length 282 Aromaticity 0.13 Instability index 40.15 Isoelectric point 9.16 Charge (pH=7) 9.36 2D Binding mode Binding energy (Kcal/mol) -10.7  Molscript Map 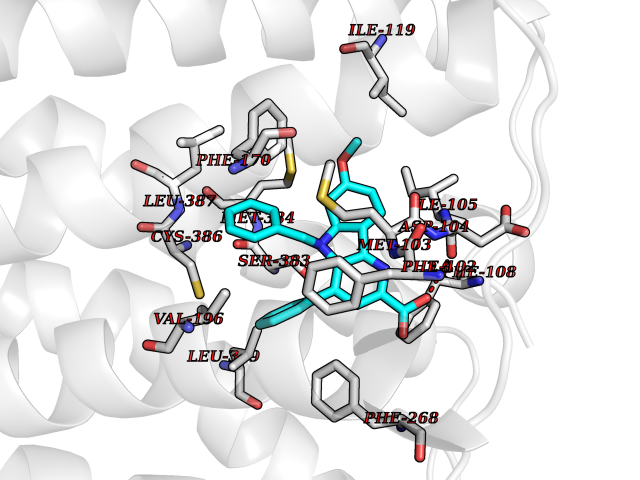 Pymol Map 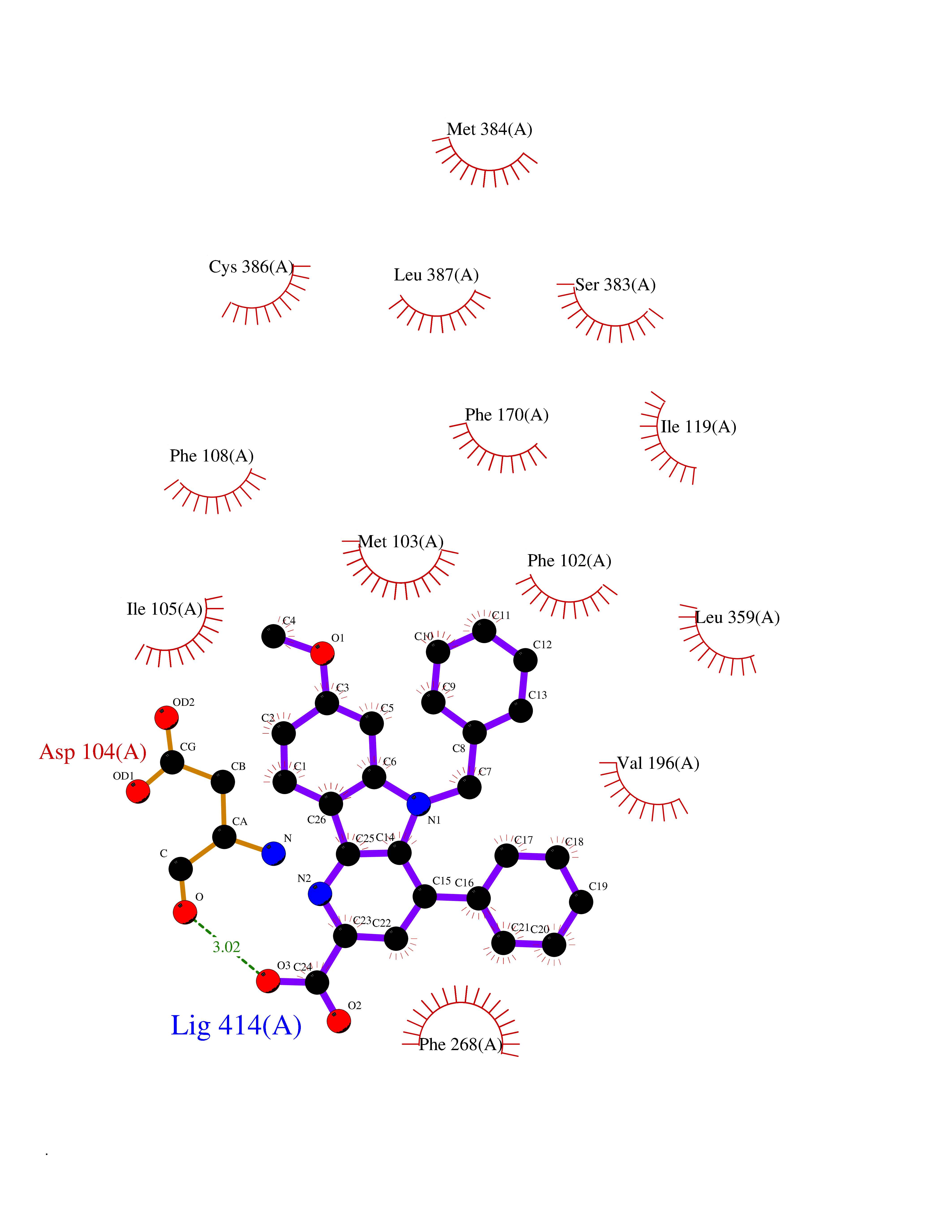 Ligplot Map 3D Binding mode Sequence ENFMDIECFMVLNPSQQLAIAVLSLTLGTFTVLENLLVLCVILHSRSLRCRPSYHFIGSLAVADLLGSVIFVYSFIDFHVFHRKDSRNVFLFKLGGVTASFTASVGSLFLAAIDRYISIHRPLAYKRIVTRPKAVVAFCLMWTIAIVIAVLPLLGWNCEKLQSVCSDIFPHIDETYLMFWIGVTSVLLLFIVYAYMYILWKADQARMDIRLAKTLVLILVVLIICWGPLLAIMVYDVFGKMNKLIKTVFAFCSMLCLLNSTVNPIIYALRSKDLRHAFRSMF Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Melatonin receptor type 1B (MTNR1B) | 6ME9 | 7.84 | |
Target general information Gen name MTNR1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1b receptor; Mel1b melatonin receptor; Mel-1B-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071; DB15133 Interacts with P28335; P48039; O76081; Q14669 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50184.9 Length 448 Aromaticity 0.11 Instability index 37.2 Isoelectric point 5.72 Charge (pH=7) -5.68 2D Binding mode Binding energy (Kcal/mol) -10.7 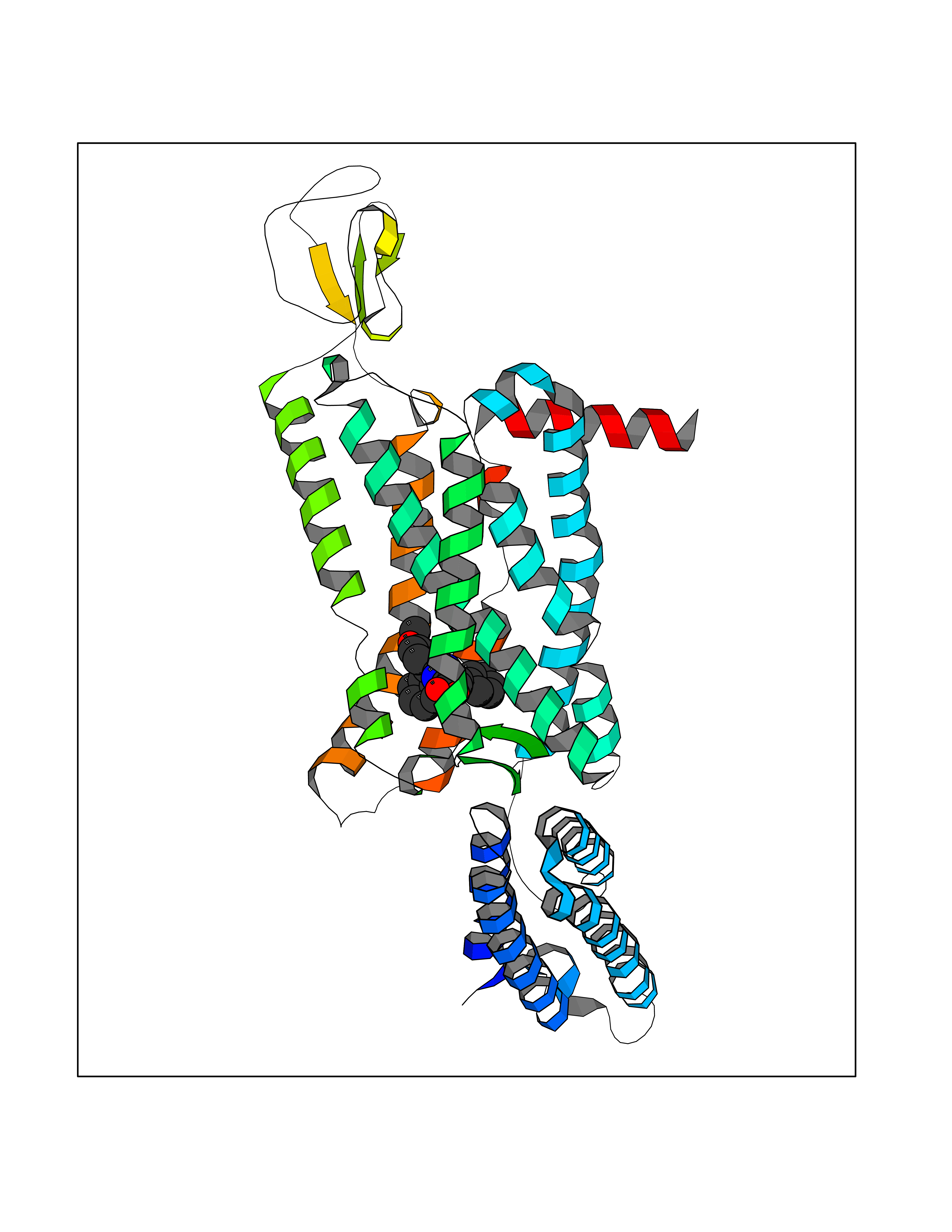 Molscript Map 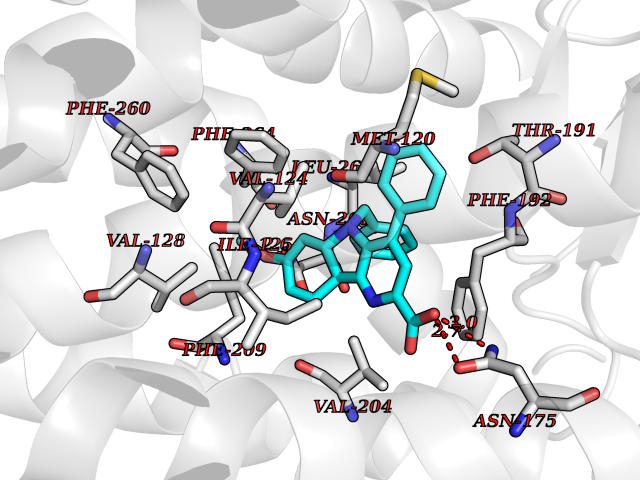 Pymol Map 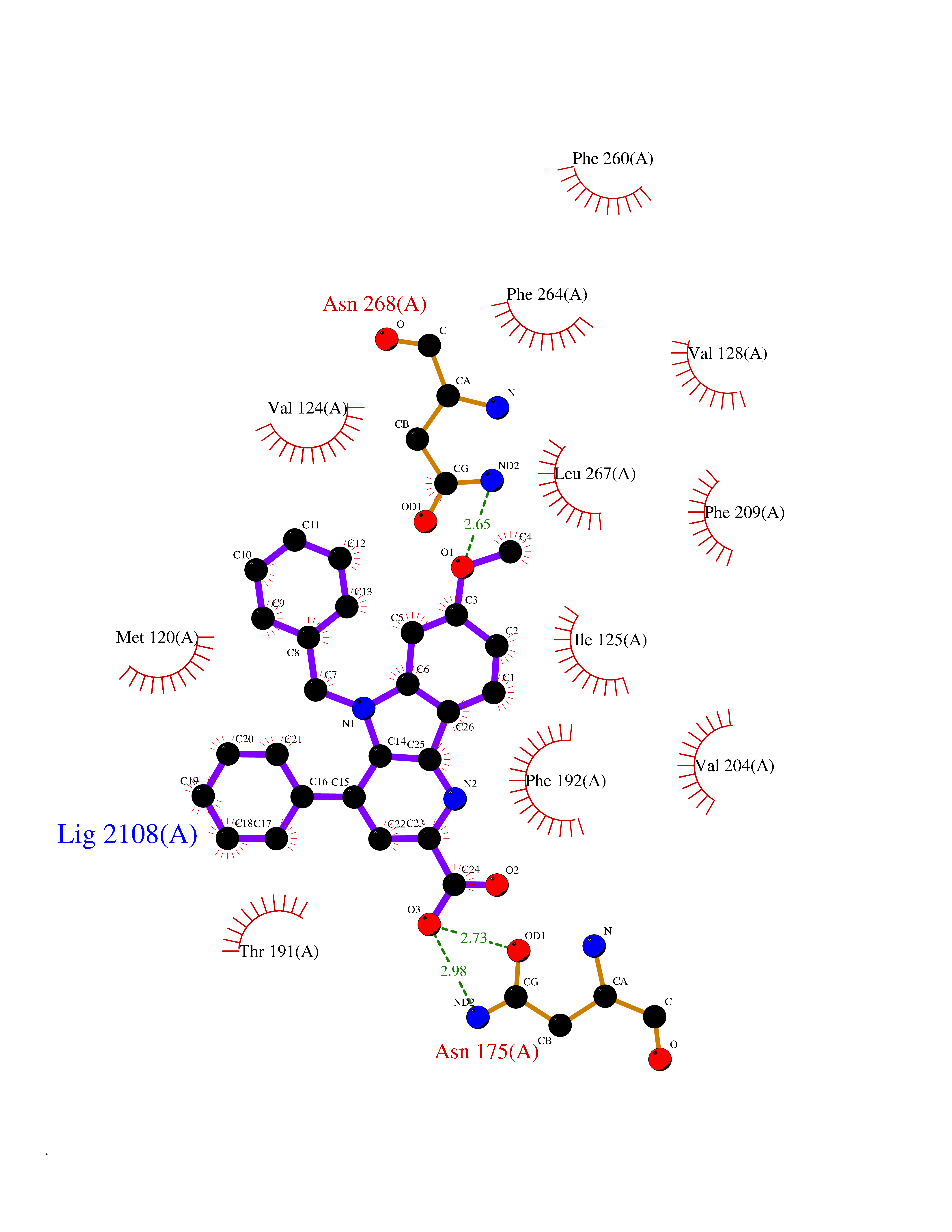 Ligplot Map 3D Binding mode Sequence ADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLGDGARPSWVAPALSAVLIVTTAVDVVGNLLVILSVLRNRKLRNAGNLFLVSLALANLVVAFYPYPLILVAIFYDGWAFGEEHCKASAFVMGLSVIGSVWNITAIAIDRYLYICHSMAYHRIYRRWHTPLHICLIWLLTVVALLPNFFVGSLEYDPRIYSCTFIQTASTQYTAAVVVIHFLLPIAVVSFCYLRIWVLVLQARMKKYTCTVCGYIYNPEDGDPDNGVNPGTDFKDIPDDWVCPLCGVGKDQFEEVECLKPSDLRSFLTMFVVFVIFAICFAPLNCIGLAVAINPQEMAPQIPEGLFVTSYLLAYFNSCLNPIVYGLLDQNFRREYKRILLALWN Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Cholesterol oxidase | 4REK | 7.82 | |
Target general information Gen name choA Organism Streptomyces sp. (strain SA-COO) Uniprot ID TTD ID NA Synonyms NA Protein family GMC oxidoreductase family Biochemical class Oxidoreductase Function Cholesterol oxidase activity.Flavin adenine dinucleotide binding.Steroid delta-isomerase activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB02332 Interacts with NA EC number 1.1.3.6; 5.3.3.1 Uniprot keywords 3D-structure; Cholesterol metabolism; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipid metabolism; Oxidoreductase; Secreted; Signal; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54367.8 Length 498 Aromaticity 0.1 Instability index 30.62 Isoelectric point 6.69 Charge (pH=7) -0.71 2D Binding mode Binding energy (Kcal/mol) -10.67 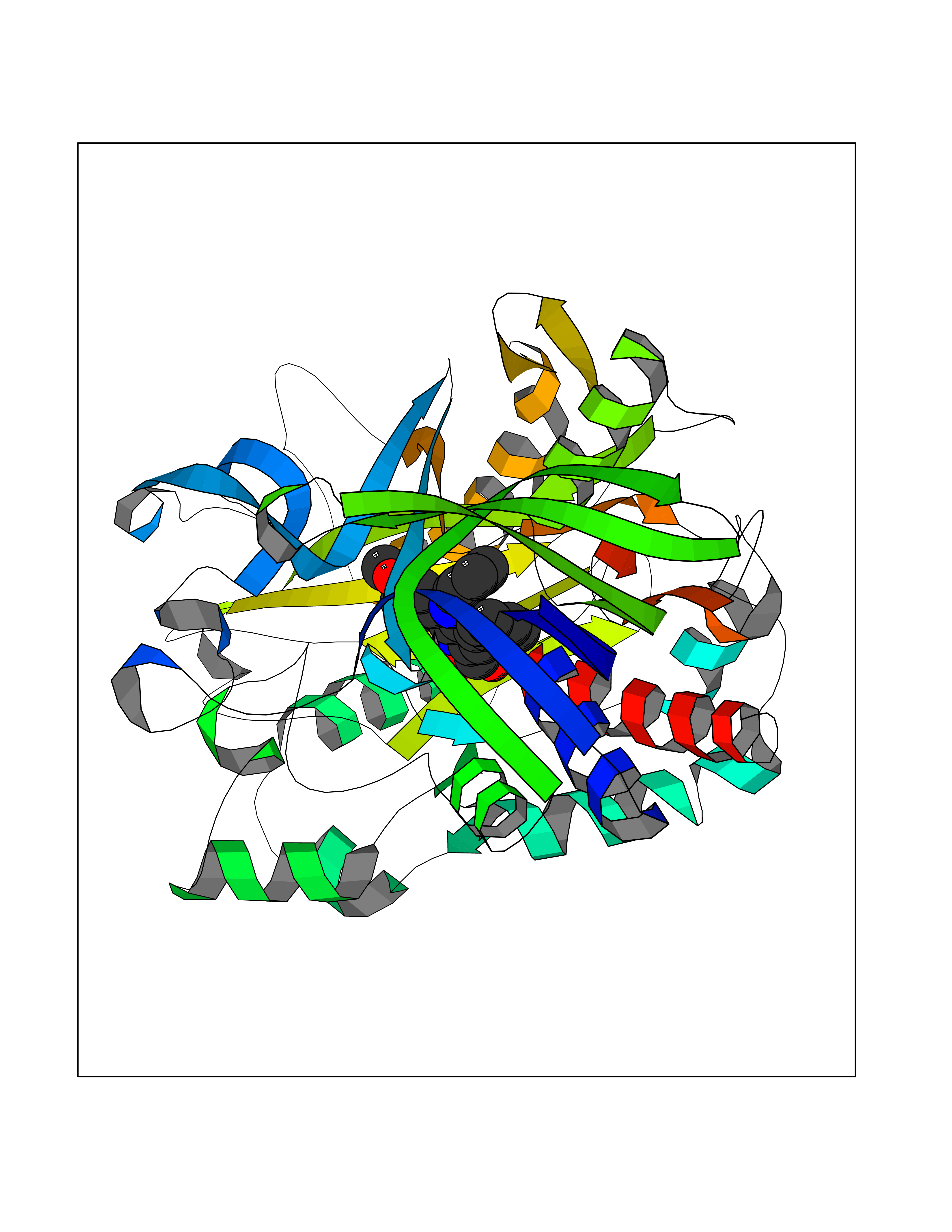 Molscript Map 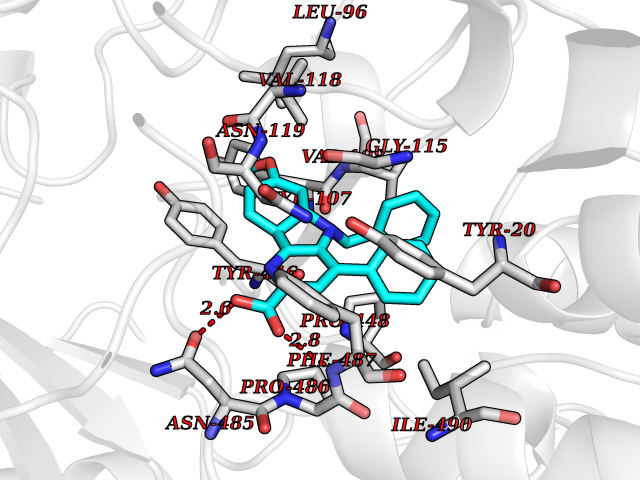 Pymol Map 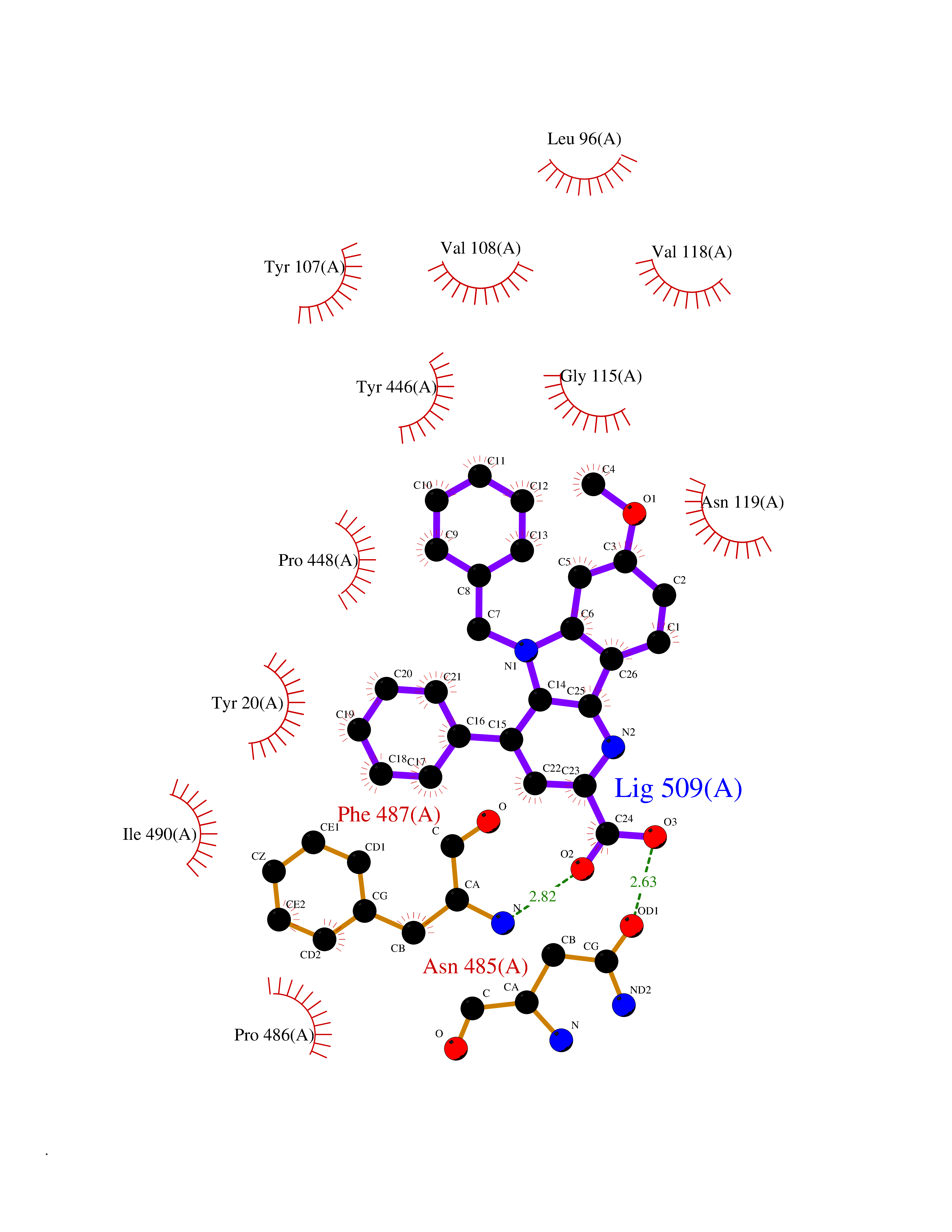 Ligplot Map 3D Binding mode Sequence GYVPAVVIGTGYGAAVSALRLGEAGVQTLMLEMGQLWNQPGPDGNIFCGMLNPDKRSSWFKNRTEAPLGSFLWLDVVNRNIDPYAGVLDRVNYDQMSVYVGRGVGGGSLVNGGMAVEPKRSYFEEILPRVDSSEMYDRYFPRANSMLRVNHIDTKWFEDTEWYKFARVSREQAGKAGLGTVFVPNVYDFGYMQREAAGEVPKSALATEVIYGNNHGKQSLDKTYLAAALGTGKVTIQTLHQVKTIRQTKDGGYALTVEQKDTDGKLLATKEISCRYLFLGAGSLGSTELLVRARDTGTLPNLNSEVGAGWGPNGNIMTARANHMWNPTGAHQSSIPALGIDAWDNSDSSVFAEIAPMPAGLETWVSLYLAITKNPQRGTFVYDAATDRAKLNWTRDQNAPAVNAAKALFDRINKANGTIYRYDLFGTQLKAFADDFCYHPLGGCVLGKATDDYGRVAGYKNLYVTDGSLIPGSVGVNPFVTITALAERNVERIIKQDV Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Dimethylglycine oxidase | 1PJ5 | 7.80 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -10.64  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Mutated oxalosuccinate decarboxylase (mIDH1) | 6ADG | 7.80 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD (mutated); Oxalosuccinate decarboxylase (mutated); NADP(+)-specific ICDH (mutated); Isocitrate dehydrogenase [NADP] cytoplasmic (mutated); IDP (mutated); IDH (mutated); Cytosolic NADP-isocitrate Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -10.65  Molscript Map 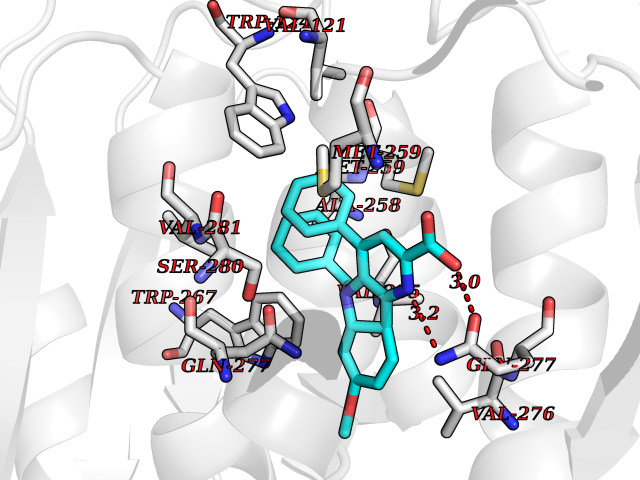 Pymol Map 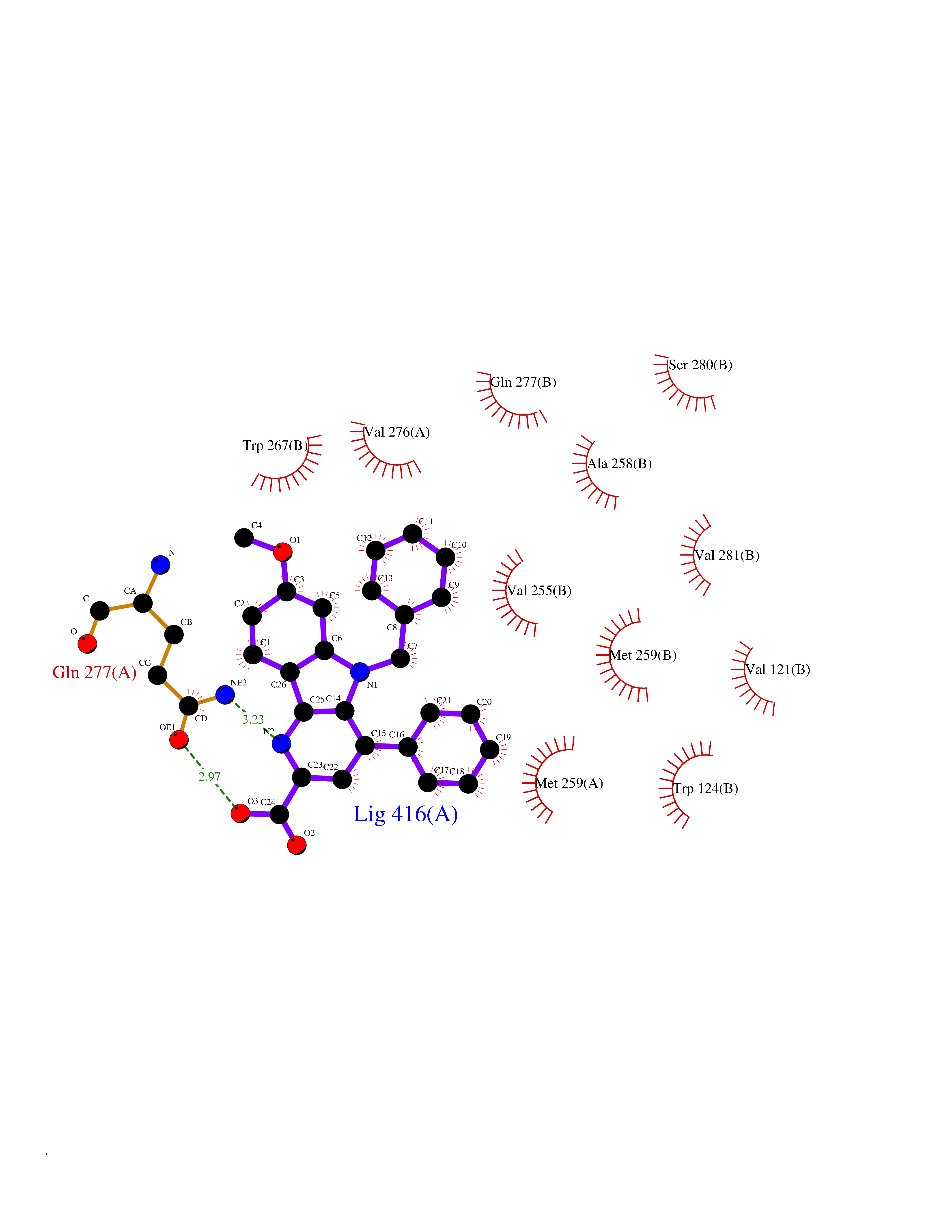 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Monomeric sarcosine oxidase | 2GF3 | 7.79 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -10.62  Molscript Map 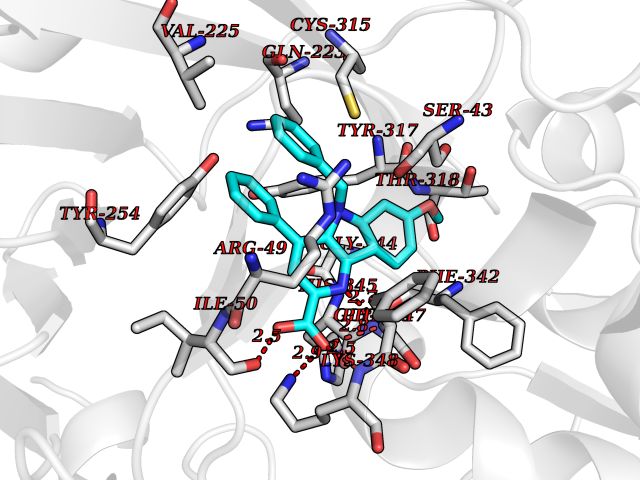 Pymol Map 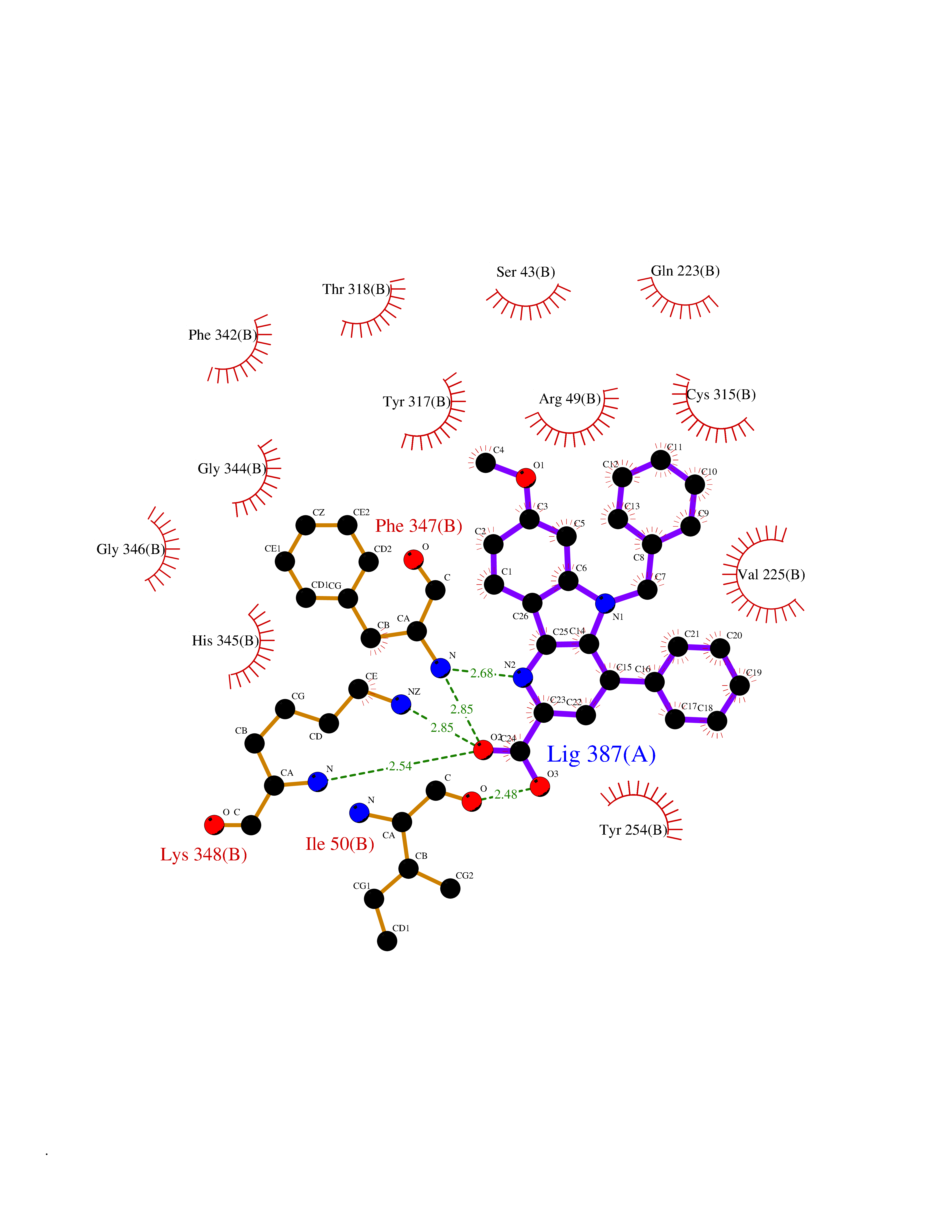 Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Aldo-keto reductase family 1 member C2 | 4XO6 | 7.79 | |
Target general information Gen name AKR1C2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DDH2 Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function Alditol:NADP+ 1-oxidoreductase activity.Bile acid binding.Carboxylic acid binding.Ketosteroid monooxygenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases 46,XY sex reversal 8 (SRXY8) [MIM:614279]: A disorder of sex development. Affected individuals have a 46,XY karyotype but present as phenotypically normal females. {ECO:0000269|PubMed:21802064}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06777; DB07768; DB01039; DB13751; DB06077; DB00959; DB00461; DB00157; DB03461; DB00776; DB12612; DB01586 Interacts with NA EC number 1.-.-.-; 1.1.1.112; 1.1.1.209; 1.1.1.357; 1.1.1.53; 1.1.1.62; 1.3.1.20 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Lipid metabolism; NADP; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36817.9 Length 324 Aromaticity 0.09 Instability index 37.64 Isoelectric point 6.86 Charge (pH=7) -0.42 2D Binding mode Binding energy (Kcal/mol) -10.62  Molscript Map 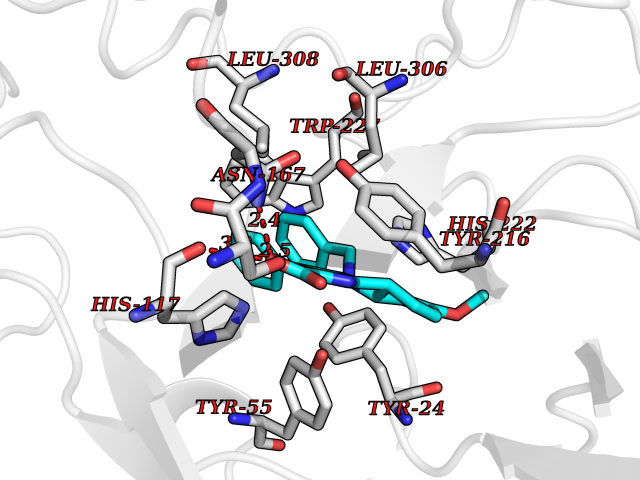 Pymol Map 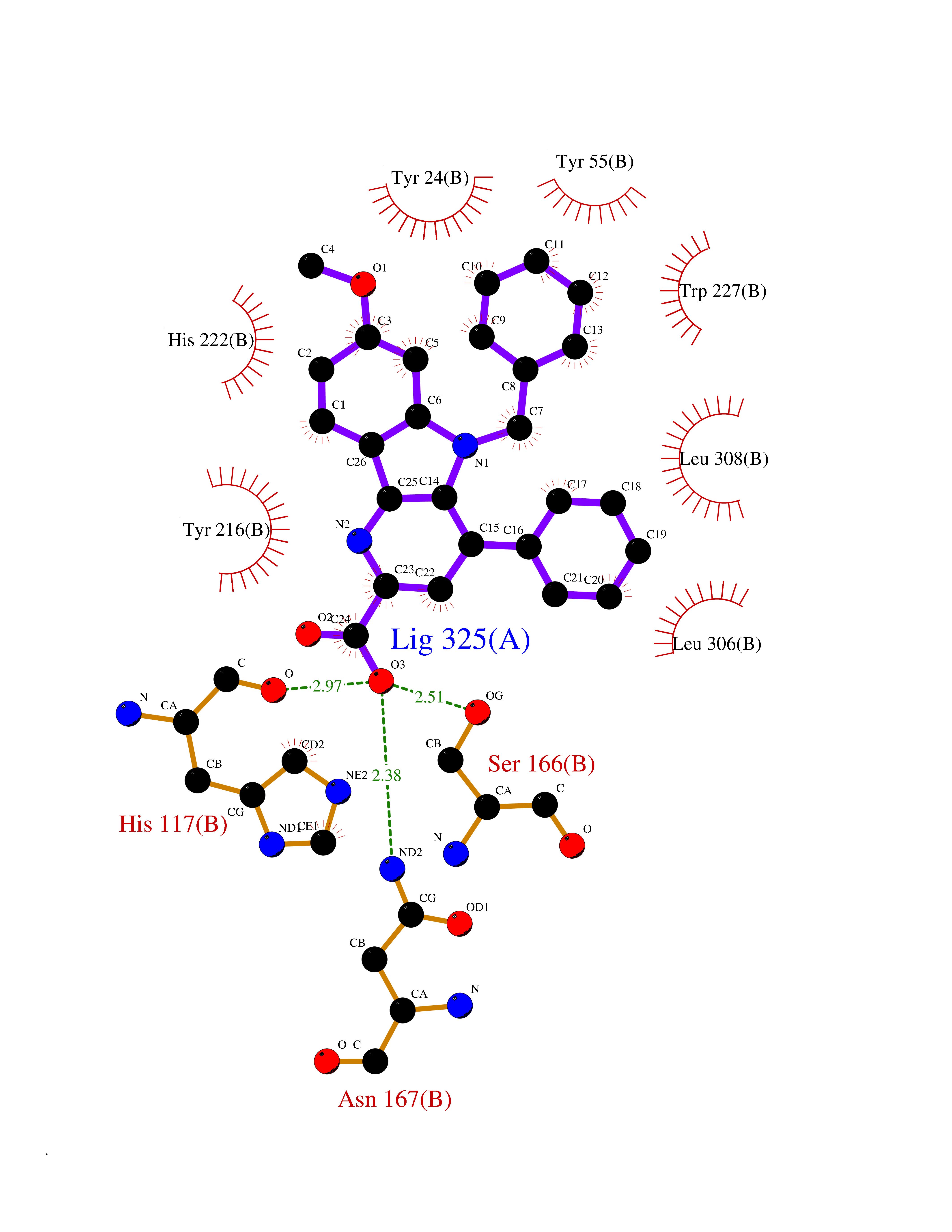 Ligplot Map 3D Binding mode Sequence VDDSKYQCVKLNDGHFMPVLGFGTYAPAEVPKSKALEAVKLAIEAGFHHIDSAHVYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWSNSHRPELVRPALERSLKNLQLDYVDLYLIHFPVSVKPGEEVIPKDENGKILFDTVDLCATWEAMEKCKDAGLAKSIGVSNFNHRLLEMILNKPGLKYKPVCNQVECHPYFNQRKLLDFCKSKDIVLVAYSALGSHREEPWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTSEEMKAIDGLNRNVRYLTLDIFAGPPNYPFSDEY Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Prolyl 3-hydroxylase OGFOD1 | 4NHY | 7.79 | |
Target general information Gen name OGFOD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TPA1;KIAA1612 Protein family TPA1 family Biochemical class Oxidoreductase / oxidoreductase inhibitor Function Iron ion binding.L-ascorbic acid binding.Peptidyl-proline 3-dioxygenase activity.Peptidyl-proline dioxygenase activity. Related diseases Long QT syndrome 10 (LQT10) [MIM:611819]: A heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. {ECO:0000269|PubMed:17592081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Atrial fibrillation, familial, 17 (ATFB17) [MIM:611819]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:23604097}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126 Interacts with Q9BSK4; P62266 EC number 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Dioxygenase; Iron; Metal-binding; Nucleus; Oxidoreductase; Proteomics identification; Reference proteome; Vitamin C Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 54223.9 Length 461 Aromaticity 0.13 Instability index 56.42 Isoelectric point 5.23 Charge (pH=7) -23.5 2D Binding mode Binding energy (Kcal/mol) -10.62  Molscript Map 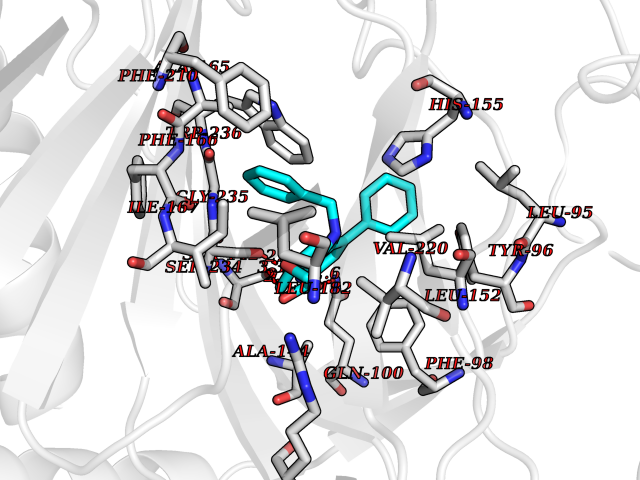 Pymol Map 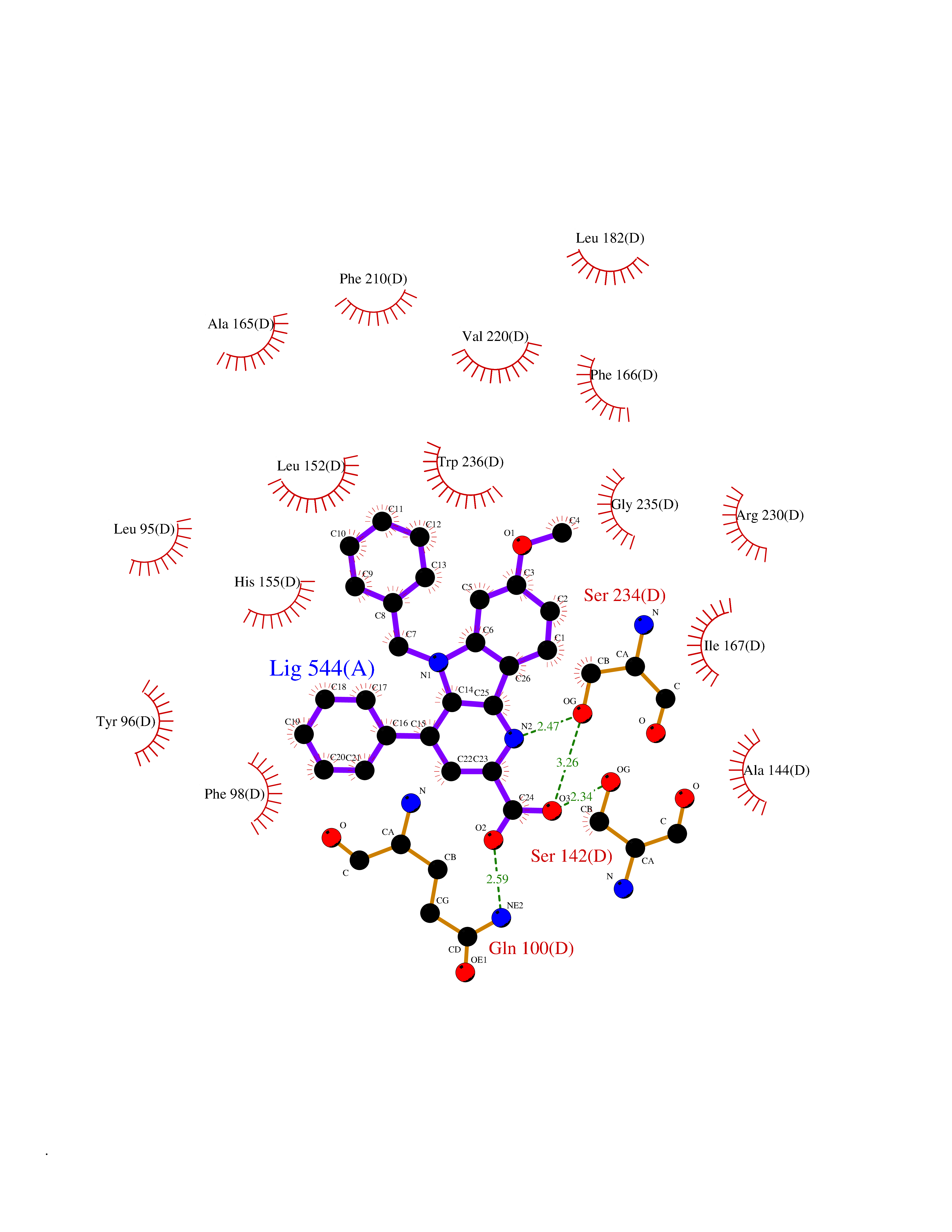 Ligplot Map 3D Binding mode Sequence AEFSDAVTEETLKKQVAEAWSRRTPFSHEVIVMDMDPFLHCVIPNFIQSQDFLEGLQKELMNLDFHEKYNDLYKFQQSDDLKKRREPHISTLRKILFEDFRSWLSDISKIDLESTIDMSCAKYEFTDALLCHDDELEGRRIAFILYLVPPWDRSMGGTLDLYSIDEHFQPKQIVKSLIPSWNKLVFFEVSPVSFHQVSEVLSEEKSRLSISGWFHGPSLTRPPNYFEPPIPRSPHIPQDHEILYDWINPTYLDMDYQVQIQEEFEESSEILLKEFLKPEKFTKVCEALEHGHVEWSSRGPPNKRFYEKAEESKLPEILKECMKLFRSEALFLLLSNFTGLKLHFLAPSSSVPMCQGELRHWKTGHYTLIHDHSKAEFALDLILYCGCEGWEPEYGGFTSYIAKGEDEELLTVNPESNSLALVYRDRETLKFVKHINHRSLEQKKTFPNRTGFWDFSFIYYE Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | CDC-like kinase 1 (CLK1) | 6KHD | 7.79 | |
Target general information Gen name CLK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity protein kinase CLK1; CLK; CDClike kinase 1 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, Lammer subfamily Biochemical class Kinase Function Phosphorylates serine- and arginine-rich (SR) proteins of the spliceosomal complex and may be a constituent of a network of regulatory mechanisms that enable SR proteins to control RNA splicing. Phosphorylates: SRSF1, SRSF3 and PTPN1. Regulates the alternative splicing of tissue factor (F3) pre-mRNA in endothelial cells and adenovirus E1A pre-mRNA. Dual specificity kinase acting on both serine/threonine and tyrosine-containing substrates. Related diseases Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Genetic variations in ALOX5AP may be associated with susceptibility to myocardial infarction. Involvement in myocardial infarction is however unclear: according to some authors (PubMed:14770184), a 4-SNP haplotype in ALOX5AP confers risk of myocardial infarction, while according to other (PubMed:17304054) ALOX5AP is not implicated in this condition. {ECO:0000269|PubMed:14770184, ECO:0000269|PubMed:17304054}. Drugs (DrugBank ID) DB06376; DB04367; DB08691; DB12010 Interacts with P60409; O76083; Q8N2M8; P49760; Q92997; P60371; P60409; Q8TBB1; O76083-2; O14492-2; A7MD48; Q07955; O75494; P84103; Q16629; P62995 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 37557.7 Length 322 Aromaticity 0.1 Instability index 37.24 Isoelectric point 6.38 Charge (pH=7) -4.53 2D Binding mode Binding energy (Kcal/mol) -10.62 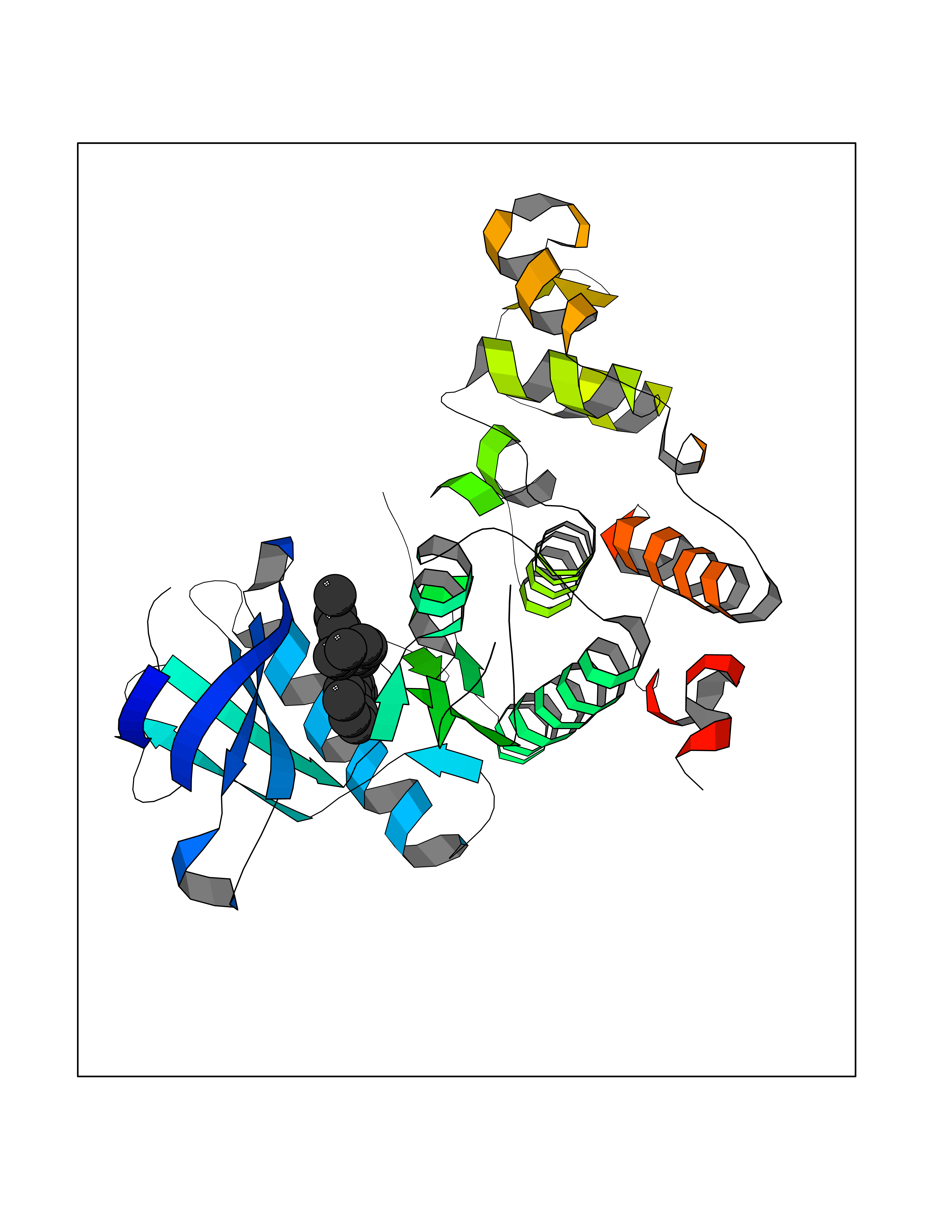 Molscript Map 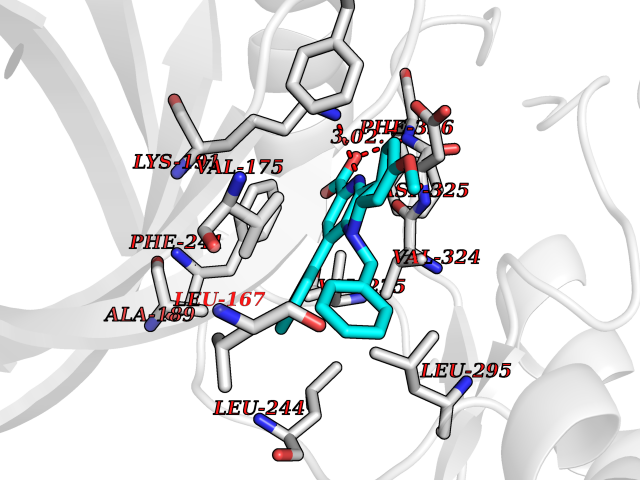 Pymol Map 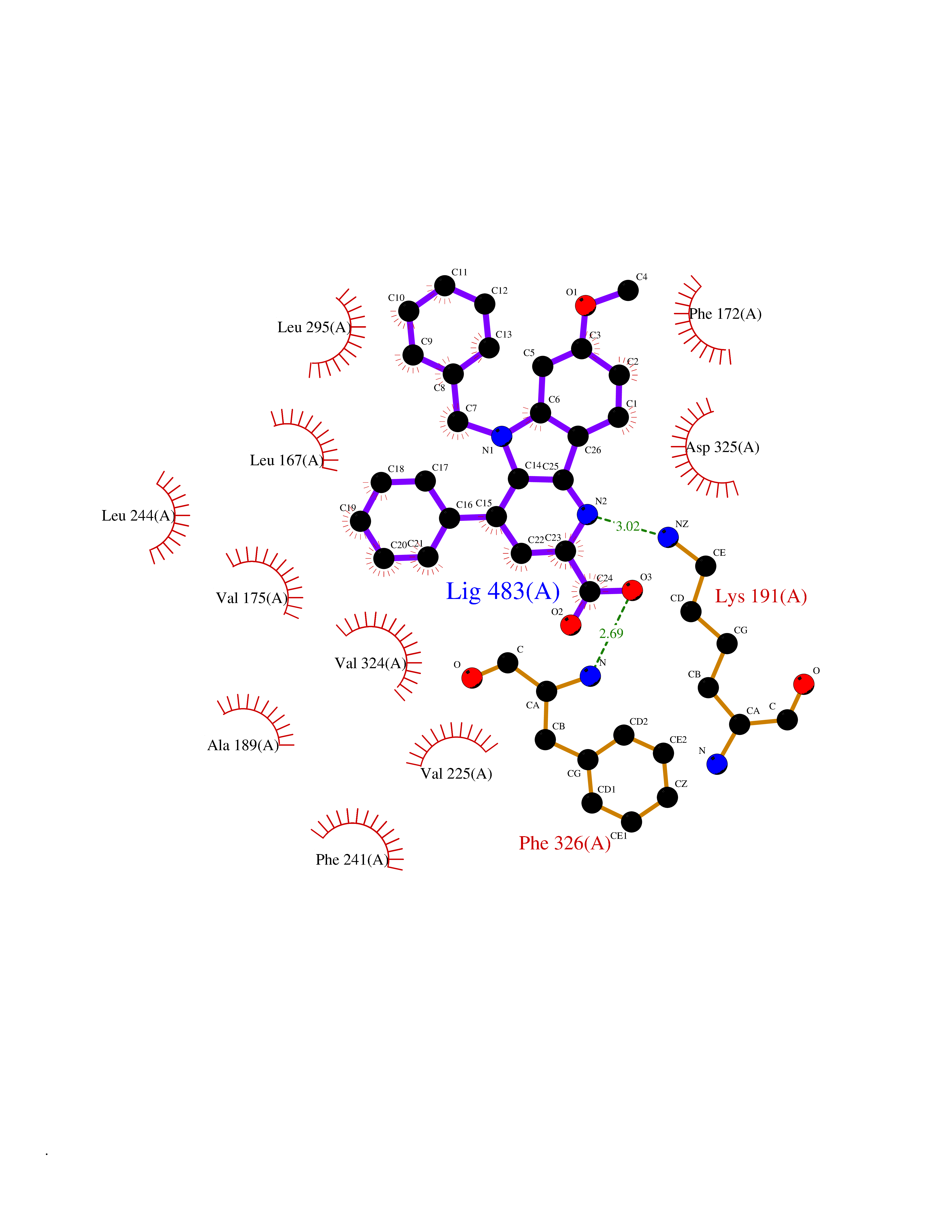 Ligplot Map 3D Binding mode Sequence LICQSGDVLSARYEIVDTLGEGAFGKVVECIDHKAGGRHVAVKIVKNVDRYCEAARSEIQVLEHLNTTDPNSTFRCVQMLEWFEHHGHICIVFELLGLSTYDFIKENGFLPFRLDHIRKMAYQICKSVNFLHSNKLTHTDLKPENILFVQSDYTEERTLINPDIKVVDFGSATYDDEHHSTLVRHYRAPEVILALGWSQPCDVWSIGCILIEYYLGFTVFPTHDSKEHLAMMERILGPLPKHMIQKTRKRKYFHHDRLDWDEHSSAGRYVSRACKPLKEFMLSQDVEHERLFDLIQKMLEYDPAKRITLREALKHPFFDLLK Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | IL-1 receptor-associated kinase 1 (IRAK1) | 6BFN | 7.79 | |
Target general information Gen name IRAK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-1 receptor-associated kinase 1; IRAK-1; IRAK Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, Pelle subfamily Biochemical class Kinase Function Involved in Toll-like receptor (TLR) and IL-1R signaling pathways. Is rapidly recruited by MYD88 to the receptor-signaling complex upon TLR activation. Association with MYD88 leads to IRAK1 phosphorylation by IRAK4 and subsequent autophosphorylation and kinase activation. Phosphorylates E3 ubiquitin ligases Pellino proteins (PELI1, PELI2 and PELI3) to promote pellino-mediated polyubiquitination of IRAK1. Then, the ubiquitin-binding domain of IKBKG/NEMO binds to polyubiquitinated IRAK1 bringing together the IRAK1-MAP3K7/TAK1-TRAF6 complex and the NEMO-IKKA-IKKB complex. In turn, MAP3K7/TAK1 activates IKKs (CHUK/IKKA and IKBKB/IKKB) leading to NF-kappa-B nuclear translocation and activation. Alternatively, phosphorylates TIRAP to promote its ubiquitination and subsequent degradation. Phosphorylates the interferon regulatory factor 7 (IRF7) to induce its activation and translocation to the nucleus, resulting in transcriptional activation of type I IFN genes, which drive the cell in an antiviral state. When sumoylated, translocates to the nucleus and phosphorylates STAT3. Serine/threonine-protein kinase that plays a critical role in initiating innate immune response against foreign pathogens. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) DB12010 Interacts with Q15306; Q92985; Q99836; Q96FA3; Q9HAT8; Q8N2H9-2; Q13526; Q86WV6; P58753; Q9Y4K3; Q8VCW4; Q5D1E7 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Host-virus interaction; Immunity; Innate immunity; Isopeptide bond; Kinase; Lipid droplet; Magnesium; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33681.4 Length 301 Aromaticity 0.09 Instability index 39.86 Isoelectric point 8.6 Charge (pH=7) 5.09 2D Binding mode Binding energy (Kcal/mol) -10.62  Molscript Map 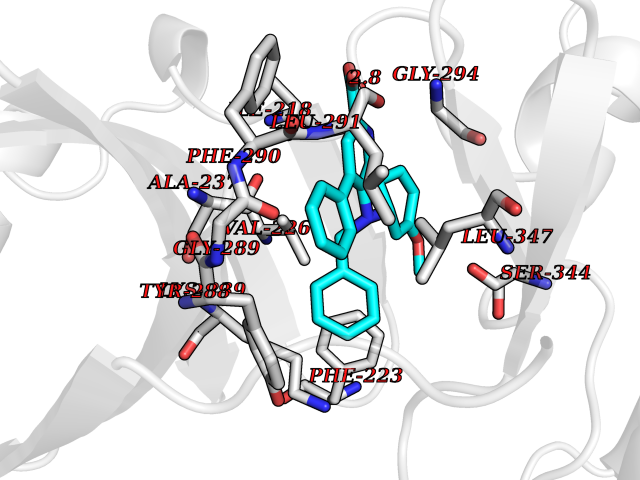 Pymol Map 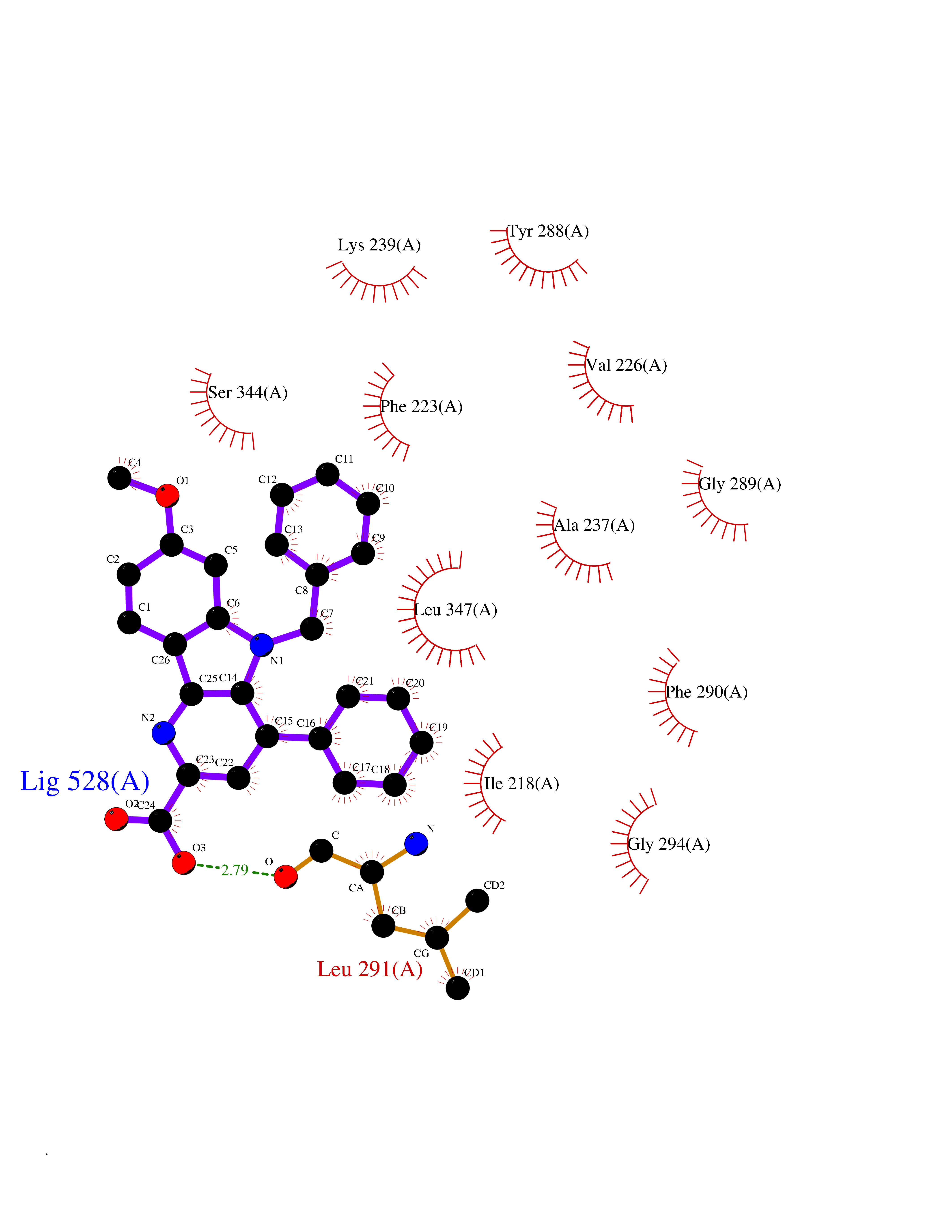 Ligplot Map 3D Binding mode Sequence SRPFPFCWPLCEISRGTHNFSEELKIGEGGFGCVYRAVMRNTVYAVKRLKEWTAVKQSFLTEVEQLSRFRHPNIVDFAGYCAQNGFYCLVYGFLPNGSLEDRLHCQTQACPPLSWPQRLDILLGTARAIQFLHQDSPSLIHGDIKSSNVLLDERLTPKLGDFGLARFSRTVRGTLAYLPEEYIKTGRLAVDTDTFSFGVVVLETLAGQRAVKTHGARTKYLKDLVEEEAEEAGVAAADAWAAPIAMQIYKKHLDPRPGPCPPELGLGLGQLACCCLHRRAKRRPPMTQVYERLEKLQAVVA Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Histidine decarboxylase (HDC) | 4E1O | 7.78 | |
Target general information Gen name HDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Human histidine decarboxylase Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the biosynthesis of histamine from histidine. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00117; DB00114 Interacts with Q86UW9 EC number EC 4.1.1.22 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 107706 Length 956 Aromaticity 0.1 Instability index 55.17 Isoelectric point 6.23 Charge (pH=7) -9.63 2D Binding mode Binding energy (Kcal/mol) -10.62  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQGSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Glycolipid transfer protein | 3RZN | 7.78 | |
Target general information Gen name GLTP Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GLTP family Biochemical class Lipid transport Function Glycolipid binding.Glycolipid transporter activity.Identical protein binding.Intermembrane lipid transfer activity.Lipid binding. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600; DB04465; DB03017; DB03203 Interacts with Q96DZ9; Q9NZD2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid transport; Proteomics identification; Reference proteome; Repeat; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23534.1 Length 206 Aromaticity 0.11 Instability index 36.45 Isoelectric point 7.08 Charge (pH=7) 0.1 2D Binding mode Binding energy (Kcal/mol) -10.61  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LAEHLLKPLPADKQIETGPFLEAVSHLPPFFDCLGSPVFTPIKADISGNITKIKAVYDTNPAKFRTLQNILEVEKEMYGAEWPKVGATLALMWLKRGLRFIQVFLQSICDGERDENHPNLIRVNATKAYEMALKKYHGWIVQKIFQAALYAAPYKSDFLKALSKGQNVTEEECLEKIRLFLVNYTATIDVIYEMYTQMNAELNYKV Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | LIM domain kinase-2 (LIMK-2) | 7QHG | 7.78 | |
Target general information Gen name LIMK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LIMK-2; LIM domain kinase 2 Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Displays serine/threonine-specific phosphorylation of myelin basic protein and histone (MBP) in vitro. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11718; DB12010 Interacts with Q16543; P08238; Q96C90; P62258 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Cytoskeleton; Kinase; LIM domain; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 32109.2 Length 283 Aromaticity 0.1 Instability index 27.28 Isoelectric point 6.01 Charge (pH=7) -3.93 2D Binding mode Binding energy (Kcal/mol) -10.61  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MDLIHGEVLGKGFFGQAIKVTHKATGKVMVMKELIRCDEETQKTFLTEVKVMRSLDHPNVLKFIGVLYKDKKLNLLTEYIEGGTLKDFLRSMDPFPWQQKVRFAKGIASGMAYLHSMCIIHRDLNSHNCLIKLDKTVVVADFGLSRLIVDRKKRYTVVGNPYWMAPEMLNGKSYDETVDIFSFGIVLCEIIGQVYADPDCLPRTLDFGLNVKLFWEKFVPTDCPPAFFPLAAICCRLEPESRPAFSKLEDSFEALSLYLGELGIPLPAELEELDHTVSMQYGL Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Fungal Scytalone dehydratase (Fung SDH1) | 3STD | 7.78 | |
Target general information Gen name Fung SDH1 Organism Pyricularia oryzae (strain 70-15 / ATCC MYA-4617 / FGSC 8958) (Rice blast fungus) (Magnaporthe oryzae) Uniprot ID TTD ID Synonyms SDH1 Protein family Scytalone dehydratase family Biochemical class Alpha-carbonic anhydrase Function Catalyzes two steps in melanin biosynthesis. From scytalone they are two dehydration steps and one reduction step to yield melanin. Related diseases CODAS syndrome (CODASS) [MIM:600373]: A rare syndrome characterized by the combination of cerebral, ocular, dental, auricular, and skeletal features. These include developmental delay, craniofacial anomalies, cataracts, ptosis, median nasal groove, delayed tooth eruption, hearing loss, short stature, delayed epiphyseal ossification, metaphyseal hip dysplasia, and vertebral coronal clefts. {ECO:0000269|PubMed:25574826, ECO:0000269|PubMed:25808063}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 4.2.1.94 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Endosome; Lyase; Melanin biosynthesis; Metal-binding; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 19102.4 Length 162 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.87 Charge (pH=7) -3.7 2D Binding mode Binding energy (Kcal/mol) -10.61  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GEITFSDYLGLMTCVYEWADSYDSKDWDRLRKVIAPTLRIDYRSFLDKLWEAMPAEEFVGMVSSKQVLGDPTLRTQHFIGGTRWEKVSEDEVIGYHQLRVPHQRYKDTTMKEVTMKGHAHSANLHWYKKIDGVWKFAGLKPDIRWGEFDFDRIFEDGRETFG Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Extracellular calcium-sensing receptor (CASR) | 5FBK | 7.77 | |
Target general information Gen name CASR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCasR; Parathyroid cell calciumreceptor; Parathyroid cell calcium-sensing receptor 1; Parathyroid calcium receptor; Parathyroid Cell calcium-sensing receptor; PCaR1; GPRC2A; CaSR Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Senses fluctuations in the circulating calcium concentration and modulates the production of parathyroid hormone (PTH) in parathyroid glands. The activity of this receptor is mediated by a G-protein that activates a phosphatidylinositol-calcium second messenger system. The G-protein-coupled receptor activity is activated by a co-agonist mechanism: aromatic amino acids, such as Trp or Phe, act concertedly with divalent cations, such as calcium or magnesium, to achieve full receptor activation. G-protein-coupled receptor that senses changes in the extracellular concentration of calcium ions and plays a key role in maintaining calcium homeostasis. Related diseases Hypocalciuric hypercalcemia, familial 1 (HHC1) [MIM:145980]: A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults. {ECO:0000269|PubMed:11762699, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:15579740, ECO:0000269|PubMed:15879434, ECO:0000269|PubMed:16598859, ECO:0000269|PubMed:16740594, ECO:0000269|PubMed:17473068, ECO:0000269|PubMed:17698911, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:19789209, ECO:0000269|PubMed:21566075, ECO:0000269|PubMed:21643651, ECO:0000269|PubMed:22114145, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25104082, ECO:0000269|PubMed:25292184, ECO:0000269|PubMed:26386835, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:7673400, ECO:0000269|PubMed:7726161, ECO:0000269|PubMed:7916660, ECO:0000269|PubMed:8636323, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9298824}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperparathyroidism, neonatal severe (NSHPT) [MIM:239200]: A disorder characterized by severe hypercalcemia, bone demineralization, and failure to thrive usually manifesting in the first 6 months of life. If untreated, NSHPT can be a devastating neurodevelopmental disorder, which in some cases is lethal without parathyroidectomy. {ECO:0000269|PubMed:14985373, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:17555508, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:8675635, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypocalcemia, autosomal dominant 1 (HYPOC1) [MIM:601198]: A disorder of mineral homeostasis characterized by blood calcium levels below normal, and low or normal serum parathyroid hormone concentrations. Disease manifestations include mild or asymptomatic hypocalcemia, paresthesias, carpopedal spasm, seizures, hypercalciuria with nephrocalcinosis or kidney stones, and ectopic and basal ganglia calcifications. Few patients manifest hypocalcemia and features of Bartter syndrome, including hypomagnesemia, hypokalemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia. {ECO:0000269|PubMed:10487661, ECO:0000269|PubMed:12050233, ECO:0000269|PubMed:12107202, ECO:0000269|PubMed:12241879, ECO:0000269|PubMed:12574188, ECO:0000269|PubMed:12915654, ECO:0000269|PubMed:15551332, ECO:0000269|PubMed:16608894, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:22789683, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25766501, ECO:0000269|PubMed:7874174, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8733126, ECO:0000269|PubMed:8813042, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253358, ECO:0000269|PubMed:9661634, ECO:0000269|PubMed:9920108}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 8 (EIG8) [MIM:612899]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Seizure types are variable, but include myoclonic seizures, absence seizures, febrile seizures, complex partial seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:18756473}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11093; DB11348; DB14481; DB01012; DB12865; DB00994; DB05695; DB05255; DB00127 Interacts with Q15363; P41180-1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 52438.9 Length 467 Aromaticity 0.12 Instability index 39.62 Isoelectric point 5.63 Charge (pH=7) -10.18 2D Binding mode Binding energy (Kcal/mol) -10.59 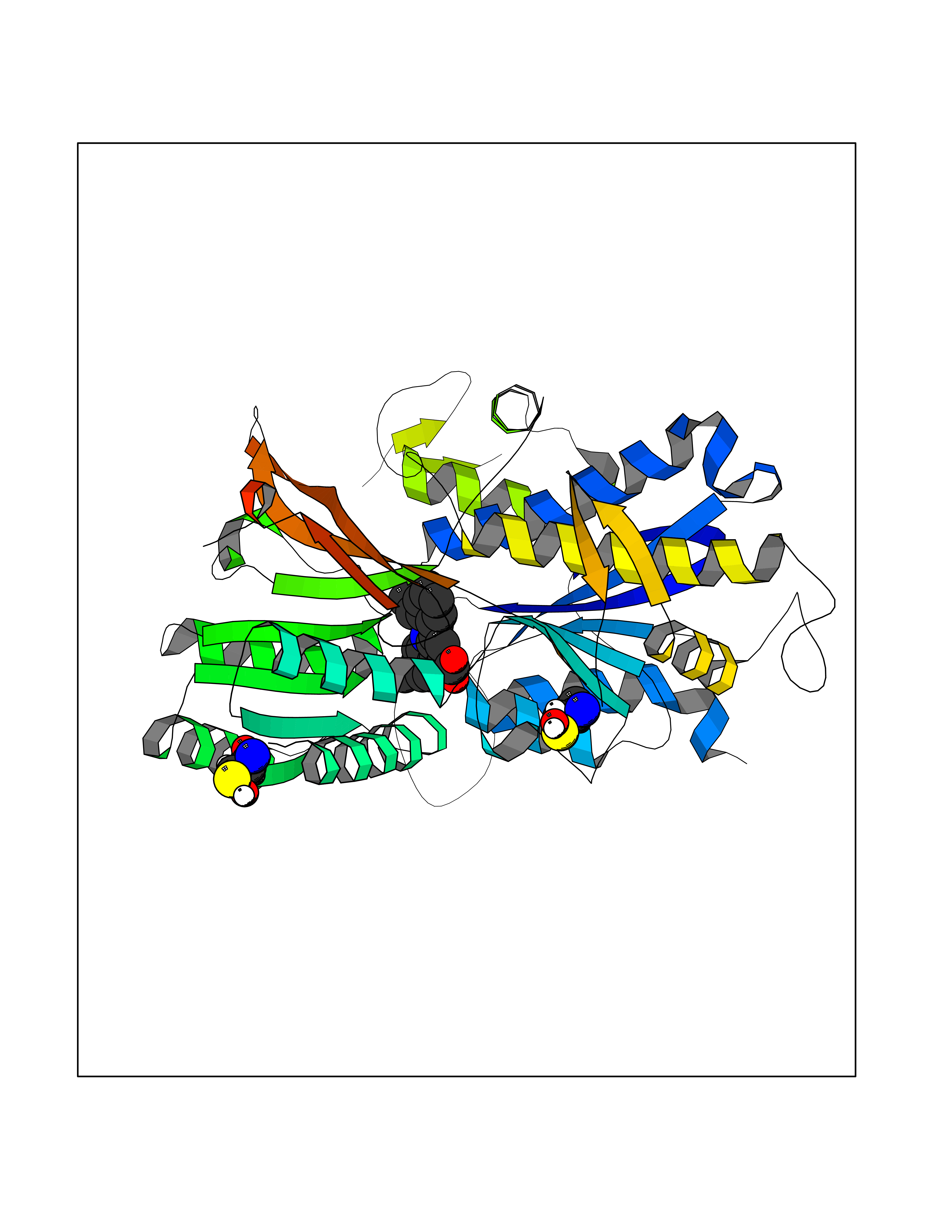 Molscript Map 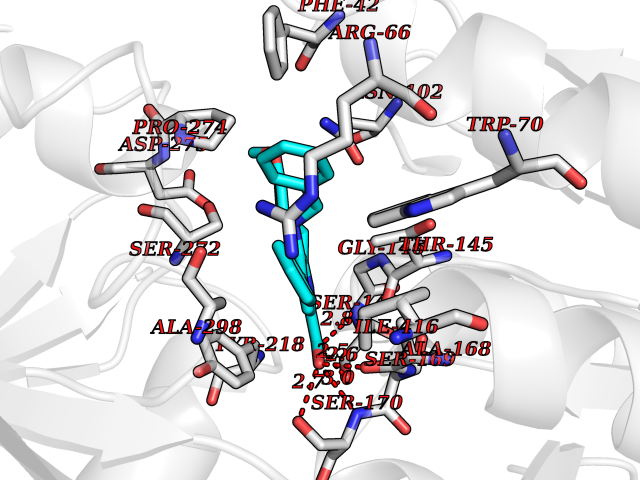 Pymol Map 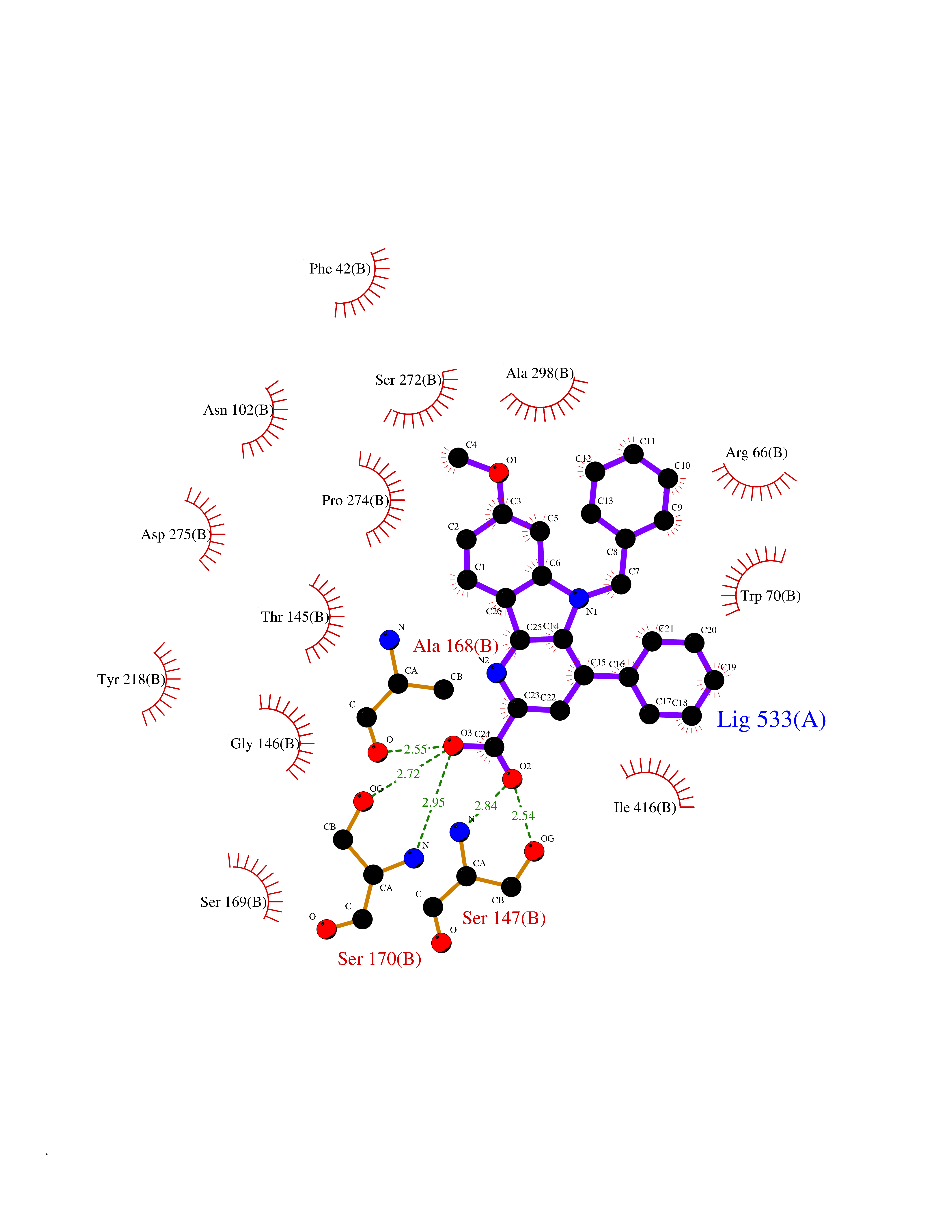 Ligplot Map 3D Binding mode Sequence GPDQRAQKKGDIILGGLFPIHFGVAAKDQDLKSRPESVECIRYNFRGFRWLQAMIFAIEEINSSPALLPNLTLGYRIFDTCNTVSKALEATLSFVAQNKIDSTIAVVGATGSGVSTAVANLLGLFYIPQVSYASSSRLLSNKNQFKSFLRTIPNDEHQATAMADIIEYFRWNWVGTIAADDDYGRPGIEKFREEAEERDIXIDFSELISQYSDEEEIQHVVEVIQNSTAKVIVVFSSGPDLEPLIKEIVRRNITGKIWLASEAWASSSLIAMPQYFHVVGGTIGFALKAGQIPGFREFLKKVHPRKSVHNGFAKEFWEETFNCHLQFRPLCTGDENISSVETPYIDYTHLRISYNVYLAVYSIAHALQDIYTCLPGRGLFTNGSCADIKKVEAWQVLKHLRHLNFTNNMGEQVTFDEXGDLVGNYSIINWHLSPEDGSIVFKEVGYYNVYAKKGERLFINEEKILWS Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Opioid receptor sigma 1 (OPRS1) | 6DJZ | 7.77 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23901 Length 212 Aromaticity 0.14 Instability index 33.12 Isoelectric point 5.61 Charge (pH=7) -5.6 2D Binding mode Binding energy (Kcal/mol) -10.6 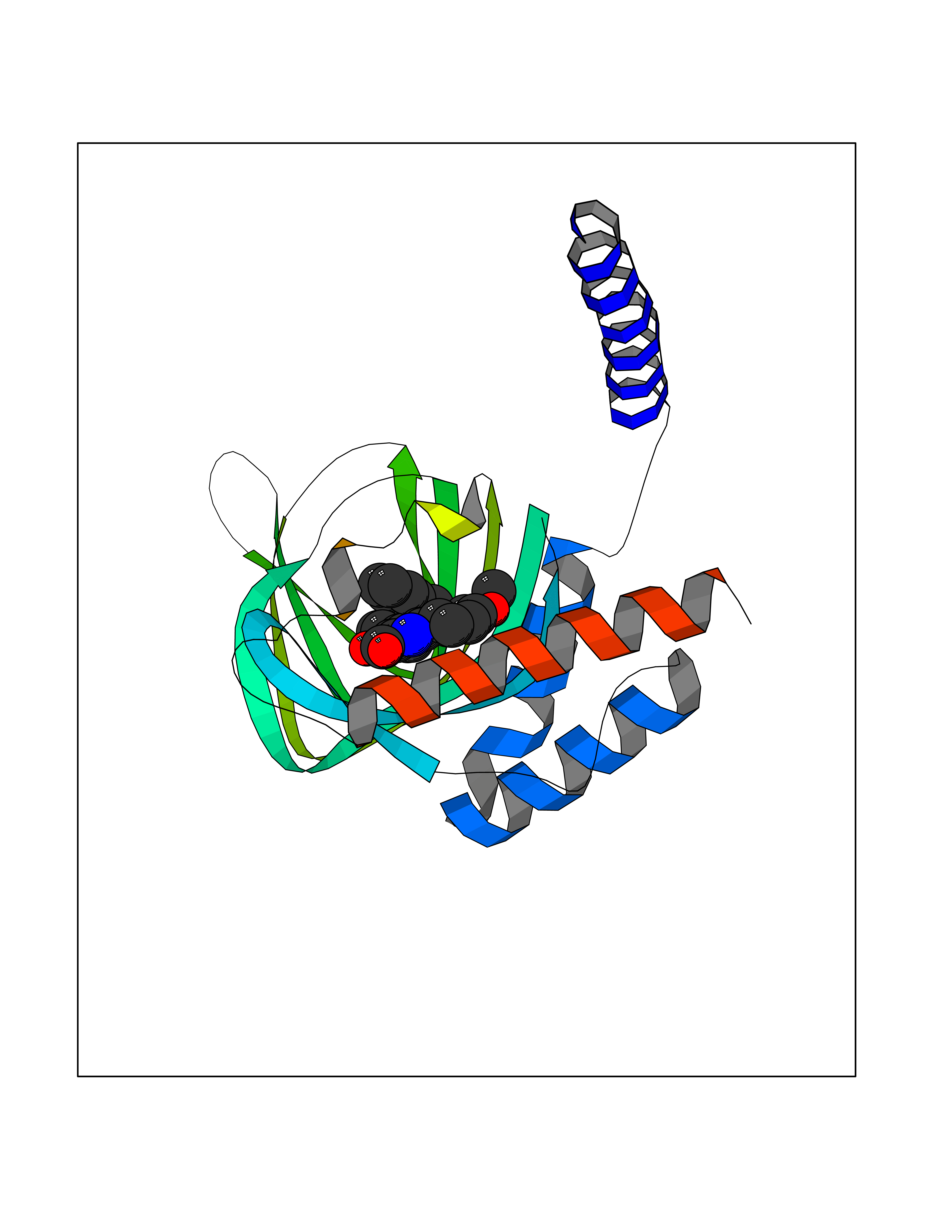 Molscript Map 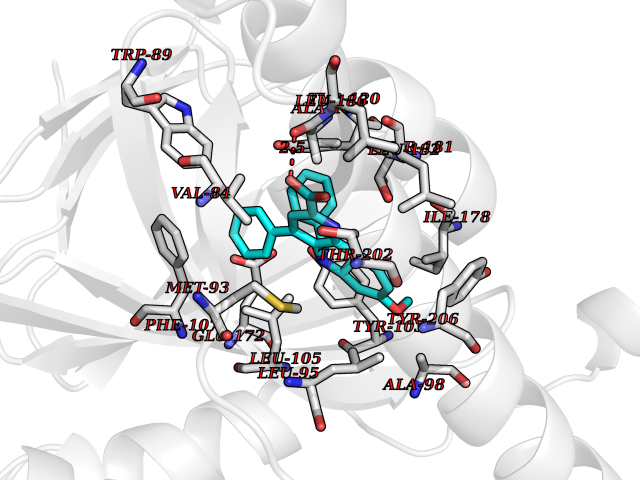 Pymol Map 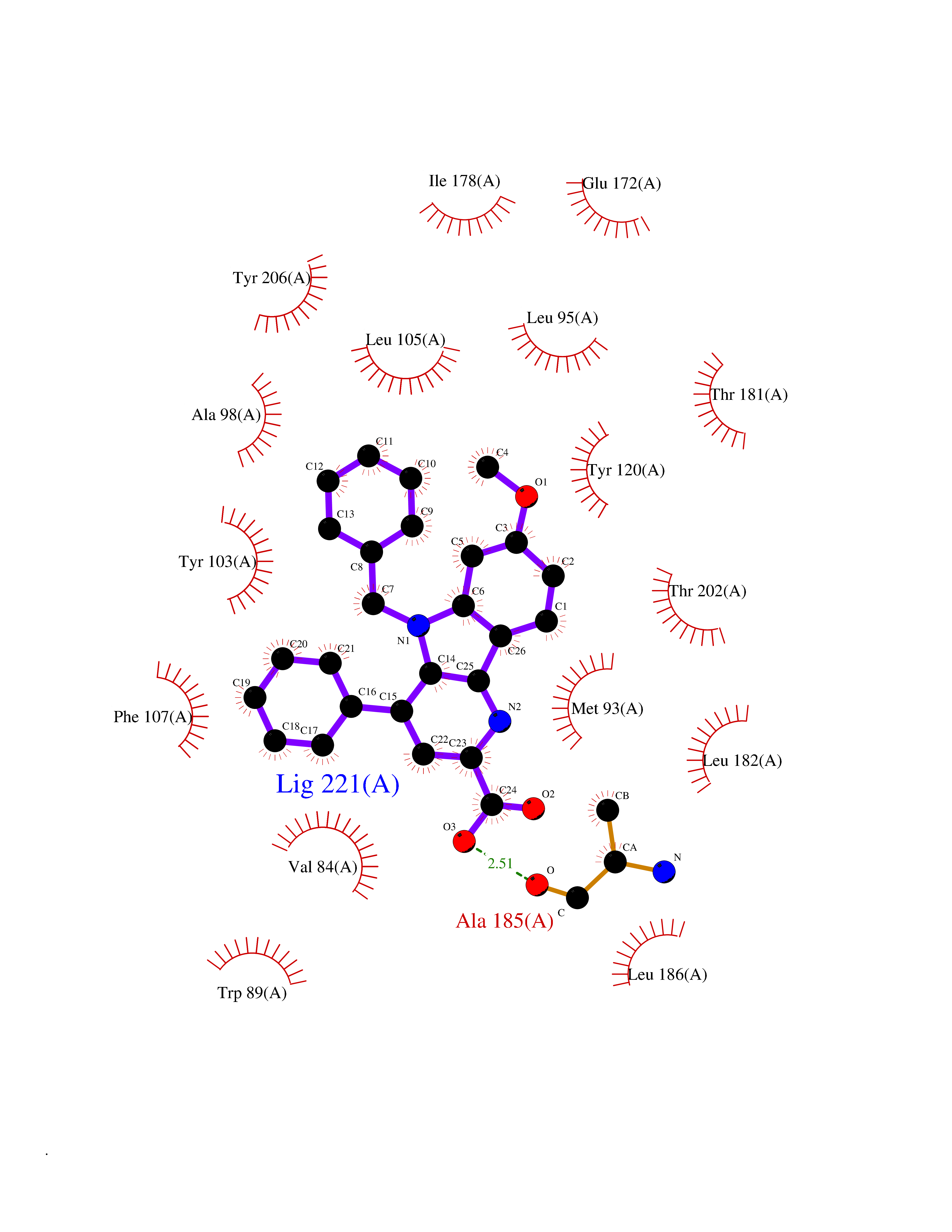 Ligplot Map 3D Binding mode Sequence RWAWAALLLAVAAVLTQVVWLWLGTQSFVFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLF Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | 4-cresol dehydrogenase [hydroxylating] flavoprotein subunit | 1WVF | 7.76 | |
Target general information Gen name pchF Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function 4-cresol dehydrogenase (hydroxylating) activity.Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on CH-OH group of donors. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.17.9.1 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase; Plasmid Protein physicochemical properties Chain ID A Molecular weight (Da) 57240.8 Length 515 Aromaticity 0.1 Instability index 30.94 Isoelectric point 6.06 Charge (pH=7) -4.42 2D Binding mode Binding energy (Kcal/mol) -10.58 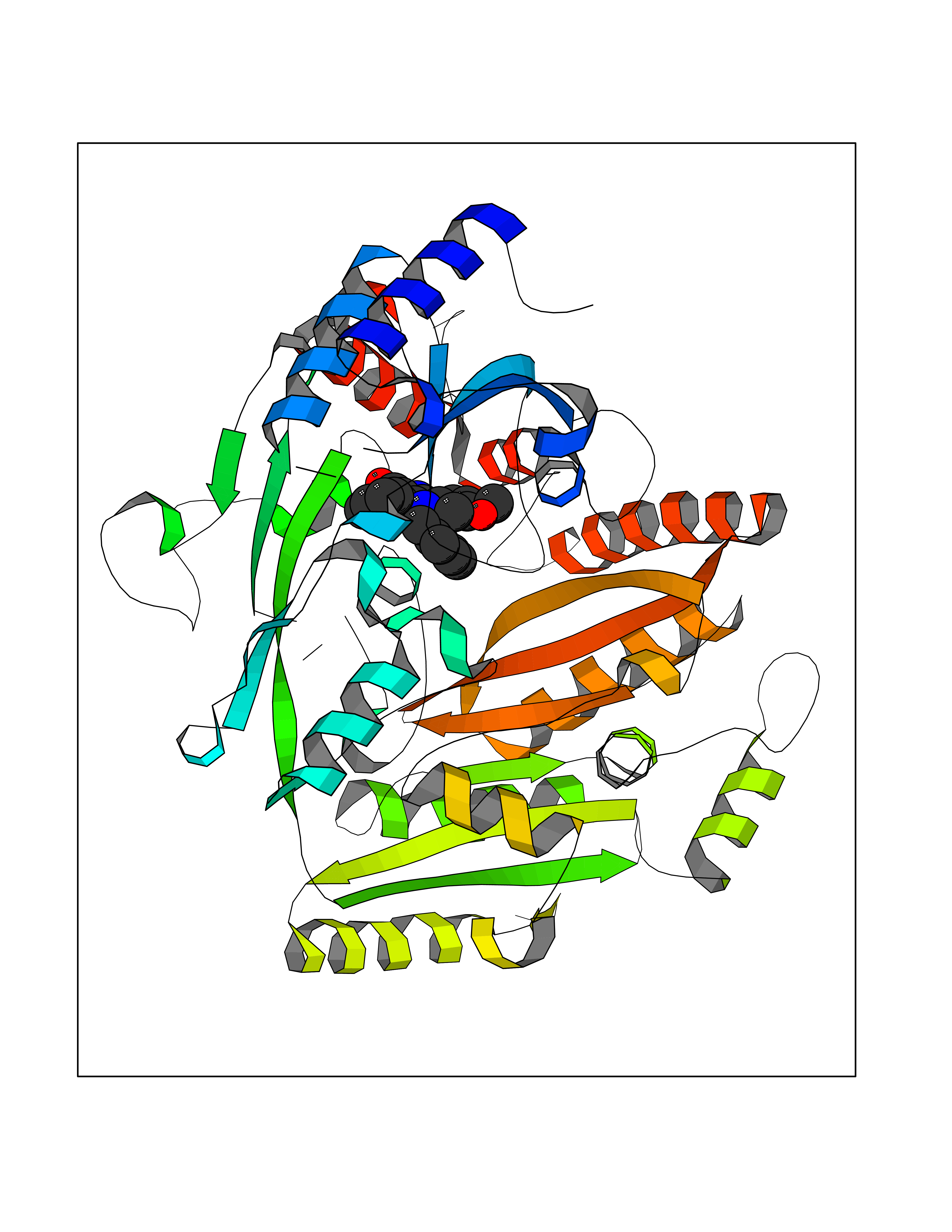 Molscript Map 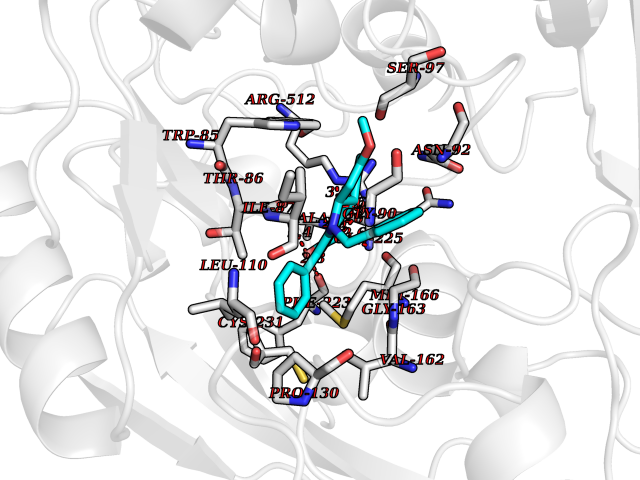 Pymol Map 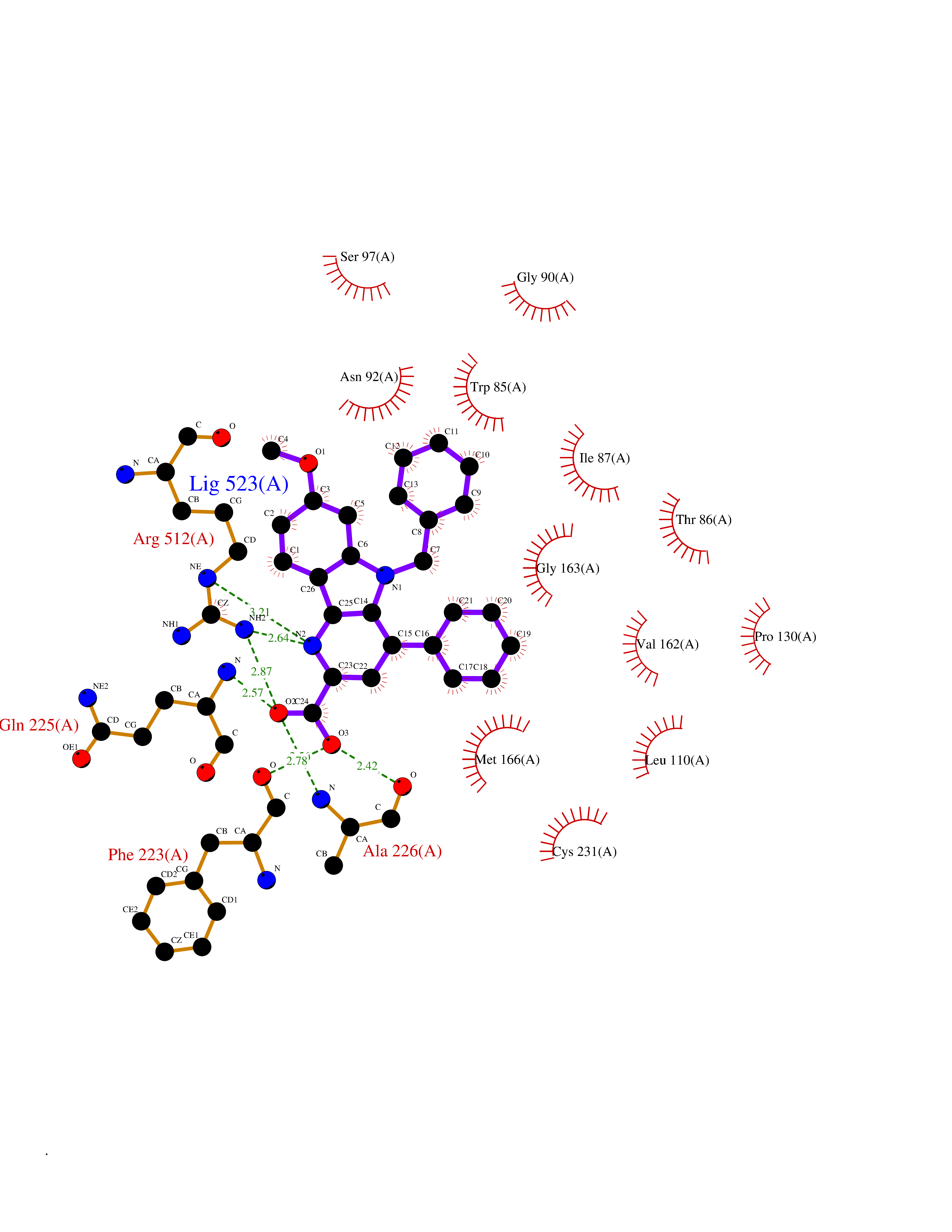 Ligplot Map 3D Binding mode Sequence AVLPKGVTQGEFNKAVQKFRALLGDDNVLVESDQLVPYNKIMMPVENAAHAPSAAVTATTVEQVQGVVKICNEHKIPIWTISTGRNFGYGSAAPVQRGQVILDLKKMNKIIKIDPEMCYALVEPGVTFGQMYDYIQENNLPVMLSFSAPSAIAGPVGNTMDRGVGYTPYGEHFMMQCGMEVVLANGDVYRTGMGGVPGSNTWQIFKWGYGPTLDGMFTQANYGICTKMGFWLMPKPPVFKPFEVIFEDEADIVEIVDALRPLRMSNTIPNSVVIASTLWEAGSAHLTRAQYTTEPGHTPDSVIKQMQKDTGMGAWNLYAALYGTQEQVDVNWKIVTDVFKKLGKGRIVTQEEAGDTQPFKYRAQLMSGVPNLQEFGLYNWRGGGGSMWFAPVSEARGSECKKQAAMAKRVLHKYGLDYVAEFIVAPRDMHHVIDVLYDRTNPEETKRADACFNELLDEFEKEGYAVYRVNTRFQDRVAQSYGPVKRKLEHAIKRAVDPNNILAPGRSGIDLNNDF Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Aldo-keto reductase family 1 member C1 | 1MRQ | 7.76 | |
Target general information Gen name AKR1C1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DDH1;DDH Protein family Aldo/keto reductase family Biochemical class Oxidoreductase Function 17-alpha,20-alpha-dihydroxypregn-4-en-3-one dehydrogenase activity.Alditol:NADP+ 1-oxidoreductase activity.Aldo-keto reductase (NADP) activity.Androsterone dehydrogenase (B-specific) activity.Bile acid binding.Carboxylic acid binding.Indanol dehydrogenase activity.Ketosteroid monooxygenase activity.Oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor.Phenanthrene 9,10-monooxygenase activity.Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase activity. Related diseases Fibrodysplasia ossificans progressiva (FOP) [MIM:135100]: A rare autosomal dominant connective tissue disorder resulting in skeletal malformations and progressive extraskeletal ossification. Heterotopic ossification begins in childhood and can be induced by trauma or may occur without warning. Bone formation is episodic and progressive, leading to a debilitating ankylosis of all major joints of the axial and appendicular skeleton, rendering movement impossible. {ECO:0000269|PubMed:16642017, ECO:0000269|PubMed:19085907, ECO:0000269|PubMed:19330033}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04674; DB00945; DB07768; DB01039; DB07931; DB06077; DB00959; DB00461; DB00157; DB03467; DB03461; DB00776; DB12612; DB00936 Interacts with P51857; P26045; Q7Z699 EC number 1.1.1.-; 1.1.1.112; 1.1.1.149; 1.1.1.209; 1.1.1.210; 1.1.1.357; 1.1.1.51; 1.1.1.53; 1.1.1.62; 1.3.1.20 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 36784.9 Length 323 Aromaticity 0.09 Instability index 42.07 Isoelectric point 8.06 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -10.58 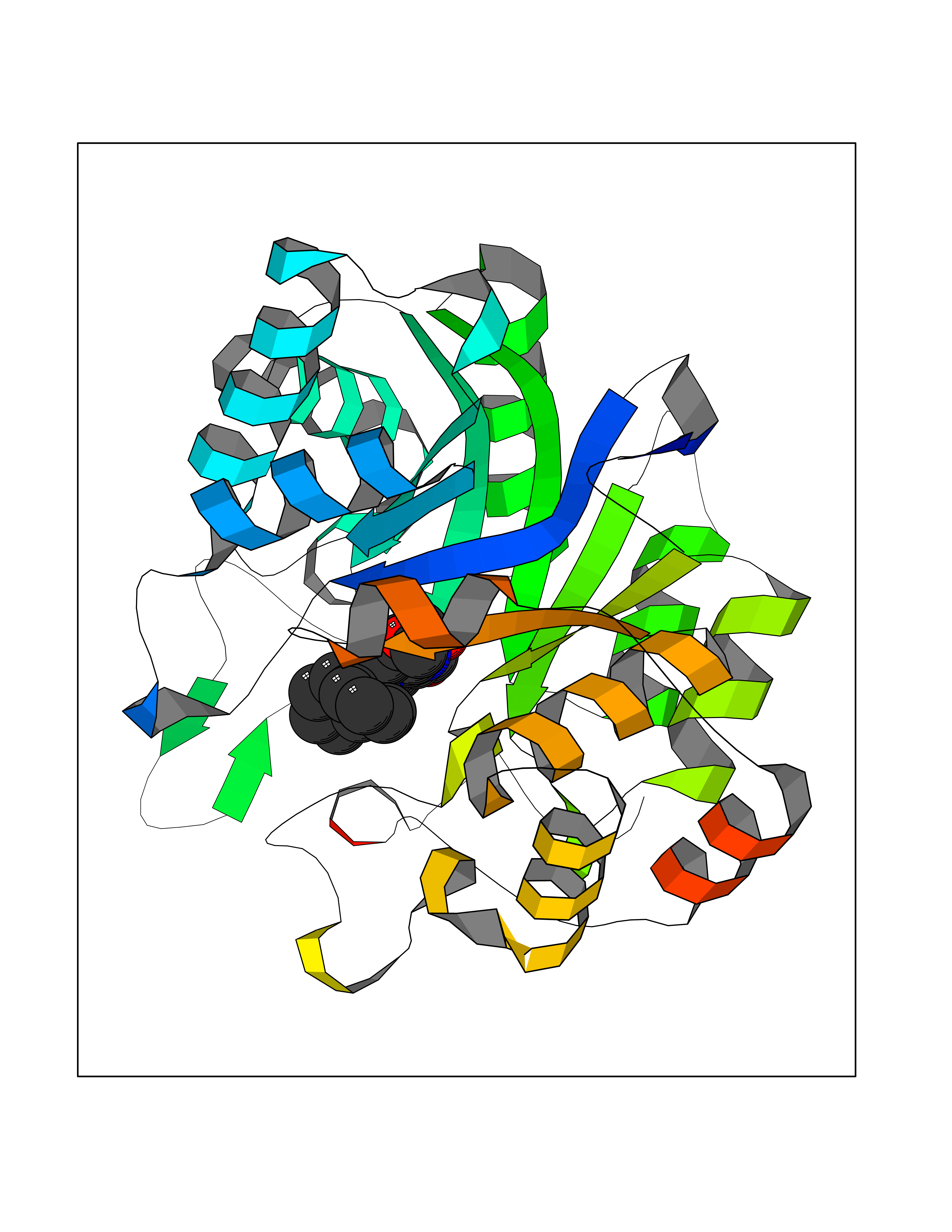 Molscript Map 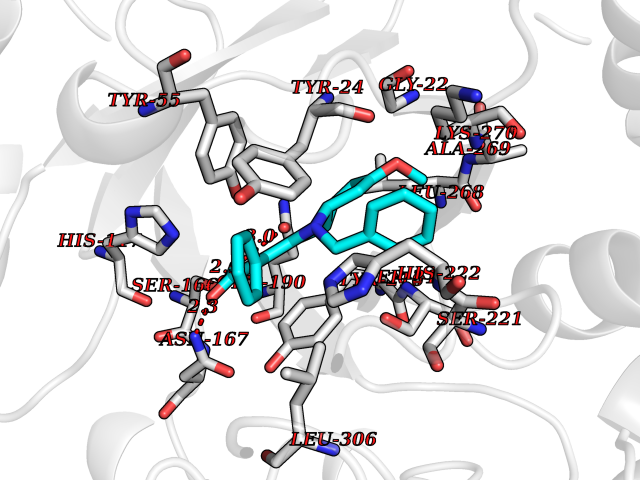 Pymol Map 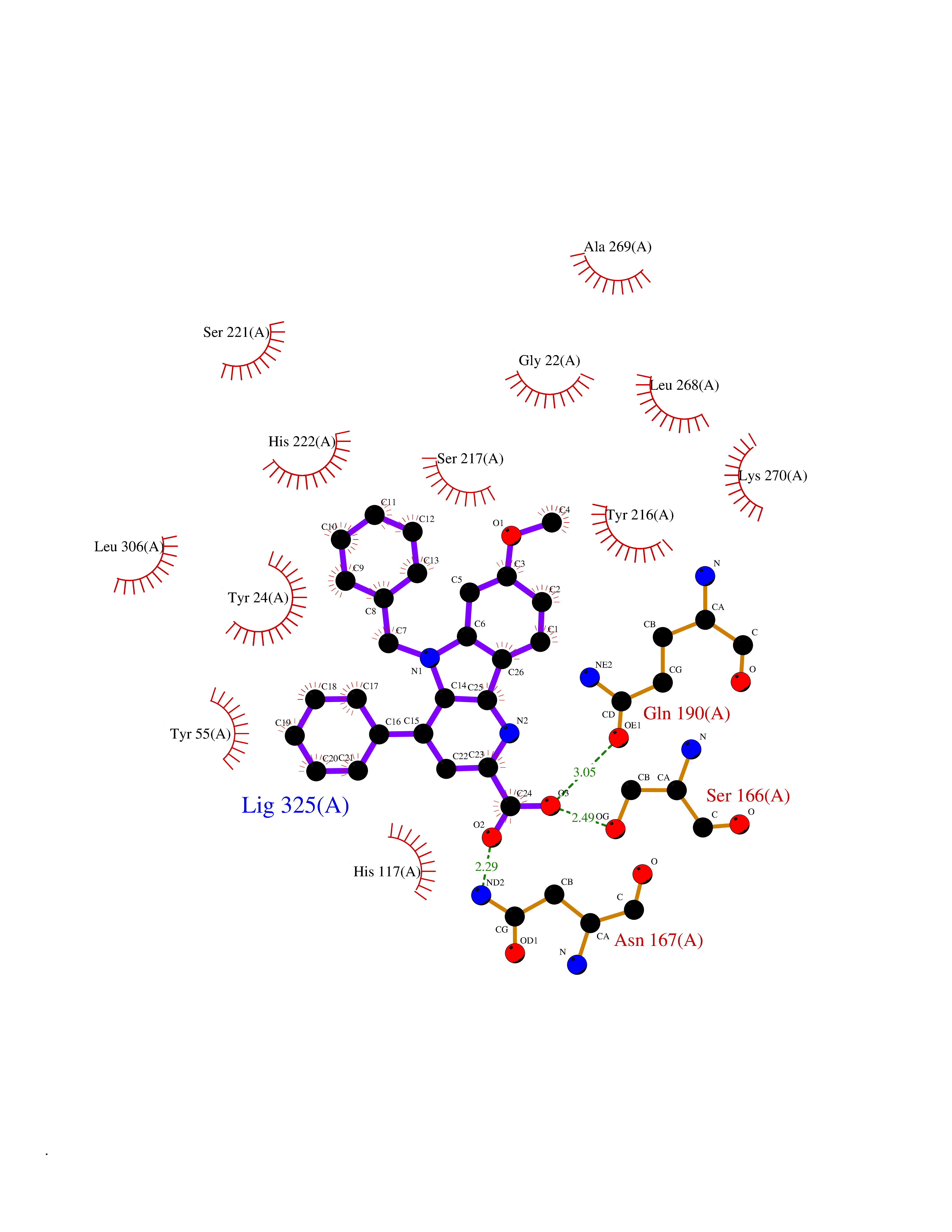 Ligplot Map 3D Binding mode Sequence QDSKYQCVKLNDGHFMPVLGFGTYAPAEVPKSKALEATKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWCNSHRPELVRPALERSLKNLQLDYVDLYLIHFPVSVKPGEEVIPKDENGKILFDTVDLCATWEAVEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNQRKLLDFCKSKDIVLVAYSALGSHREEPWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTSEEMKAIDGLNRNVRYLTLDIFAGPPNYPFSDEY Hydrogen bonds contact Hydrophobic contact | ||||