Job Results:
Ligand
Structure
Job ID
7eae9f0de32fdedcd28c1404448b5ea8
Job name
NA
Time
2025-04-07 15:33:07
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Glutamate carboxypeptidase III (NAALAD2) | 3FED | 4.30 | |
Target general information Gen name NAALAD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAALADase II; NAALAD2 Protein family Peptidase M28 family, M28B subfamily Biochemical class Peptidase Function Has N-acetylated-alpha-linked-acidic dipeptidase (NAALADase) activity. Also exhibits a dipeptidyl-peptidase IV type activity. Inactivate the peptide neurotransmitter N- acetylaspartylglutamate. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHD4; Q6NTF9-3; B2RUZ4; O76024 EC number EC 3.4.17.21 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Carboxypeptidase; Cell membrane; Dipeptidase; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Multifunctional enzyme; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 77761.6 Length 690 Aromaticity 0.12 Instability index 39.42 Isoelectric point 8.48 Charge (pH=7) 4.65 2D Binding mode Binding energy (Kcal/mol) -5.87  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SIRWKLVSEMKAENIKSFLRSFTKLPHLAGTEQNFLLAKKIQTQWKKFGLDSAKLVHYDVLLSYPNETNANYISIVDEHETEIFKTSPPPDGYENVTNIVPPYNAFSAQGMPEGDLVYVNYARTEDFFKLEREMGINCTGKIVIARYGKIFRGNKVKNAMLAGAIGIILYSDPADYFAPEVQPYPKGWNLPGTAAQRGNVLNLNGAGDPLTPGYPAKEYTFRLDVEEGVGIPRIPVHPIGYNDAEILLRYLGGIAPPDKSWKGALNVSYSIGPGFTGSSFRKVRMHVYNINKITRIYNVVGTIRGSVEPDRYVILGGHRDSWVFGAIDPTSGVAVLQEIARSFGKLMSKGWRPRRTIIFASWDAEEFGLLGSTEWAEENVKILQERSIAYINSDSSIEGNYTLRVDCTPLLYQLVYKLTKEIPSPDDGFESKSLYESWLEKDPSPENKNLPRINKLGSGSDFEAYFQRLGIASGRARYTKNKKTDKYSSYPVYHTIYETFELVEKFYDPTFKKQLSVAQLRGALVYELVDSKIIPFNIQDYAEALKNYAASIYNLSKKHDQQLTDHGVSFDSLFSAVKNFSEAASDFHKRLIQVDLNNPIAVRMMNDQLMLLERAFIDPLGLPGKLFYRHIIFAPSSHNKYAGESFPGIYDAIFDIENKANSRLAWKEVKKHISIAAFTIQAAAGTLKEV Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Fumarate reductase flavoprotein subunit | 1Y0P | 4.29 | |
Target general information Gen name fccA Organism Shewanella frigidimarina Uniprot ID TTD ID NA Synonyms fcc3 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.Fumarate reductase (menaquinone).Metal ion binding.Nucleic acid binding.Succinate dehydrogenase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04734; DB03147; DB01677; DB03343 Interacts with NA EC number 1.3.2.4 Uniprot keywords 3D-structure; Electron transport; FAD; Flavoprotein; Heme; Iron; Metal-binding; Oxidoreductase; Periplasm; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 60177.2 Length 568 Aromaticity 0.06 Instability index 27.7 Isoelectric point 6 Charge (pH=7) -8.64 2D Binding mode Binding energy (Kcal/mol) -5.85  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ADNLAEFHVQNQECDSCHTPDGELSNDSLTYENTQCVSCHGTLAEVAETTKHEHYNAHASHFPGEVACTSCHSAHEKSMVYCDSCHSFDFNMPYAKKWLRDEPTIAELAKDKSERQAALASAPHDTVDVVVVGSGGAGFSAAISATDSGAKVILIEKEPVIGGNAKLAAGGMNAAWTDQQKAKKITDSPELMFEDTMKGGQNINDPALVKVLSSHSKDSVDWMTAMGADLTDVGMMGGASVNRAHRPTGGAGVGAHVVQVLYDNAVKRNIDLRMNTRGIEVLKDDKGTVKGILVKGMYKGYYWVKADAVILATGGFAKNNERVAKLDPSLKGFISTNQPGAVGDGLDVAENAGGALKDMQYIQAHPTLSVKGGVMVTEAVRGNGAILVNREGKRFVNEITTRDKASAAILAQTGKSAYLIFDDSVRKSLSKIDKYIGLGVAPTADSLVKLGKMEGIDGKALTETVARYNSLVSSGKDTDFERPNLPRALNEGNYYAIEVTPGVHHTMGGVMIDTKAEVMNAKKQVIPGLYGAGEVTGGVHGANRLGGNAISDIITFGRLAGEEAAKYS Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Tyrosine-protein kinase ABL1 (ABL) | 5HU9 | 4.29 | |
Target general information Gen name ABL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p150; Proto-oncogene tyrosine-protein kinase ABL1; Proto-oncogene c-Abl; JTK7; C-ABL; Abl; Abelson tyrosine-protein kinase 1; Abelson murine leukemia viral oncogene homolog 1 Protein family Protein kinase superfamily, Tyr protein kinase family, ABL subfamily Biochemical class Kinase Function Coordinates actin remodeling through tyrosine phosphorylation of proteins controlling cytoskeleton dynamics like WASF3 (involved in branch formation); ANXA1 (involved in membrane anchoring); DBN1, DBNL, CTTN, RAPH1 and ENAH (involved in signaling); or MAPT and PXN (microtubule-binding proteins). Phosphorylation of WASF3 is critical for the stimulation of lamellipodia formation and cell migration. Involved in the regulation of cell adhesion and motility through phosphorylation of key regulators of these processes such as BCAR1, CRK, CRKL, DOK1, EFS or NEDD9. Phosphorylates multiple receptor tyrosine kinases and more particularly promotes endocytosis of EGFR, facilitates the formation of neuromuscular synapses through MUSK, inhibits PDGFRB-mediated chemotaxis and modulates the endocytosis of activated B-cell receptor complexes. Other substrates which are involved in endocytosis regulation are the caveolin (CAV1) and RIN1. Moreover, ABL1 regulates the CBL family of ubiquitin ligases that drive receptor down-regulation and actin remodeling. Phosphorylation of CBL leads to increased EGFR stability. Involved in late-stage autophagy by regulating positively the trafficking and function of lysosomal components. ABL1 targets to mitochondria in response to oxidative stress and thereby mediates mitochondrial dysfunction and cell death. In response to oxidative stress, phosphorylates serine/threonine kinase PRKD2 at 'Tyr-717'. ABL1 is also translocated in the nucleus where it has DNA-binding activity and is involved in DNA-damage response and apoptosis. Many substrates are known mediators of DNA repair: DDB1, DDB2, ERCC3, ERCC6, RAD9A, RAD51, RAD52 or WRN. Activates the proapoptotic pathway when the DNA damage is too severe to be repaired. Phosphorylates TP73, a primary regulator for this type of damage-induced apoptosis. Phosphorylates the caspase CASP9 on 'Tyr-153' and regulates its processing in the apoptotic response to DNA damage. Phosphorylates PSMA7 that leads to an inhibition of proteasomal activity and cell cycle transition blocks. ABL1 acts also as a regulator of multiple pathological signaling cascades during infection. Several known tyrosine-phosphorylated microbial proteins have been identified as ABL1 substrates. This is the case of A36R of Vaccinia virus, Tir (translocated intimin receptor) of pathogenic E. coli and possibly Citrobacter, CagA (cytotoxin-associated gene A) of H. pylori, or AnkA (ankyrin repeat-containing protein A) of A. phagocytophilum. Pathogens can highjack ABL1 kinase signaling to reorganize the host actin cytoskeleton for multiple purposes, like facilitating intracellular movement and host cell exit. Finally, functions as its own regulator through autocatalytic activity as well as through phosphorylation of its inhibitor, ABI1. Regulates T-cell differentiation in a TBX21-dependent manner. Phosphorylates TBX21 on tyrosine residues leading to an enhancement of its transcriptional activator activity. Non-receptor tyrosine-protein kinase that plays a role in many key processes linked to cell growth and survival such as cytoskeleton remodeling in response to extracellular stimuli, cell motility and adhesion, receptor endocytosis, autophagy, DNA damage response and apoptosis. Related diseases Leukemia, chronic myeloid (CML) [MIM:608232]: A clonal myeloproliferative disorder of a pluripotent stem cell with a specific cytogenetic abnormality, the Philadelphia chromosome (Ph), involving myeloid, erythroid, megakaryocytic, B-lymphoid, and sometimes T-lymphoid cells, but not marrow fibroblasts. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: A chromosomal aberration involving ABL1 has been found in patients with chronic myeloid leukemia. Translocation t(9;22)(q34;q11) with BCR. The translocation produces a BCR-ABL found also in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). {ECO:0000269|PubMed:3021337}.; DISEASE: A chromosomal aberration involving ABL1 is found in a form of acute lymphoblastic leukemia (PubMed:15361874). Translocation t(9;9)(q34;q34) with NUP214 (PubMed:15361874). {ECO:0000269|PubMed:15361874}.; DISEASE: Congenital heart defects and skeletal malformations syndrome (CHDSKM) [MIM:617602]: An autosomal dominant disorder characterized by congenital heart disease with atrial and ventricular septal defects, variable skeletal abnormalities, and failure to thrive. Skeletal defects include pectus excavatum, scoliosis, and finger contractures. Some patient exhibit joint laxity. {ECO:0000269|PubMed:28288113}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08043; DB08583; DB07831; DB08350; DB12597; DB00171; DB06616; DB12267; DB01254; DB12010; DB00619; DB13749; DB08231; DB03878; DB04868; DB08339; DB08901; DB12323; DB08896; DB14989; DB05184 Interacts with Q8IZP0; Q9NYB9; O14672; P10275; Q13315; Q4KMG0; P46108; P46109; P35222; P00533; P04626; Q03468; Q14315; P36888; P05107; P10721; Q38SD2; Q92918; Q7Z434; O43196; P15941; P15941-12; P16333; O43900; Q13905; Q86UR5; Q13671; P31947; Q15464; O75751; P37840; Q9BX66; O60504-2; Q07890; P12931; P51692; Q9Y4G6; P11387; P04637; P15498; Q9Y6W5; P62258; P61981; P63104; O35158; P37840; P48165; Q15323; P37840 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Autophagy; Cell adhesion; Chromosomal rearrangement; Cytoplasm; Cytoskeleton; Disease variant; DNA damage; DNA repair; DNA-binding; Endocytosis; Kinase; Lipoprotein; Magnesium; Manganese; Membrane; Metal-binding; Mitochondrion; Myristate; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31264.6 Length 270 Aromaticity 0.12 Instability index 37.99 Isoelectric point 5.42 Charge (pH=7) -7.67 2D Binding mode Binding energy (Kcal/mol) -5.86  Molscript Map 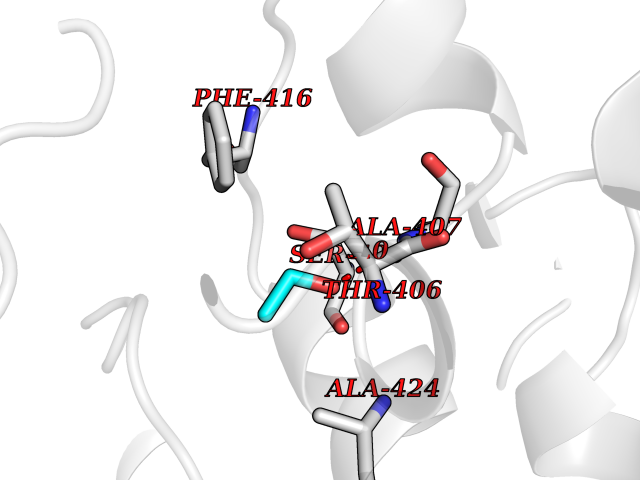 Pymol Map 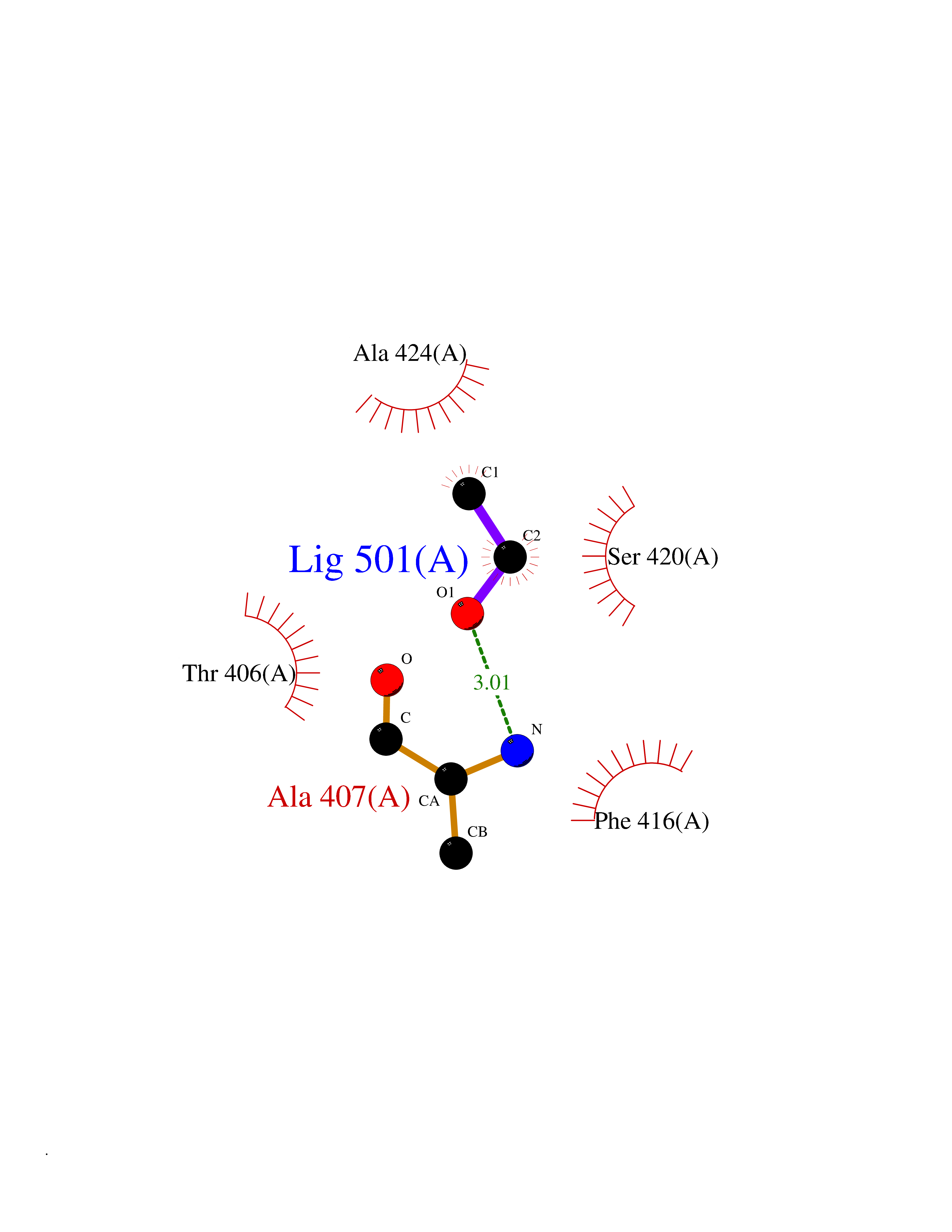 Ligplot Map 3D Binding mode Sequence AMGSSPNYDKWEMERTDITMKHKLGGGQYGEVYEGVWKKYSLTVAVKTLKEDTMEVEEFLKEAAVMKEIKHPNLVQLLGVCTREPPFYIITEFMTYGNLLDYLRECNRQEVNAVVLLYMATQISSAMEYLEKKNFIHRDLAARNCLVGENHLVKVADFGLSRLMTAHAGAKFPIKWTAPESLAYNKFSIKSDVWAFGVLLWEIATYGMSPYPGIDLSQVYELLEKDYRMERPEGCPEKVYELMRACWQWNPSDRPSFAEIHQAFETMFQE Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Ecto-5'-nucleotidase (CD73) | 4H2G | 4.29 | |
Target general information Gen name NT5E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NT5; CD73 antigen; 5'-nucleotidase; 5'-NT Protein family 5'-nucleotidase family Biochemical class Phosphoric monoester hydrolase Function Exhibits AMP-, NAD-, and NMN-nucleosidase activities. Hydrolyzes extracellular nucleotides into membrane permeable nucleosides. Related diseases Calcification of joints and arteries (CALJA) [MIM:211800]: A condition characterized by adult-onset calcification of the lower extremity arteries, including the iliac, femoral and tibial arteries, and hand and foot capsule joints. Age of onset has been reported as early as the second decade of life, usually involving intense joint pain or calcification in the hands. {ECO:0000269|PubMed:21288095, ECO:0000269|PubMed:24887587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00987; DB00806 Interacts with Q9Y225-2; Q8WWF5 EC number EC 3.1.3.5 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 24417.6 Length 219 Aromaticity 0.09 Instability index 40.43 Isoelectric point 5.49 Charge (pH=7) -5.75 2D Binding mode Binding energy (Kcal/mol) -5.85 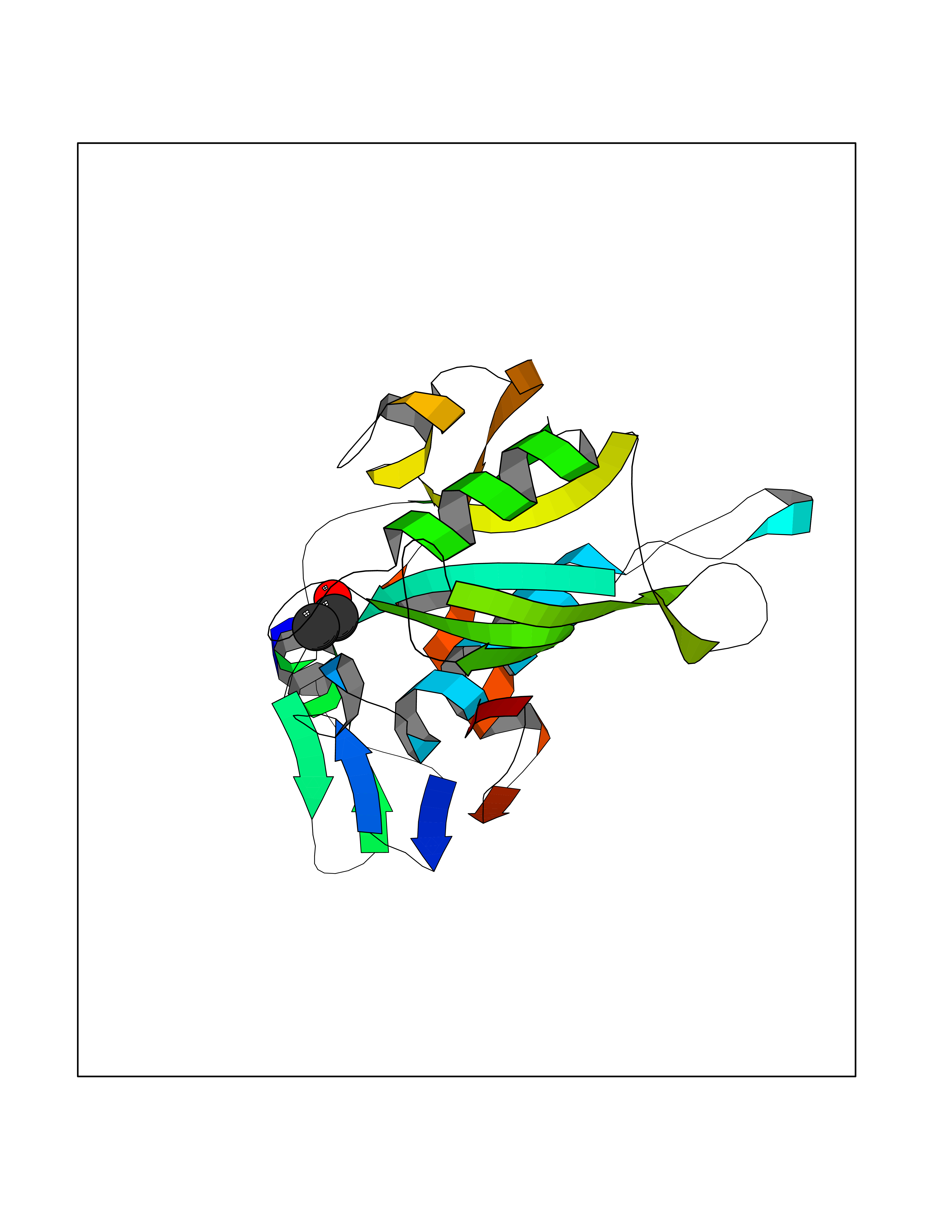 Molscript Map 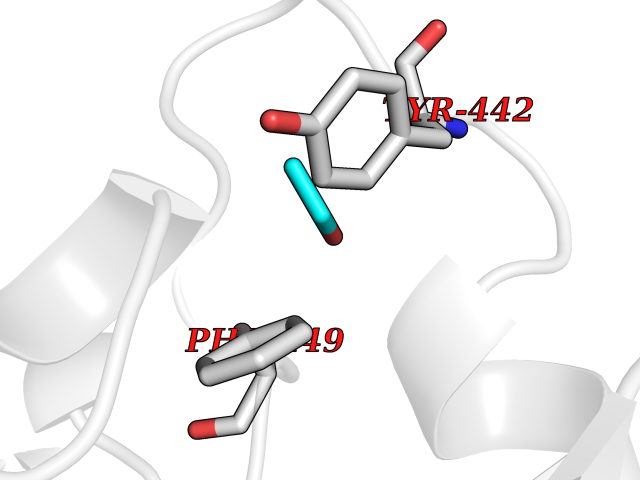 Pymol Map  Ligplot Map 3D Binding mode Sequence LDDYSTQELGKTIVYLDGSSQSCRFRECNMGNLICDAMINNNLRHADEMFWNHVSMCILNGGGIRSPIDERNDGTITWENLAAVLPFGGTFDLVQLKGSTLKKAFEHSVHRYGQSTGEFLQVGGIHVVYDLSRKPGDRVVKLDVLCTACAVPSYDPLKMDEVYKVILPNFLANGGDGFQMIKDELLRHDSGDQDINVVSTYISKMKVIYPAVEGRIKFS Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | UDP-galactopyranose mutase | 1I8T | 4.29 | |
Target general information Gen name glf Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b2036;JW2021;yefE Protein family UDP-galactopyranose/dTDP-fucopyranose mutase family Biochemical class Isomerase Function Flavin adenine dinucleotide binding.UDP-galactopyranose mutase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03147 Interacts with P11868 EC number 5.4.99.9 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipopolysaccharide biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42965.3 Length 367 Aromaticity 0.14 Instability index 32.48 Isoelectric point 6.62 Charge (pH=7) -1.52 2D Binding mode Binding energy (Kcal/mol) -5.85  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MYDYIIVGSGLFGAVCANELKKLNKKVLVIEKRNHIGGNAYTEDCEGIQIHKYGAHIFHTNDKYIWDYVNDLVEFNRFTNSPLAIYKDKLFNLPFNMNTFHQMWGVKDPQEAQNIINAQKKKYGDKVPENLEEQAISLVGEDLYQALIKGYTEKQWGRSAKELPAFIIKRIPVRFTFDNNYFSDRYQGIPVGGYTKLIEKMLEGVDVKLGIDFLKDKDSLASKAHRIIYTGPIDQYFDYRFGALEYRSLKFETERHEFPNFQGNAVINFTDANVPYTRIIEHKHFDYVETKHTVVTKEYPLEWKVGDEPYYPVNDNKNMELFKKYRELASREDKVIFGGRLAEYKYYDMHQVISAALYQVKNIMSTD Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Choloylglycine hydrolase | 2BJF | 4.29 | |
Target general information Gen name cbh Organism Clostridium perfringens (strain 13 / Type A) Uniprot ID TTD ID NA Synonyms CPE0709 Protein family Peptidase C59 family Biochemical class Hydrolase Function Choloylglycine hydrolase activity. Related diseases Mucopolysaccharidosis 3B (MPS3B) [MIM:252920]: A form of mucopolysaccharidosis type 3, an autosomal recessive lysosomal storage disease due to impaired degradation of heparan sulfate. MPS3 is characterized by severe central nervous system degeneration, but only mild somatic disease. Onset of clinical features usually occurs between 2 and 6 years; severe neurologic degeneration occurs in most patients between 6 and 10 years of age, and death occurs typically during the second or third decade of life. {ECO:0000269|PubMed:10094189, ECO:0000269|PubMed:11068184, ECO:0000269|PubMed:11153910, ECO:0000269|PubMed:11286389, ECO:0000269|PubMed:11793481, ECO:0000269|PubMed:11836372, ECO:0000269|PubMed:12202988, ECO:0000269|PubMed:14984474, ECO:0000269|PubMed:15933803, ECO:0000269|PubMed:16151907, ECO:0000269|PubMed:28101780, ECO:0000269|PubMed:9443875, ECO:0000269|PubMed:9443878, ECO:0000269|PubMed:9832037, ECO:0000269|PubMed:9950362}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2V (CMT2V) [MIM:616491]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2V is an autosomal dominant sensory neuropathy with late onset. The main clinical feature is recurrent leg pain that progresses to constant painful paraesthesias in the feet and later the hands. As it evolves, some patients develop a mild sensory ataxia. {ECO:0000269|PubMed:25818867}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB03619; DB01956 Interacts with NA EC number 2.3.1.-; 3.5.1.-; 3.5.1.24 Uniprot keywords 3D-structure; Direct protein sequencing; Hydrolase; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 34075.3 Length 300 Aromaticity 0.11 Instability index 34.81 Isoelectric point 5.15 Charge (pH=7) -7.9 2D Binding mode Binding energy (Kcal/mol) -5.86  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence CTGLALETKDGLHLFGRNMDIEYSFNQSIIFIPRNFKCVNKSNKKELTTKYAVLGMGTIFDDYPTFADGMNEKGLGCAGLNFPVYVSYSKEDIEGKTNIPVYNFLLWVLANFSSVEEVKEALKNANIVDIPISENIPNTTLHWMISDITGKSIVVEQTKEKLNVFDNNIGVLTNSPTFDWHVANLNQYVGLRYNGLVGLPGDFTPASRFIRVAFLRDAMIKNDKDSIDLIEFFHILNNVAMVRGSTRTVEEKSDLTQYTSCMCLEKGIYYYNTYENNQINAIDMNKENLDGNEIKTYKYN Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Epithelial discoidin domain receptor 1 (DDR1) | 4BKJ | 4.29 | |
Target general information Gen name DDR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase CAK; Tyrosine kinase DDR; TRKE; TRK E; RTK6; Protein-tyrosine kinase RTK-6; Protein-tyrosine kinase 3A; PTK3A; NTRK4; NEP; Mammary carcinoma kinase 10; MCK-10; HGK2; Epithelial Protein family Protein kinase superfamily, Tyr protein kinase family, Insulin receptor subfamily Biochemical class Kinase Function Collagen binding triggers a signaling pathway that involves SRC and leads to the activation of MAP kinases. Regulates remodeling of the extracellular matrix by up-regulation of the matrix metalloproteinases MMP2, MMP7 and MMP9, and thereby facilitates cell migration and wound healing. Required for normal blastocyst implantation during pregnancy, for normal mammary gland differentiation and normal lactation. Required for normal ear morphology and normal hearing. Promotes smooth muscle cell migration, and thereby contributes to arterial wound healing. Also plays a role in tumor cell invasion. Phosphorylates PTPN11. Tyrosine kinase that functions as cell surface receptor for fibrillar collagen and regulates cell attachment to the extracellular matrix, remodeling of the extracellular matrix, cell migration, differentiation, survival and cell proliferation. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB00619; DB15822 Interacts with Q16832; O43639; Q06124; Q9UHD9 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calcium; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; Kinase; Lactation; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Pregnancy; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34061.1 Length 297 Aromaticity 0.1 Instability index 42.8 Isoelectric point 6.32 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -5.85  Molscript Map 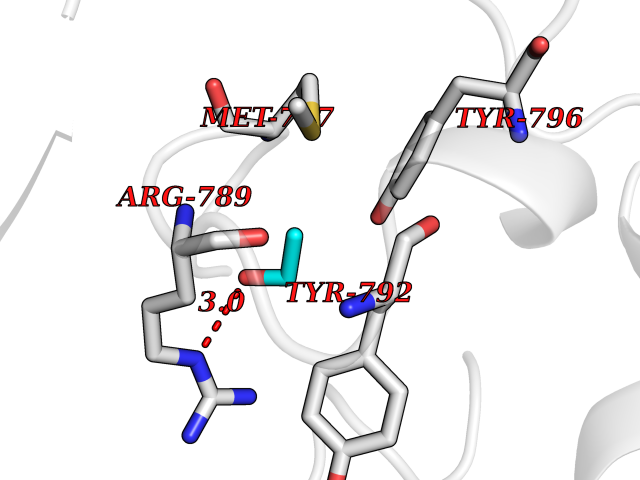 Pymol Map 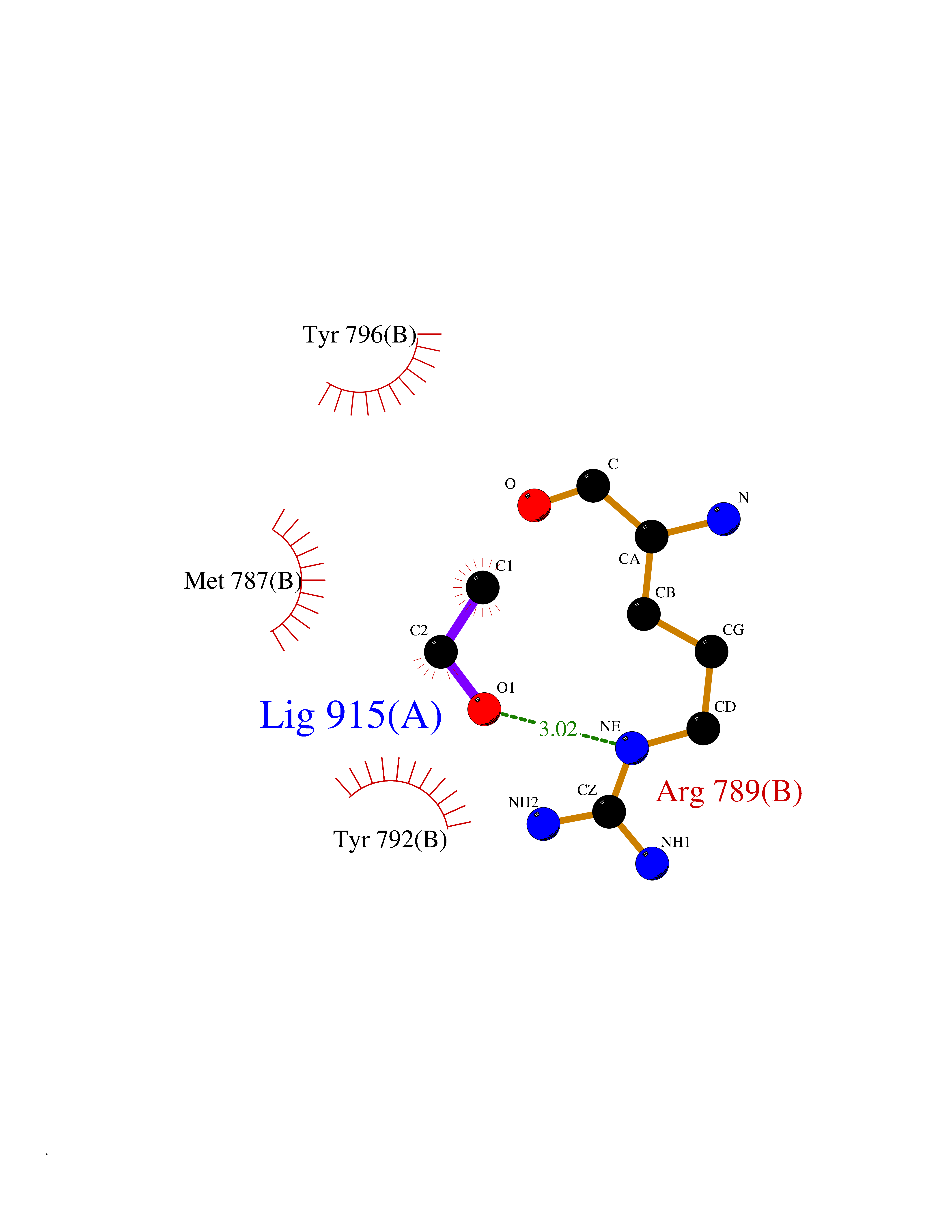 Ligplot Map 3D Binding mode Sequence MPRVDFPRSRLRFKEKLGEGQFGEVHLCEVDSPQDLVSLDFPLNVRKGHPLLVAVKILRPDATKNARNDFLKEVKIMSRLKDPNIIRLLGVCVQDDPLCMITDYMENGDLNQFLSAHQLEDKGPTISYPMLLHVAAQIASGMRYLATLNFVHRDLATRNCLVGENFTIKIADFGMSRNLYAGDYYRAVLPIRWMAWECILMGKFTTASDVWAFGVTLWEVLMLCRAQPFGQLTDEQVIENAGEFFRDQGRQVYLSRPPACPQGLYELMLRCWSRESEQRPPFSQLHRFLAEDALNTV Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Peptidyl-prolyl cis-trans isomerase G | 2GW2 | 4.29 | |
Target general information Gen name PPIG Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Isomerase Function Cyclosporin A binding.Peptidyl-prolyl cis-trans isomerase activity.RNA binding. Related diseases Intellectual developmental disorder, autosomal dominant 6, with or without seizures (MRD6) [MIM:613970]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD6 additional features may include seizures, hypotonia, abnormal movements, such as dystonia, and autistic features. {ECO:0000269|PubMed:20890276, ECO:0000269|PubMed:23033978, ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24863970, ECO:0000269|PubMed:25356899, ECO:0000269|PubMed:27839871, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 27 (DEE27) [MIM:616139]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. {ECO:0000269|PubMed:24272827, ECO:0000269|PubMed:27839871, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberrations involving GRIN2B has been found in patients with intellectual disability. Translocations t(9;12)(p23;p13.1) and t(10;12)(q21.1;p13.1) with a common breakpoint in 12p13.1. Drugs (DrugBank ID) DB00172 Interacts with Q8N7W2-2; Q8NHQ1; O75553; Q9UI36-2; Q96C98; Q8NC69; P17931; Q6NVH9; Q15365; Q9UL42; Q96CD2; Q14498; Q16637; Q12800; Q9NVV9; PRO_0000037309 [P0C6X7] EC number 5.2.1.8 Uniprot keywords 3D-structure; Alternative splicing; Isomerase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Rotamase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 19125.4 Length 173 Aromaticity 0.1 Instability index 26.46 Isoelectric point 7.14 Charge (pH=7) 0.24 2D Binding mode Binding energy (Kcal/mol) -5.86 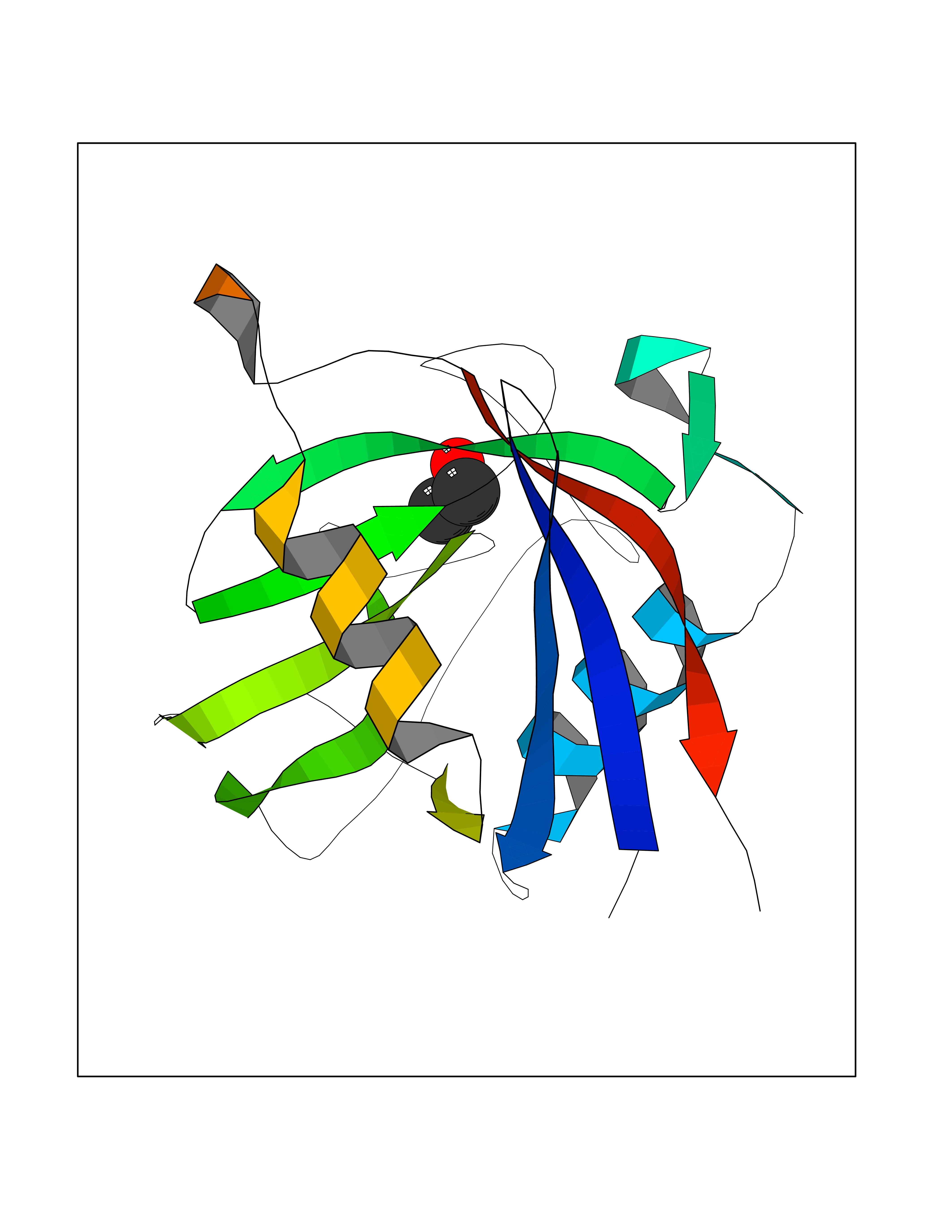 Molscript Map 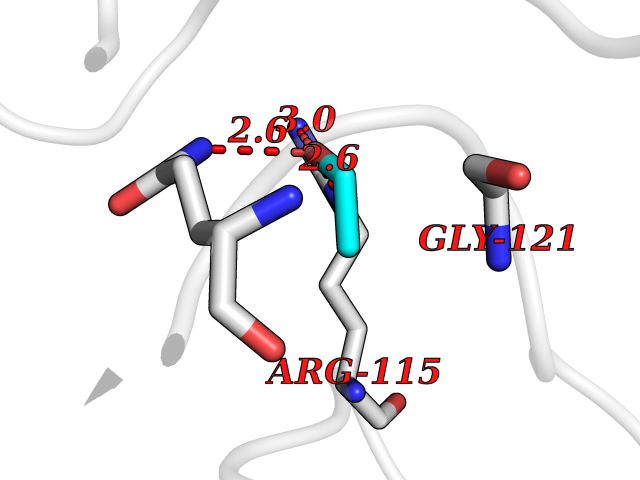 Pymol Map 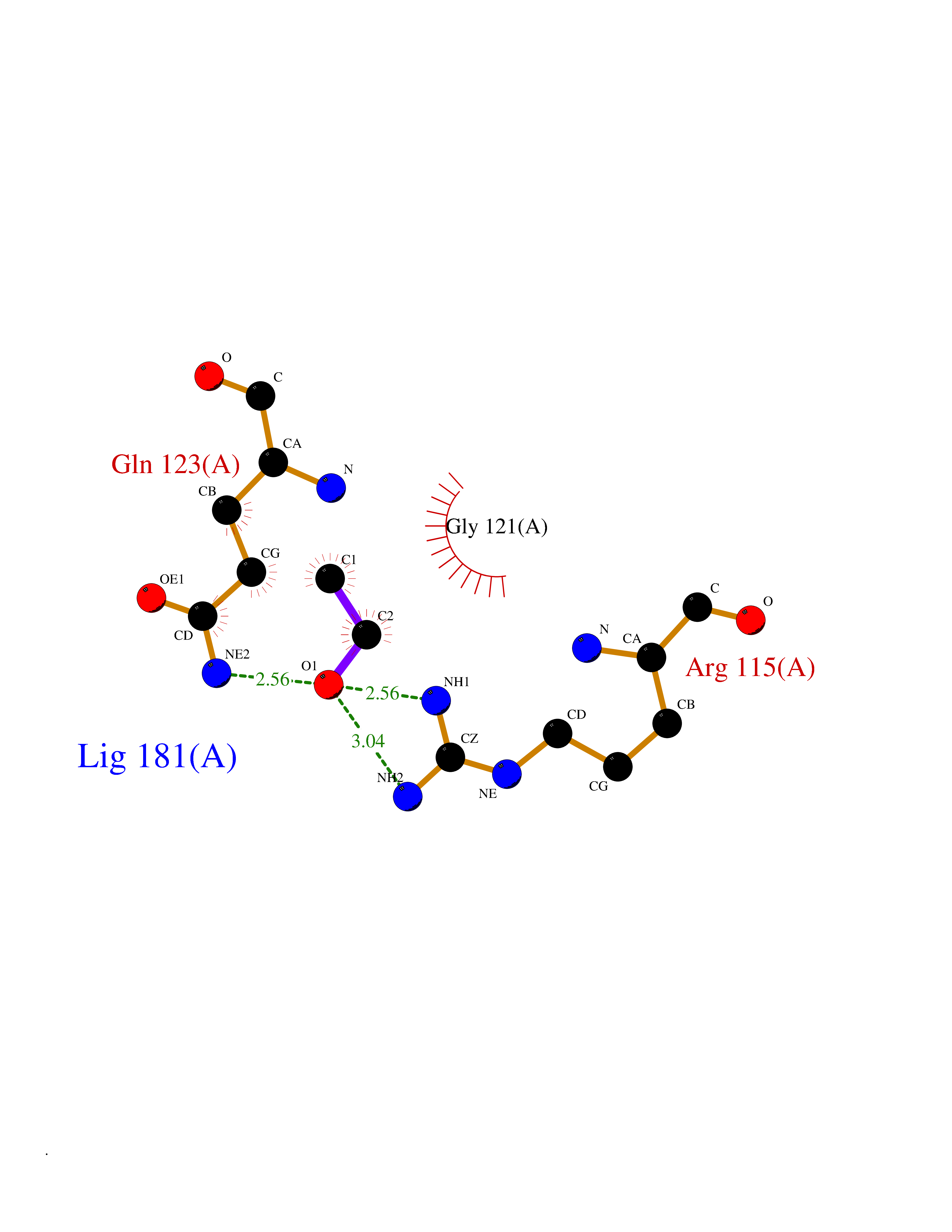 Ligplot Map 3D Binding mode Sequence RPRCFFDIAINNQPAGRVVFELFSDVCPKTCENFRCLCTGEKGTGKSTQKPLHYKSCLFHRVVKDFMVQGGDFSEGNGRGGESIYGGFFEDESFAVKHNAAFLLSMANRGKDTNGSQFFITTKPTPHLDGHHVVFGQVISGQEVVREIENQKTDAASKPFAEVRILSCGELIP Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Cathepsin D (CTSD) | 4OC6 | 4.29 | |
Target general information Gen name CTSD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CPSD; CD Protein family Peptidase A1 family Biochemical class Peptidase Function Plays a role in APP processing following cleavage and activation by ADAM30 which leads to APP degradation. Involved in the pathogenesis of several diseases such as breast cancer and possibly Alzheimer disease. Acid protease active in intracellular protein breakdown. Related diseases Ceroid lipofuscinosis, neuronal, 10 (CLN10) [MIM:610127]: A form of neuronal ceroid lipofuscinosis with onset at birth or early childhood. Neuronal ceroid lipofuscinoses are progressive neurodegenerative, lysosomal storage diseases characterized by intracellular accumulation of autofluorescent liposomal material, and clinically by seizures, dementia, visual loss, and/or cerebral atrophy. {ECO:0000269|PubMed:16670177, ECO:0000269|PubMed:16685649, ECO:0000269|PubMed:21990111}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03028; DB03096; DB07542; DB08740; DB02216 Interacts with P05067; Q9P1A6-3; I6L9I8; Q9H6S3; Q7Z602; P28799; PRO_0000012695 [P28799]; PRO_0000012696 [P28799]; PRO_0000012697 [P28799]; PRO_0000012698 [P28799]; PRO_0000012699 [P28799]; PRO_0000012700 [P28799]; PRO_0000012701 [P28799]; P68431; Q9Y6F6-3; Q12756; Q5TA79; Q86VF5-3; O15130-2; Q96LB9; P09565; Q9C004; Q8NBJ7; Q9BQG1; P28347-2; P45880; Q15007-2; O00308; Q5W0Z9-4; Q6ZNH5 EC number EC 3.4.23.5 Uniprot keywords 3D-structure; Alzheimer disease; Aspartyl protease; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Neurodegeneration; Neuronal ceroid lipofuscinosis; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37264.2 Length 341 Aromaticity 0.1 Instability index 32.32 Isoelectric point 5.6 Charge (pH=7) -4.86 2D Binding mode Binding energy (Kcal/mol) -5.86  Molscript Map 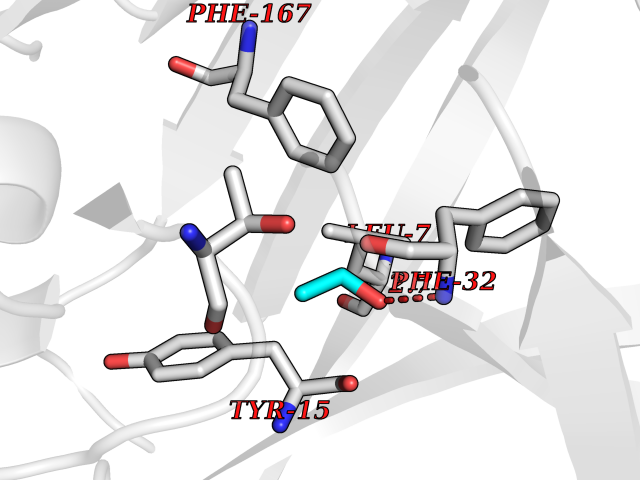 Pymol Map 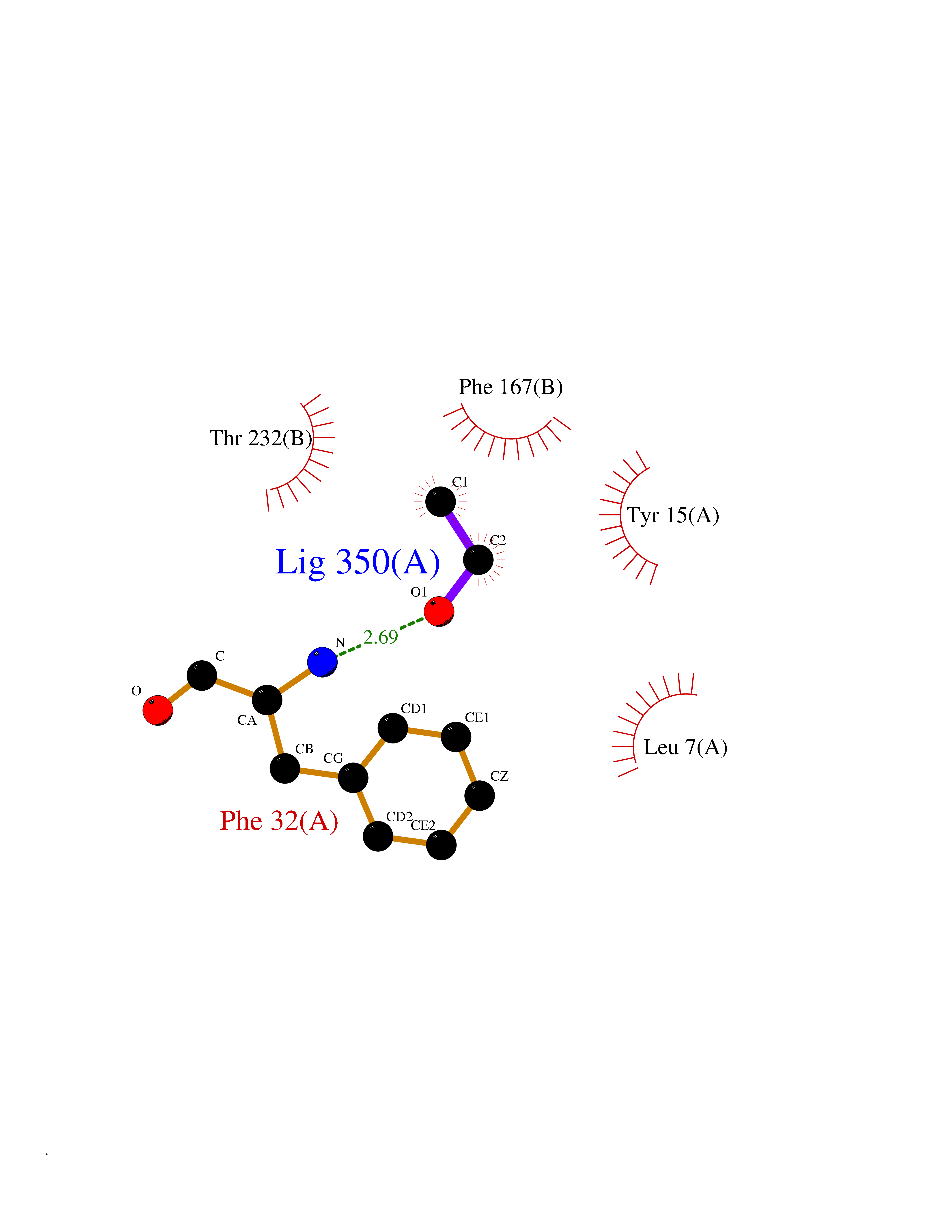 Ligplot Map 3D Binding mode Sequence GPIPEVLKNYMDAQYYGEIGIGTPPQCFTVVFDTGSSNLWVPSIHCKLLDIACWIHHKYNSDKSSTYVKNGTSFDIHYGSGSLSGYLSQDTVSVPCQSGGVKVERQVFGEATKQPGITFIAAKFDGILGMAYPRISVNNVLPVFDNLMQQKLVDQNIFSFYLSRDPDAQPGGELMLGGTDSKYYKGSLSYLNVTRKAYWQVHLDQVEVASGLTLCKEGCEAIVDTGTSLMVGPVDEVRELQKAIGAVPLIQGEYMIPCEKVSTLPAITLKLGGKGYKLSPEDYTLKVSQAGKTLCLSGFMGMDIPPPSGPLWILGDVFIGRYYTVFDRDNNRVGFAEAARL Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | NAPE-hydrolyzing phospholipase D (NAPE-PLD) | 4QN9 | 4.29 | |
Target general information Gen name NAPEPLD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAPE-PLD; N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D; N-acyl phosphatidylethanolamine phospholipase D; C7orf18 Protein family NAPE-PLD family Biochemical class NA Function Hydrolyzes N-acyl-phosphatidylethanolamines (NAPEs) to produce N-acylethanolamines (NAEs) and phosphatidic acid. Responsible for the generation of these bioactive fatty acid ethanolamides (FAEs), including anandamide (N-arachidonoylethanolamine), the ligand of cannabinoid and vanilloid receptors. As a regulator of lipid metabolism in the adipose tissue, mediates the crosstalk between adipocytes, gut microbiota and immune cells to control body temperature and weight. In particular, regulates energy homeostasis by promoting cold-induced brown or beige adipocyte differentiation program to generate heat from fatty acids and glucose (By similarity). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) DB14009 Interacts with Q6IQ20 EC number EC 3.1.4.54 Uniprot keywords 3D-structure; Acetylation; Endosome; Golgi apparatus; Hydrolase; Lipid degradation; Lipid metabolism; Membrane; Metal-binding; Nucleus; Phospholipid degradation; Phospholipid metabolism; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 74256.5 Length 643 Aromaticity 0.13 Instability index 48.34 Isoelectric point 5.65 Charge (pH=7) -17.61 2D Binding mode Binding energy (Kcal/mol) -5.85  Molscript Map 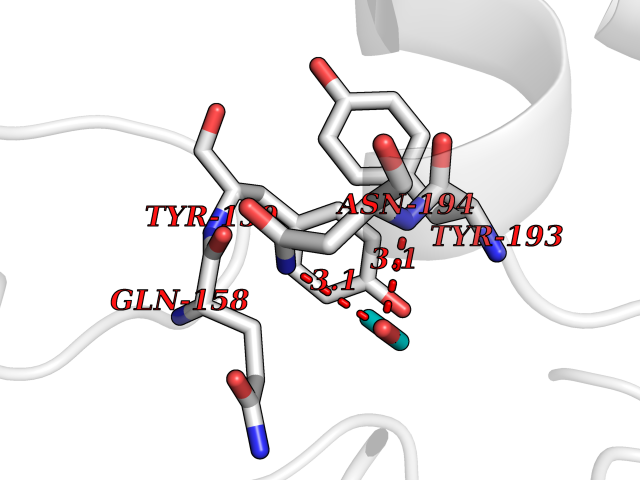 Pymol Map 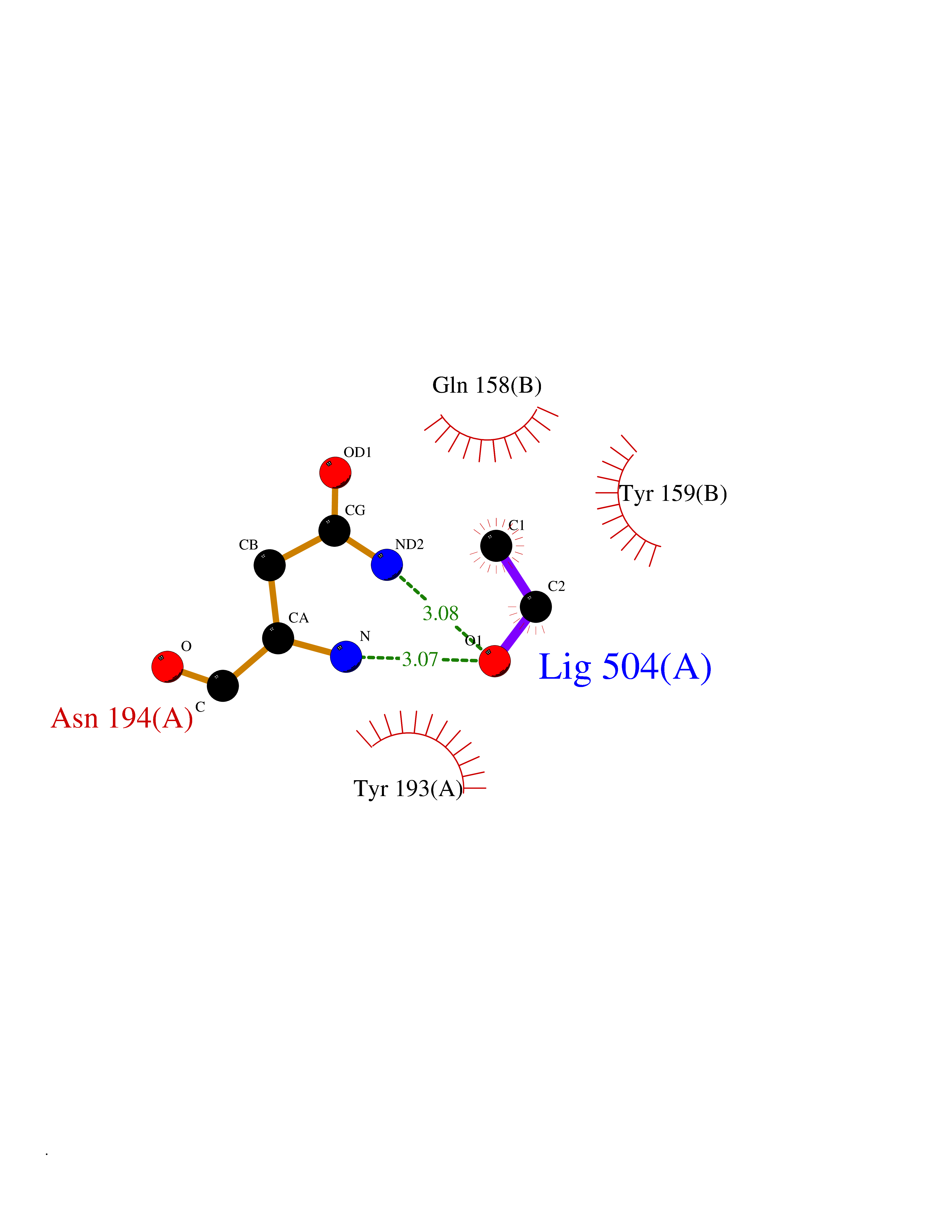 Ligplot Map 3D Binding mode Sequence SKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNNSKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNND Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Ubiquitin carboxyl-terminal hydrolase 2 (USP2) | 5XU8 | 4.29 | |
Target general information Gen name USP2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-specific-processing protease 2; Ubiquitin thioesterase 2; UBP41; Deubiquitinating enzyme 2; 41 kDa ubiquitin-specific protease Protein family Peptidase C19 family, USP2 subfamily Biochemical class Peptidase Function Isoform 1 and isoform 4 possess both ubiquitin-specific peptidase and isopeptidase activities. Deubiquitinates MDM2 without reversing MDM2-mediated p53/TP53 ubiquitination and thus indirectly promotes p53/TP53 degradation and limits p53 activity. Has no deubiquitinase activity against p53/TP53. Prevents MDM2-mediated degradation of MDM4. Plays a role in the G1/S cell-cycle progression in normal and cancer cells. Regulates the circadian clock by modulating its intrinsic circadian rhythm and its capacity to respond to external cues. Associates with clock proteins and deubiquitinates core clock component PER1 but does not affect its overall stability. Regulates the nucleocytoplasmic shuttling and nuclear retention of PER1 and its repressive role on the clock transcription factors CLOCK and ARNTL/BMAL1. Plays a role in the regulation of myogenic differentiation of embryonic muscle cells. Hydrolase that deubiquitinates polyubiquitinated target proteins such as MDM2, MDM4 and CCND1. Related diseases Defects in AKT2 are a cause of susceptibility to breast cancer (BC). AKT2 promotes metastasis of tumor cells without affecting the latency of tumor development. May play a role in glioblastoma cell survival (PubMed:20167810). {ECO:0000269|PubMed:20167810}.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:15166380, ECO:0000269|PubMed:19164855}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Hypoinsulinemic hypoglycemia with hemihypertrophy (HIHGHH) [MIM:240900]: A disorder characterized by hypoglycemia, low insulin levels, low serum levels of ketone bodies and branched-chain amino acids, left-sided hemihypertrophy, neonatal macrosomia, reduced consciousness and hypoglycemic seizures. {ECO:0000269|PubMed:21979934}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NYB9-2; P12814; P35609; Q08043; Q86U10; Q86V38; P56945; Q8TD16-2; Q96CA5; A2RRN7; Q13137; Q9H257-2; Q96JN2-2; Q2TAC2; A6NC98; Q96MT8-3; Q8NHQ1; Q9BSW2; Q8N4Y2-3; Q8WTU0; O75140-2; Q9NRI5-2; Q8N9I9; Q9H596; Q8WWB3; Q5JST6; Q9NRA8; O00471; Q96B26; P57678; Q08379; Q9NYA3; A6NEM1; Q6PI77; Q14451-3; Q4V328; Q9NSC5; Q9UJC3; Q96ED9-2; Q8IYA8; Q9UKT9; Q5TA45; Q96N16; O75564-2; Q674X7-2; Q9BVG8; Q9BVG8-5; P19012; Q7Z3Y8; Q15323; Q14525; O76011; Q92764; Q6A162; Q9UBR4-2; Q969G2; Q03252; Q9BRK4; Q00987; Q9UJV3-2; Q5VZ52; Q13084; Q5JR59; Q5JR59-3; Q15742; Q9GZM8; I6L9F6; P07196; O43482; Q96CV9; Q4G0R1; Q9NRD5; Q58EX7; Q8ND90; Q16633; Q9GZV8; Q6MZQ0; Q15276; Q8HWS3; Q59EK9-3; P60903; O14492-2; O60504; Q99932-2; A6NLX3; P51692; Q86VP1; Q8WW24; Q9UBB9; Q08117-2; Q03169; Q13077; Q12933; Q9Y4K3; P36406; P14373; Q86XT4; Q15654; Q8N6Y0; Q70EL1-9; Q9UK41-2; Q8N1B4; O96006; Q9NZV7; Q9UGI0; P05067; P54253; G5E9A7; Q01658; Q00403; Q9Y5Q9; P04792; O43464; P42858; Q8WXH2; O60333-2; A0A6Q8PF08; O60260-5; P60891; Q9Y3C5; Q7Z333; P37840; P00441; Q7Z699; Q13148; O76024 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; Cell cycle; Cytoplasm; Hydrolase; Membrane; Metal-binding; Myogenesis; Nucleus; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 37785.5 Length 327 Aromaticity 0.11 Instability index 42.45 Isoelectric point 8.23 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -5.85  Molscript Map 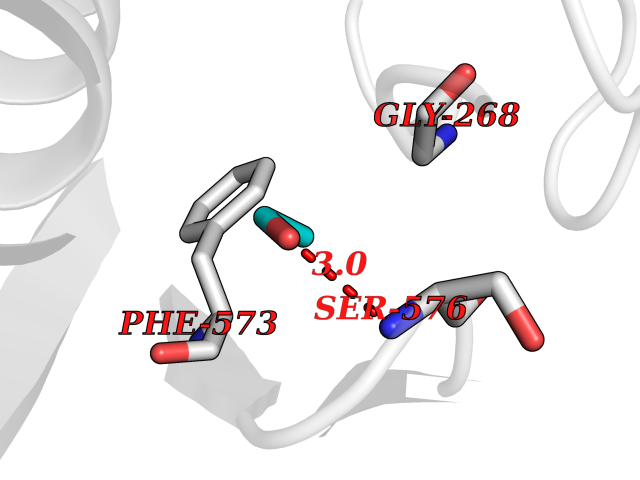 Pymol Map 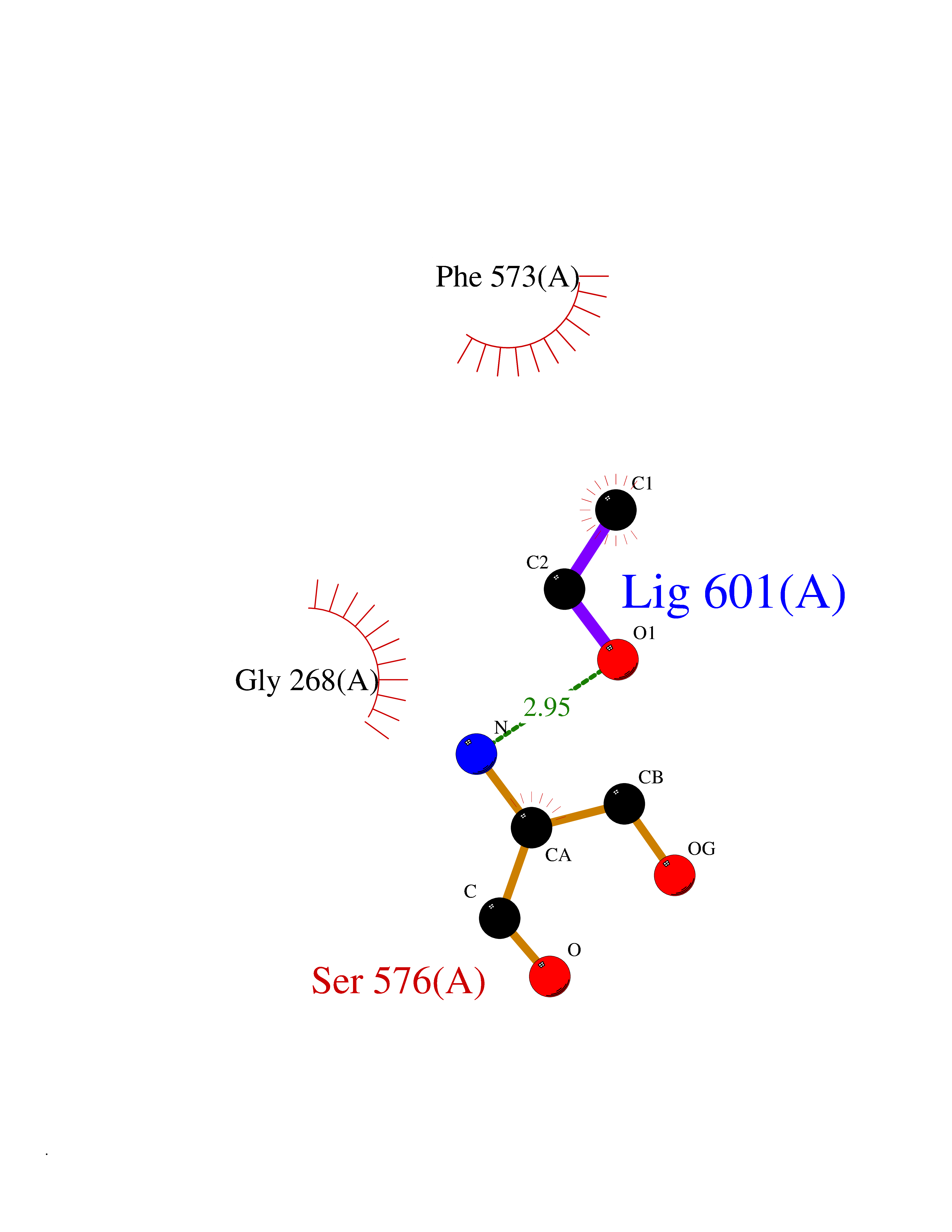 Ligplot Map 3D Binding mode Sequence QGLAGLRNLGNTCFMNSILQCLSNTRELRDYCLQRLYMRDLHHGSNAHTALVEEFAKLIQTIWTSSPNDVVSPSEFKTQIQRYAPRFVGYNQQDAQEFLRFLLDGLHNEVNRVNLDHLPDDEKGRQMWRKYLEREDSRIGDLFVGQLKSSLTCTDCGYCSTVFDPFWDLSLPIAKRGYPEVTLMDCMRLFTKEDVLDGDEKPTCCRCRGRKRCIKKFSIQRFPKILVLHLKRFSESRIRTSKLTTFVNFPLRDLDLREFASENTNHAVYNLYAVSNHSGTTMGGHYTAYCRSPGTGEWHTFNDSSVTPMSSSQVRTSDAYLLFYELA Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Cerebroside-sulfatase (ARSA) | 1E2S | 4.29 | |
Target general information Gen name ARSA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cerebrosidesulfatase; Arylsulfatase A component C; ASA; ARSA Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Hydrolyzes cerebroside sulfate. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB03821; DB01800; DB01141; DB04786 Interacts with P50995; Q6P5X5; Q13554-3; O60826; Q96D98; Q9H0I2; Q12951-2; Q16512; P28069; O75360; Q9BQY4; Q15645; O95231 EC number EC 3.1.6.8 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Ichthyosis; Leukodystrophy; Lipid metabolism; Lysosome; Metachromatic leukodystrophy; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID P Molecular weight (Da) 51173.7 Length 481 Aromaticity 0.09 Instability index 47.42 Isoelectric point 5.59 Charge (pH=7) -12.84 2D Binding mode Binding energy (Kcal/mol) -5.85  Molscript Map 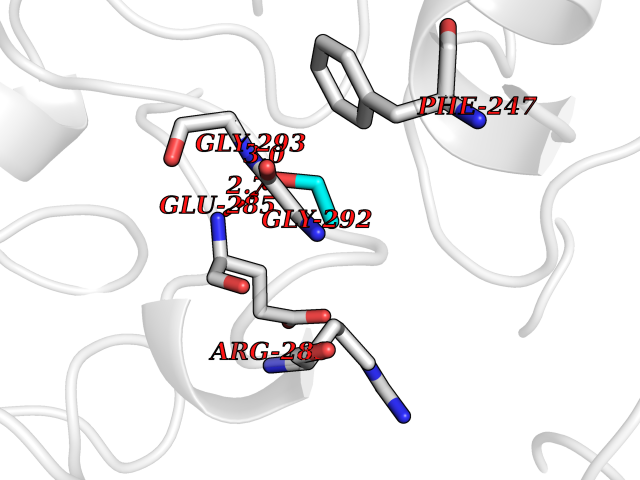 Pymol Map 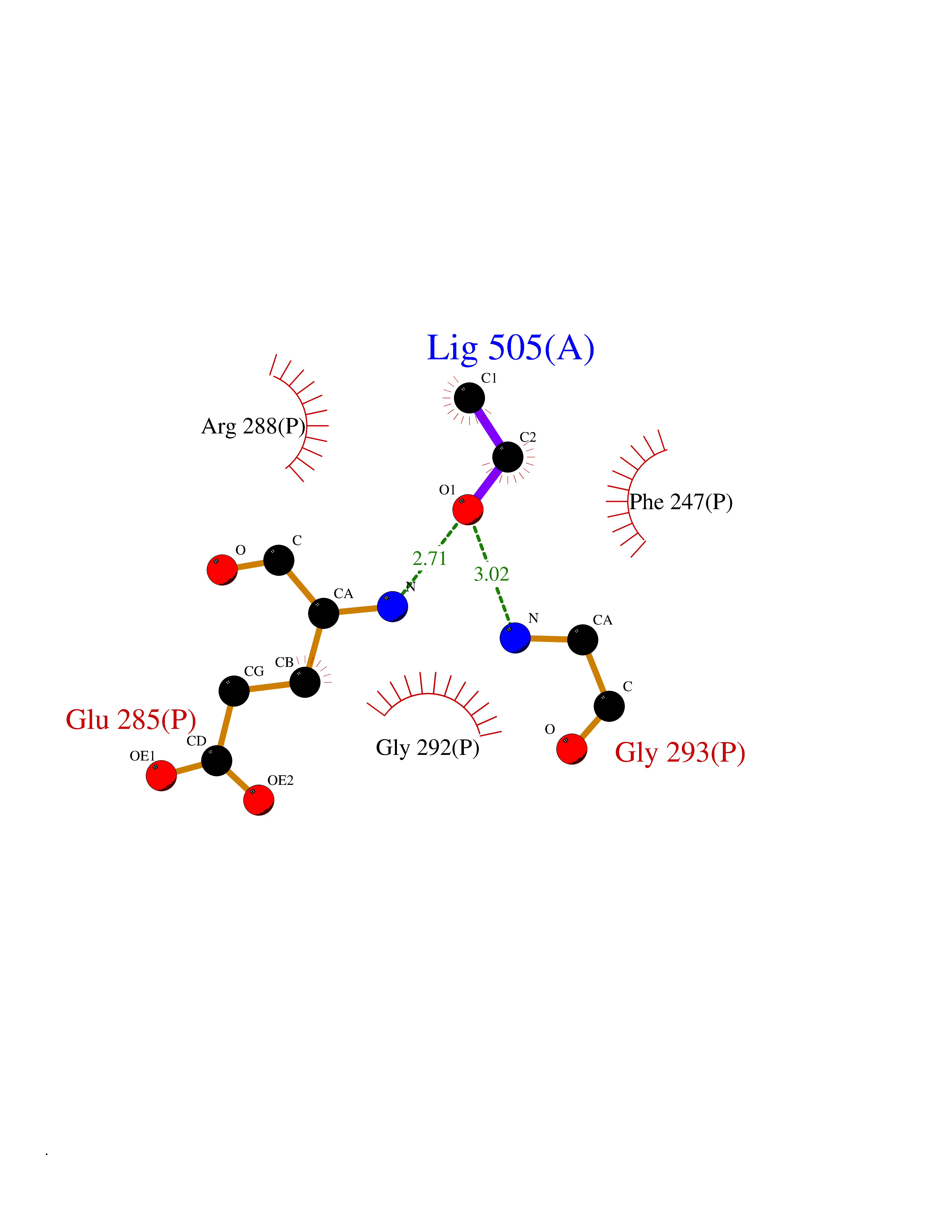 Ligplot Map 3D Binding mode Sequence RPPNIVLIFADDLGYGDLGCYGHPSSTTPNLDQLAAGGLRFTDFYVPVSLATPSRAALLTGRLPVRMGMYPGVLVPSSRGGLPLEEVTVAEVLAARGYLTGMAGKWHLGVGPEGAFLPPHQGFHRFLGIPYSHDQGPCQNLTCFPPATPCDGGCDQGLVPIPLLANLSVEAQPPWLPGLEARYMAFAHDLMADAQRQDRPFFLYYASHHTHYPQFSGQSFAERSGRGPFGDSLMELDAAVGTLMTAIGDLGLLEETLVIFTADNGPETMRMSRGGCSGLLRCGKGTTYEGGVREPALAFWPGHIAPGVTHELASSLDLLPTLAALAGAPLPNVTLDGFDLSPLLLGTGKSPRQSLFFYPSYPDEVRGVFAVRTGKYKAHFFTQGSAHSDTTADPACHASSSLTAHEPPLLYDLSKDPGENYNLLGATPEVLQALKQLQLLKAQLDAAVTFGPSQVARGEDPALQICCHPGCTPRPACCHCP Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Prothrombin | 4UD9 | 4.28 | |
Target general information Gen name F2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase S1 family Biochemical class Hydrolase Function Calcium ion binding.Growth factor activity.Heparin binding.Lipopolysaccharide binding.Receptor binding.Serine-type endopeptidase activity.Thrombospondin receptor activity. Related diseases Factor II deficiency (FA2D) [MIM:613679]: A very rare blood coagulation disorder characterized by mucocutaneous bleeding symptoms. The severity of the bleeding manifestations correlates with blood factor II levels. {ECO:0000269|PubMed:1349838, ECO:0000269|PubMed:1354985, ECO:0000269|PubMed:1421398, ECO:0000269|PubMed:14962227, ECO:0000269|PubMed:2719946, ECO:0000269|PubMed:3242619, ECO:0000269|PubMed:3567158, ECO:0000269|PubMed:3771562, ECO:0000269|PubMed:3801671, ECO:0000269|PubMed:6405779, ECO:0000269|PubMed:7792730, ECO:0000269|PubMed:7865694}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to thrombin defect (THPH1) [MIM:188050]: A multifactorial disorder of hemostasis characterized by abnormal platelet aggregation in response to various agents and recurrent thrombi formation. {ECO:0000269|PubMed:2825773}. The disease is caused by variants affecting the gene represented in this entry. A common genetic variation in the 3-prime untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased risk of venous thrombosis.; DISEASE: Pregnancy loss, recurrent, 2 (RPRGL2) [MIM:614390]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11506076}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07211; DB07796; DB07016; DB07521; DB06850; DB07091; DB06845; DB07088; DB07131; DB07095; DB07515; DB07897; DB06878; DB06947; DB08624; DB06869; DB06929; DB07400; DB04771; DB04772; DB02287; DB07277; DB07550; DB07549; DB07548; DB07105; DB04722; DB07366; DB08254; DB01725; DB08062; DB07639; DB07461; DB07120; DB07190; DB07741; DB07353; DB07508; DB07809; DB08546; DB08061; DB07718; DB03136; DB02723; DB07440; DB07376; DB06861; DB06866; DB06865; DB03865; DB06841; DB07934; DB08422; DB07659; DB07660; DB07658; DB13151; DB00025; DB11166; DB00278; DB01766; DB07083; DB00006; DB00100; DB13152; DB09228; DB09130; DB03159; DB06911; DB06996; DB06919; DB07027; DB07133; DB07143; DB07005; DB06695; DB00055; DB01225; DB05714; DB12831; DB03847; DB07278; DB01767; DB06404; DB09332; DB00001; DB13998; DB04136; DB00170; DB06838; DB13999; DB06868; DB06942; DB06936; DB07165; DB07527; DB07522; DB07665; DB07946; DB06859; DB06853; DB06858; DB07279; DB08187; DB04591; DB07944; DB07128; DB12598; DB01123; DB04786; DB05777; DB04697; DB09109; DB14738; DB04898; DB01593; DB14487; DB08152 Interacts with P05067; P07204; Q846V4; PRO_0000032489 [P01008] EC number 3.4.21.5 Uniprot keywords 3D-structure; Acute phase; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID H Molecular weight (Da) 29321.6 Length 254 Aromaticity 0.1 Instability index 39.57 Isoelectric point 8.56 Charge (pH=7) 4.16 2D Binding mode Binding energy (Kcal/mol) -5.84 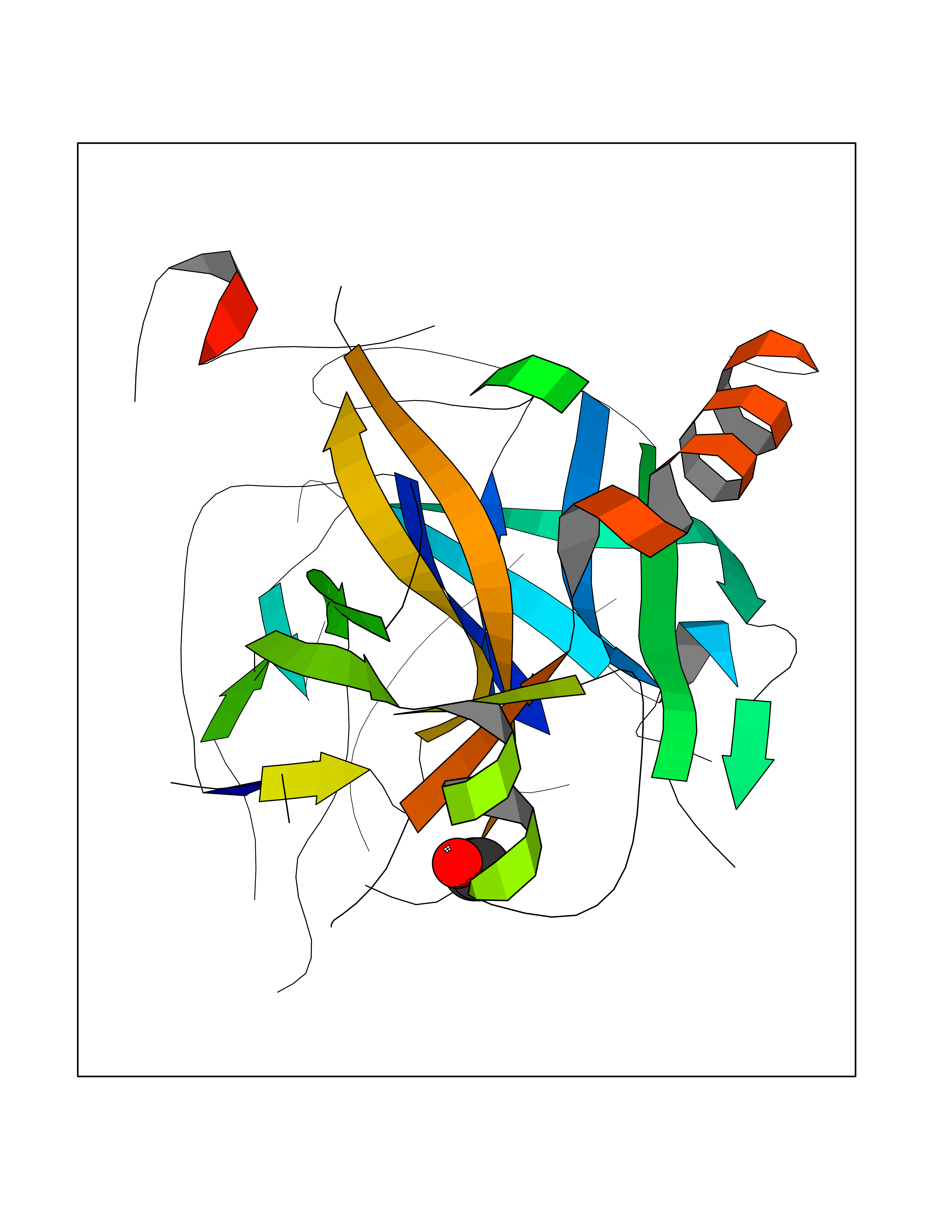 Molscript Map 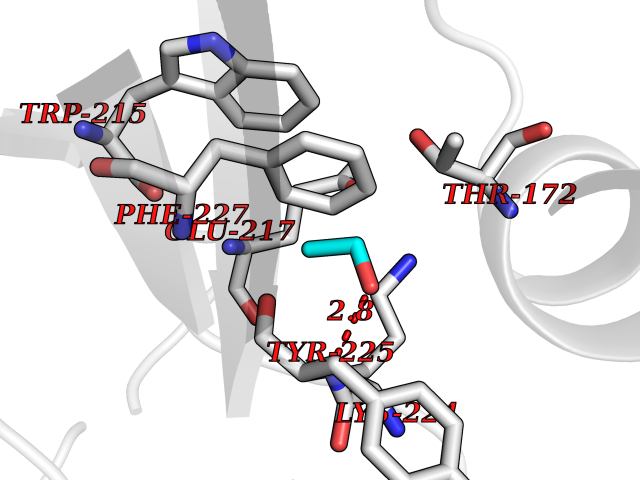 Pymol Map 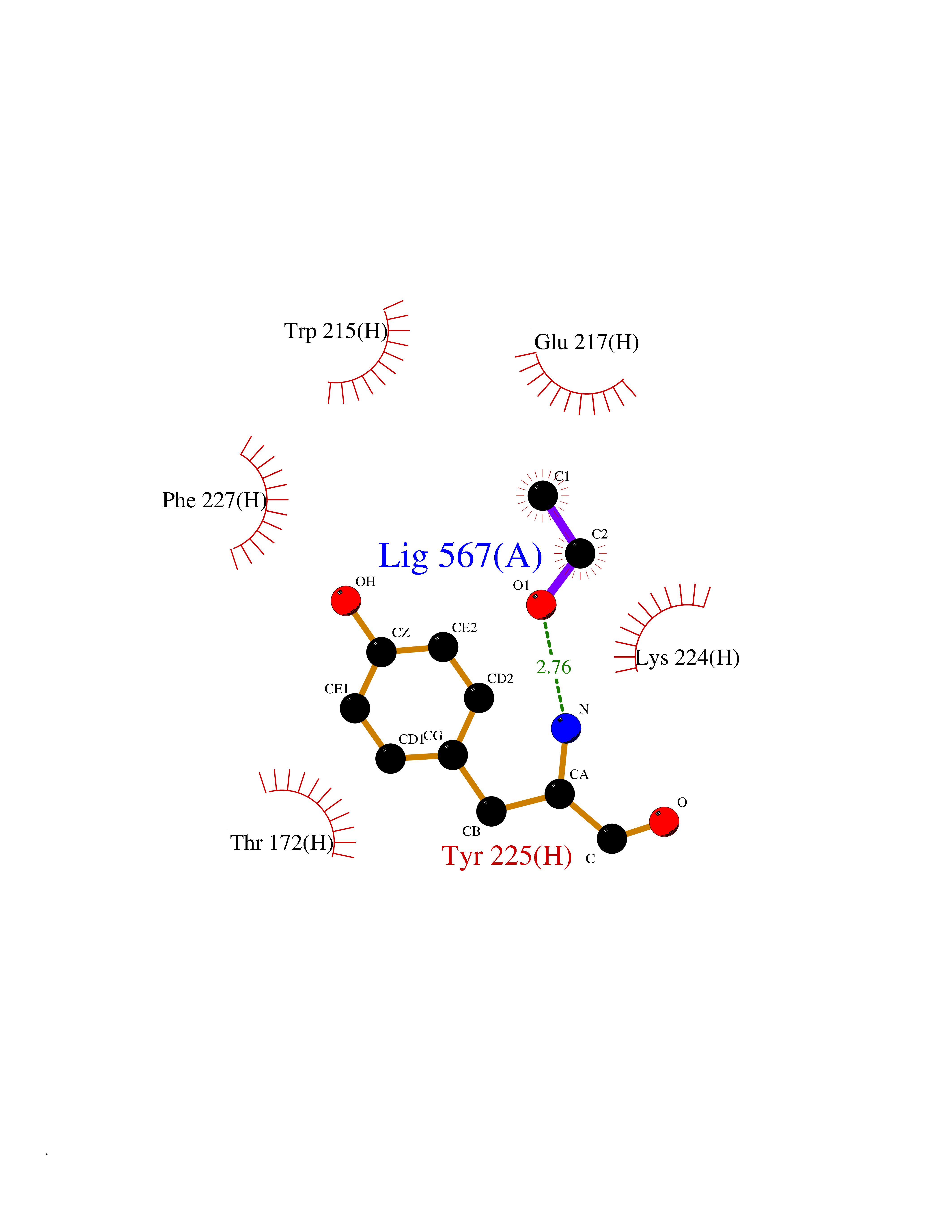 Ligplot Map 3D Binding mode Sequence IVEGSDAEIGMSPWQVMLFRSPQELLCGASLISDRWVLTAAHCLLTENDLLVRIGKHSRTRYRNIEKISMLEKIYIHPRYNWENLDRDIALMKLKKPVAFSDYIHPVCLPDRETLLQAGYKGRVTGWGNLKETWGQPSVLQVVNLPIVERPVCKDSTRIRITDNMFCAYKKRGDACEGDSGGPFVMKSNNRWYQMGIVSWGEGCRDGKYGFYTHVFRLKKWIQKVIDQFGGDFEEIPEELQCGLRPLFEKKSLE Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Peptidyl-prolyl cis-trans isomerase H | 1MZW | 4.28 | |
Target general information Gen name PPIH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYP20;CYPH Protein family Cyclophilin-type PPIase family, PPIase H subfamily Biochemical class Isomerase Function Cyclosporin A binding.Peptidyl-prolyl cis-trans isomerase activity.Ribonucleoprotein complex binding. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00172 Interacts with Q9NP55; Q7L591-3; P42858; Q8TBB1; Q92802; Q9GZT8; Q99633; O43172; Q9UHD9; P98170; PRO_0000037309 [P0C6X7] EC number 5.2.1.8 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chaperone; Cytoplasm; Direct protein sequencing; Isomerase; mRNA processing; mRNA splicing; Nucleus; Proteomics identification; Reference proteome; Rotamase; Spliceosome Protein physicochemical properties Chain ID A Molecular weight (Da) 22309.4 Length 204 Aromaticity 0.09 Instability index 31.68 Isoelectric point 8.62 Charge (pH=7) 3 2D Binding mode Binding energy (Kcal/mol) -5.83 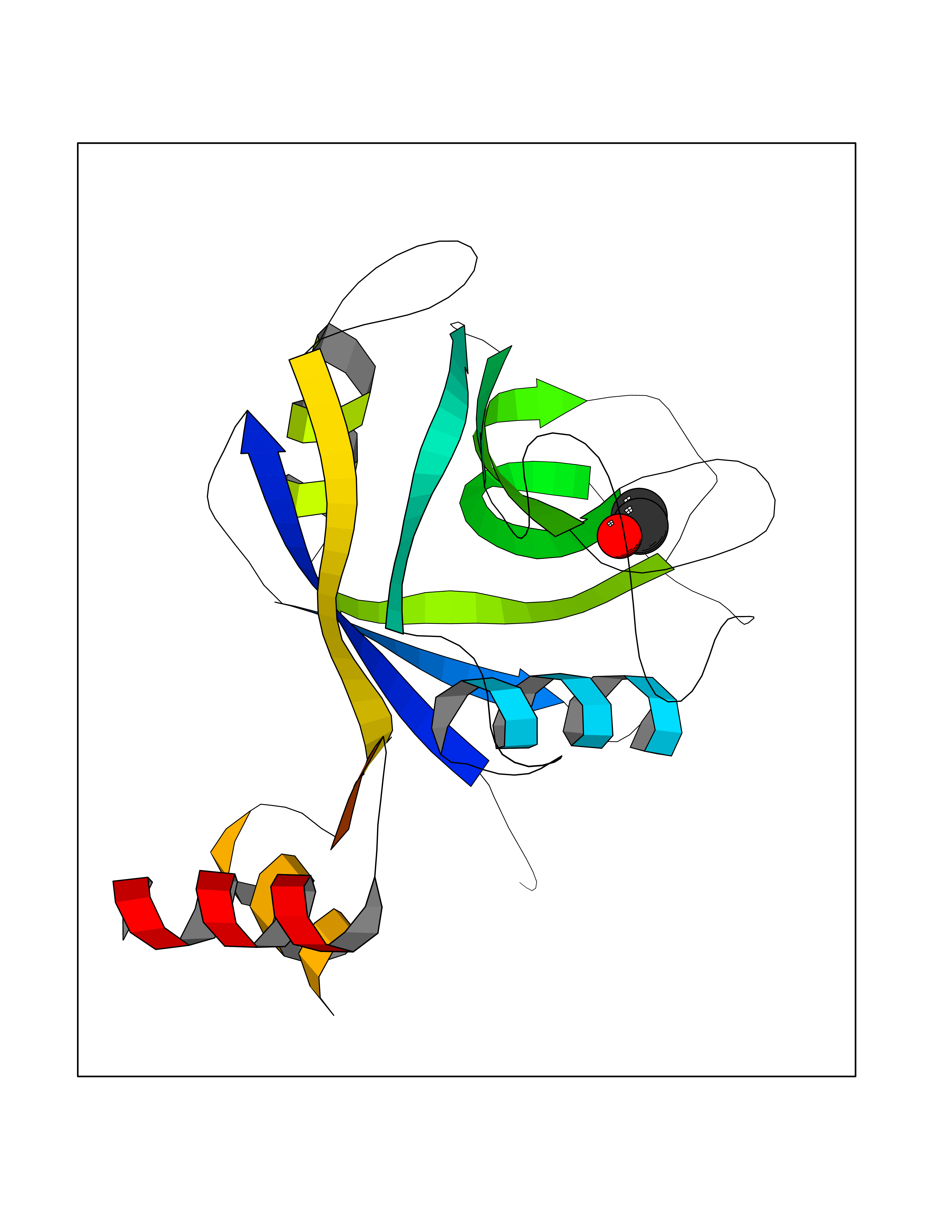 Molscript Map 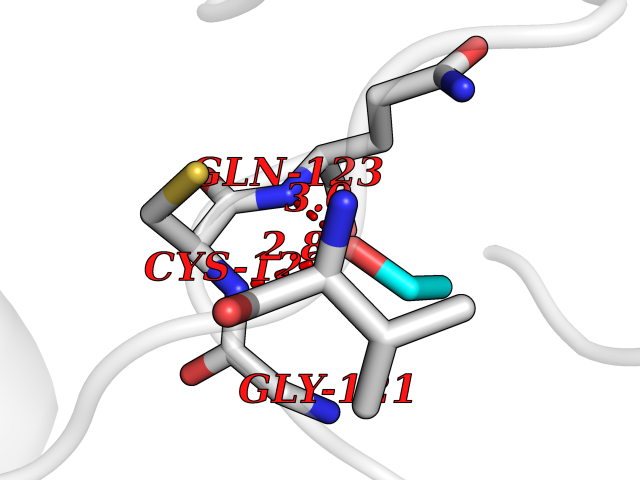 Pymol Map 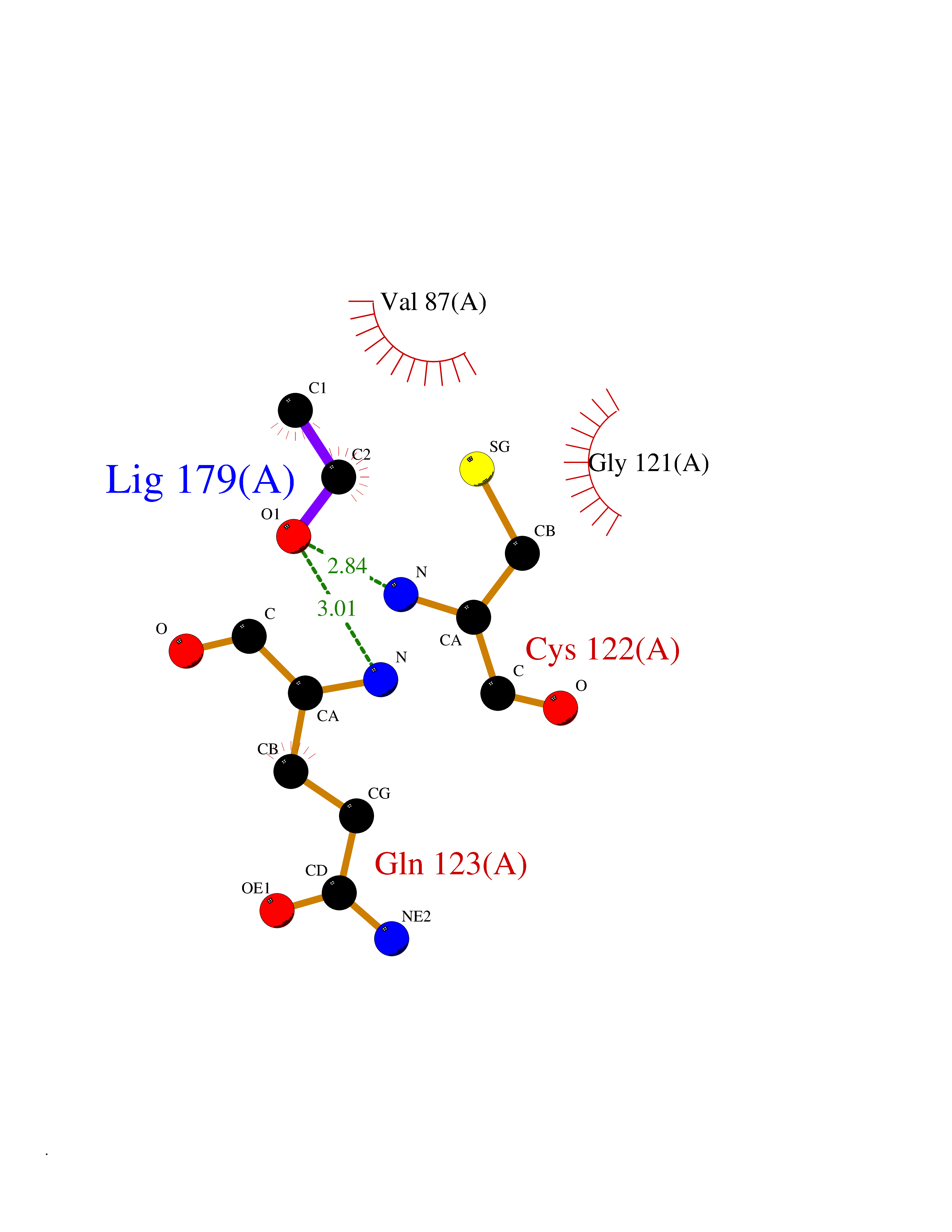 Ligplot Map 3D Binding mode Sequence NSSPVNPVVFFDVSIGGQEVGRMKIELFADVVPKTAENFRQFCTGEFRKDGVPIGYKGSTFHRVIKDFMIQGGDFVNGDGTGVASIYRGPFADENFKLRHSAPGLLSMANSGPSTNGCQFFITCSKCDWLDGKHVVFGKIIDGLLVMRKIENVPTGPNNKPKLPVVISQCGEMEVKASLRALGEPITLFGEGPAERRERLRNIL Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Neutrophil collagenase | 4QKZ | 4.28 | |
Target general information Gen name MMP8 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CLG1 Protein family Peptidase M10A family Biochemical class Hydrolase / hydrolase inhibitor Function Metalloendopeptidase activity.Serine-type endopeptidase activity.Zinc ion binding. Related diseases Protoporphyria, erythropoietic, 1 (EPP1) [MIM:177000]: An autosomal recessive form of porphyria with onset usually before age 10 years. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. Erythropoietic protoporphyria is marked by excessive protoporphyrin in erythrocytes, plasma, liver and feces, and by widely varying photosensitive skin changes ranging from a burning or pruritic sensation to erythema, edema and wheals. {ECO:0000269|PubMed:10942404, ECO:0000269|PubMed:11375302, ECO:0000269|PubMed:12063482, ECO:0000269|PubMed:12601550, ECO:0000269|PubMed:1376018, ECO:0000269|PubMed:15286165, ECO:0000269|PubMed:17196862, ECO:0000269|PubMed:1755842, ECO:0000269|PubMed:7910885, ECO:0000269|PubMed:8757534, ECO:0000269|PubMed:9211198, ECO:0000269|PubMed:9585598, ECO:0000269|PubMed:9740232}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07772; DB07713; DB07397; DB02326; DB03207; DB02953; DB08476; DB07900; DB03622; DB03880; DB08028; DB03636; DB00786; DB08403; DB06971 Interacts with NA EC number 3.4.24.34 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18096.6 Length 163 Aromaticity 0.12 Instability index 27.13 Isoelectric point 4.64 Charge (pH=7) -11.95 2D Binding mode Binding energy (Kcal/mol) -5.84 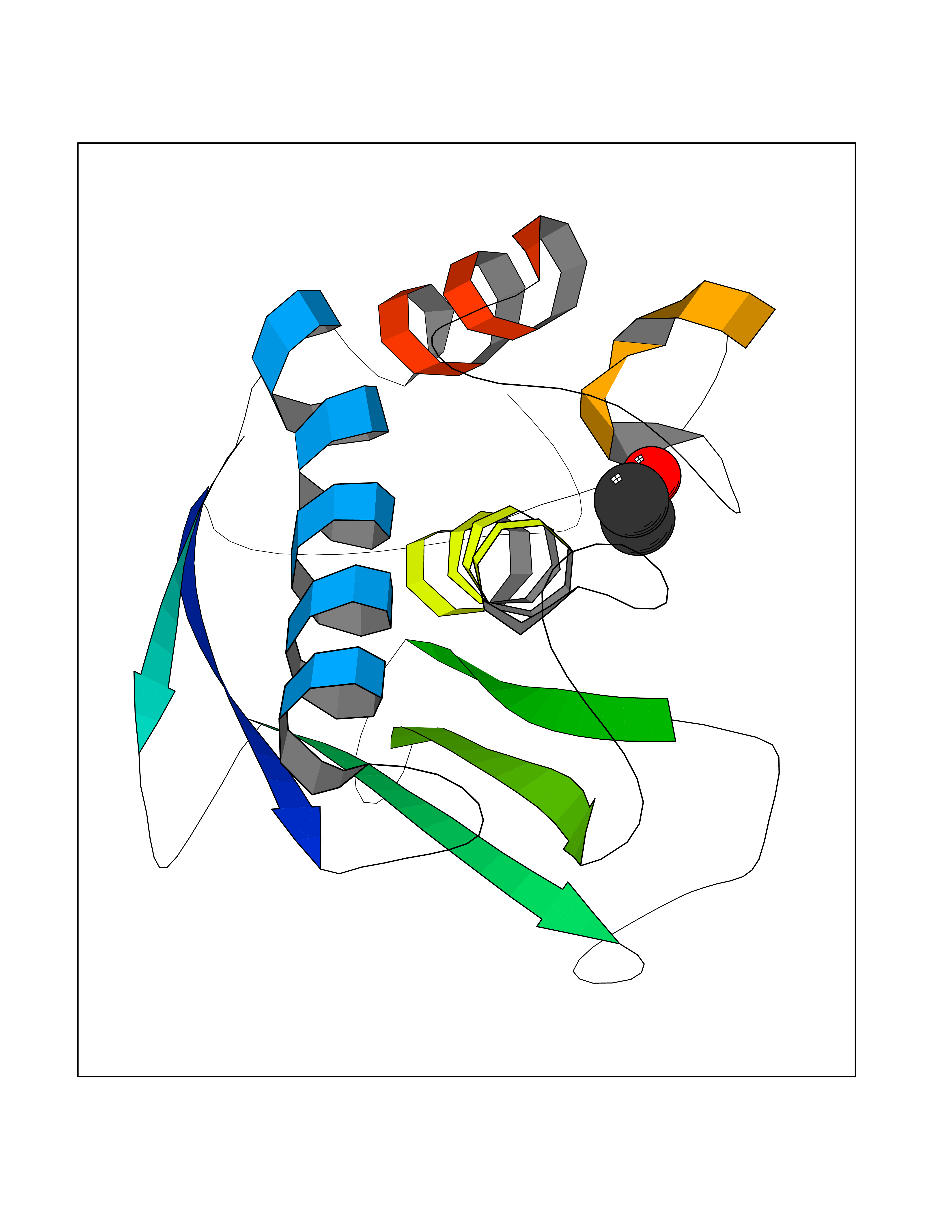 Molscript Map 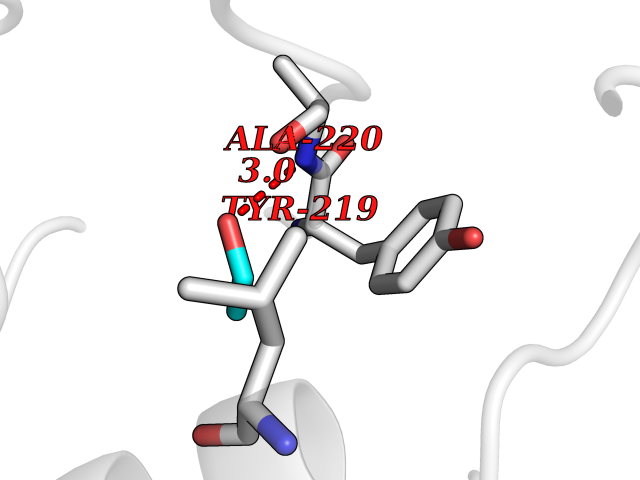 Pymol Map 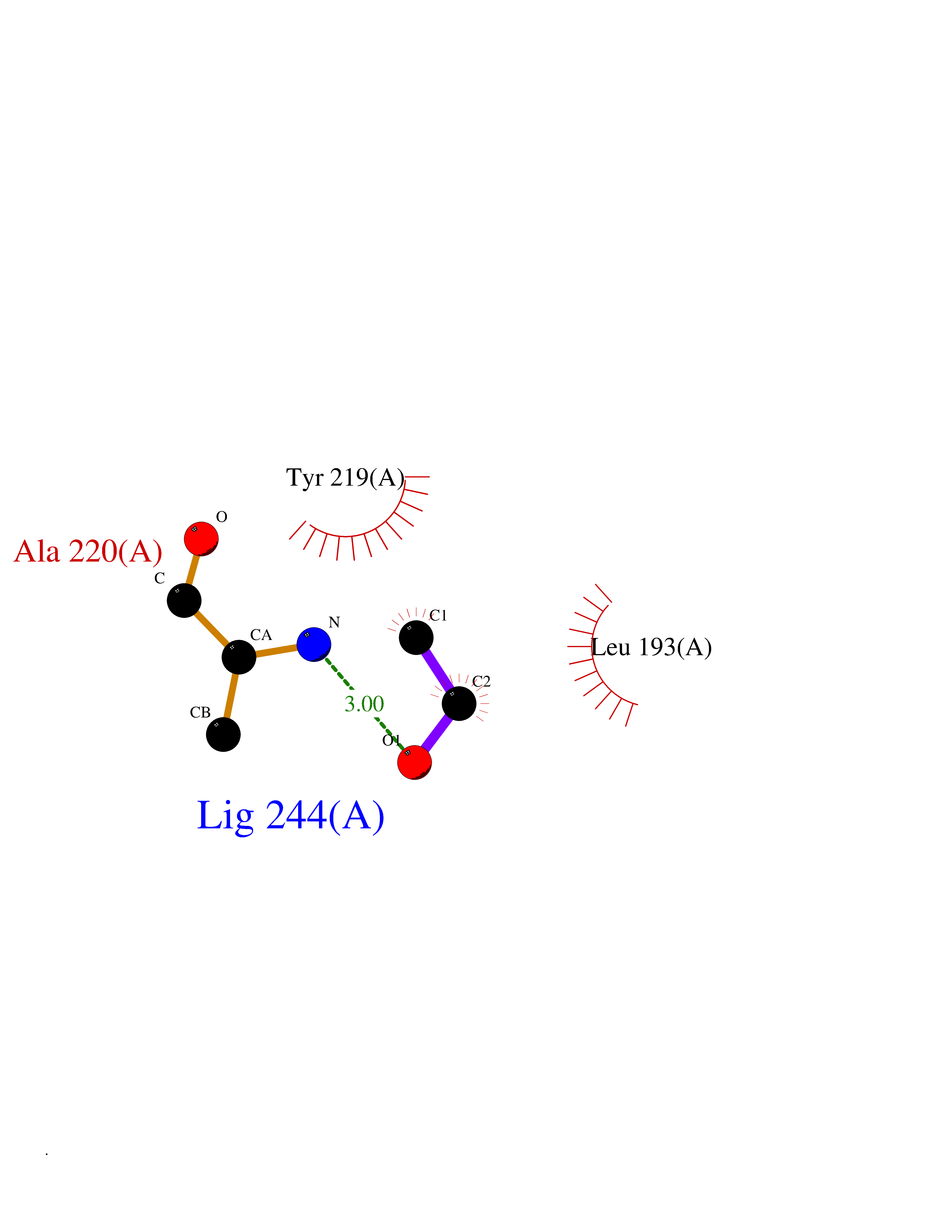 Ligplot Map 3D Binding mode Sequence MLTPGNPKWERTNLTYRIRNYTPQLSEAEVERAIKDAFELWSVASPLIFTRISQGEADINIAFYQRDHGDNSPFDGPNGILAHAFQPGQGIGGDAHFDAEETWTNTSANYNLFLVAAHEFGHSLGLAHSSDPGALMYPNYAFRETSNYSLPQDDIDGIQAIYG Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Lactaldehyde dehydrogenase | 2IMP | 4.28 | |
Target general information Gen name aldA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms ald;JW1412;b1415 Protein family Aldehyde dehydrogenase family Biochemical class Oxidoreductase Function Glycolaldehyde dehydrogenase activity.Identical protein binding.Lactaldehyde dehydrogenase activity.NAD binding.Succinate-semialdehyde dehydrogenase (NAD+) activity. Related diseases 3-ketothiolase deficiency (3KTD) [MIM:203750]: An autosomal recessive inborn error of isoleucine catabolism characterized by intermittent ketoacidotic attacks associated with unconsciousness. Some patients die during an attack or are mentally retarded. Urinary excretion of 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid, triglylglycine, butanone is increased. It seems likely that the severity of this disease correlates better with the environmental or acquired factors than with the ACAT1 genotype. {ECO:0000269|PubMed:1346617, ECO:0000269|PubMed:1715688, ECO:0000269|PubMed:7728148, ECO:0000269|PubMed:9744475}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03619 Interacts with NA EC number 1.2.1.21; 1.2.1.22 Uniprot keywords 3D-structure; Direct protein sequencing; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 48991.4 Length 451 Aromaticity 0.08 Instability index 26.75 Isoelectric point 5.14 Charge (pH=7) -11.76 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map 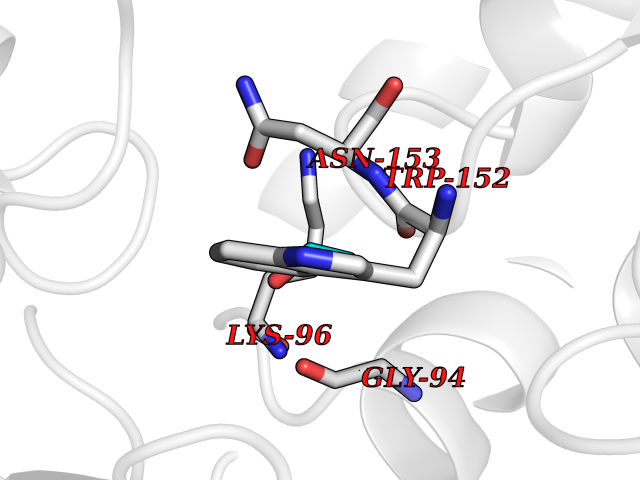 Pymol Map 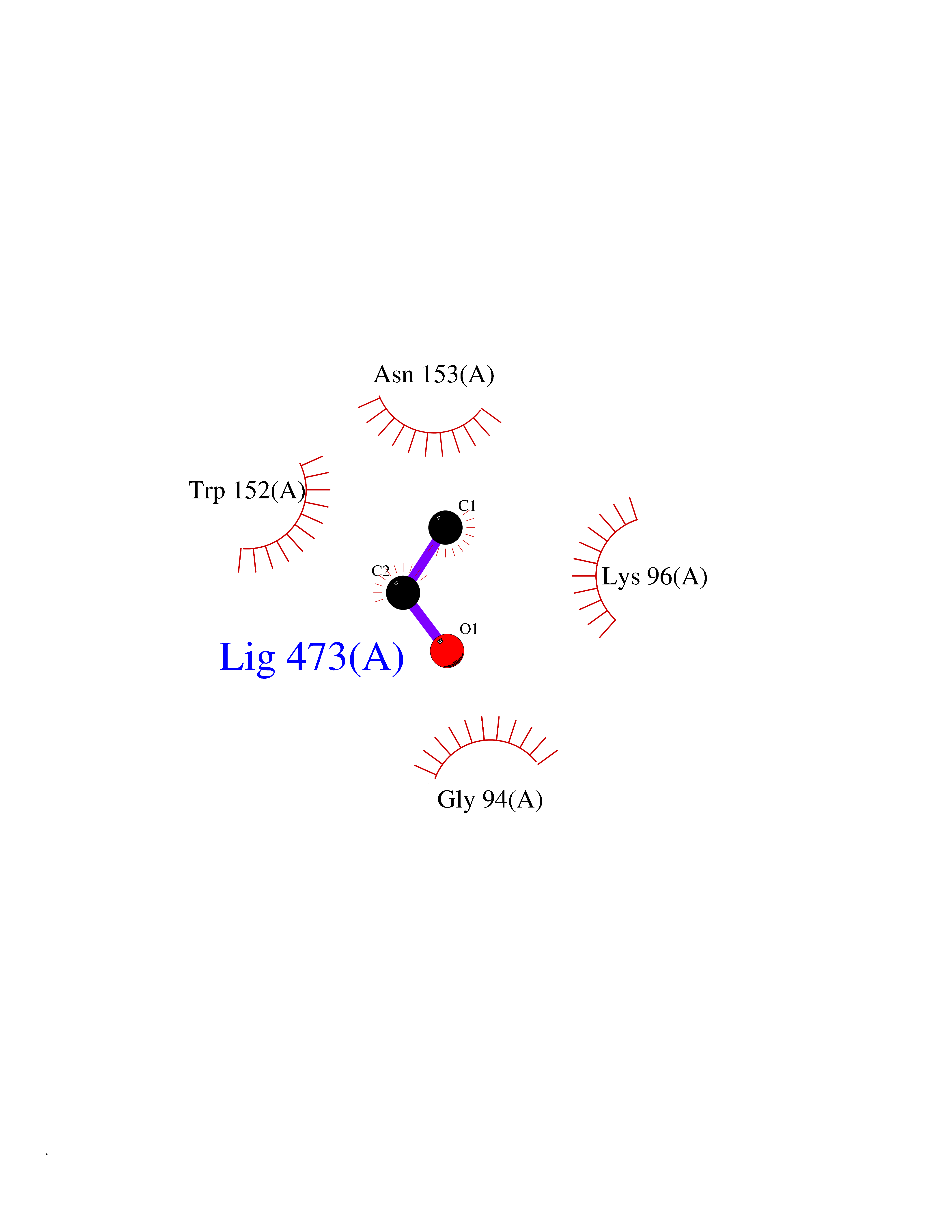 Ligplot Map 3D Binding mode Sequence VPVQHPMYIDGQFVTWRGDAWIDVVNPATEAVISRIPDGQAEDARKAIDAAERAQPEWEALPAIERASWLRKISAGIRERASEISALIVEEGGKIQQLAEVEVAFTADYIDYMAEWARRYRALGVTTGILPWNFPFFLIARKMAPALLTGNTIVIKPSEFTPNNAIAFAKIVDEIGLPRGVFNLVLGRGETVGQELAGNPKVAMVSMTGSVSAGEKIMATAAKNITKVXLELGGKAPAIVMDDADLELAVKAIVDSRVINSGQVCNCAERVYVQKGIYDQFVNRLGEAMQAVQFGNPAERNDIAMGPLINAAALERVEQKVARAVEEGARVAFGGKAVEGKGYYYPPTLLLDVRQEMSIMHEETFGPVLPVVAFDTLEDAISMANDSDYGLTSSIYTQNLNVAMKAIKGLKFGETYINRENFEAMQGFHAGWRKSGIGGADGKHGLHEYLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Serum paraoxonase/arylesterase 1 | 1V04 | 4.28 | |
Target general information Gen name PON1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PON Protein family Paraoxonase family Biochemical class Hydrolase Function Acyl-L-homoserine-lactone lactonohydrolase activity.Aryldialkylphosphatase activity.Arylesterase activity.Calcium ion binding.Phospholipid binding.Protein homodimerization activity. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB01327; DB09130; DB01395; DB14598; DB14600; DB14596; DB00218; DB01085; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number 3.1.1.2; 3.1.1.81; 3.1.8.1 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disulfide bond; Glycoprotein; HDL; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 37232.8 Length 332 Aromaticity 0.11 Instability index 35.09 Isoelectric point 5.06 Charge (pH=7) -17.08 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map 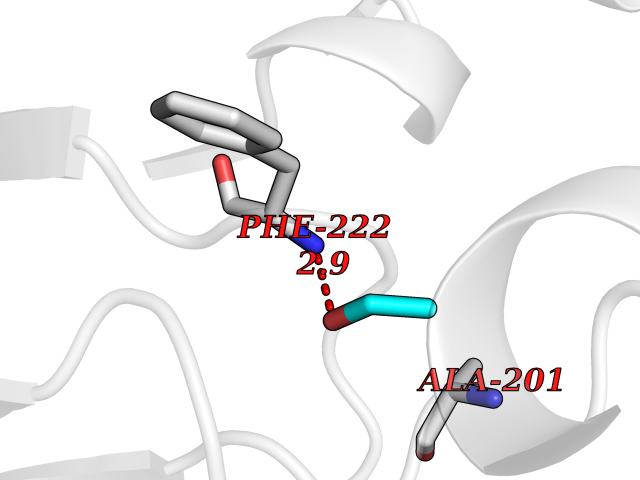 Pymol Map 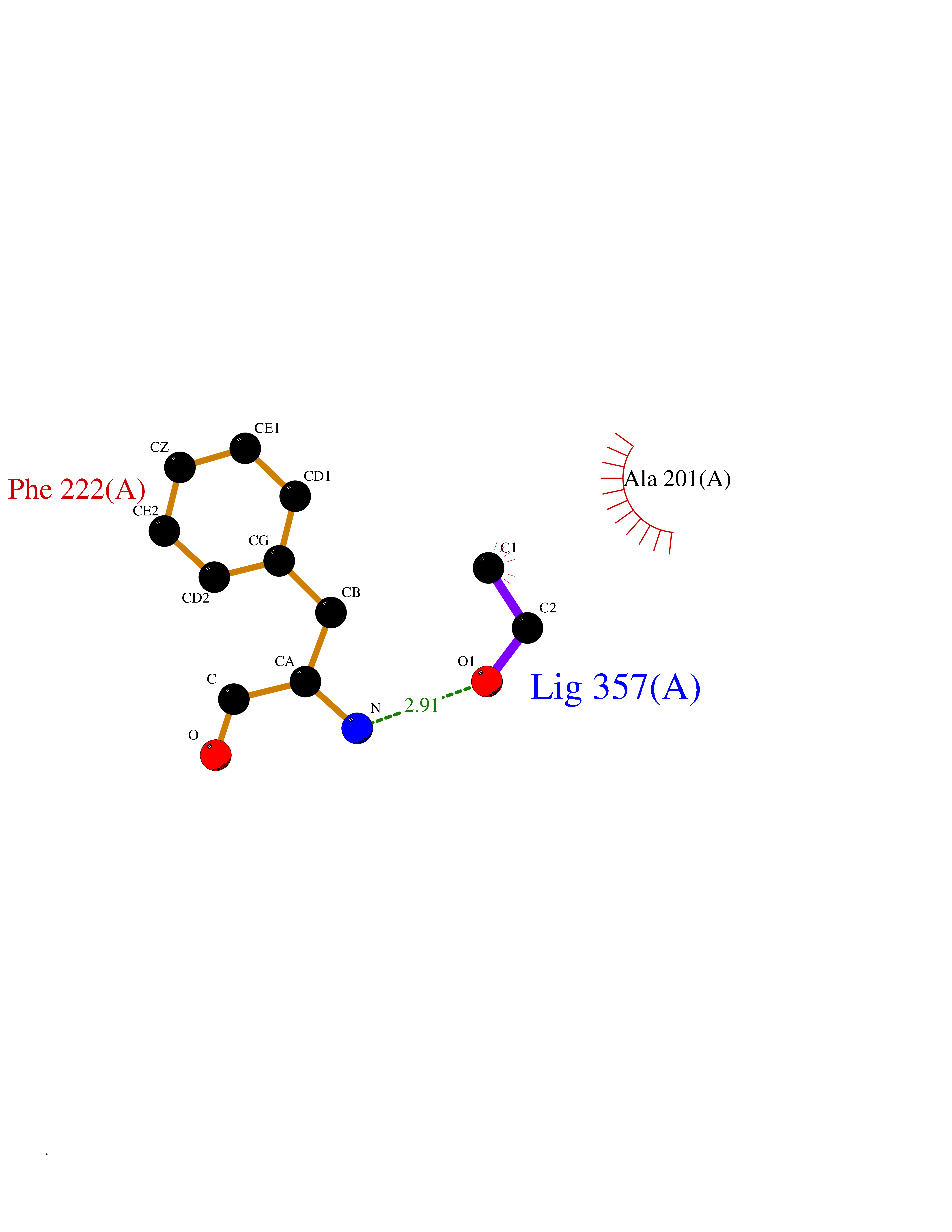 Ligplot Map 3D Binding mode Sequence LFDRQKSSFQTRFNVHREVTPVELPNCNLVKGIDNGSEDLEILPNGLAFISSGLKYDKSGKILLMDLNEKEPAVSELEIIGNTLDISSFNPHGISTFIDDDNTVYLLVVNHPGSSSTVEVFKFQEEEKSLLHLKTIRHKLLPSVNDIVAVGPEHFYATNDHYFIDPYLKSWEMHLGLAWSFVTYYSPNDVRVVAEGFDFANGINISPDGKYVYIAELLAHKIHVYEKHANWTLTPLRVLSFDTLVDNISVDPVTGDLWVGCHPNGMRIFFYDAENPPGSEVLRIQDILSEEPKVTVVYAENGTVLQGSTVAAVYKGKLLIGTVFHKALYCDL Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Nitric-oxide synthase brain (NOS1) | 5ADF | 4.28 | |
Target general information Gen name NOS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase NOS1; Nitric oxide synthase, brain; Neuronal NOS; NOS, type I; NOS type I; NNOS; NC-NOS; N-NOS; BNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function In the brain and peripheral nervous system, NO displays many properties of a neurotransmitter. Probably has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such SRR. Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB02143; DB02727; DB01997; DB03892; DB02207; DB03710; DB00155; DB00843; DB00997; DB03147; DB03247; DB01942; DB01221; DB02077; DB01821; DB09241; DB03144; DB03449; DB02044; DB02644; DB08019; DB08018; DB02027; DB03461; DB04223; DB06096; DB02991; DB03707 Interacts with Q08AM6 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calmodulin-binding; Cell membrane; Cell projection; FAD; Flavoprotein; FMN; Heme; Iron; Membrane; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34875.7 Length 299 Aromaticity 0.1 Instability index 42.94 Isoelectric point 5.96 Charge (pH=7) -6.25 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map 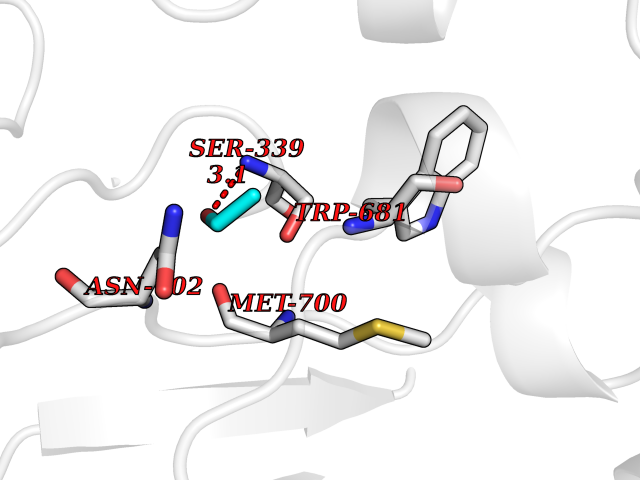 Pymol Map 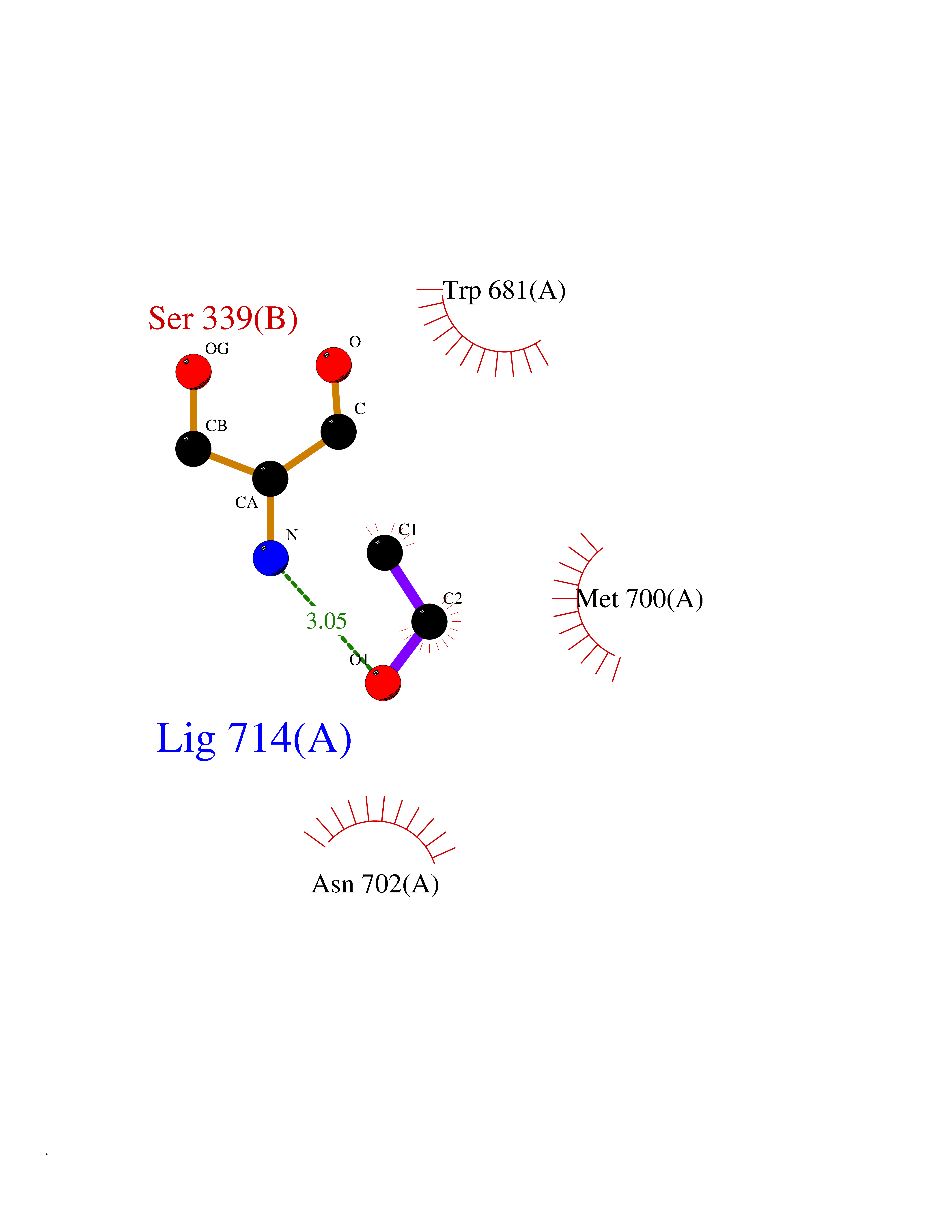 Ligplot Map 3D Binding mode Sequence CPRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLTPSFEYQ Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Angiopoietin 1 receptor (TEK) | 3BEA | 4.28 | |
Target general information Gen name TEK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hTIE2; VMCM1; VMCM; Tyrosine-protein kinase receptor TIE-2; Tyrosine-protein kinase receptor TEK; Tyrosine kinase with Ig and EGF homology domains-2; Tunica interna endothelial cell kinase; TIE2; P140 Protein family Protein kinase superfamily, Tyr protein kinase family, Tie subfamily Biochemical class Kinase Function Has anti-inflammatory effects by preventing the leakage of proinflammatory plasma proteins and leukocytes from blood vessels. Required for normal angiogenesis and heart development during embryogenesis. Required for post-natal hematopoiesis. After birth, activates or inhibits angiogenesis, depending on the context. Inhibits angiogenesis and promotes vascular stability in quiescent vessels, where endothelial cells have tight contacts. In quiescent vessels, ANGPT1 oligomers recruit TEK to cell-cell contacts, forming complexes with TEK molecules from adjoining cells, and this leads to preferential activation of phosphatidylinositol 3-kinase and the AKT1 signaling cascades. In migrating endothelial cells that lack cell-cell adhesions, ANGT1 recruits TEK to contacts with the extracellular matrix, leading to the formation of focal adhesion complexes, activation of PTK2/FAK and of the downstream kinases MAPK1/ERK2 and MAPK3/ERK1, and ultimately to the stimulation of sprouting angiogenesis. ANGPT1 signaling triggers receptor dimerization and autophosphorylation at specific tyrosine residues that then serve as binding sites for scaffold proteins and effectors. Signaling is modulated by ANGPT2 that has lower affinity for TEK, can promote TEK autophosphorylation in the absence of ANGPT1, but inhibits ANGPT1-mediated signaling by competing for the same binding site. Signaling is also modulated by formation of heterodimers with TIE1, and by proteolytic processing that gives rise to a soluble TEK extracellular domain. The soluble extracellular domain modulates signaling by functioning as decoy receptor for angiopoietins. TEK phosphorylates DOK2, GRB7, GRB14, PIK3R1; SHC1 and TIE1. Tyrosine-protein kinase that acts as cell-surface receptor for ANGPT1, ANGPT2 and ANGPT4 and regulates angiogenesis, endothelial cell survival, proliferation, migration, adhesion and cell spreading, reorganization of the actin cytoskeleton, but also maintenance of vascular quiescence. Related diseases Dominantly inherited venous malformations (VMCM) [MIM:600195]: An error of vascular morphogenesis characterized by dilated, serpiginous channels. {ECO:0000269|PubMed:10369874, ECO:0000269|PubMed:19079259, ECO:0000269|PubMed:19888299, ECO:0000269|PubMed:8980225}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Somatic mutations of TEK are associated with solitary and multiple sporadic venous malformations. {ECO:0000269|PubMed:19079259}.; DISEASE: May play a role in a range of diseases with a vascular component, including neovascularization of tumors, psoriasis and inflammation.; DISEASE: Glaucoma 3, primary congenital, E (GLC3E) [MIM:617272]: An autosomal dominant form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:27270174}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00415; DB12010; DB08221; DB08901; DB08896; DB14840; DB11800; DB05294 Interacts with Q15389; O15123; O15123-1; Q16678; Q05209; P23467; P08575; Q12913; Q15262; Q16827 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell junction; Cell membrane; Cytoplasm; Cytoskeleton; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Glaucoma; Glycoprotein; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 34965.9 Length 310 Aromaticity 0.11 Instability index 43.57 Isoelectric point 8.39 Charge (pH=7) 3.42 2D Binding mode Binding energy (Kcal/mol) -5.83 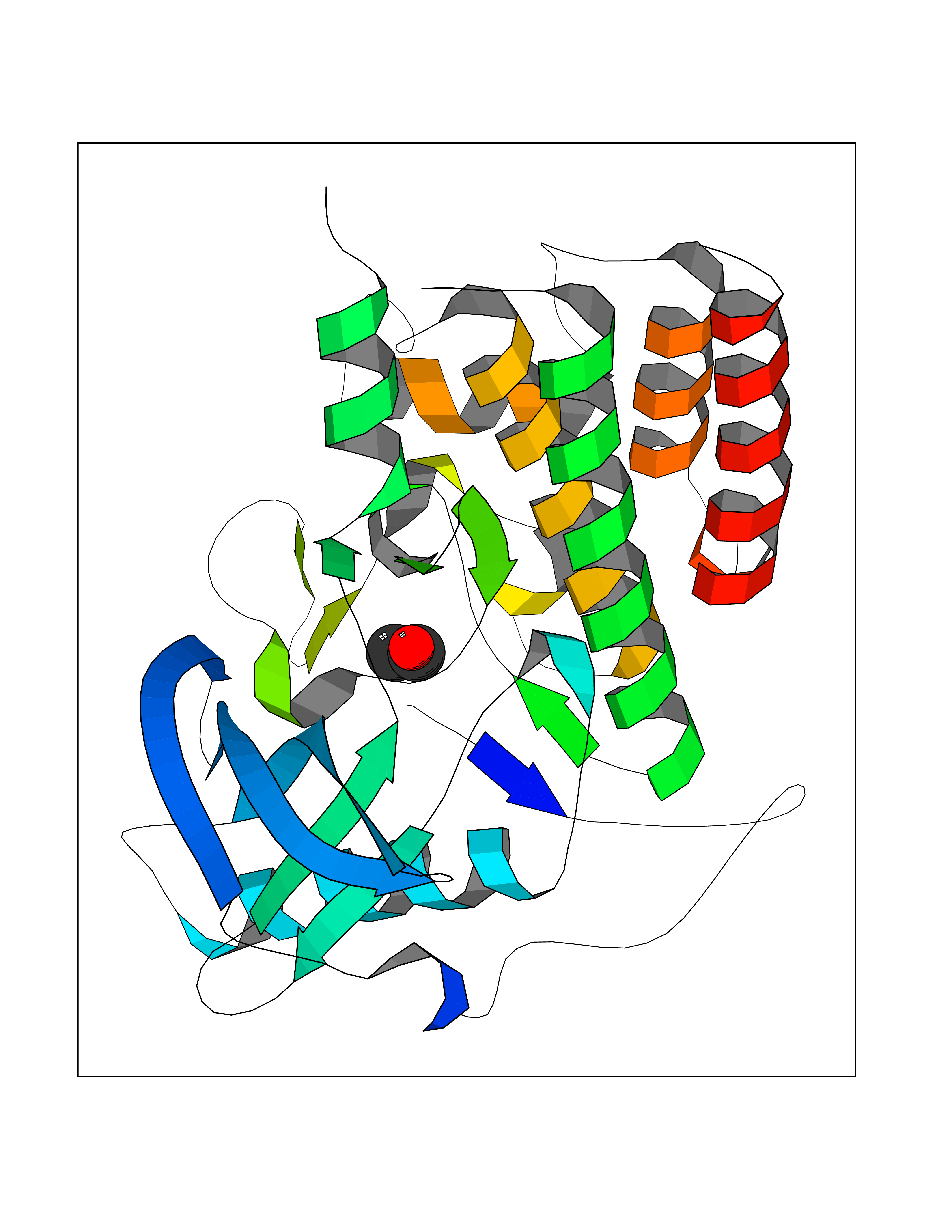 Molscript Map 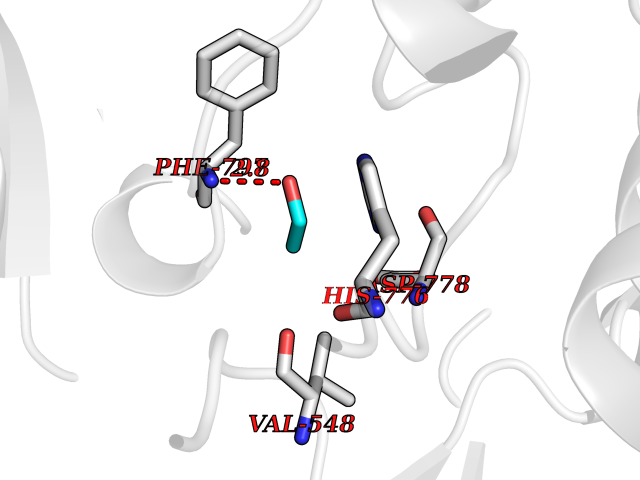 Pymol Map 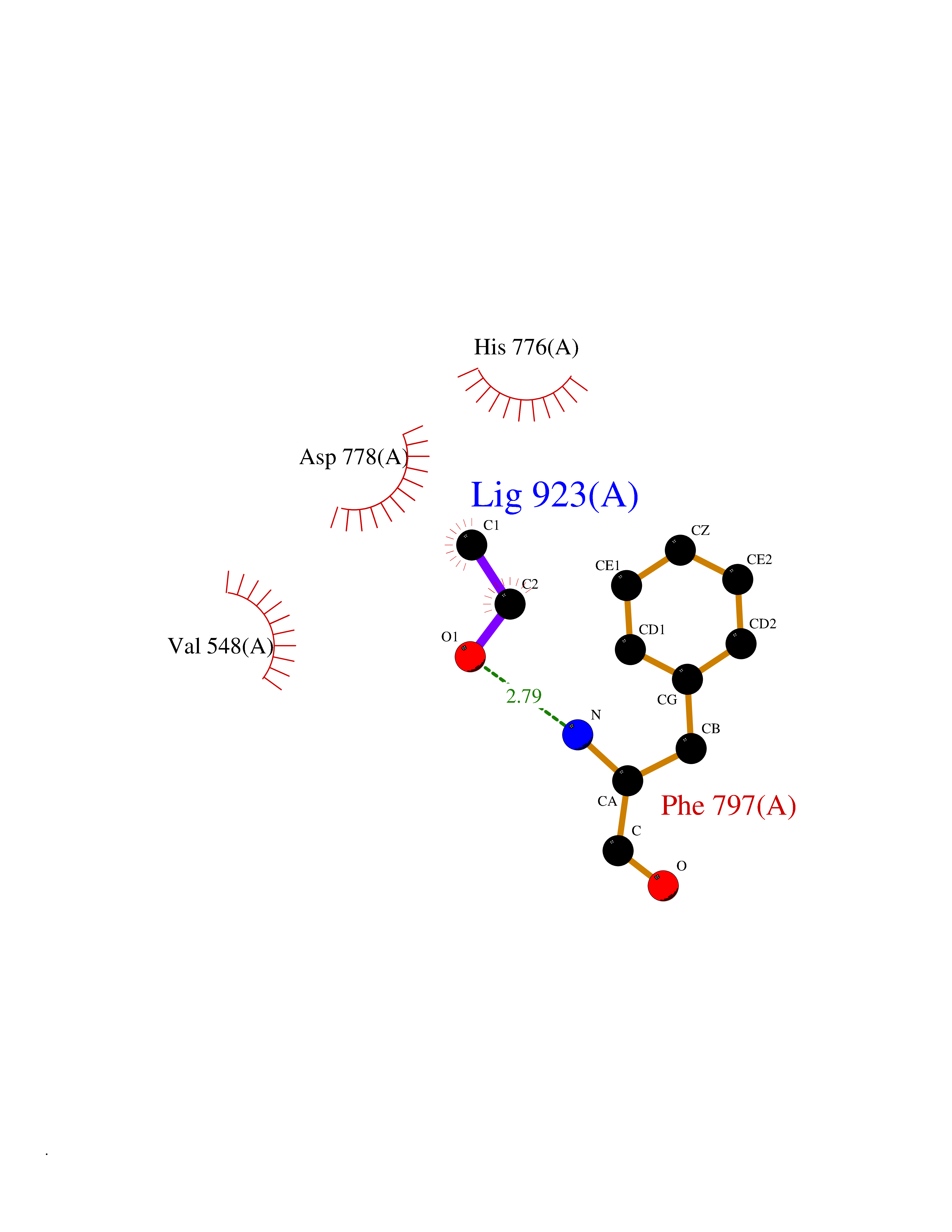 Ligplot Map 3D Binding mode Sequence QVRWKIIESYEGNSYTFIDPTQLPYNEKWEFPRNNLQFGKTLGAGAFGKVVEATAFGLGKEDAVLKVAVKMLKSTAHADEKEALMSELKIMSHLGQHENIVNLLGACTHGGPVLVITEYCCYGDLLNFLRRKSRVLSTLSTRDLLHFSSQVAQGMAFLASKNCIHRDVAARNVLLTNGHVAKIGDFGLARDIMNDSNYIVKGNARLPVKWMAPESIFDCVYTVQSDVWSYGILLWEIFSLGLNPYPGILVNSKFYKLVKDGYQMAQPAFAPKNIYSIMQACWALEPTHRPTFQQICSFLQEQAQEDRRER Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | cAMP-specific 3',5'-cyclic phosphodiesterase 4C | 2QYM | 4.28 | |
Target general information Gen name PDE4C Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DPDE1 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Hydrolase Function 3',5'-cyclic-AMP phosphodiesterase activity.Metal ion binding. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01427; DB00201; DB05219; DB00651; DB06246; DB05266; DB01088; DB01791; DB01656; DB01954; DB09283 Interacts with Q96D03; O15499; P26718; P50221; Q6FHY5; Q9UJX0; P26367; P30626; P59817; P30626 EC number 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell projection; Cilium; Hydrolase; Manganese; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 33042.2 Length 291 Aromaticity 0.08 Instability index 23.45 Isoelectric point 4.91 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -5.83 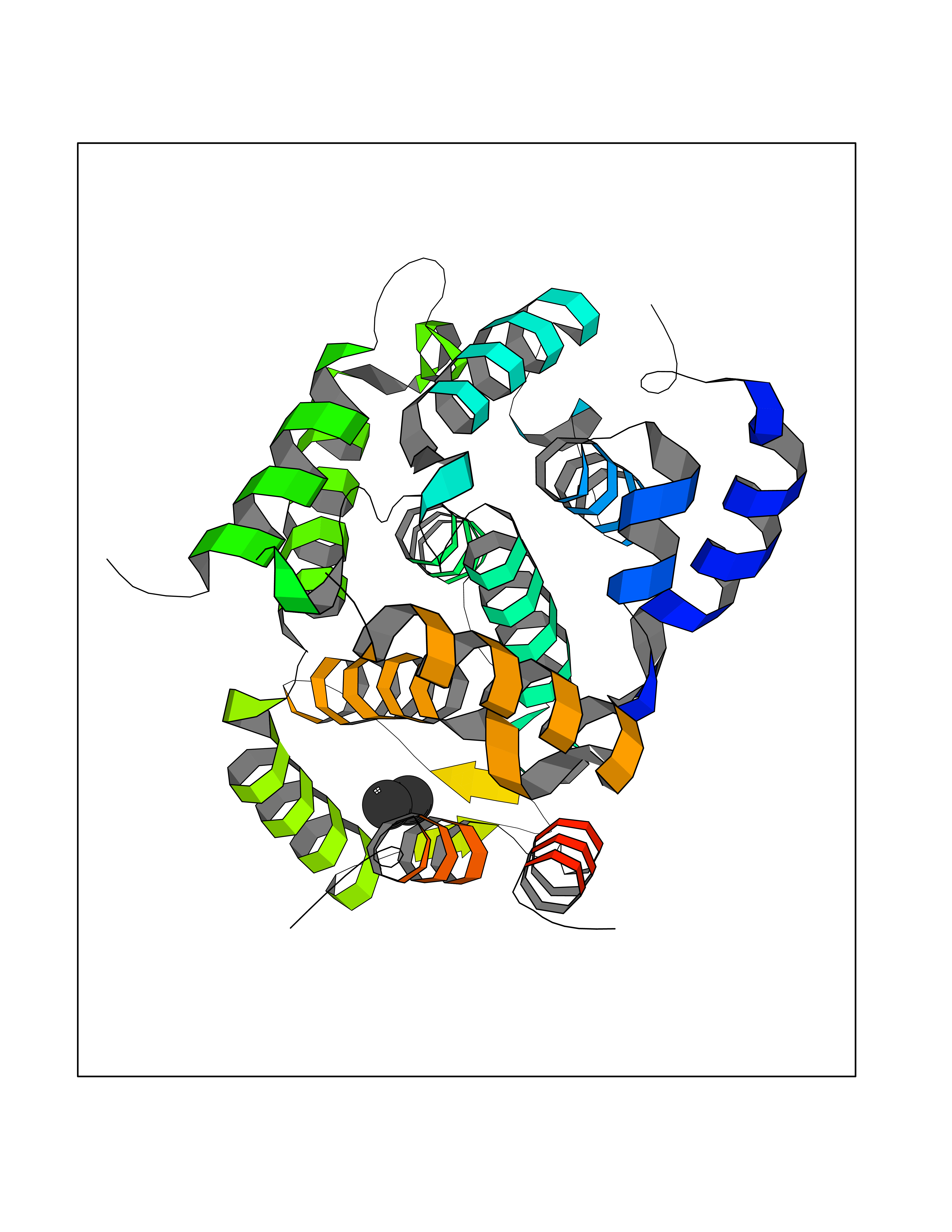 Molscript Map 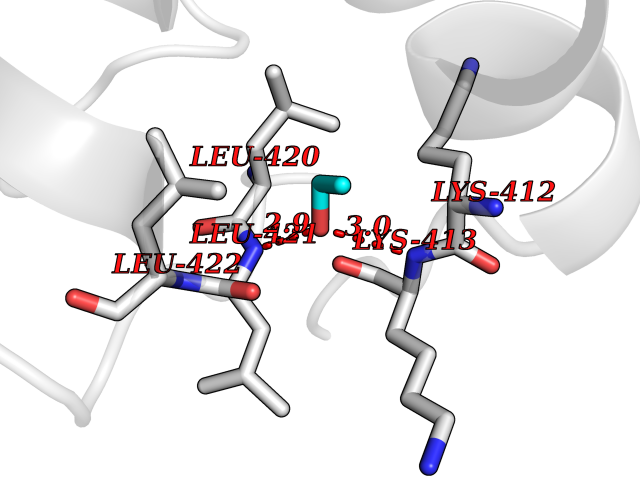 Pymol Map 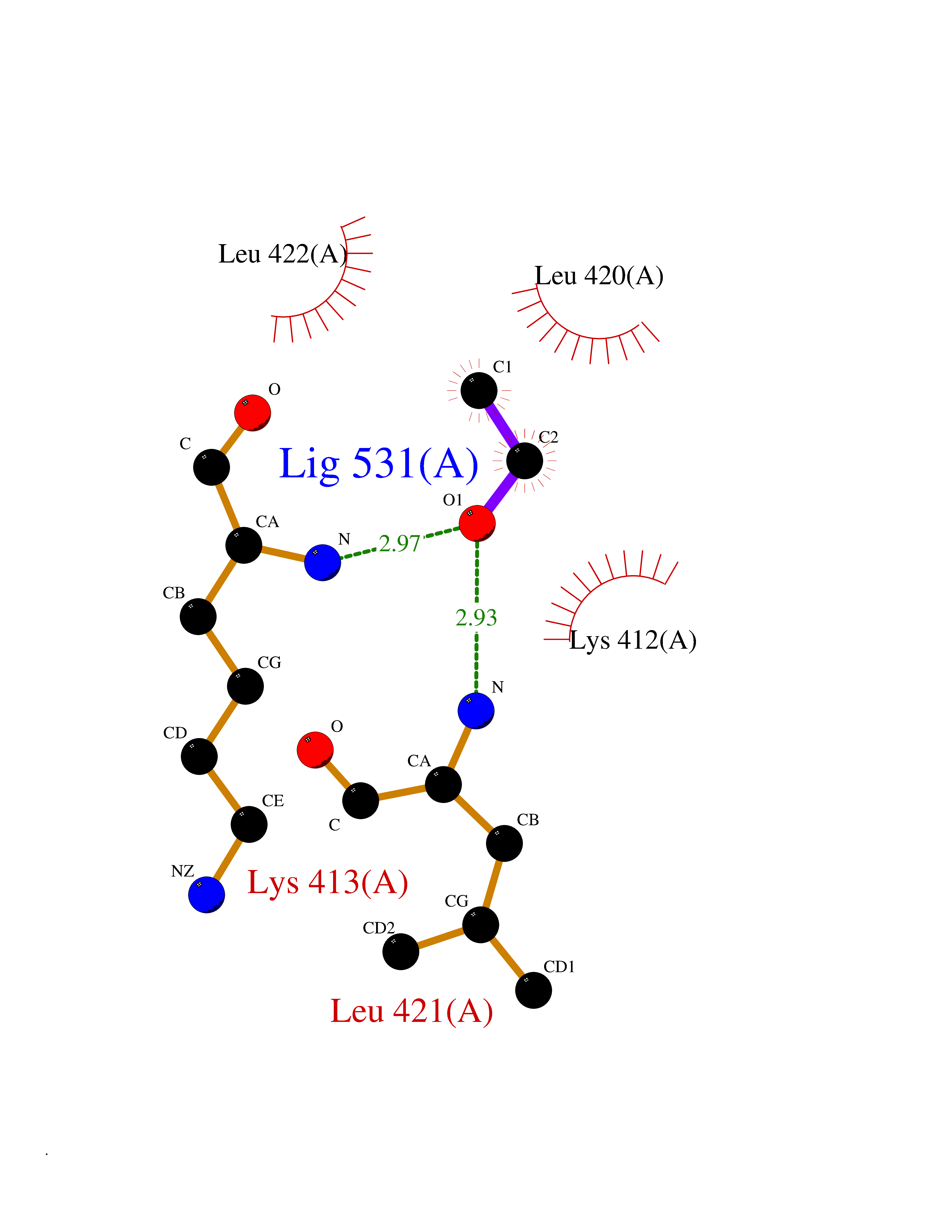 Ligplot Map 3D Binding mode Sequence VPRFGVQTDQEEQLAKELEDTNKWGLDVFKVAELSGNRPLTAIIFSIFQERDLLKTFQIPADTLATYLLMLEGHYHANVAYHNSLHAADVAQSTHVLLATPALEAVFTDLEILAALFASAIHDVDHPGVSNQNDASVLENHHLAVGFKLLQAENCDIFQNLSAKQRLSLRRMVIDMVLATDMSKHMNLLADLKTMVETKKVTSLGVLLLDNYSDRIQVLQNLVHCADLSNPTKPLPLYRQWTDRIMAEFFQQQVGFIDYIAHPLWETWADLVHPDAQDLLDTLEDNREWYQ Hydrogen bonds contact Hydrophobic contact | ||||