Job Results:
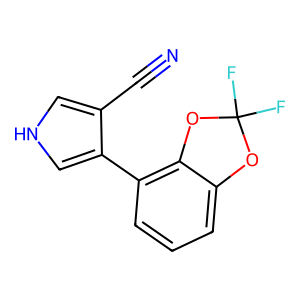
Ligand
Structure
Job ID
9af75475a67248e060ab578f15db3d0b
Job name
NA
Time
2025-04-03 17:54:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Macrophage colony-stimulating factor 1 receptor (CSF1R) | 2I1M | 6.07 | |
Target general information Gen name CSF1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Proto-oncogene c-Fms; M-CSF-R; FMS; CSF-1R; CSF-1-R; CSF-1 receptor; CD115 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Promotes the release of proinflammatory chemokines in response to IL34 and CSF1, and thereby plays an important role in innate immunity and in inflammatory processes. Plays an important role in the regulation of osteoclast proliferation and differentiation, the regulation of bone resorption, and is required for normal bone and tooth development. Required for normal male and female fertility, and for normal development of milk ducts and acinar structures in the mammary gland during pregnancy. Promotes reorganization of the actin cytoskeleton, regulates formation of membrane ruffles, cell adhesion and cell migration, and promotes cancer cell invasion. Activates several signaling pathways in response to ligand binding. Phosphorylates PIK3R1, PLCG2, GRB2, SLA2 and CBL. Activation of PLCG2 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, that then lead to the activation of protein kinase C family members, especially PRKCD. Phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, leads to activation of the AKT1 signaling pathway. Activated CSF1R also mediates activation of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1, and of the SRC family kinases SRC, FYN and YES1. Activated CSF1R transmits signals both via proteins that directly interact with phosphorylated tyrosine residues in its intracellular domain, or via adapter proteins, such as GRB2. Promotes activation of STAT family members STAT3, STAT5A and/or STAT5B. Promotes tyrosine phosphorylation of SHC1 and INPP5D/SHIP-1. Receptor signaling is down-regulated by protein phosphatases, such as INPP5D/SHIP-1, that dephosphorylate the receptor and its downstream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as cell-surface receptor for CSF1 and IL34 and plays an essential role in the regulation of survival, proliferation and differentiation of hematopoietic precursor cells, especially mononuclear phagocytes, such as macrophages and monocytes. Related diseases Aberrant expression of CSF1 or CSF1R can promote cancer cell proliferation, invasion and formation of metastases. Overexpression of CSF1 or CSF1R is observed in a significant percentage of breast, ovarian, prostate, and endometrial cancers.; DISEASE: Aberrant expression of CSF1 or CSF1R may play a role in inflammatory diseases, such as rheumatoid arthritis, glomerulonephritis, atherosclerosis, and allograft rejection.; DISEASE: Leukoencephalopathy, hereditary diffuse, with spheroids 1 (HDLS1) [MIM:221820]: An autosomal dominant adult-onset rapidly progressive neurodegenerative disorder characterized by variable behavioral, cognitive, and motor changes. Patients often die of dementia within 6 years of onset. Brain imaging shows patchy abnormalities in the cerebral white matter, predominantly affecting the frontal and parietal lobes. {ECO:0000269|PubMed:22197934, ECO:0000269|PubMed:23408870, ECO:0000269|PubMed:24336230, ECO:0000269|PubMed:24532199}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brain abnormalities, neurodegeneration, and dysosteosclerosis (BANDDOS) [MIM:618476]: An autosomal recessive disease with variable manifestations. Main features are brain malformations with calcifying leukoencephalopathy, progressive neurodegeneration, and bone sclerotic features. The age at onset ranges from infancy to early adulthood. Neurologic features include loss of previous motor and language skills, cognitive impairment, spasticity, and focal seizures. Brain imaging shows periventricular white matter abnormalities and calcifications, large cisterna magna or Dandy-Walker malformation, and sometimes agenesis of the corpus callosum. {ECO:0000269|PubMed:30982608, ECO:0000269|PubMed:30982609}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07167; DB07202; DB12147; DB12010; DB00619; DB06080; DB12978; DB01268 Interacts with P09603; Q15375; P29323; Q6ZMJ4-1 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Inflammatory response; Innate immunity; Kinase; Membrane; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35082.9 Length 311 Aromaticity 0.11 Instability index 44.6 Isoelectric point 8.13 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -8.28 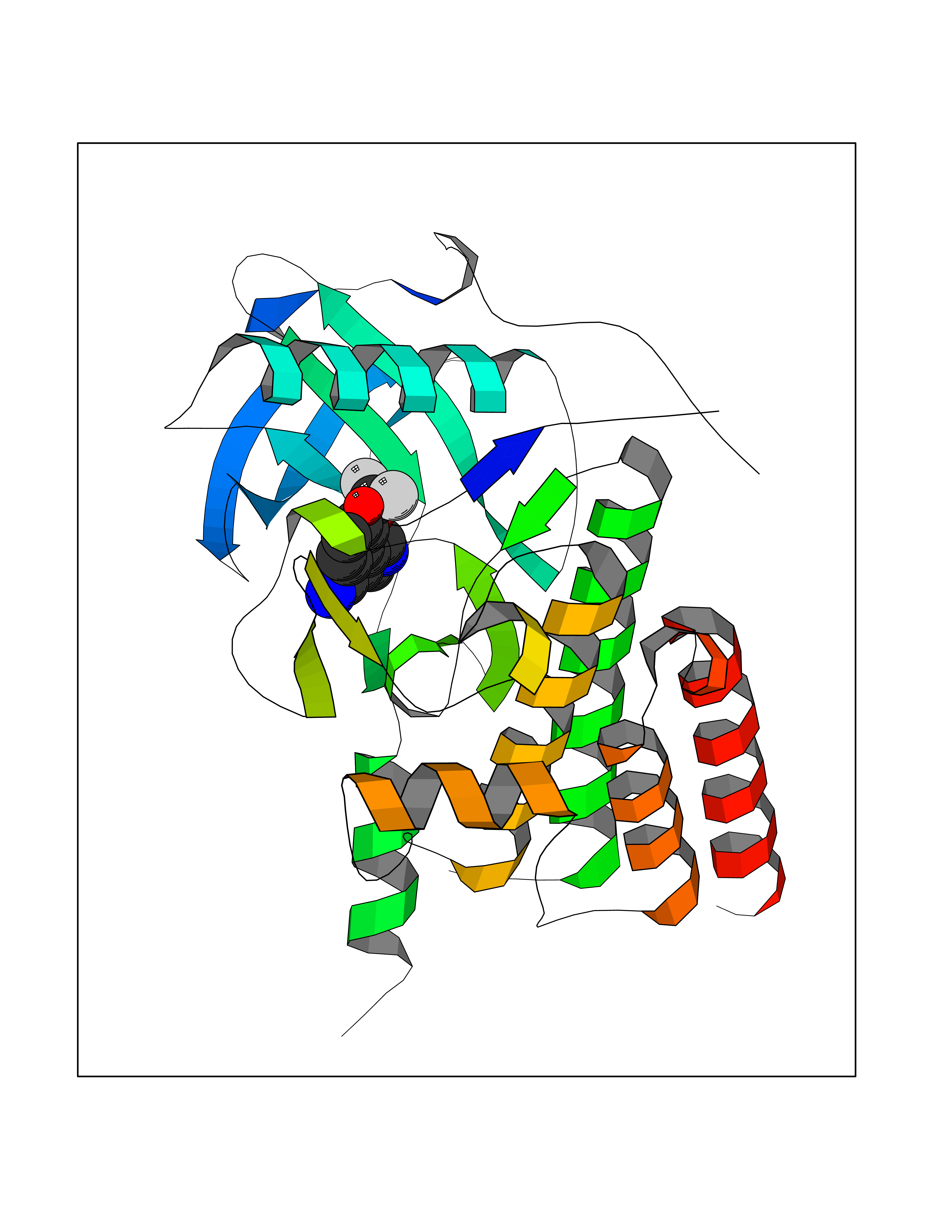 Molscript Map 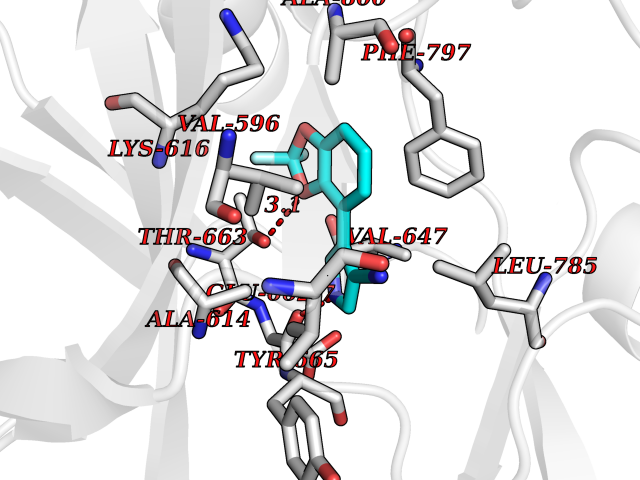 Pymol Map 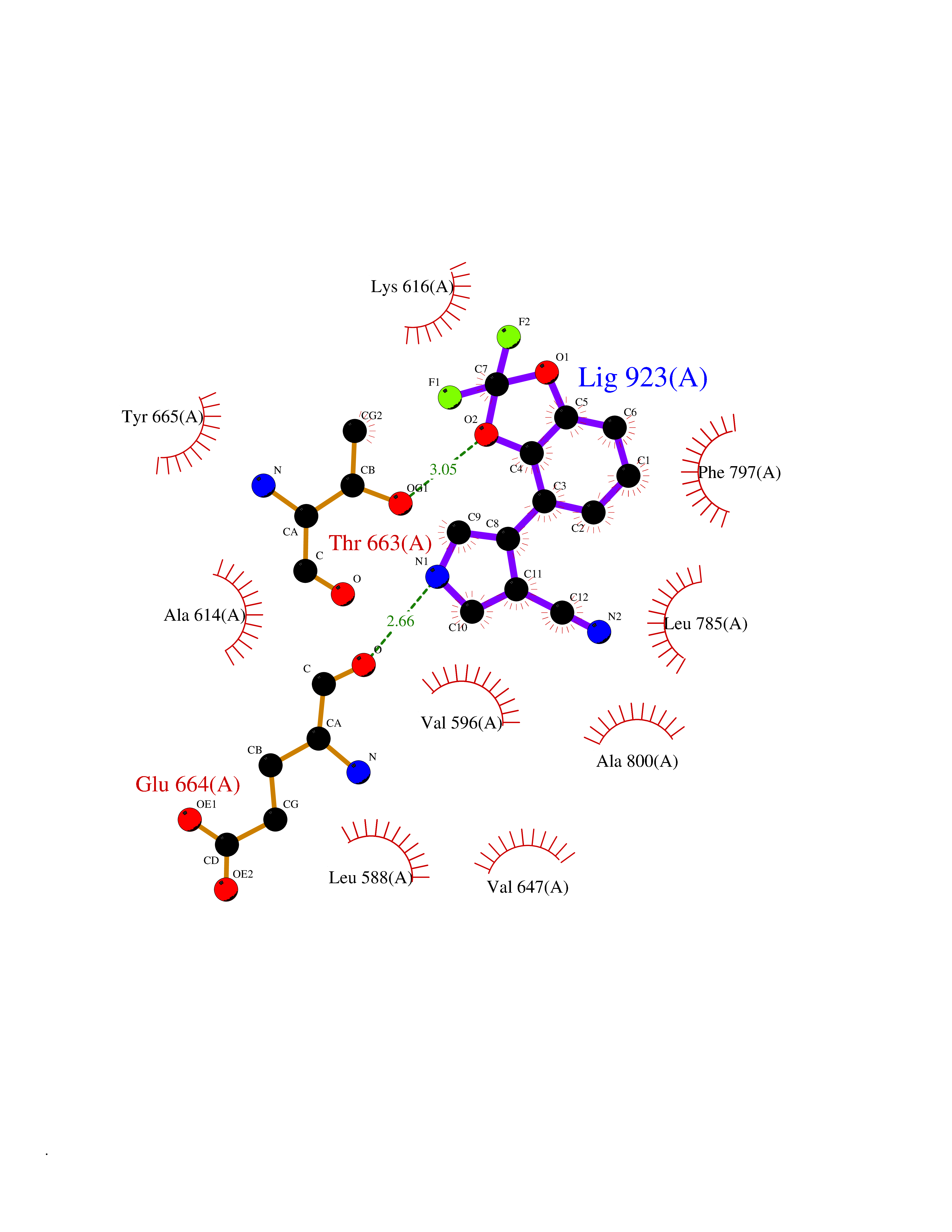 Ligplot Map 3D Binding mode Sequence QVRWKIIESYNSYTFIDPTQLPYNEKWEFPRNNLQFGKTLGAGAFGKVVEATAFGLGKEDAVLKVAVKMLKSTAHADEKEALMSELKIMSHLGQHENIVNLLGACTHGGPVLVITEYCCYGDLLNFLRRKSRVLETDSTASTRDLLHFSSQVAQGMAFLASKNCIHRDVAARNVLLTNGHVAKIGDFGLARDIMNDSNYIVKGNARLPVKWMAPESIFDCVYTVQSDVWSYGILLWEIFSLGLNPYPGILVNSKFYKLVKDGYQMAQPAFAPKNIYSIMQACWALEPTHRPTFQQICSFLQEQAQEDRRER Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Bacterial Flavohemoglobin (Bact hmp) | 1GVH | 6.07 | |
Target general information Gen name Bact hmp Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Nitric oxide dioxygenase; NOD; NO oxygenase; Hemoglobin-like protein; HMP; Ferrisiderophore reductase B; Dihydropteridine reductase Protein family Globin family, Two-domain flavohemoproteins subfamily; Flavoprotein pyridine nucleotide cytochrome reductase family Biochemical class Paired donor oxygen oxidoreductase Function Various electron acceptors arealso reduced by HMP in vitro, including dihydropterine, ferrisiderophores, ferric citrate, cytochrome c, nitrite, S-nitrosoglutathione, and alkylhydroperoxides. However, it is unknown if these reactions are of any biological significance in vivo. Related diseases Ovarian dysgenesis 1 (ODG1) [MIM:233300]: An autosomal recessive disease characterized by primary amenorrhea, variable development of secondary sex characteristics, poorly developed streak ovaries, and high serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). {ECO:0000269|PubMed:10551778, ECO:0000269|PubMed:11889179, ECO:0000269|PubMed:12571157, ECO:0000269|PubMed:12915623, ECO:0000269|PubMed:7553856, ECO:0000269|PubMed:9769327, ECO:0000269|PubMed:9851774}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ovarian hyperstimulation syndrome (OHSS) [MIM:608115]: Disorder which occurs either spontaneously or most often as an iatrogenic complication of ovarian stimulation treatments for in vitro fertilization. The clinical manifestations vary from abdominal distention and discomfort to potentially life-threatening, massive ovarian enlargement and capillary leak with fluid sequestration. Pathologic features of this syndrome include the presence of multiple serous and hemorrhagic follicular cysts lined by luteinized cells, a condition called hyperreactio luteinalis. {ECO:0000269|PubMed:12930927, ECO:0000269|PubMed:12930928, ECO:0000269|PubMed:15080154, ECO:0000269|PubMed:16278261, ECO:0000269|PubMed:17721928, ECO:0000269|PubMed:24058690, ECO:0000269|PubMed:25581598}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number EC 1.14.12.17 Uniprot keywords 3D-structure; Cytoplasm; Detoxification; Direct protein sequencing; FAD; Flavoprotein; Heme; Iron; Metal-binding; NAD; NADP; Oxidoreductase; Oxygen transport; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 43867.1 Length 396 Aromaticity 0.1 Instability index 28.85 Isoelectric point 5.48 Charge (pH=7) -12.32 2D Binding mode Binding energy (Kcal/mol) -8.28 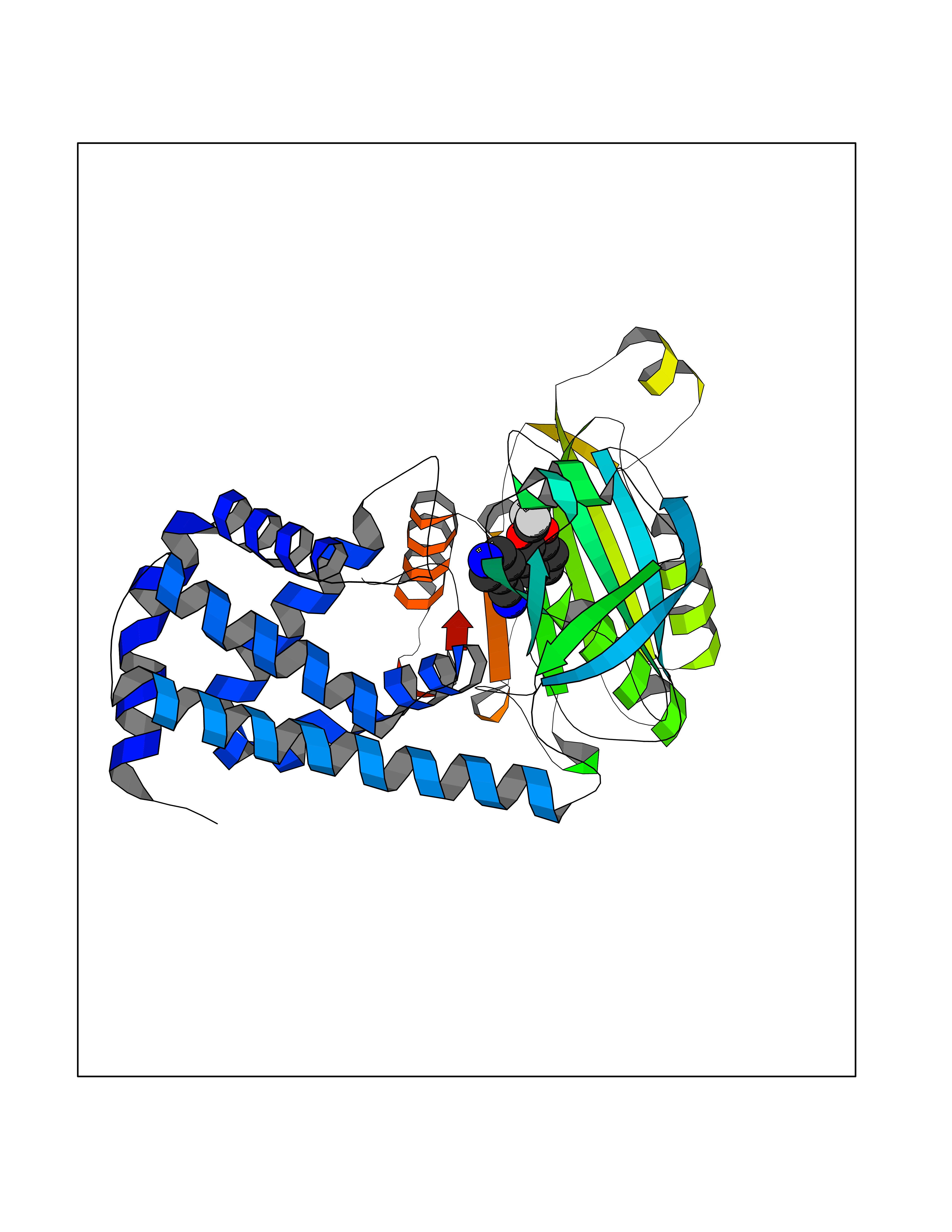 Molscript Map 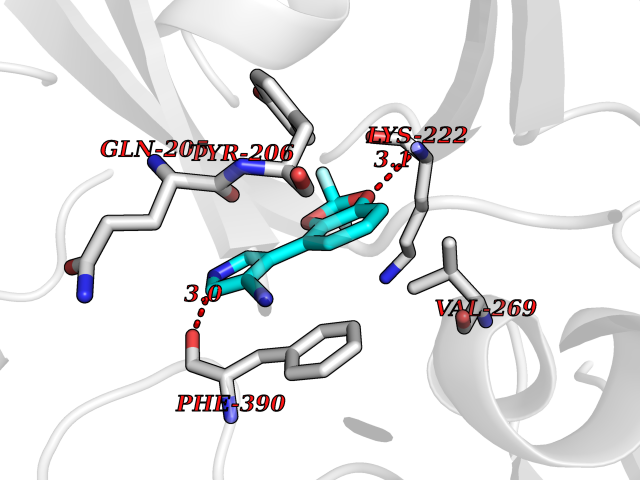 Pymol Map 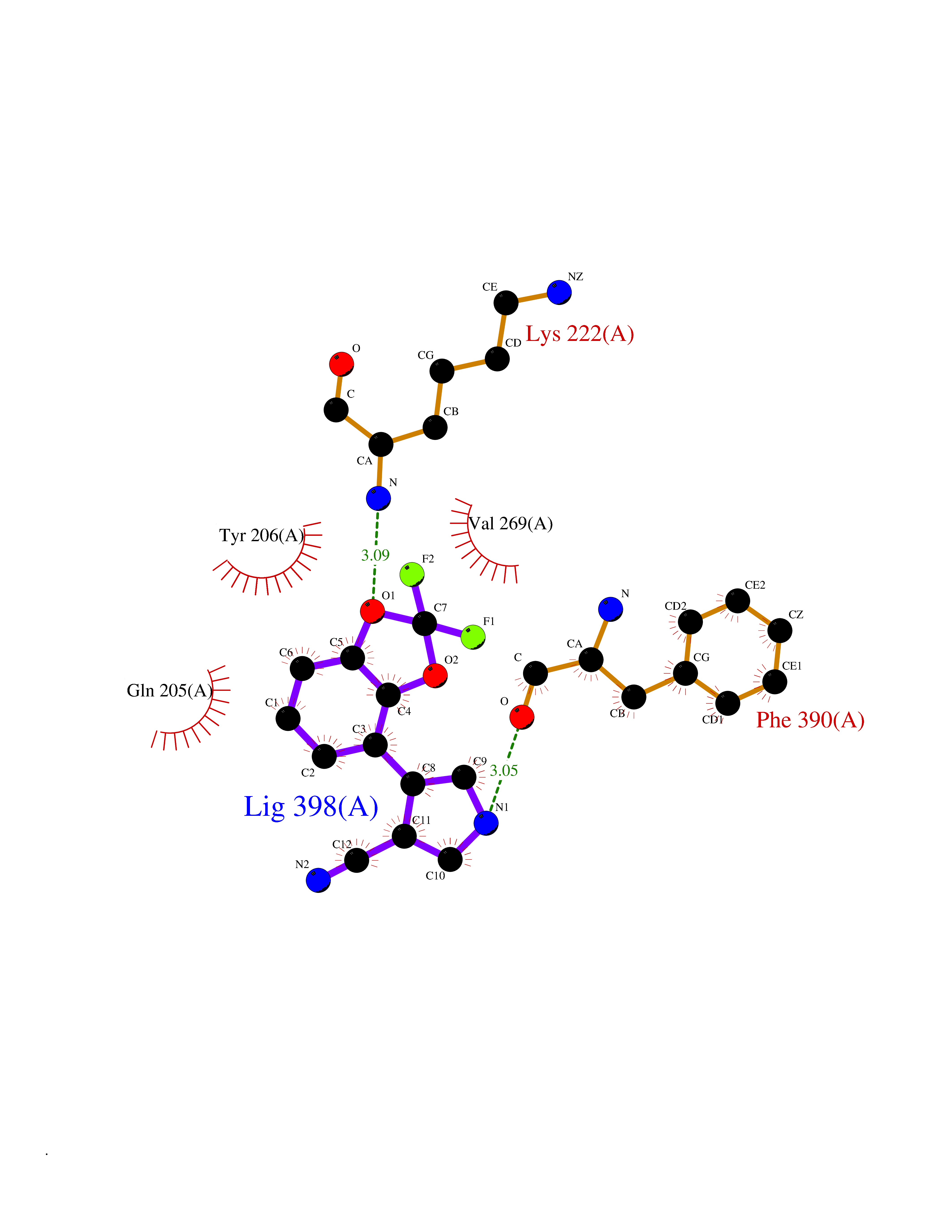 Ligplot Map 3D Binding mode Sequence MLDAQTIATVKATIPLLVETGPKLTAHFYDRMFTHNPELKEIFNMSNQRNGDQREALFNAIAAYASNIENLPALLPAVEKIAQKHTSFQIKPEQYNIVGEHLLATLDEMFSPGQEVLDAWGKAYGVLANVFINREAEIYNENASKAGGWEGTRDFRIVAKTPRSALITSFELEPVDGGAVAEYRPGQYLGVWLKPEGFPHQEIRQYSLTRKPDGKGYRIAVKREEGGQVSNWLHNHANVGDVVKLVAPAGDFFMAVADDTPVTLISAGVGQTPMLAMLDTLAKAGHTAQVNWFHAAENGDVHAFADEVKELGQSLPRFTAHTWYRQPSEADRAKGQFDSEGLMDLSKLEGAFSDPTMQFYLCGPVGFMQFTAKQLVDLGVKQENIHYECFGPHKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Cytochrome P450 1B1 (CYP1B1) | 3PM0 | 6.07 | |
Target general information Gen name CYP1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CYPIB1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, retinoid and xenobiotics. Preferentially oxidizes 17beta-estradiol to the carcinogenic 4-hydroxy derivative, and a variety of procarcinogenic compounds to their activated forms, including polycyclic aromatic hydrocarbons. Promotes angiogenesis by removing cellular oxygenation products, thereby decreasing oxidative stress, release of antiangiogenic factor THBS2, then allowing endothelial cells migration, cell adhesion and capillary morphogenesis. These changes are concommitant with the endothelial nitric oxide synthase activity and nitric oxide synthesis. Plays an important role in the regulation of perivascular cell proliferation, migration, and survival through modulation of the intracellular oxidative state and NF-kappa-B expression and/or activity, during angiogenesis. Contributes to oxidative homeostasis and ultrastructural organization and function of trabecular meshwork tissue through modulation of POSTN expression. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) DB02342; DB00613; DB06732; DB00443; DB00121; DB01222; DB00201; DB09061; DB14737; DB01254; DB00694; DB01248; DB00997; DB00470; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB07776; DB00499; DB01645; DB01381; DB00741; DB01064; DB01026; DB00448; DB14009; DB01065; DB00170; DB00959; DB01204; DB14011; DB03467; DB00338; DB01229; DB14631; DB00635; DB01087; DB00396; DB00818; DB04216; DB02709; DB00675; DB00624; DB13946; DB00277; DB12245; DB11155 Interacts with Q02763 EC number EC 1.14.14.- Uniprot keywords 3D-structure; Disease variant; Endoplasmic reticulum; Fatty acid metabolism; Glaucoma; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Mitochondrion; Monooxygenase; Oxidoreductase; Peters anomaly; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51875.9 Length 459 Aromaticity 0.1 Instability index 34.16 Isoelectric point 8.64 Charge (pH=7) 4.89 2D Binding mode Binding energy (Kcal/mol) -8.28 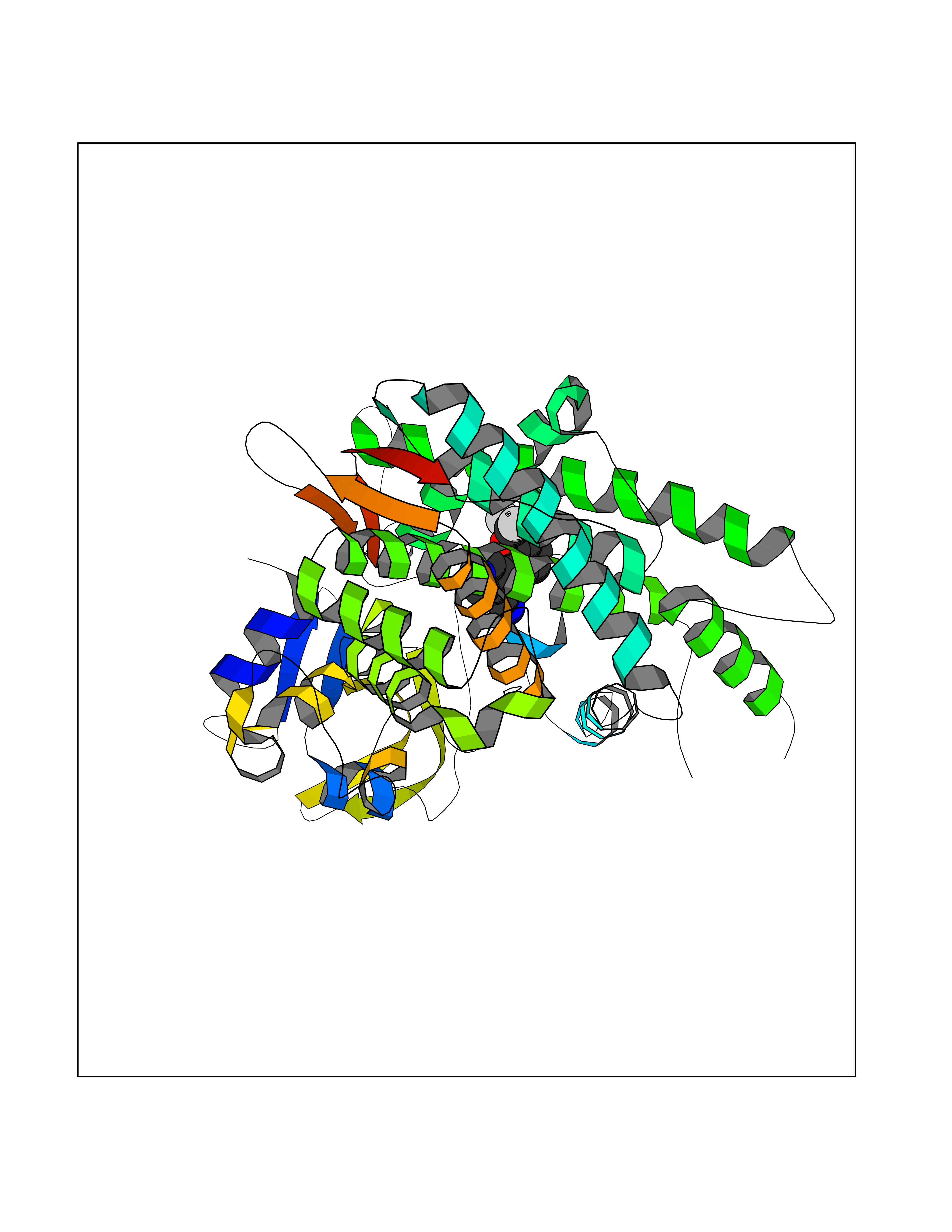 Molscript Map 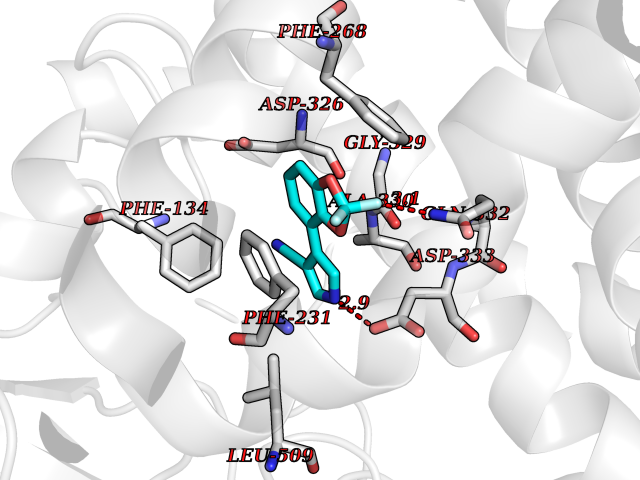 Pymol Map 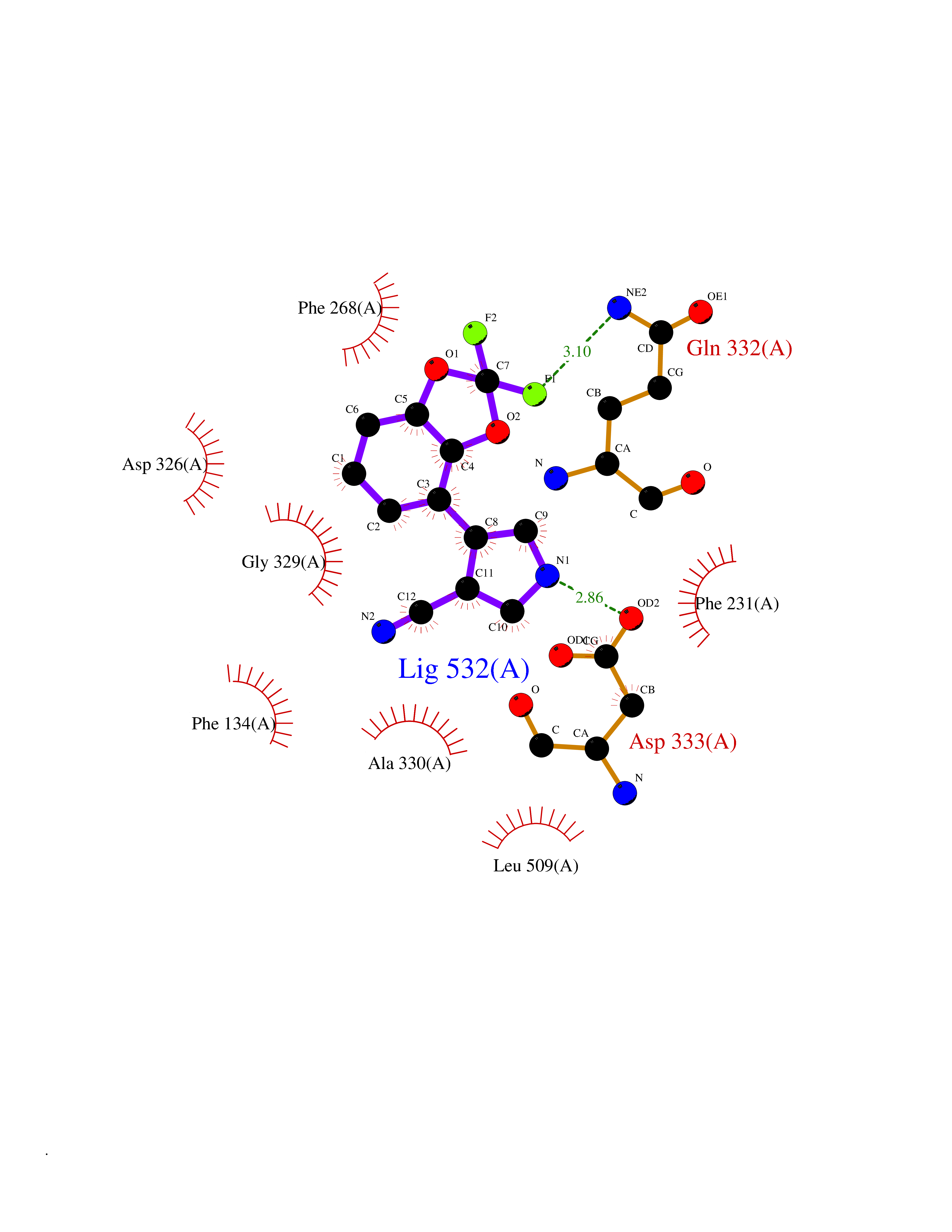 Ligplot Map 3D Binding mode Sequence QAAHLSFARLARRYGDVFQIRLGSCPIVVLNGERAIHQALVQQGSAFADRPSFASFRVVSGGRSMAFGHYSEHWKVQRRAAHSMMRNFFTRQPRSRQVLEGHVLSEARELVALLVRGSADGAFLDPRPLTVVAVANVMSAVCFGCRYSHDDPEFRELLSHNEEFGRTVGAGSLVDVMPWLQYFPNPVRTVFREFEQLNRNFSNFILDKFLRHCESLRPGAAPRDMMDAFILSAEKKAAGDGARLDLENVPATITDIFGASQDTLSTALQWLLLLFTRYPDVQTRVQAELDQVVGRDRLPCMGDQPNLPYVLAFLYEAMRFSSFVPVTIPHATTANTSVLGYHIPKDTVVFVNQWSVNHDPLKWPNPENFDPARFLDKDGLINKDLTSRVMIFSVGKRRCIGEELSKMQLFLFISILAHQCDFRANPNEPAKMNFSYGLTIKPKSFKVNVTLRESMELLD Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Cytochrome P450 1A2 | 2HI4 | 6.06 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -8.26  Molscript Map 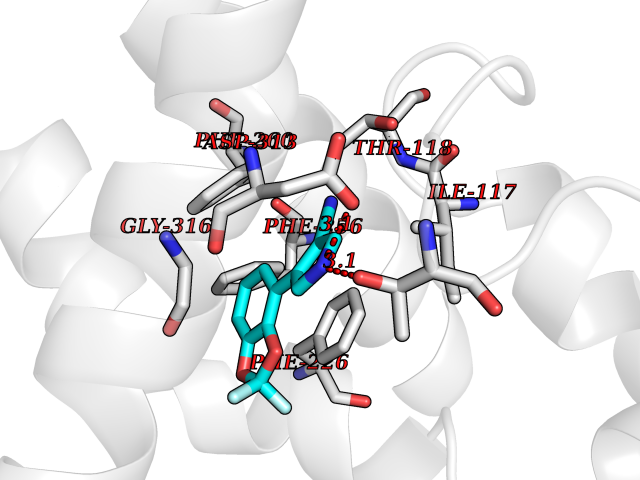 Pymol Map 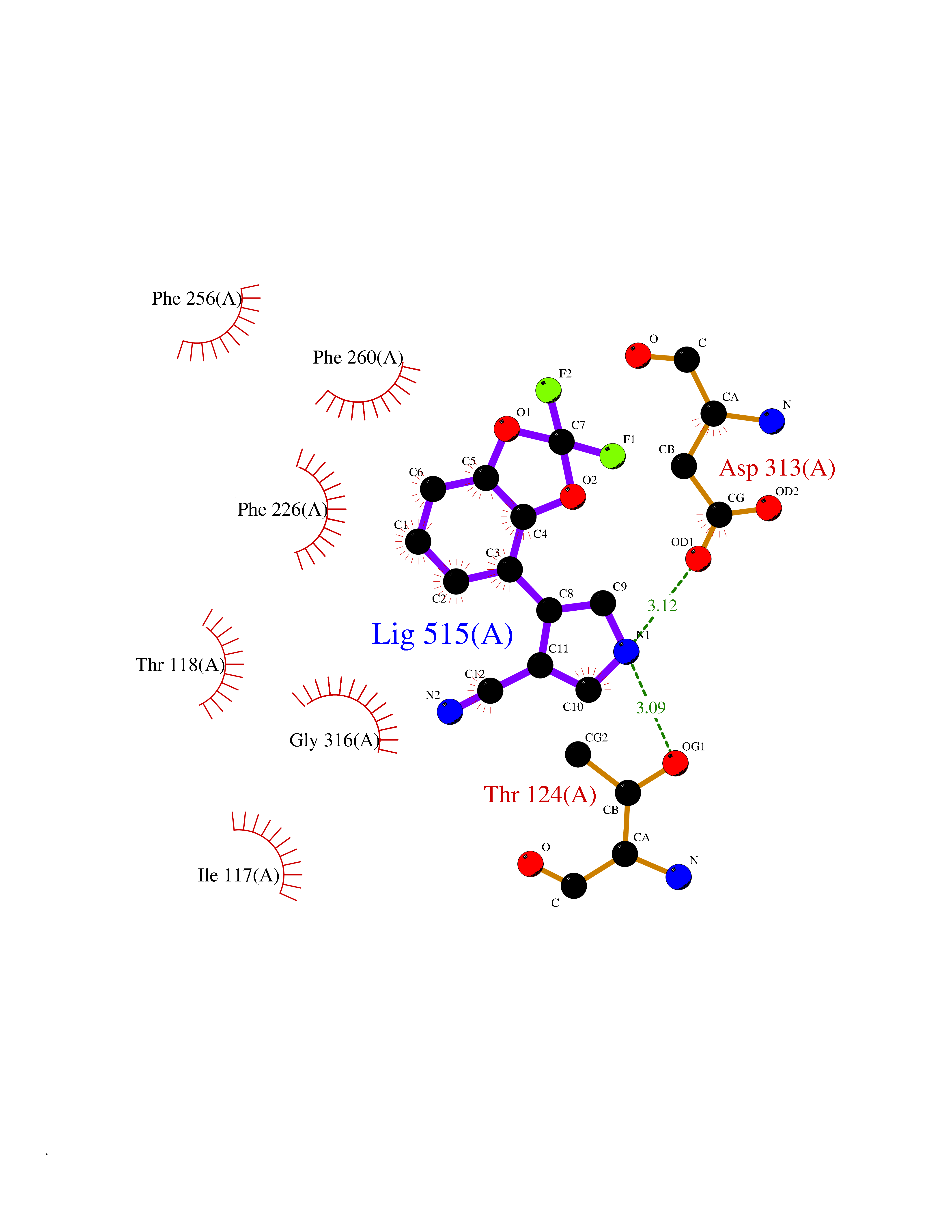 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Peptostreptococcal albumin-binding protein | 2VDB | 6.06 | |
Target general information Gen name pab Organism Finegoldia magna (Peptostreptococcus magnus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Protein binding Function Binds serum albumin. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03600; DB00788 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell wall; Peptidoglycan-anchor; Secreted; Signal Protein physicochemical properties Chain ID B Molecular weight (Da) 21751.6 Length 189 Aromaticity 0.1 Instability index 49.17 Isoelectric point 5.44 Charge (pH=7) -6.7 2D Binding mode Binding energy (Kcal/mol) -8.27  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVADESAENCDKSLHTLFGDKLCTVATLEMADCCAKQEPERNECFLQHKDDNPNLPRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELRDEGKASSAKQRLK Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Sodium/glucose cotransporter 1 (SGLT1) | 7WMV | 6.06 | |
Target general information Gen name SLC5A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 5 member 1; Na(+)/glucose cotransporter 1; NAGT; High affinity sodium-glucose cotransporter Protein family Sodium:solute symporter (SSF) (TC 2.A.21) family Biochemical class Solute:sodium symporter Function Efficient substrate transport in mammalian kidney is provided by the concerted action of a low affinity high capacity and a high affinity low capacity Na(+)/glucose cotransporter arranged in series along kidney proximal tubules. Actively transports glucose into cells by Na(+) cotransport with a Na(+) to glucose coupling ratio of 2:1. Related diseases Congenital glucose/galactose malabsorption (GGM) [MIM:606824]: Intestinal monosaccharide transporter deficiency. It is an autosomal recessive disorder manifesting itself within the first weeks of life. It is characterized by severe diarrhea and dehydration which are usually fatal unless glucose and galactose are eliminated from the diet. {ECO:0000269|PubMed:10036327, ECO:0000269|PubMed:11406349, ECO:0000269|PubMed:2008213, ECO:0000269|PubMed:8195156, ECO:0000269|PubMed:8563765}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00766; DB01914; DB09341; DB05018; DB12713 Interacts with P00533 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Sodium; Sodium transport; Sugar transport; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 66451.3 Length 602 Aromaticity 0.12 Instability index 35.84 Isoelectric point 8.3 Charge (pH=7) 4.43 2D Binding mode Binding energy (Kcal/mol) -8.26  Molscript Map 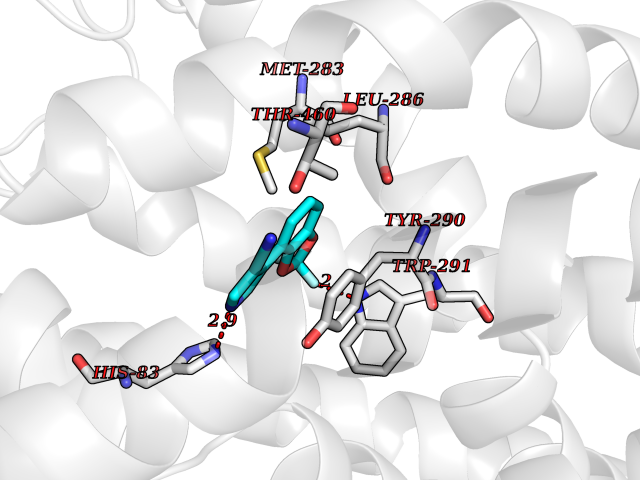 Pymol Map 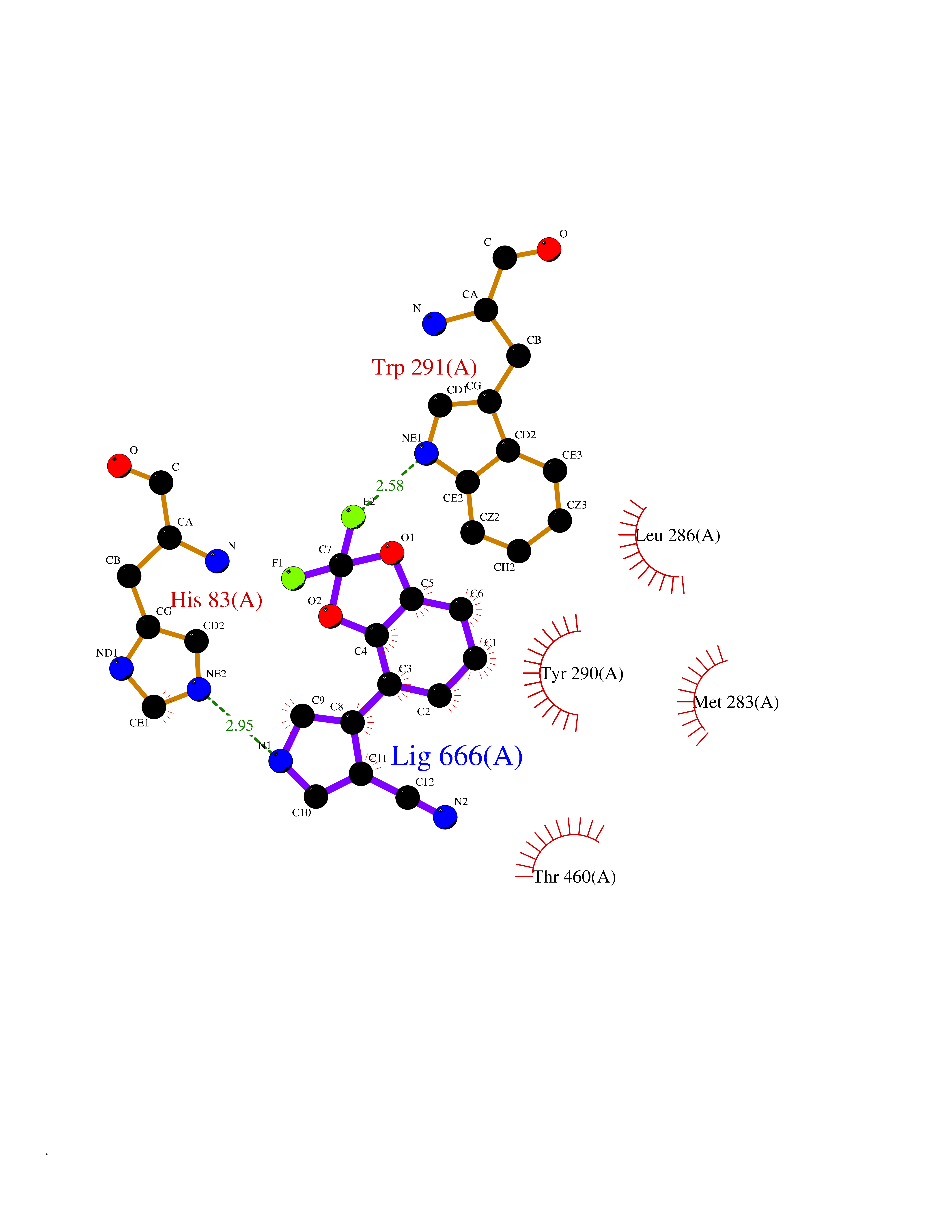 Ligplot Map 3D Binding mode Sequence ETHELIRNAADISIIVIYFVVVMAVGLWAMFSTNRGTVGGFFLAGRSMVWWPIGASLFASNIGSGHFVGLAGTGAASGIAIGGFEWNALVLVVVLGWLFVPIYIKAGVVTMPEYLRKRFGGQRIQVYLSLLSLLLYIFTKISADIFSGAIFINLALGLNLYLAIFLLLAITALYTITGGLAAVIYTDTLQTVIMLVGSLILTGFAFHEVGGYDAFMEKYMKAIPTIVSDGNTTFQEKCYTPRADSFHIFRDPLTGDLPWPGFIFGMSILTLWYWCTDQVIVQRCLSAKNMSHVKGGCILCGYLKLMPMFIMVMPGMISRILYTEKIACVVPSECEKYCGTKVGCTNIAYPTLVVELMPNGLRGLMLSVMLASLMSSLTSIFNSASTLFTMDIYAKVRKRASEKELMIAGRLFILVLIGISIAWVPIVQSAQSGQLFDYIQSITSYLGPPIAAVFLLAIFWKRVNEPGAFWGLILGLLIGISRMITEFAYGTGSCMEPSNCPTIICGVHYLYFAIILFAISFITIVVISLLTKPIPDVHLYRLCWSLRNSKEERIDLDATEEEEKAMKMKMTDTSEKPLWRTVLNVNGIILVTVAVFCHAYFA Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Flavodoxin/ferredoxin--NADP reductase | 1FDR | 6.05 | |
Target general information Gen name fpr Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms mvrA;b3924;JW3895 Protein family Ferredoxin--NADP reductase type 1 family Biochemical class Flavoprotein Function FAD binding.Ferredoxin-NADP+ reductase activity.Oxidoreductase activity. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.18.1.2; 1.19.1.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; NADP; Nucleotide-binding; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 27346.2 Length 244 Aromaticity 0.08 Instability index 30.68 Isoelectric point 7.25 Charge (pH=7) 0.42 2D Binding mode Binding energy (Kcal/mol) -8.26  Molscript Map 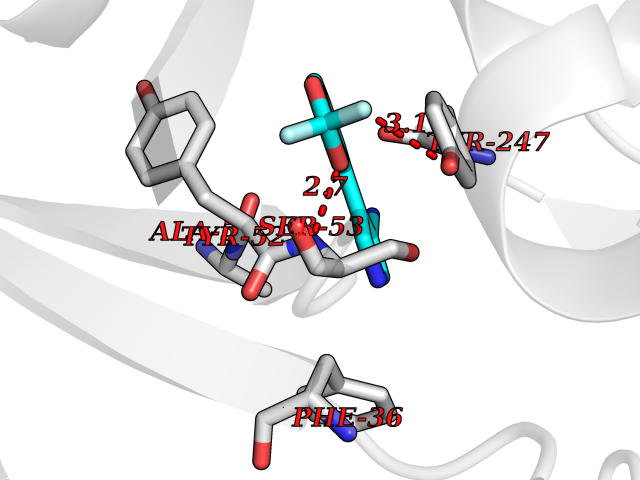 Pymol Map 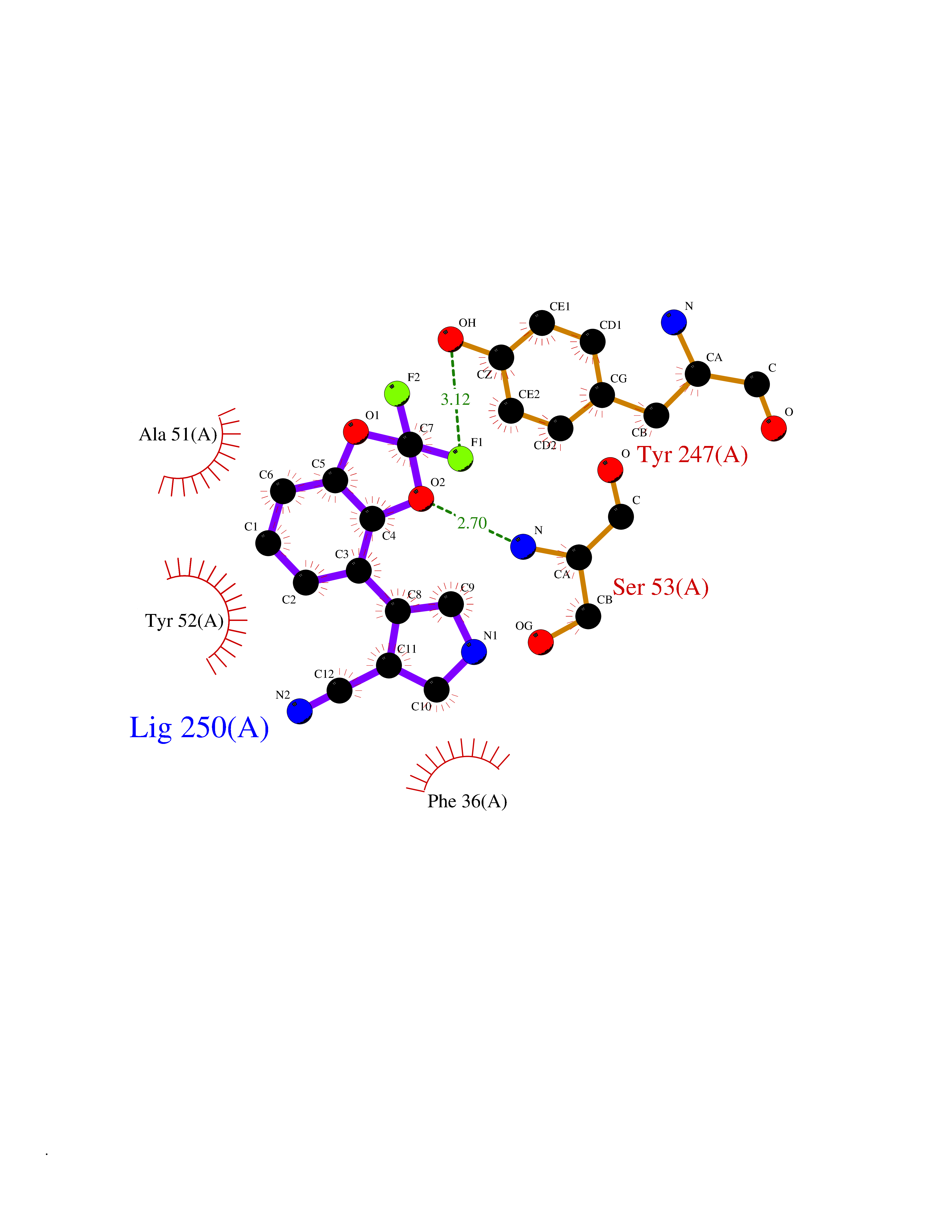 Ligplot Map 3D Binding mode Sequence ADWVTGKVTKVQNWTDALFSLTVHAPVLPFTAGQFTKLGLEIRVQRAYSYVNSPDNPDLEFYLVTVPDGKLSPRLAALKPGDEVQVVSEAAGFFVLDEVPHCETLWMLATGTAIGPYLSILRLGKDLDRFKNLVLVHAARYAADLSYLPLMQELEKRYEGKLRIQTVVSRETAAGSLTGRIPALIESGELESTIGLPMNKETSHVMLCGNPQMVRDTQQLLKETRQMTKHLRRRPGHMTAEHYW Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Caspase-7 (CASP7) | 1SHJ | 6.05 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -8.25  Molscript Map 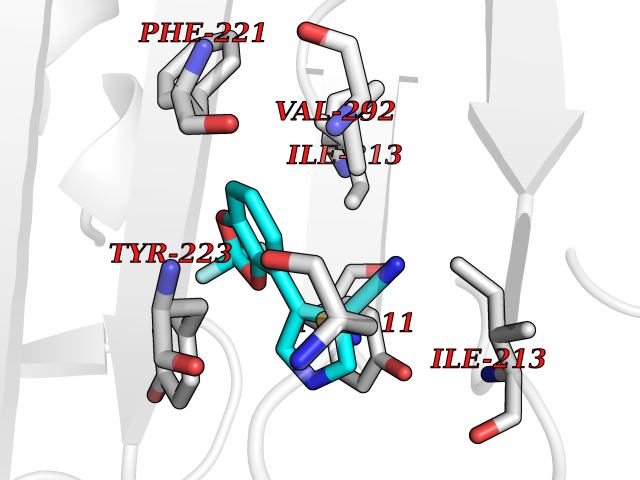 Pymol Map 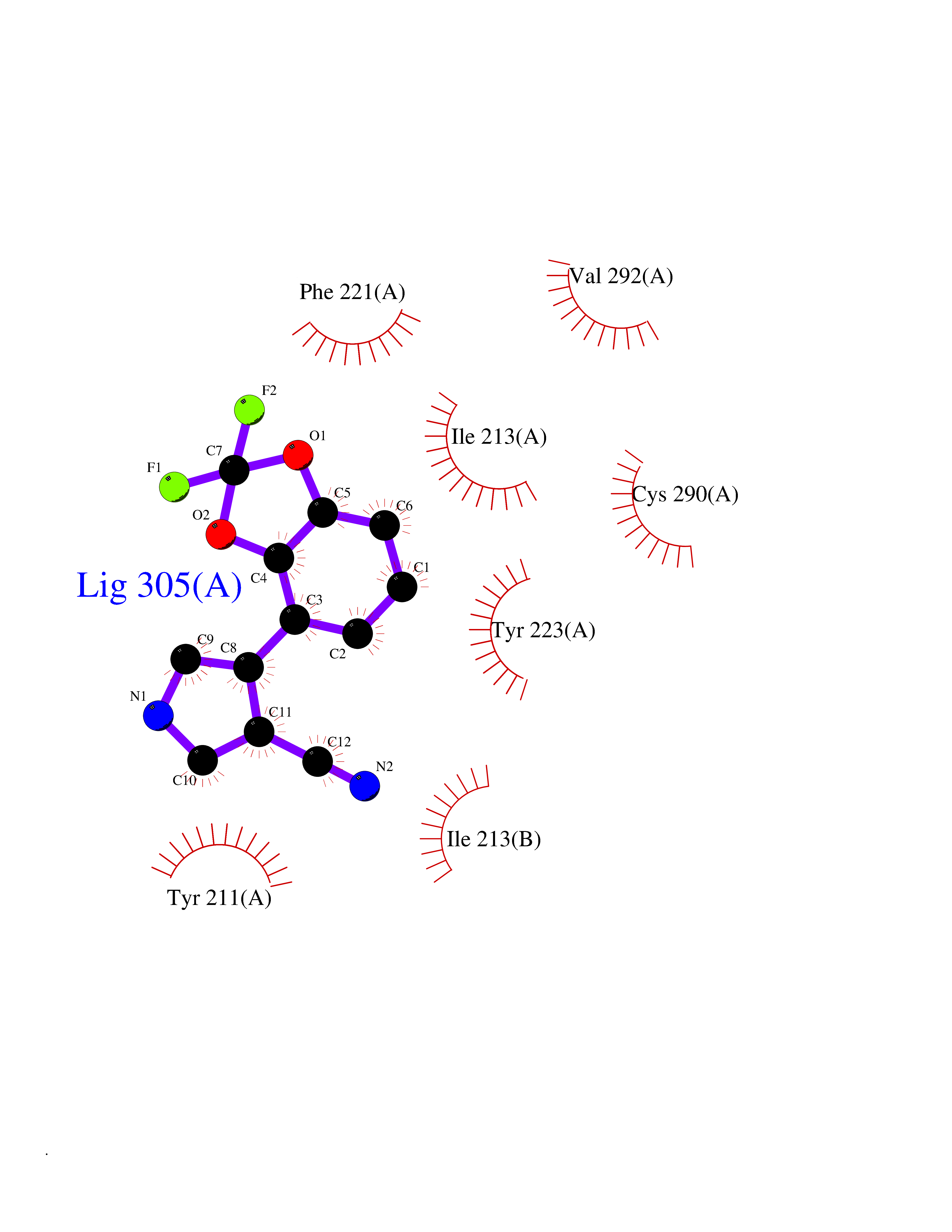 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Monoglyceride lipase (MAGL) | 3PE6 | 6.04 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -8.25 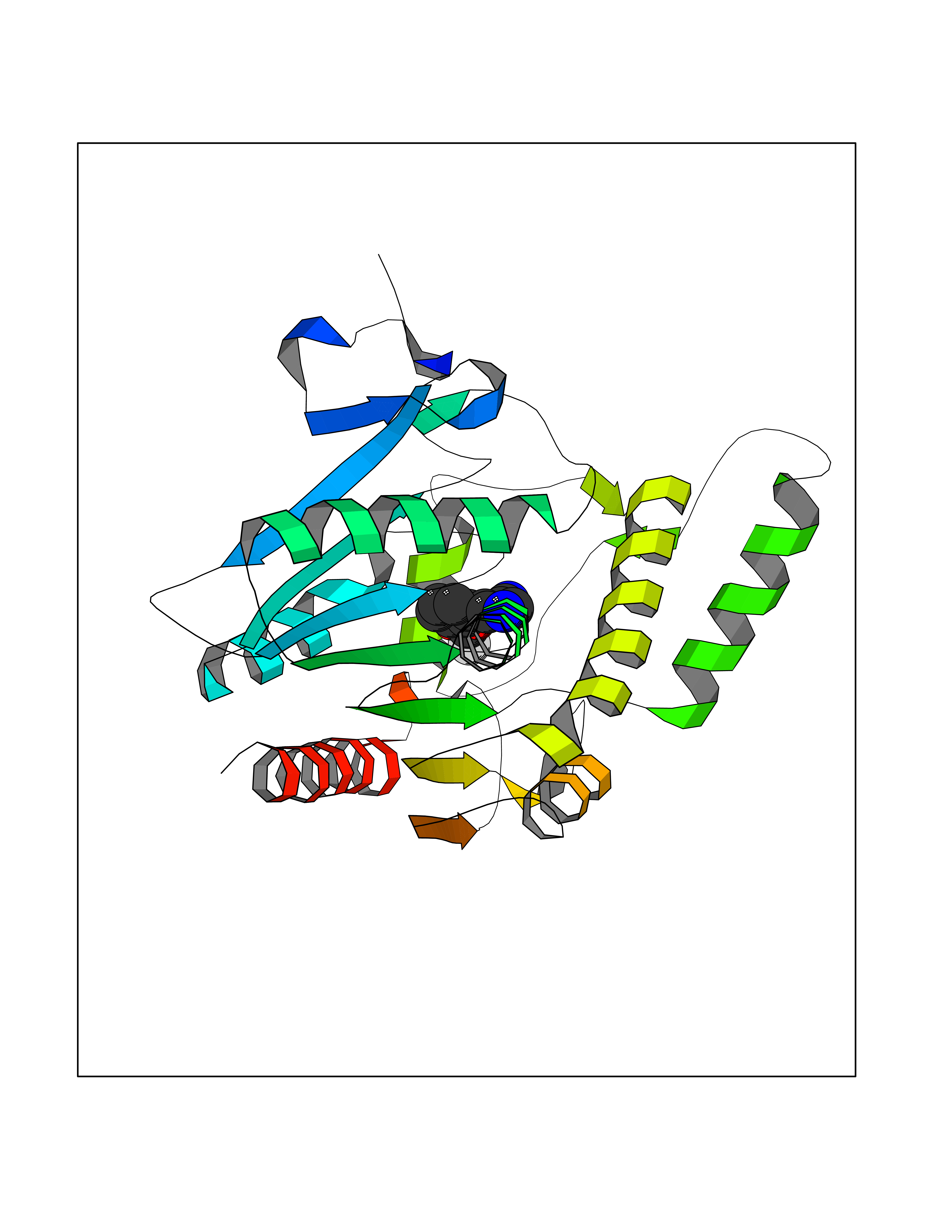 Molscript Map 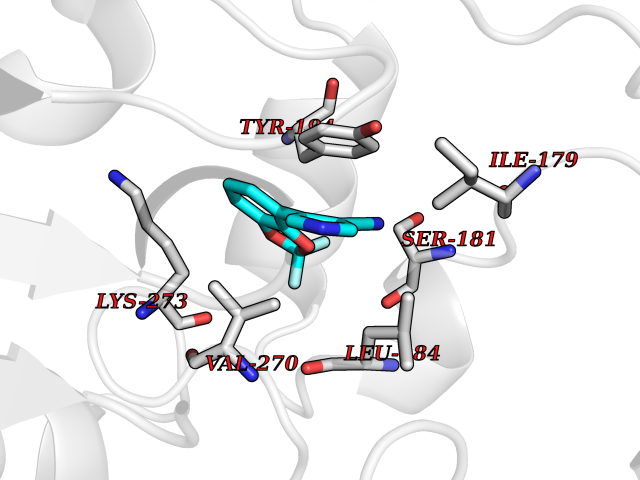 Pymol Map 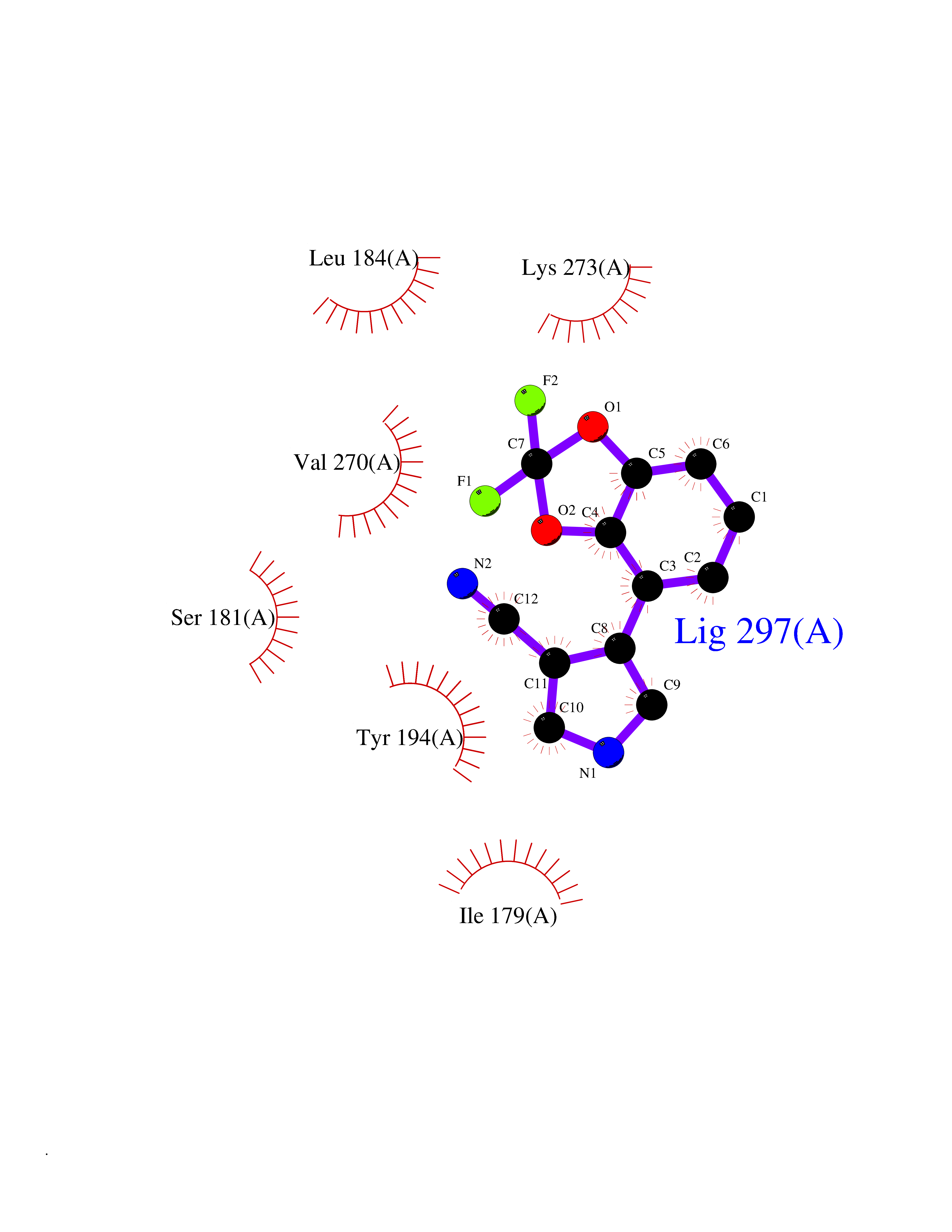 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Fatty acid synthase (FASN) | 3TJM | 6.03 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -8.23  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 6.03 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -8.22  Molscript Map 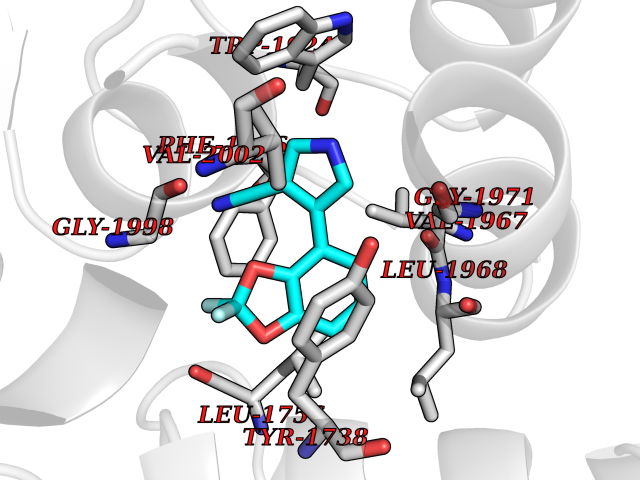 Pymol Map 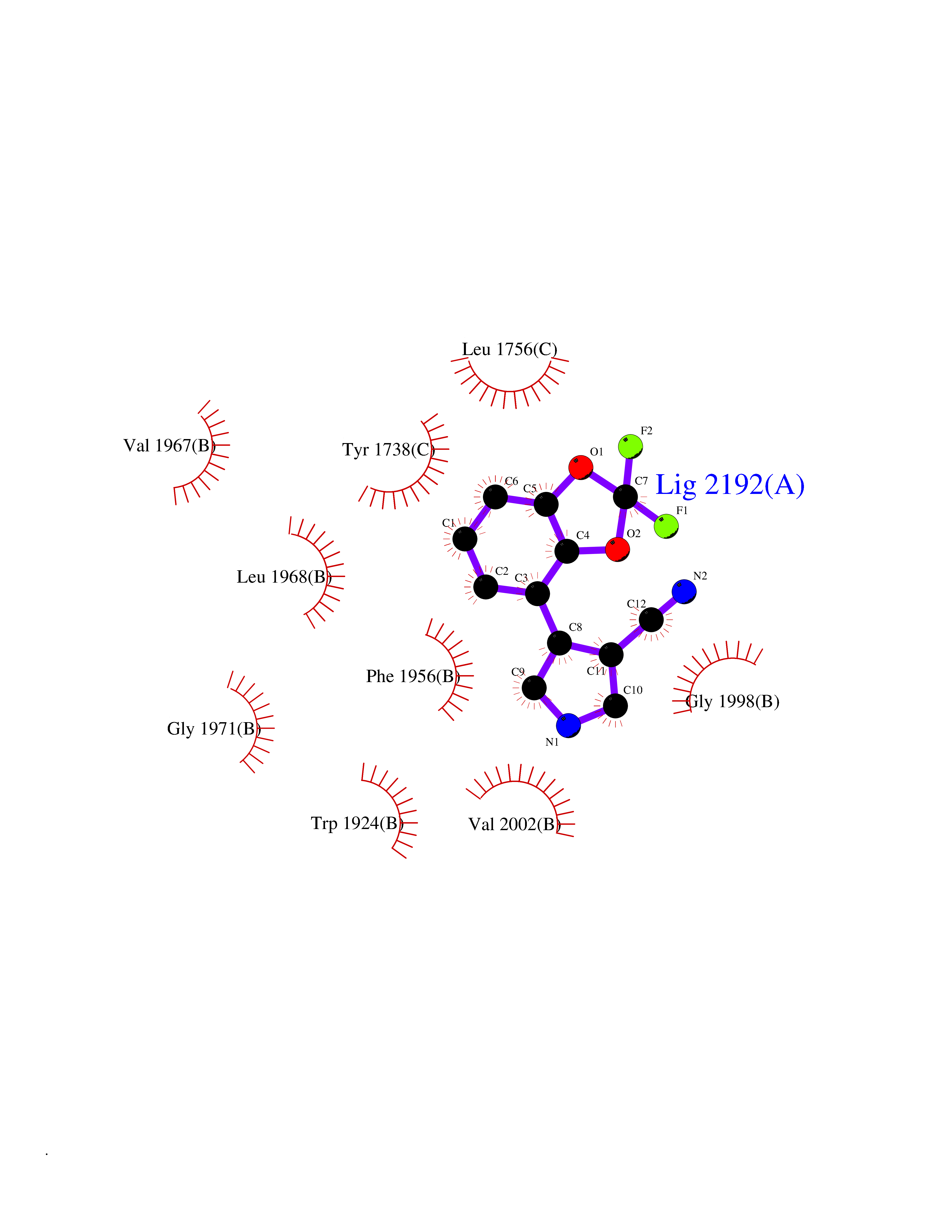 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Aldehyde oxidoreductase | 4USA | 6.03 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -8.23  Molscript Map 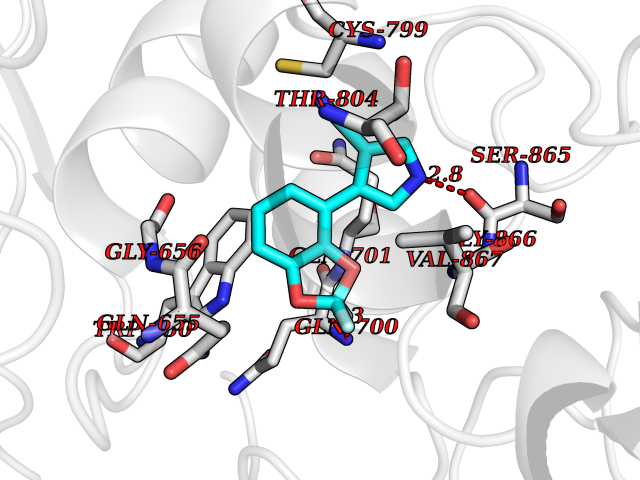 Pymol Map 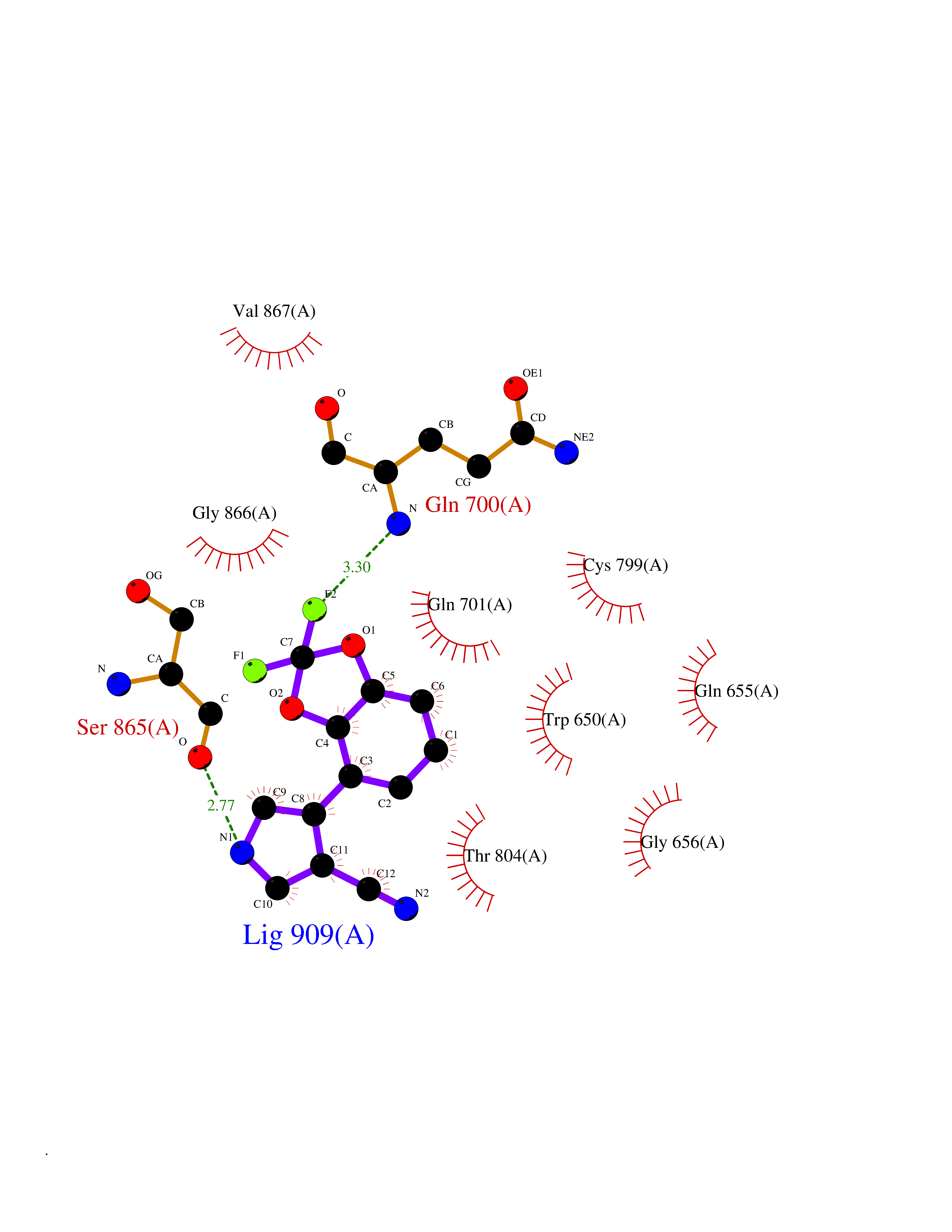 Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | BUB1 mitotic checkpoint serine/threonine kinase (BUB1) | 6F7B | 6.03 | |
Target general information Gen name BUB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hBUB1; Mitotic checkpoint serine/threonine-protein kinase BUB1; BUB1L; BUB1A Protein family Protein kinase superfamily, Ser/Thr protein kinase family, BUB1 subfamily Biochemical class Kinase Function Has a key role in the assembly of checkpoint proteins at the kinetochore, being required for the subsequent localization of CENPF, BUB1B, CENPE and MAD2L1. Required for the kinetochore localization of PLK1. Required for centromeric enrichment of AUKRB in prometaphase. Plays an important role in defining SGO1 localization and thereby affects sister chromatid cohesion. Acts as a substrate for anaphase-promoting complex or cyclosome (APC/C) in complex with its activator CDH1 (APC/C-Cdh1). Necessary for ensuring proper chromosome segregation and binding to BUB3 is essential for this function. Can regulate chromosome segregation in a kinetochore-independent manner. Can phosphorylate BUB3. The BUB1-BUB3 complex plays a role in the inhibition of APC/C when spindle-assembly checkpoint is activated and inhibits the ubiquitin ligase activity of APC/C by phosphorylating its activator CDC20. This complex can also phosphorylate MAD1L1. Kinase activity is essential for inhibition of APC/CCDC20 and for chromosome alignment but does not play a major role in the spindle-assembly checkpoint activity. Mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. Serine/threonine-protein kinase that performs 2 crucial functions during mitosis: it is essential for spindle-assembly checkpoint signaling and for correct chromosome alignment. Related diseases Microcephaly 30, primary, autosomal recessive (MCPH30) [MIM:620183]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age, sex and ethnically matched mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. MCPH30 is characterized by small head, poor overall growth, and global developmental delay with variably impaired intellectual development. Affected individuals may also have variable congenital anomalies, including atrial septal defect, dysmorphic facial features, tracheal stenosis, and anomalies of the skin and teeth. {ECO:0000269|PubMed:35044816}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95376; O60566; O43684; P46108; Q8NG31; Q8NG31-2; Q9GZQ8; P03070 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell division; Centromere; Chromosome; Chromosome partition; Host-virus interaction; Intellectual disability; Kinase; Kinetochore; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39564.8 Length 343 Aromaticity 0.12 Instability index 31.17 Isoelectric point 8.49 Charge (pH=7) 4.7 2D Binding mode Binding energy (Kcal/mol) -8.22 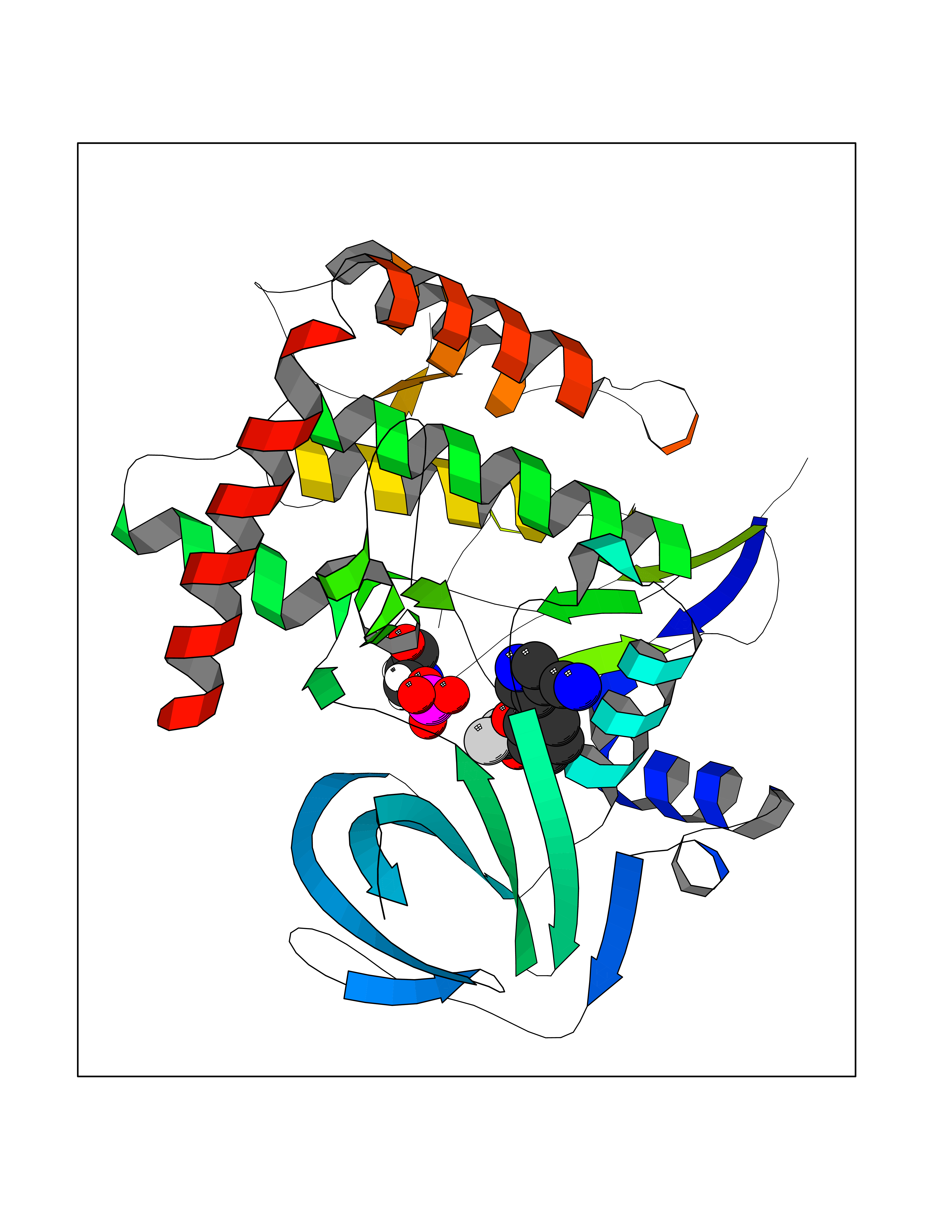 Molscript Map 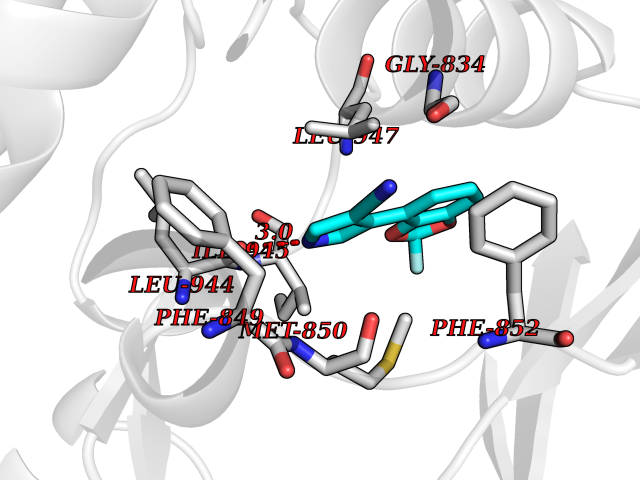 Pymol Map 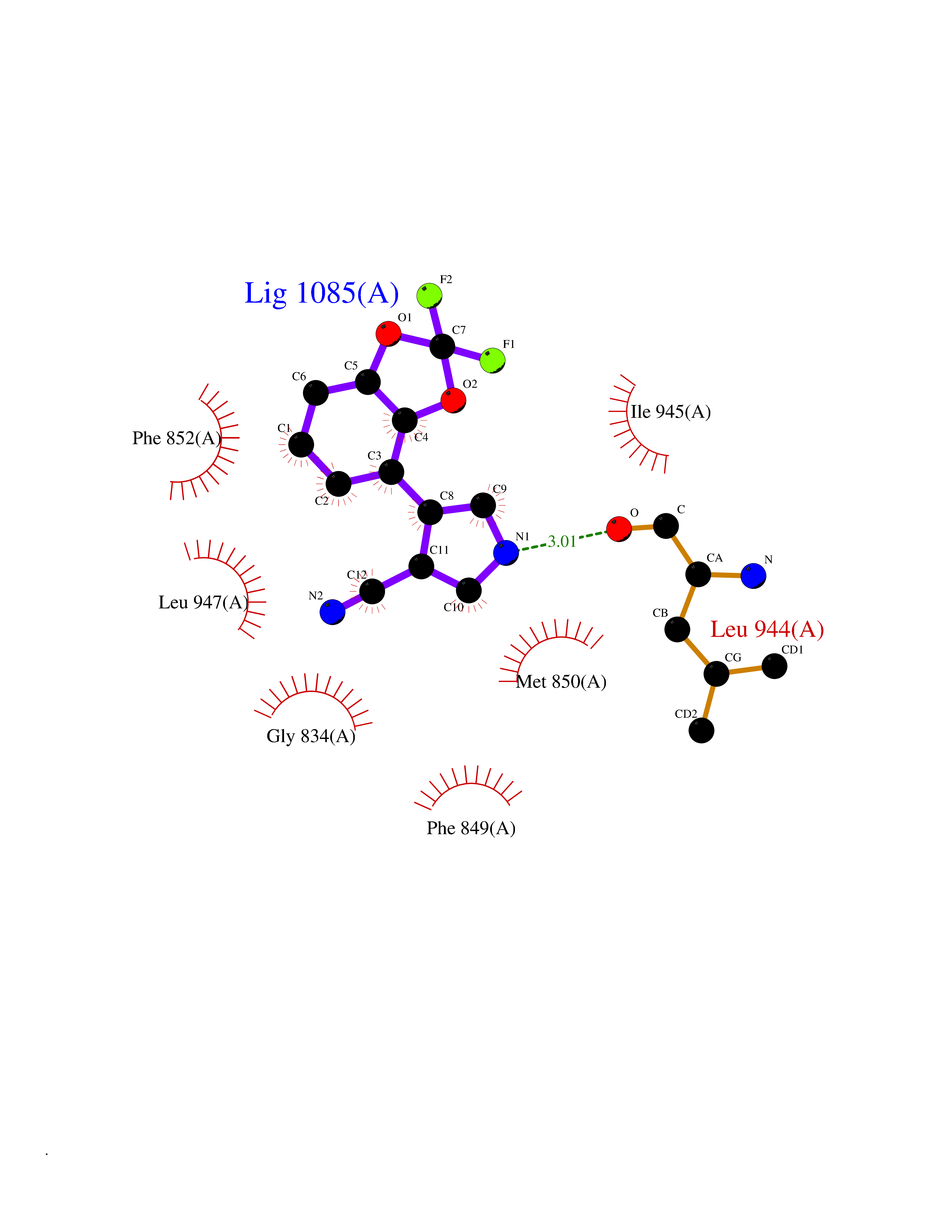 Ligplot Map 3D Binding mode Sequence APNFIVGNPWDDKLIFKLLSGLSKPVSSYPNTFEWQCKLPAIKPKTEFQLGSKLVYVHHLLGEGAFAQVYEATQKNKQKFVLKVQKPANPWEFYIGTQLMERLKPSMQHMFMKFYSAHLFQNGSVLVGELYSYGTLLNAINLYKNTPEKVMPQGLVISFAMRMLYMIEQVHDCEIIHGDIKPDNFILGNGFLEQDDEDDLSAGLALIDLGQSIDMKLFPKGTIFTAKCETXGFQCVEMLSNKPWNYQIDYFGVAATVYCMLFGTYMKVKNEECKPEGLFRRLPHLDMWNEFFHVMLNIPDCHHLPSLDLLRQKLKKVFQQHYTNKIRALRNRLIVLLLECKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 6.03 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -8.22  Molscript Map 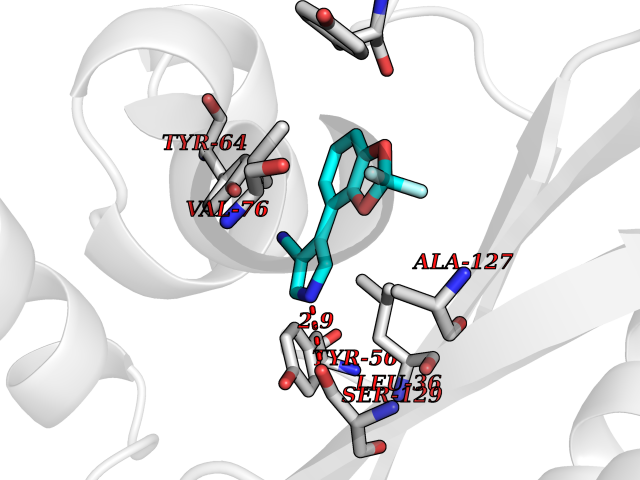 Pymol Map 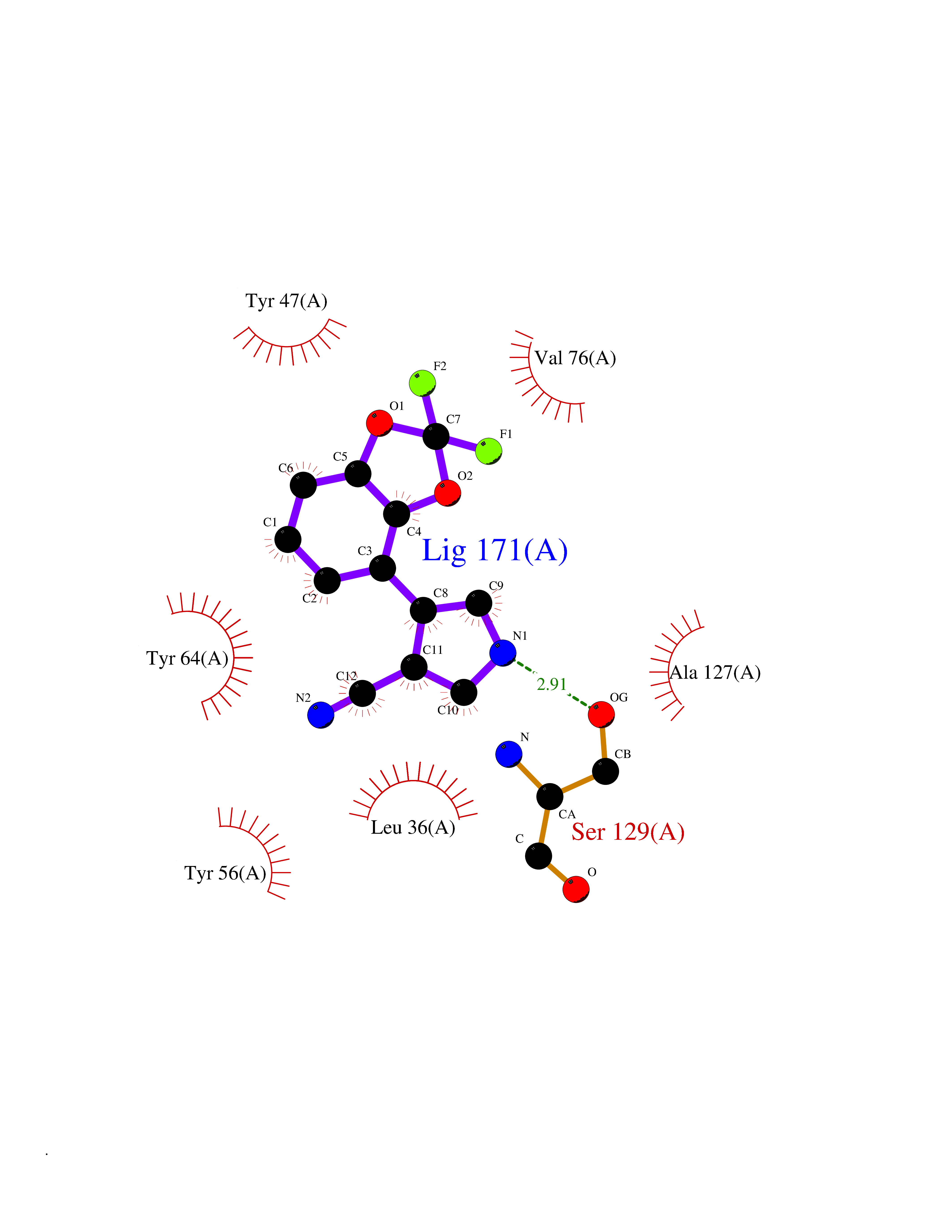 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Hyperpolarization cyclic nucleotide-gated channel 2 (HCN2) | 3U10 | 6.03 | |
Target general information Gen name HCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 2; Brain cyclic nucleotide-gated channel 2; BCNG2; BCNG-2 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) and in neurons (Ih). Can also transport ammonium in the distal nephron. Produces a large instantaneous current. Modulated by intracellular chloride ions and pH; acidic pH shifts the activation to more negative voltages. Hyperpolarization-activated ion channel exhibiting weak selectivity for potassium over sodium ions. Related diseases Epilepsy, idiopathic generalized 17 (EIG17) [MIM:602477]: A form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. Both autosomal dominant and autosomal recessive EIG17 inheritance have been reported. {ECO:0000269|PubMed:22131395, ECO:0000269|PubMed:29064616}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Febrile seizures, familial, 2 (FEB2) [MIM:602477]: Seizures associated with febrile episodes in childhood without any evidence of intracranial infection or defined pathologic or traumatic cause. It is a common condition, affecting 2-5% of children aged 3 months to 5 years. The majority are simple febrile seizures (generally defined as generalized onset, single seizures with a duration of less than 30 minutes). Complex febrile seizures are characterized by focal onset, duration greater than 30 minutes, and/or more than one seizure in a 24 hour period. The likelihood of developing epilepsy following simple febrile seizures is low. Complex febrile seizures are associated with a moderately increased incidence of epilepsy. FEB2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:24324597}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02527; DB02315; DB09083 Interacts with Q9UL51; Q4ACU6-1 EC number NA Uniprot keywords 3D-structure; Ammonia transport; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Palmitate; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23672.9 Length 202 Aromaticity 0.12 Instability index 38.05 Isoelectric point 8.85 Charge (pH=7) 4.11 2D Binding mode Binding energy (Kcal/mol) -8.22  Molscript Map 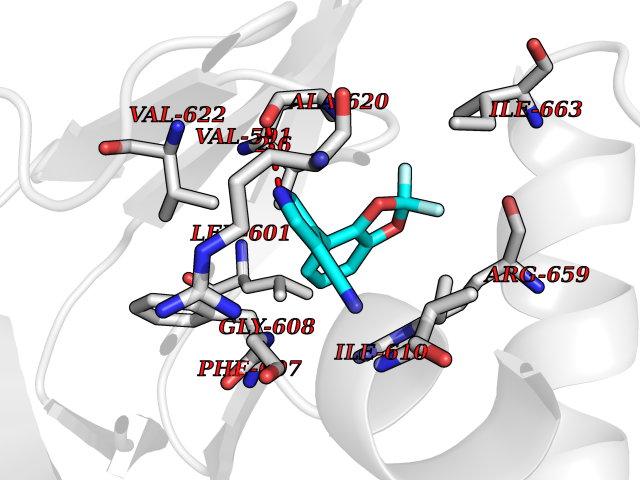 Pymol Map 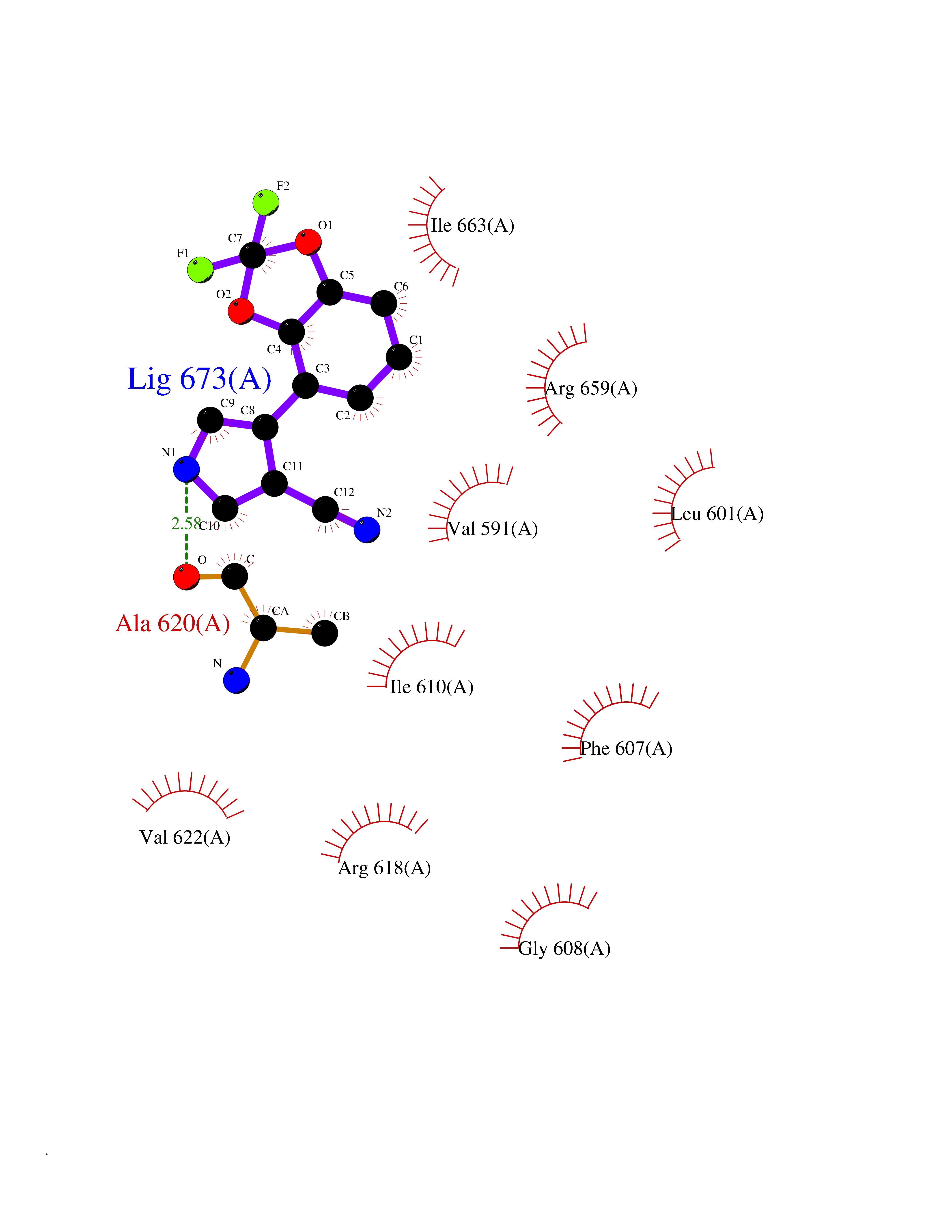 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPADFRQKIHDYYEHRYQGKMFDEDSILGELNGPLREEIVNFNCRKLVASMPLFANADPNFVTAMLTKLKFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKEMKLSDGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVAIDRLDRIGKKNSILL Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 6.03 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -8.22 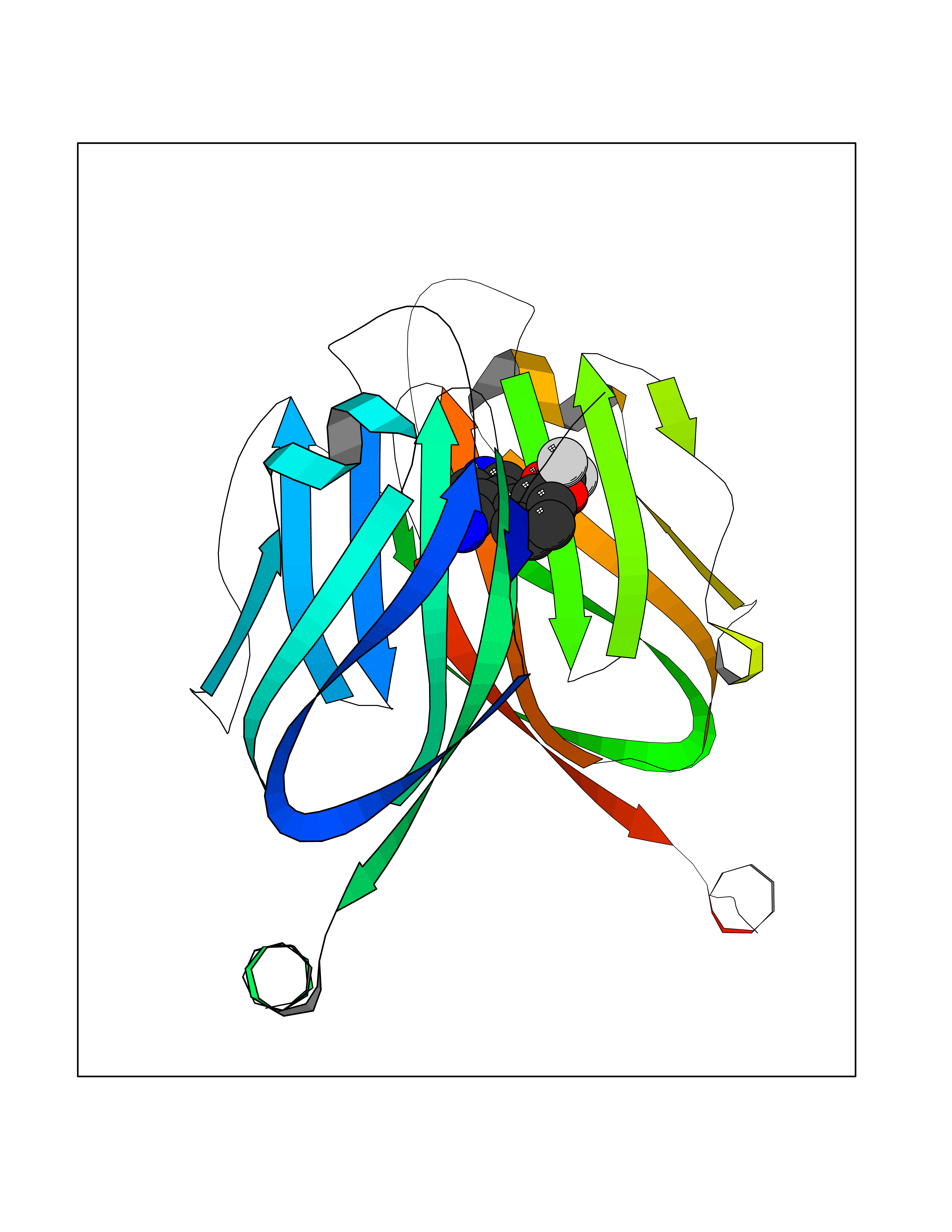 Molscript Map 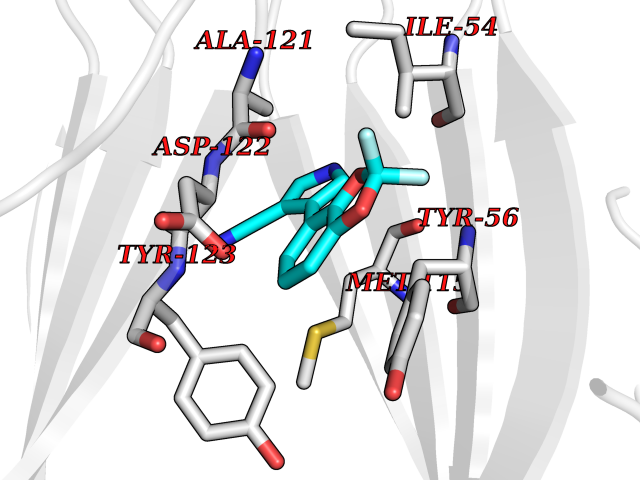 Pymol Map 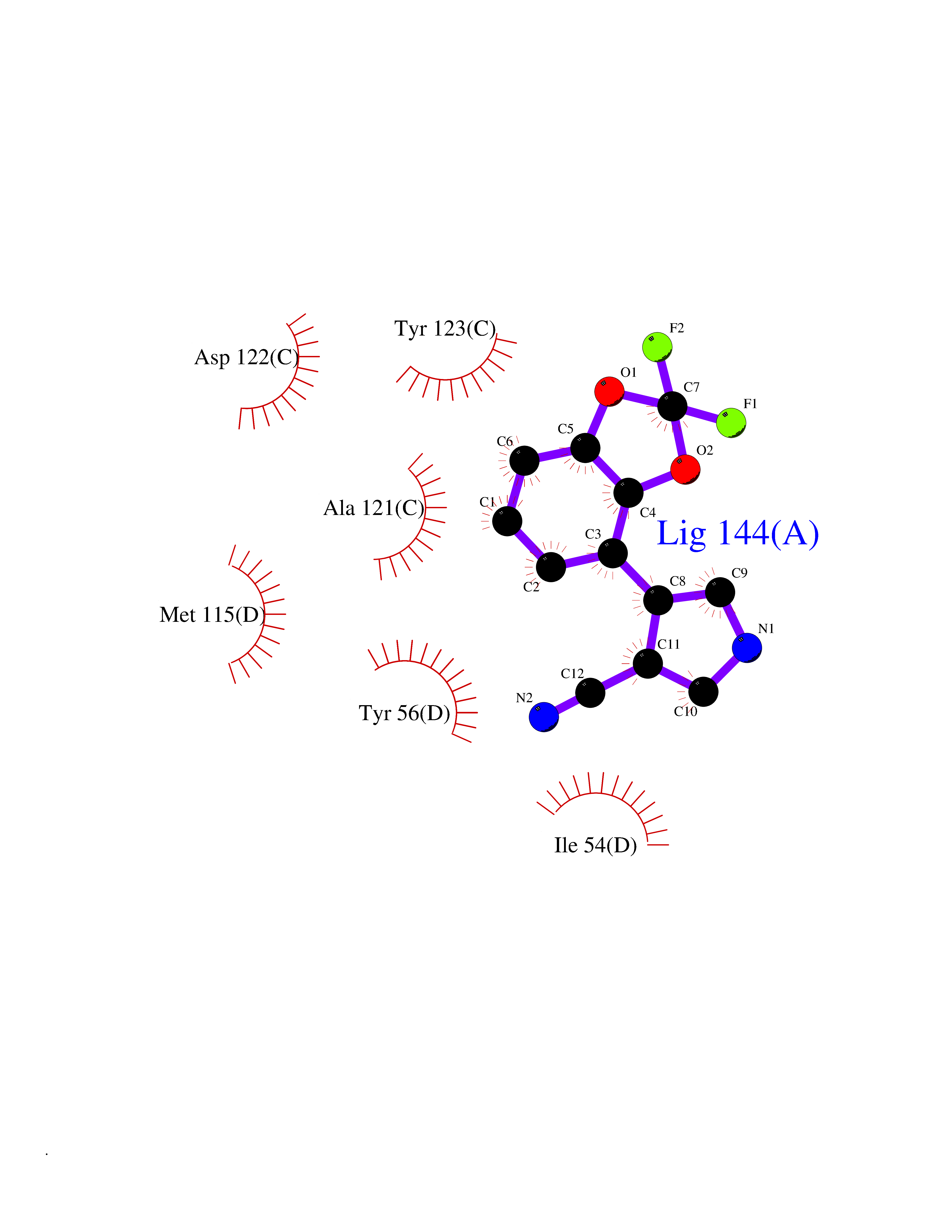 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 6.02 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -8.21  Molscript Map 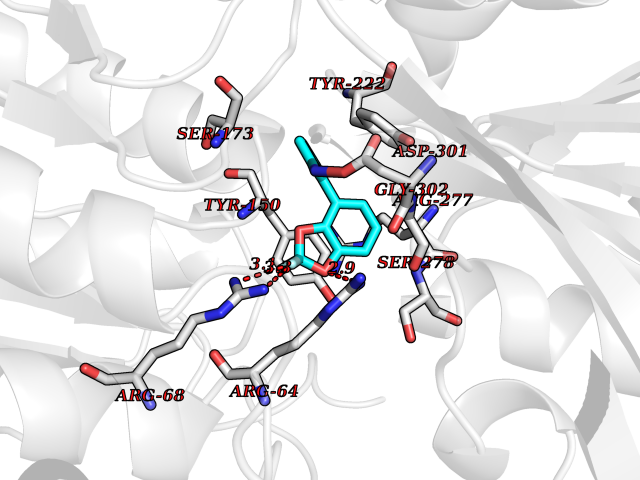 Pymol Map 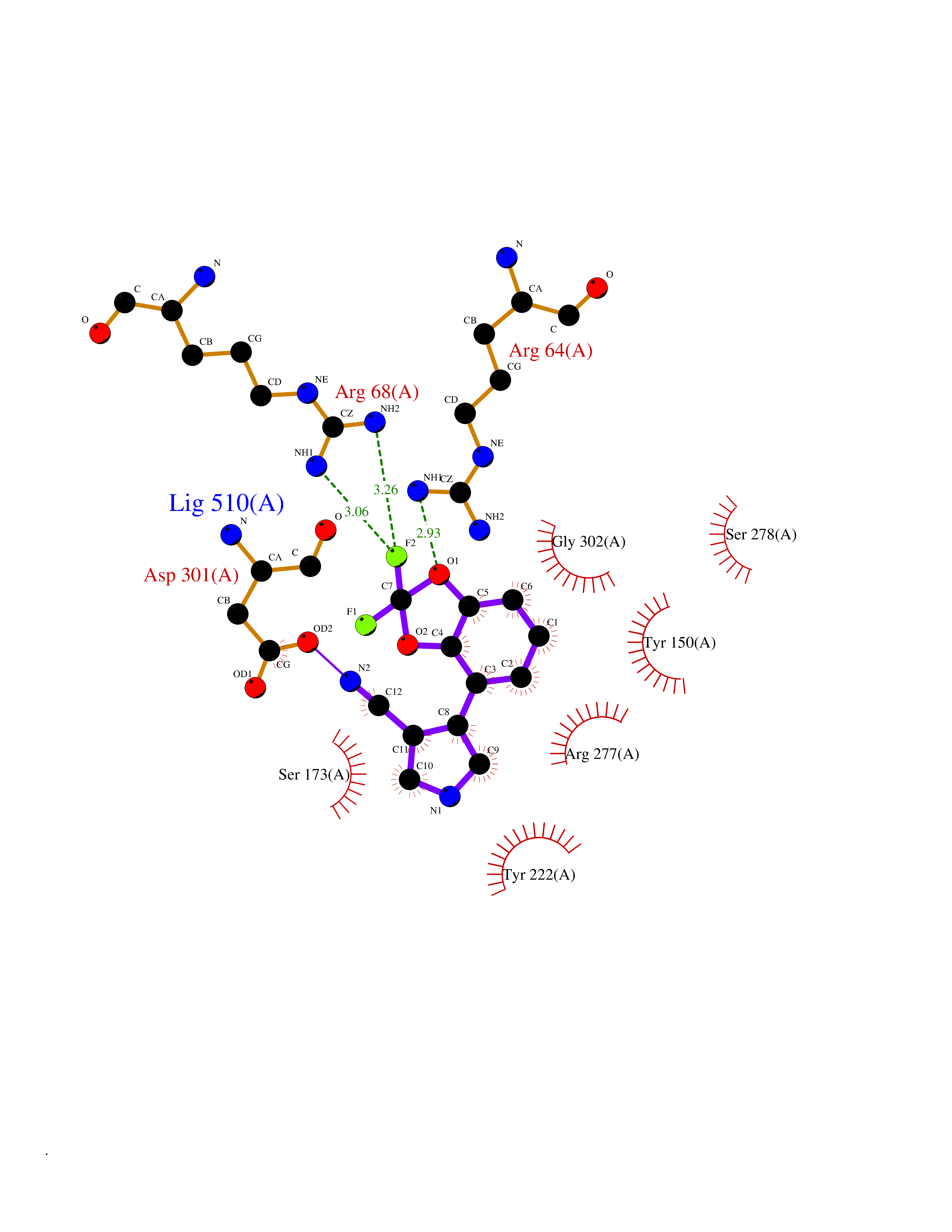 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Leucine carboxyl methyltransferase 1 | 3IEI | 6.01 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -8.2  Molscript Map 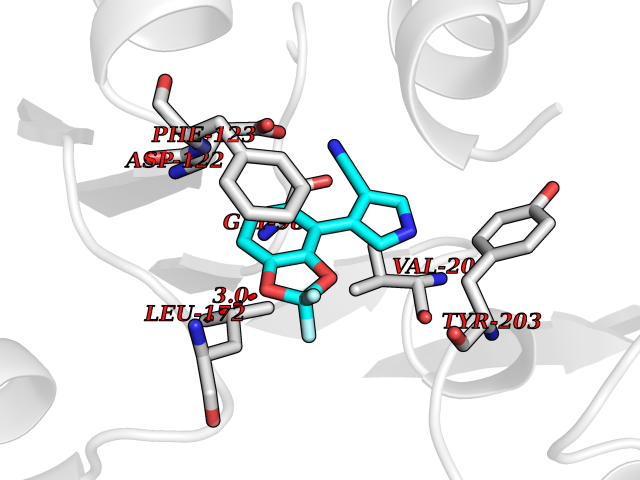 Pymol Map 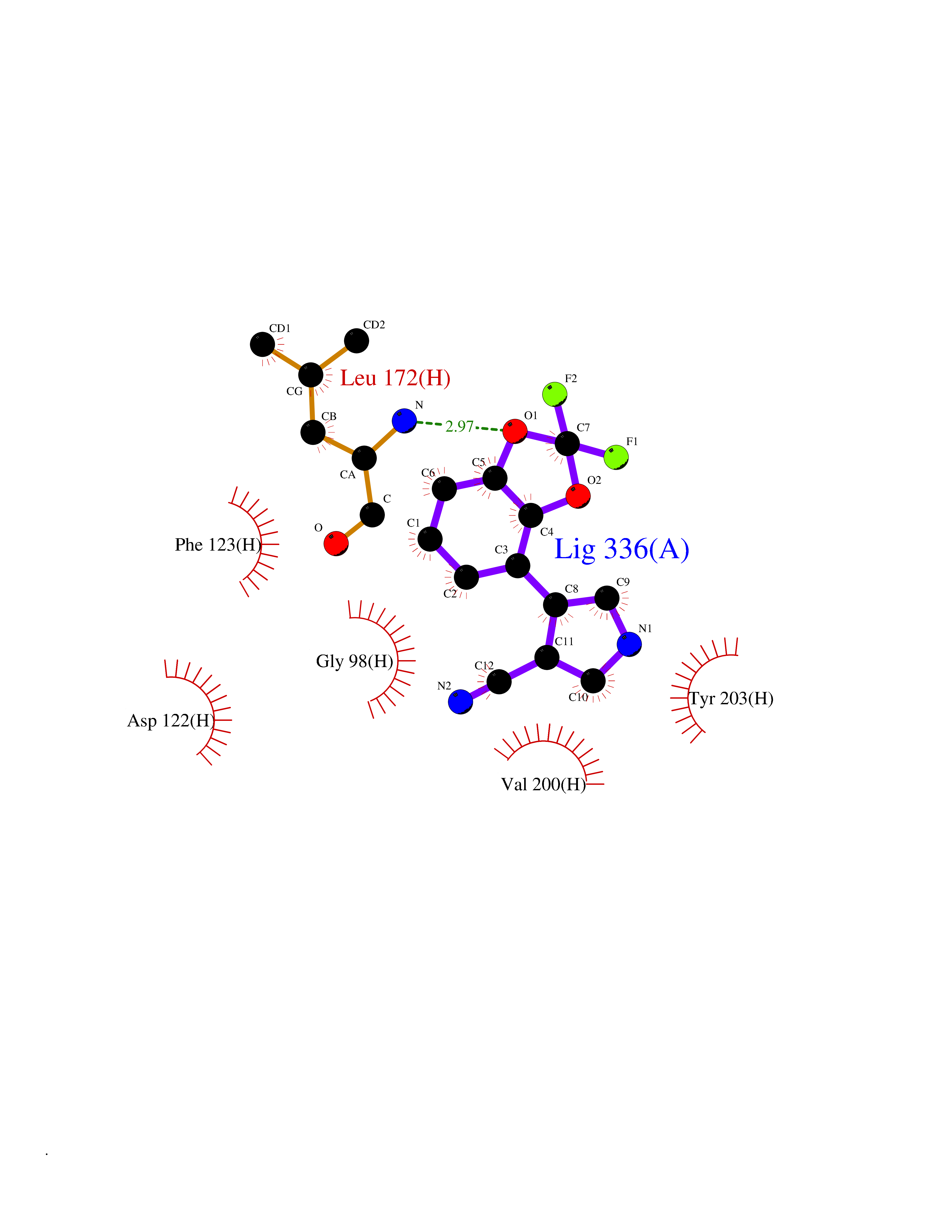 Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | p53-binding protein Mdm4 (MDM4) | 6Q9Y | 6.01 | |
Target general information Gen name MDM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Mdmx; Mdm2-like p53-binding protein; Double minute 4 protein Protein family MDM2/MDM4 family Biochemical class MDM2/MDM4 family Function Inhibits p53/TP53- and TP73/p73-mediated cell cycle arrest and apoptosis by binding its transcriptional activation domain. Inhibits degradation of MDM2. Can reverse MDM2-targeted degradation of TP53 while maintaining suppression of TP53 transactivation and apoptotic functions. Related diseases Bone marrow failure syndrome 6 (BMFS6) [MIM:618849]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS6 is an autosomal dominant form characterized by intermittent neutropenia, lymphopenia, or anemia associated with hypocellular bone marrow, and increased susceptibility to cancer. {ECO:0000269|PubMed:32300648}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NX04; P10415; Q7Z479; O95971; P48729; Q00987; Q13064; P41227; P06400; Q9Y4L5; P23297; P29034; P33763; P04271; P31947; P04637; P62837; Q93009; O14972; P61964; P62258; P61981; P63104; Q9BRR0; A0A0S2Z6X0; Q3YBA8; P03255-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19722 Length 173 Aromaticity 0.08 Instability index 50.78 Isoelectric point 8.48 Charge (pH=7) 2.27 2D Binding mode Binding energy (Kcal/mol) -8.2  Molscript Map 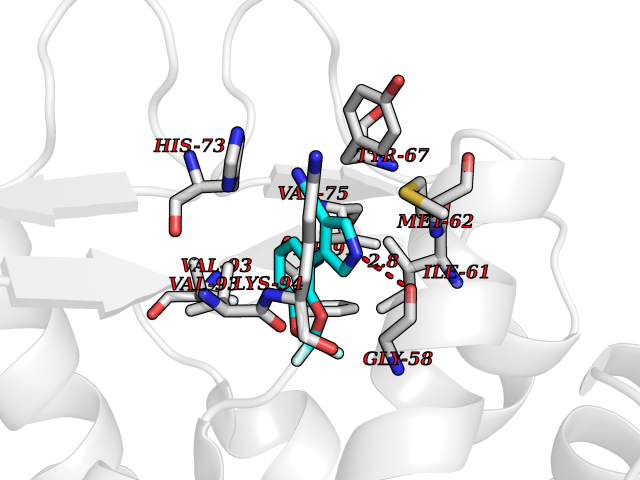 Pymol Map 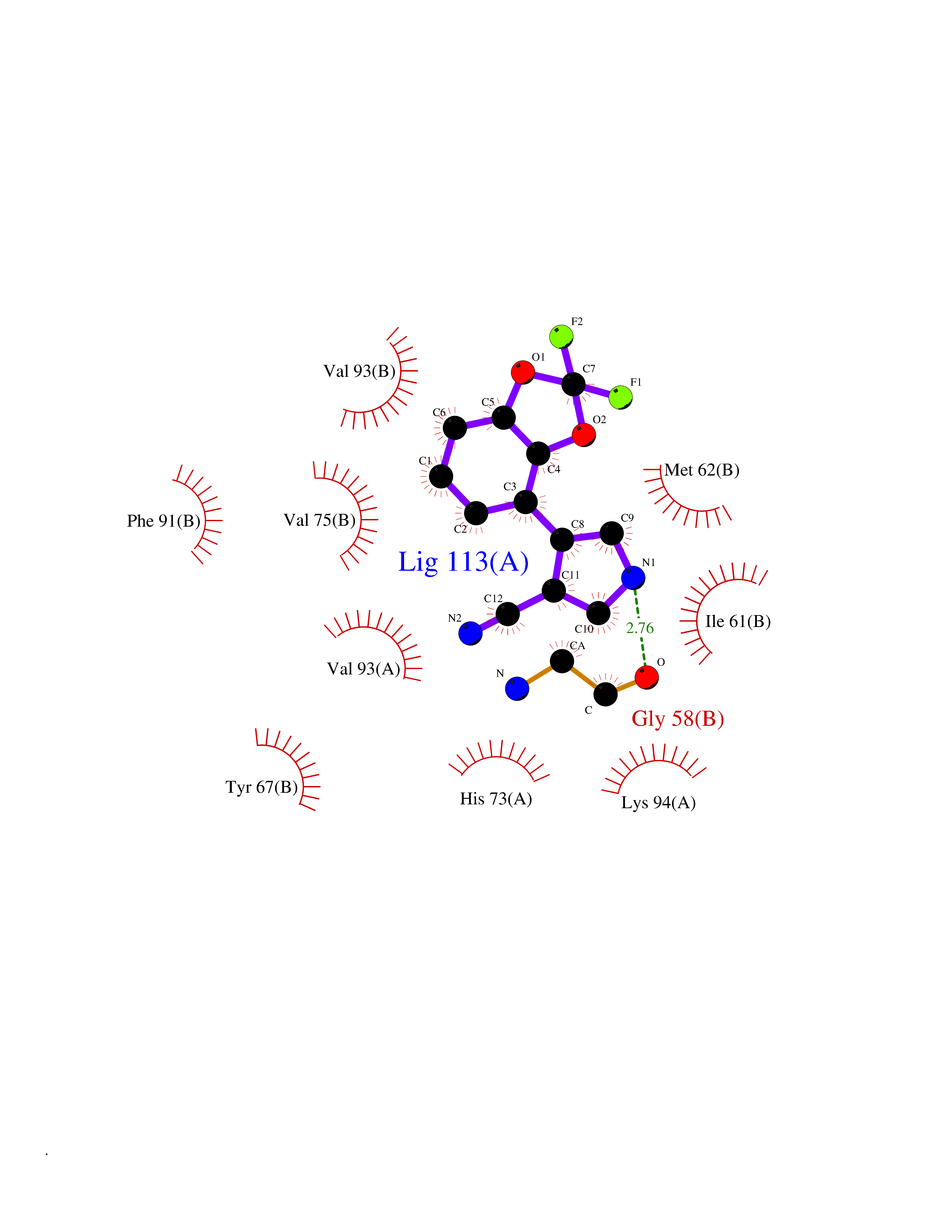 Ligplot Map 3D Binding mode Sequence QVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLAQINQVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLA Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 6.01 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -8.2 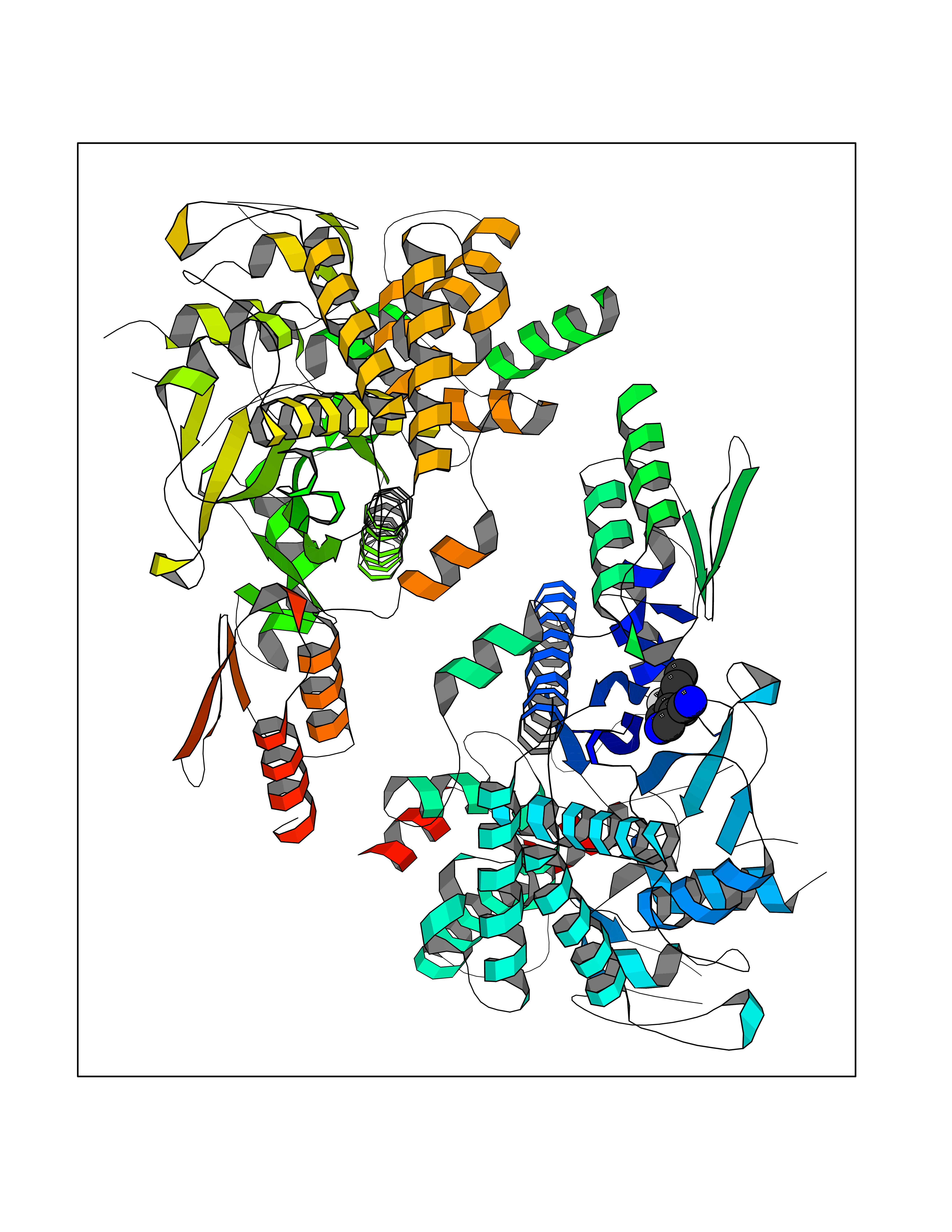 Molscript Map 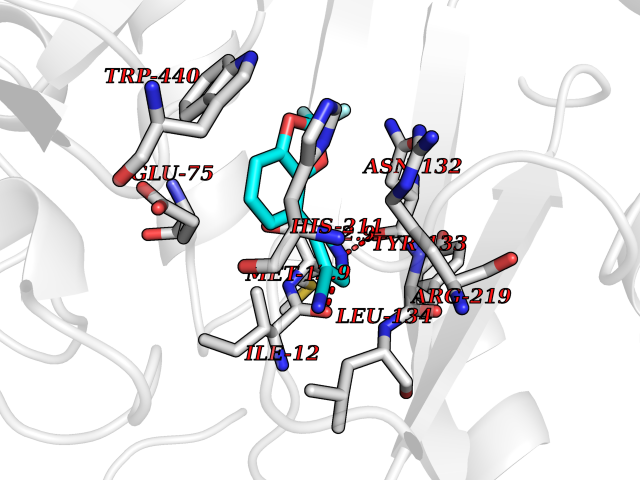 Pymol Map 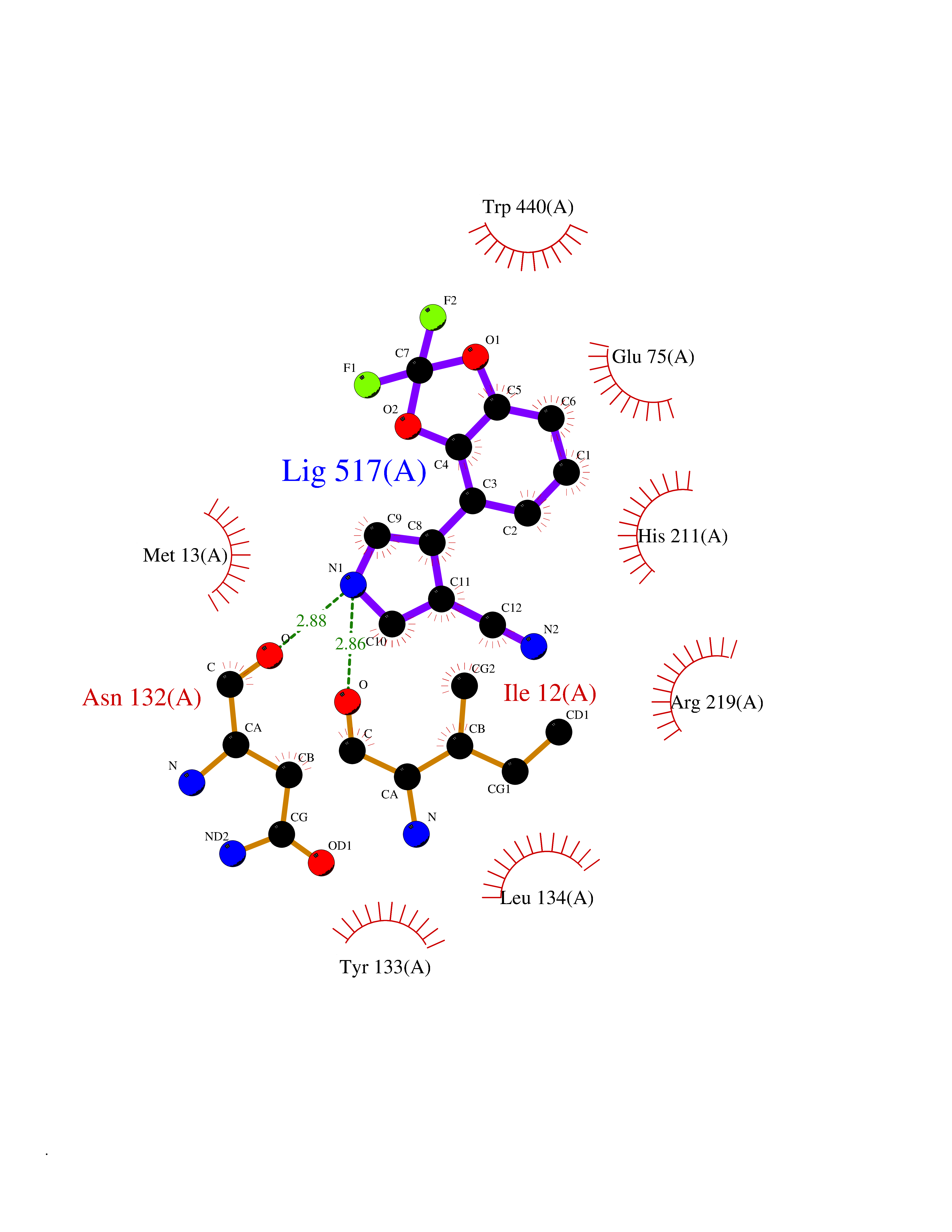 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||