Job Results:
Ligand
Structure
Job ID
0fdb6be2dae96620ab05e05b4d1adc2b
Job name
NA
Time
2025-04-03 17:50:26
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Hypoxia-inducible factor 2 alpha (HIF-2A) | 5TBM | 7.11 | |
Target general information Gen name EPAS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms bHLHe73; PASD2; PAS domain-containing protein 2; Member of PAS protein 2; MOP2; Hypoxia-inducible factor 2-alpha; HLF; HIF2A; HIF2-alpha; HIF-2-alpha; HIF-1-alpha-like factor; Endothelial PAS domain-c Protein family NA Biochemical class NA Function Heterodimerizes with ARNT; heterodimer binds to core DNA sequence 5'-TACGTG-3' within the hypoxia response element (HRE) of target gene promoters. Regulates the vascular endothelial growth factor (VEGF) expression and seems to be implicated in the development of blood vessels and the tubular system of lung. May also play a role in the formation of the endothelium that gives rise to the blood brain barrier. Potent activator of the Tie-2 tyrosine kinase expression. Activation requires recruitment of transcriptional coactivators such as CREBBP and probably EP300. Interaction with redox regulatory protein APEX seems to activate CTAD. Transcription factor involved in the induction of oxygen regulated genes. Related diseases Erythrocytosis, familial, 4 (ECYT4) [MIM:611783]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal platelet and leukocyte counts. {ECO:0000269|PubMed:18184961, ECO:0000269|PubMed:18378852, ECO:0000269|PubMed:19208626, ECO:0000269|PubMed:22367913}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15463; DB12255 Interacts with P27540; Q96RK4; O00327-8; Q8WYA1-3; Q9GZT9; P60228; O60573; P09467; P61244; Q9BWF3-1; P08047; Q9Y2K6; P40818 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; Congenital erythrocytosis; Developmental protein; Differentiation; Disease variant; DNA-binding; Host-virus interaction; Hydroxylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 12249.8 Length 106 Aromaticity 0.11 Instability index 40.77 Isoelectric point 5.25 Charge (pH=7) -5.82 2D Binding mode Binding energy (Kcal/mol) -9.7  Molscript Map 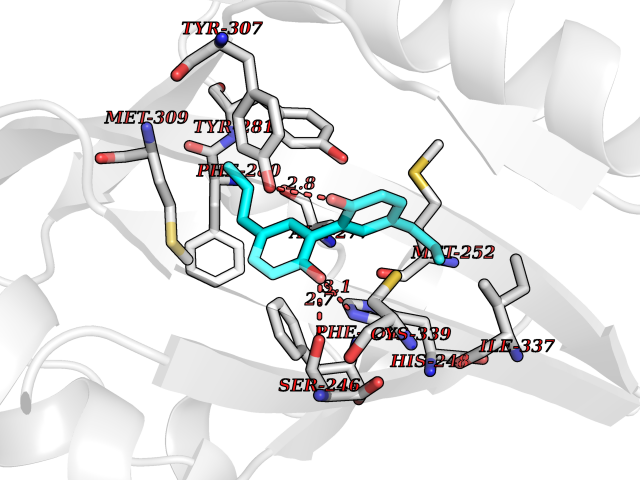 Pymol Map 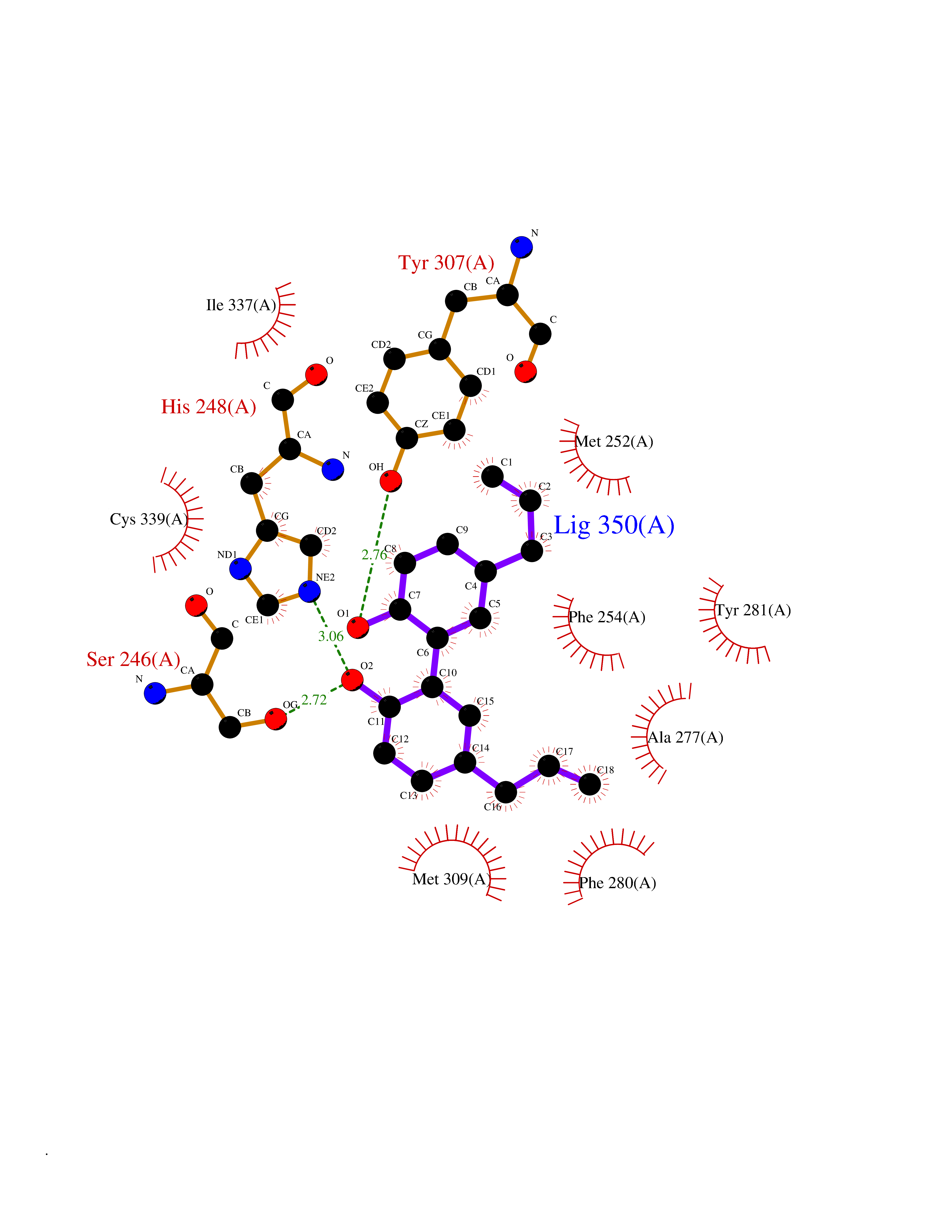 Ligplot Map 3D Binding mode Sequence LDSKTFLSEHSMDMKFTYCDDRITELIGYHPEELLGRSAYEFYHALDSENMTKSHQNLCTKGQVVSGQYRMLAKHGGYVWLETQGTVIYNPPQCIMCVNYVLSEIE Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Neprilysin | 1R1H | 7.10 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -9.68  Molscript Map 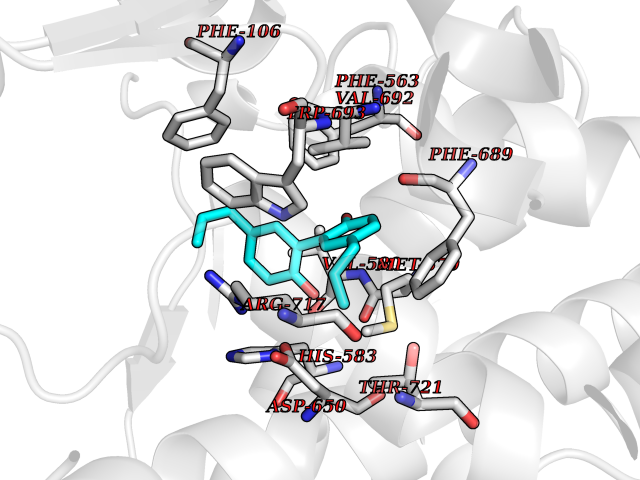 Pymol Map 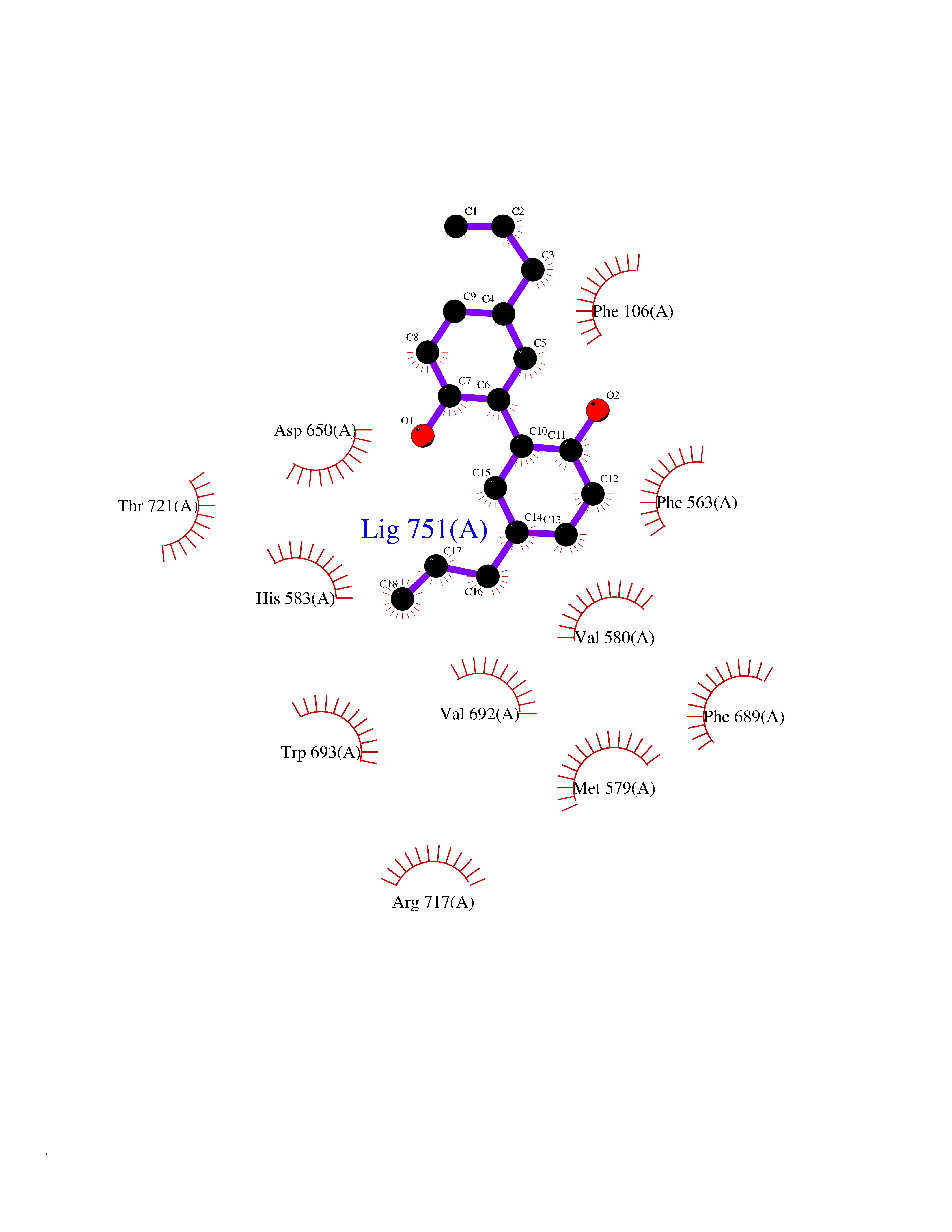 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 7.10 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -9.69 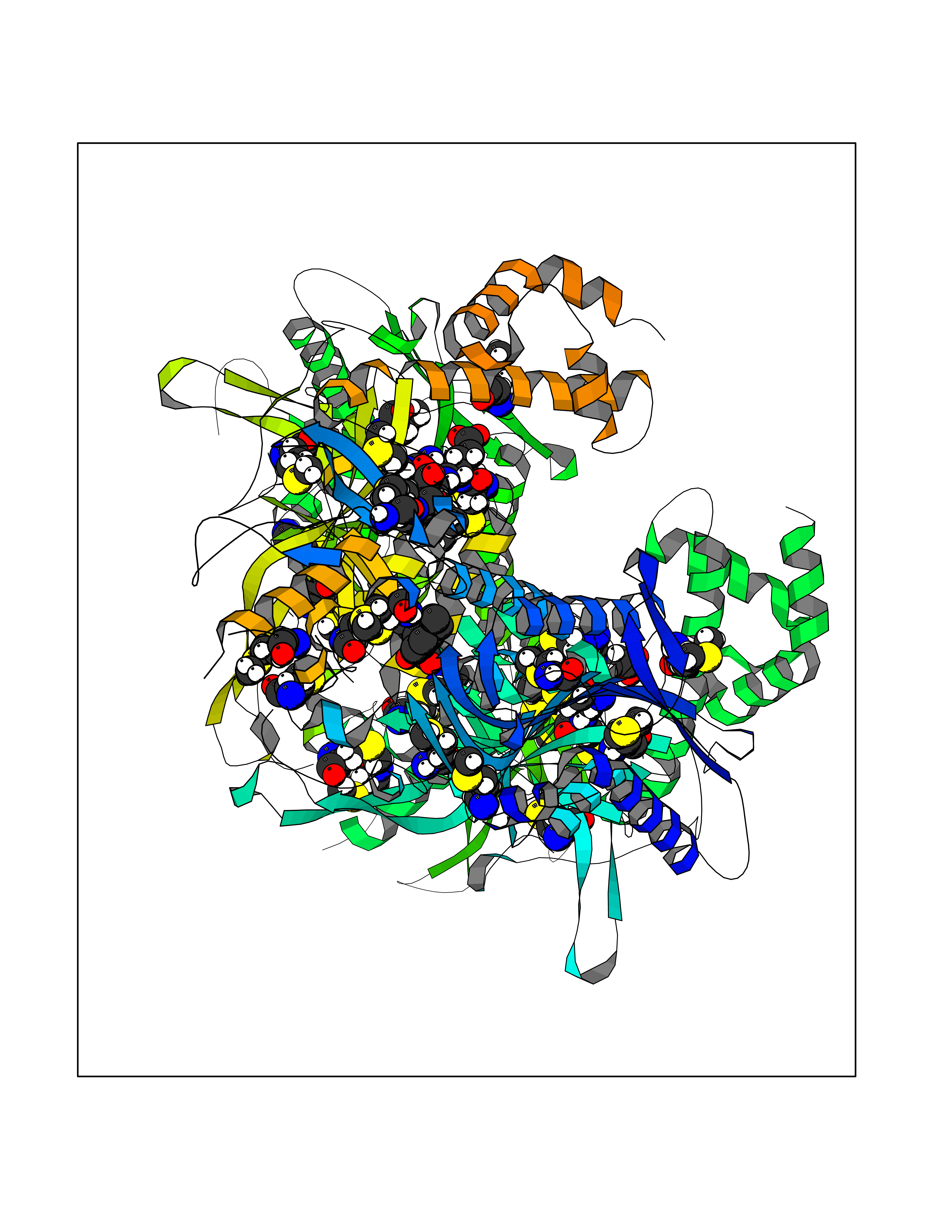 Molscript Map  Pymol Map 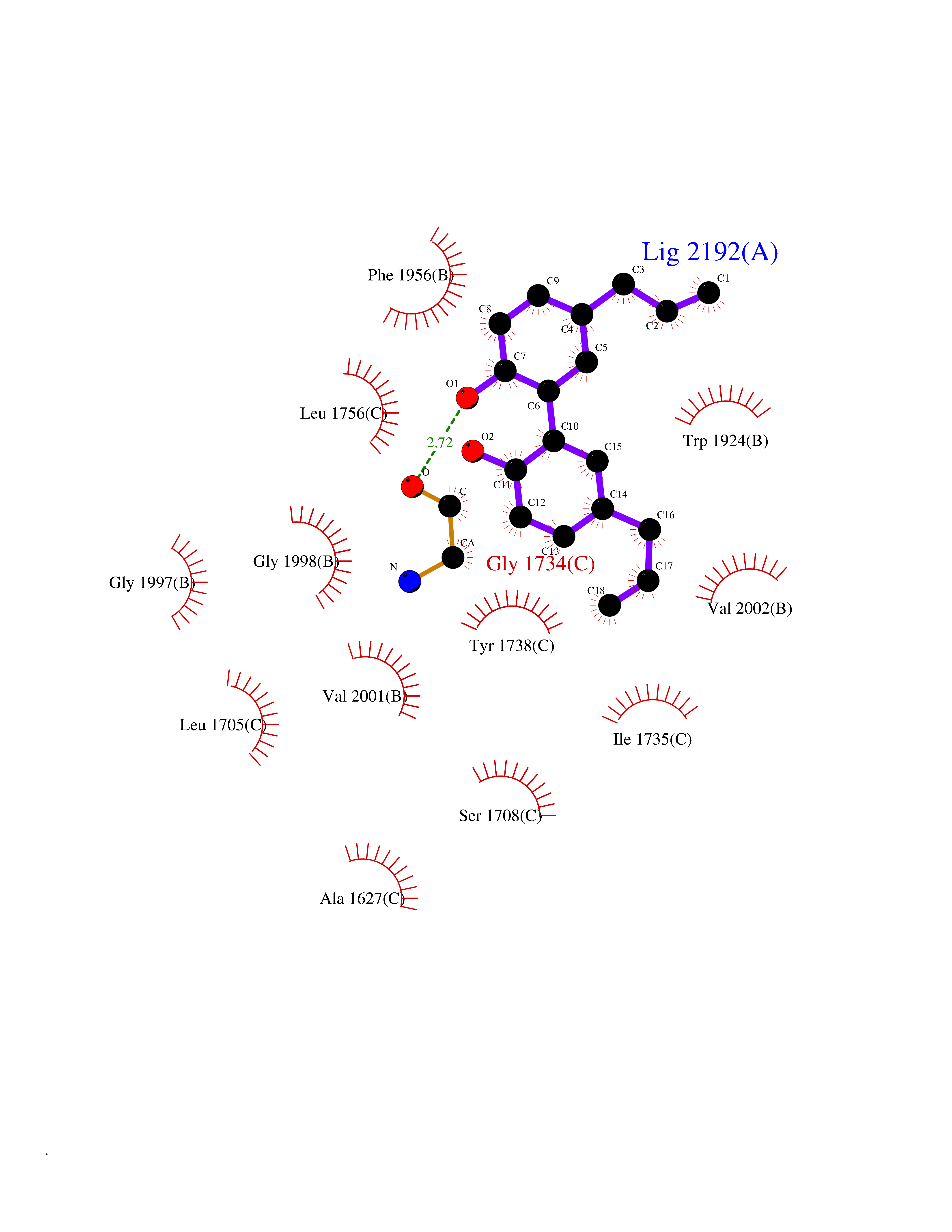 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Folate receptor alpha (FOLR1) | 4LRH | 7.10 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -9.69 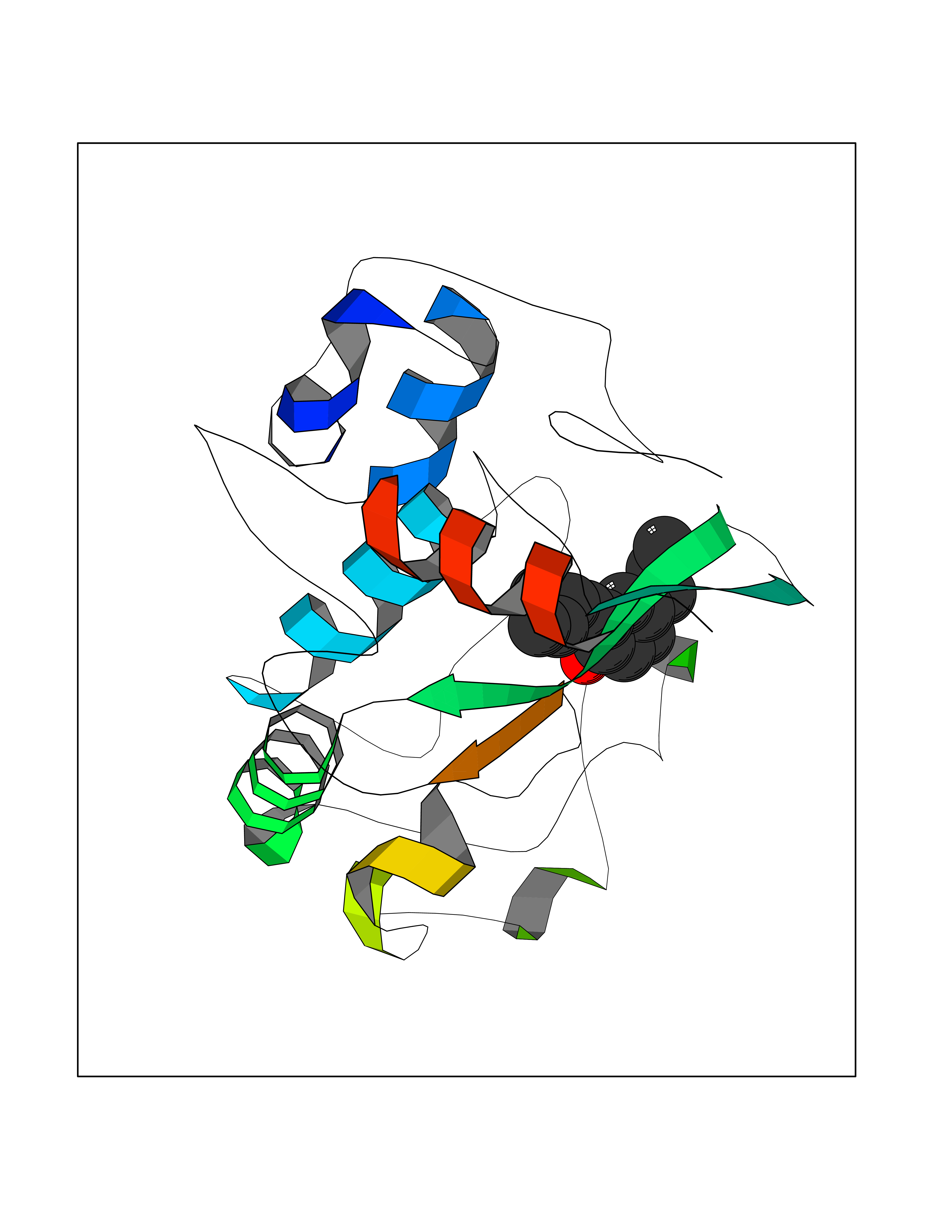 Molscript Map 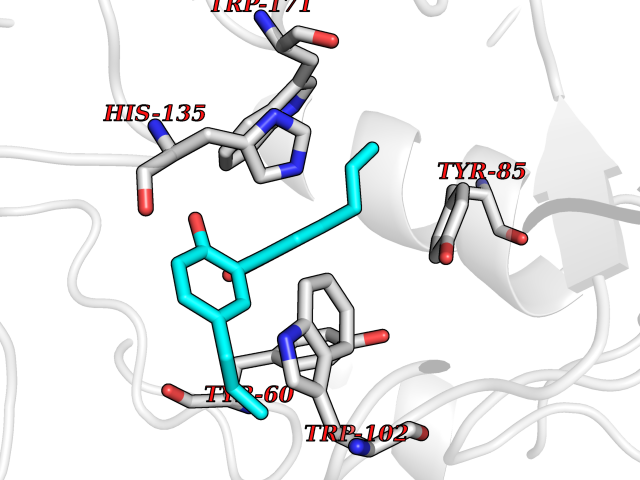 Pymol Map 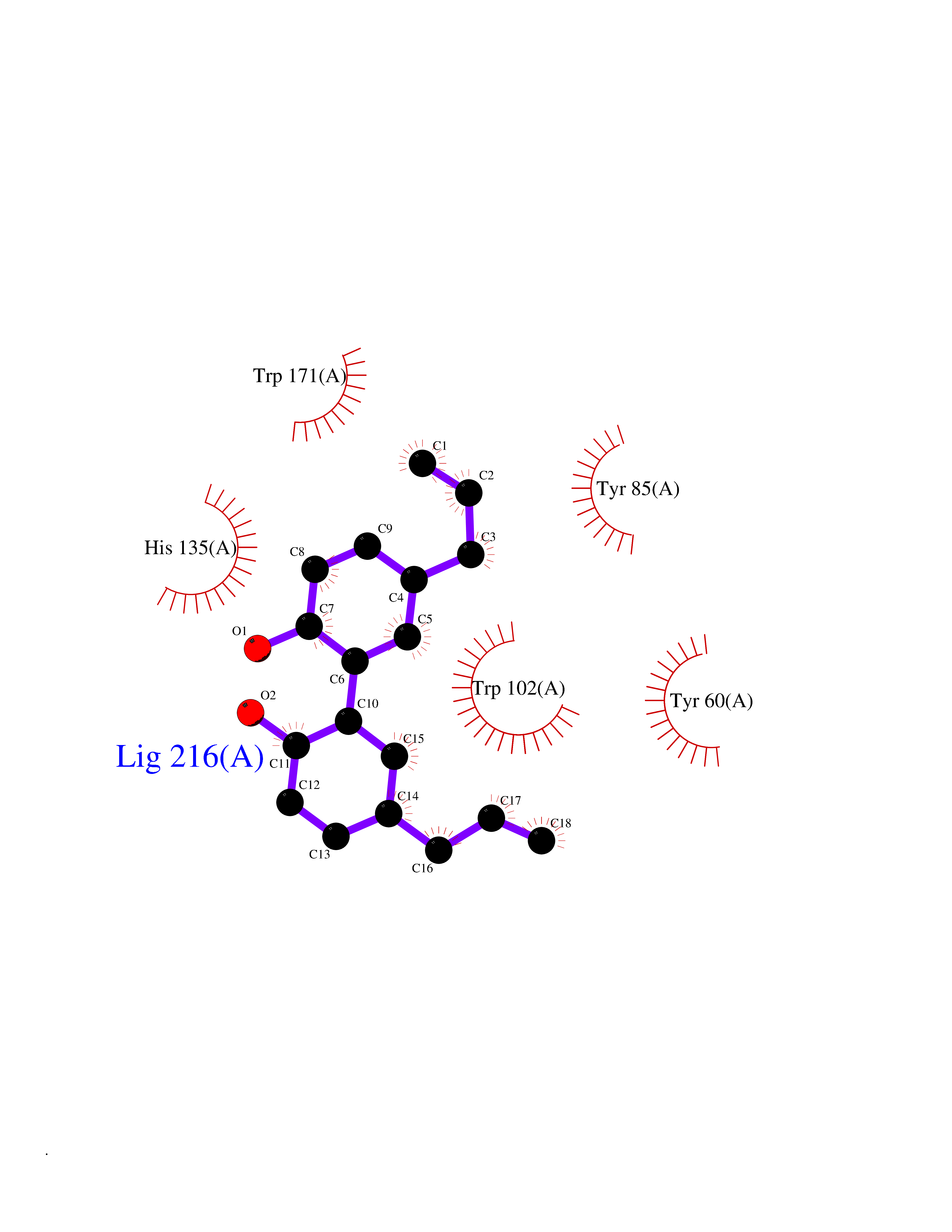 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Angiopoietin-related protein 4 (ANGPTL4) | 6U73 | 7.09 | |
Target general information Gen name ANGPTL4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ171/PRO197; PSEC0166; PP1158; PGAR; Hepatic fibrinogen/angiopoietin-related protein; HFARP; Angiopoietin-like protein PP1158; Angiopoietin-like protein 4; Angiopoietin-like 4; ARP4 Protein family NA Biochemical class Fibrinogen protein Function May also play a role in regulating glucose homeostasis and insulin sensitivity. Inhibits proliferation, migration, and tubule formation of endothelial cells and reduces vascular leakage. Upon heterologous expression, inhibits the adhesion of endothelial cell to the extracellular matrix (ECM), and inhibits the reorganization of the actin cytoskeleton, formation of actin stress fibers and focal adhesions in endothelial cells that have adhered to ANGPTL4-containing ECM (in vitro). Depending on context, may modulate tumor-related angiogenesis. Mediates inactivation of the lipoprotein lipase LPL, and thereby plays a role in the regulation of triglyceride clearance from the blood serum and in lipid metabolism. Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9BY76; P05556; P18084 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Coiled coil; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 24429.1 Length 216 Aromaticity 0.12 Instability index 40.6 Isoelectric point 8.51 Charge (pH=7) 2.46 2D Binding mode Binding energy (Kcal/mol) -9.66 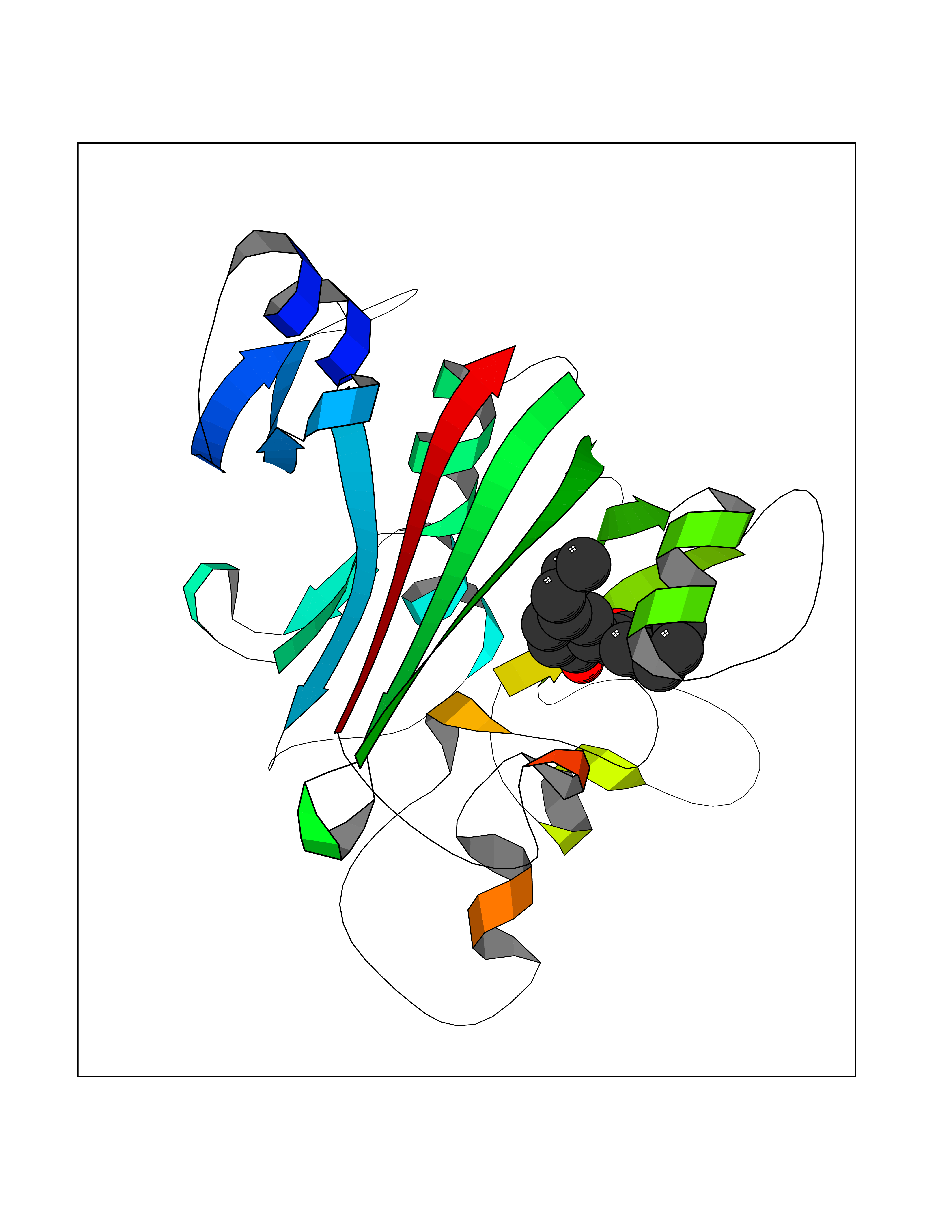 Molscript Map 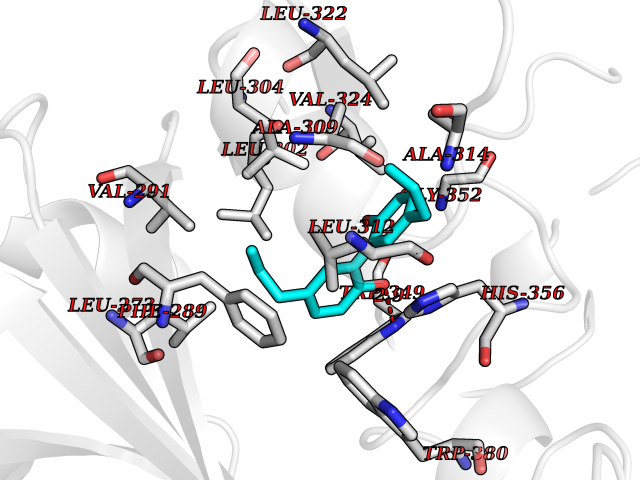 Pymol Map 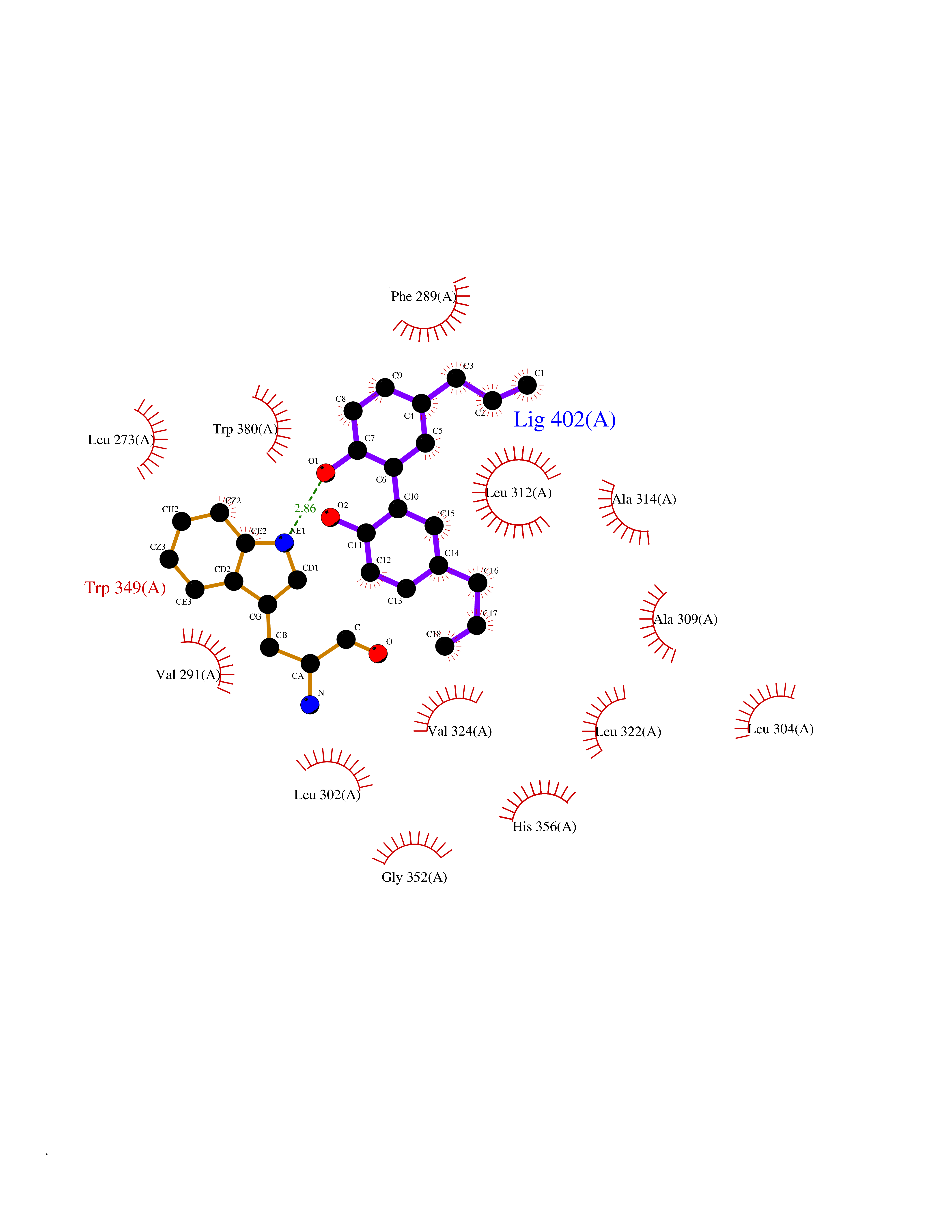 Ligplot Map 3D Binding mode Sequence PRDCQELFQVGERQSGLFEIQPQGSPPFLVNCKMTSDGGWTVIQRRHDGSVDFNRPWEAYKAGFGDPHGEFWLGLEKVHSITGDRNSRLAVQLRDWDGNAELLQFSVHLGGEDTAYSLQLTAPVAGQLGATTVPPSGLSVPFSTWDQDHDLRRDKNCAKSLSGGWWFGTCSHSNLNGQYFRSIPQQRQKLKKGIFWKTWRGRYYPLQATTMLIQPM Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Monoamine oxidase type A (MAO-A) | 2Z5Y | 7.08 | |
Target general information Gen name MAOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoamine oxidase A; Amine oxidase [flavin-containing] A Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function MAOA preferentially oxidizes biogenic amines such as 5-hydroxytryptamine (5-HT), norepinephrine and epinephrine. Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. Related diseases Brunner syndrome (BRNRS) [MIM:300615]: A form of X-linked non-dysmorphic mild intellectual disability. Male patients are affected by borderline intellectual deficit and exhibit abnormal behavior, including disturbed regulation of impulsive aggression. Obligate female carriers have normal intelligence and behavior. {ECO:0000269|PubMed:8211186}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00918; DB00182; DB06698; DB04889; DB13876; DB01445; DB06774; DB00215; DB04017; DB09130; DB05205; DB07641; DB00988; DB01363; DB00668; DB12329; DB01175; DB03147; DB14914; DB00614; DB01381; DB07919; DB04818; DB01247; DB00601; DB01577; DB00805; DB01442; DB01171; DB08804; DB00952; DB04820; DB00184; DB04821; DB06412; DB01626; DB00780; DB00191; DB00388; DB00397; DB09244; DB04850; DB00721; DB01168; DB00571; DB00852; DB09363; DB00140; DB00953; DB06654; DB01037; DB01104; DB00669; DB14569; DB09042; DB00624; DB13943; DB13944; DB13946; DB09245; DB00752; DB15328; DB09185; DB04832; DB00315; DB00909 Interacts with P27338 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine metabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Intellectual disability; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurotransmitter degradation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 58195.3 Length 513 Aromaticity 0.11 Instability index 34.97 Isoelectric point 7.98 Charge (pH=7) 2.87 2D Binding mode Binding energy (Kcal/mol) -9.66 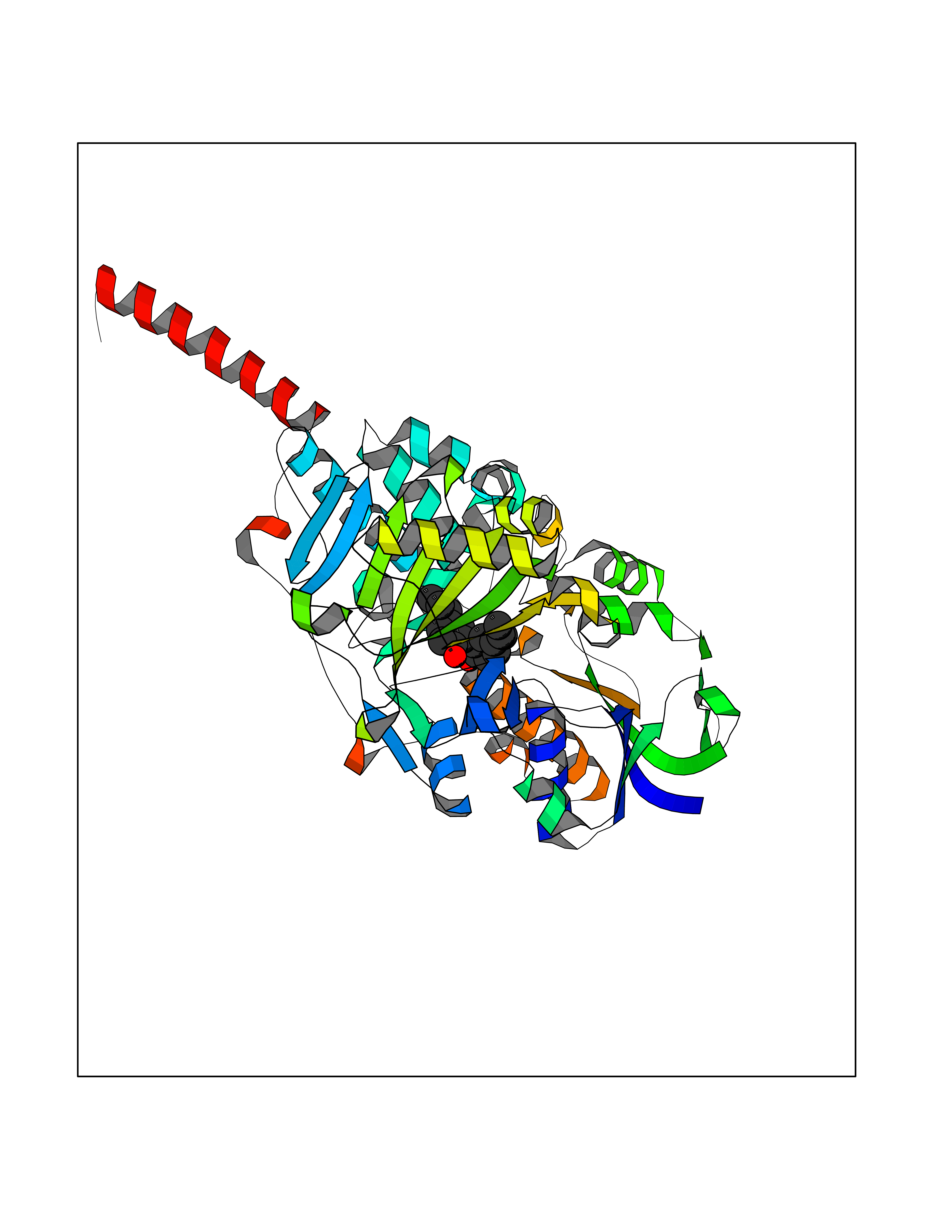 Molscript Map 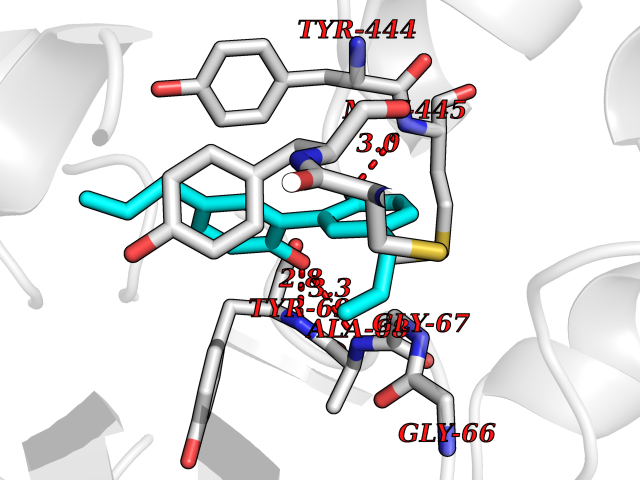 Pymol Map 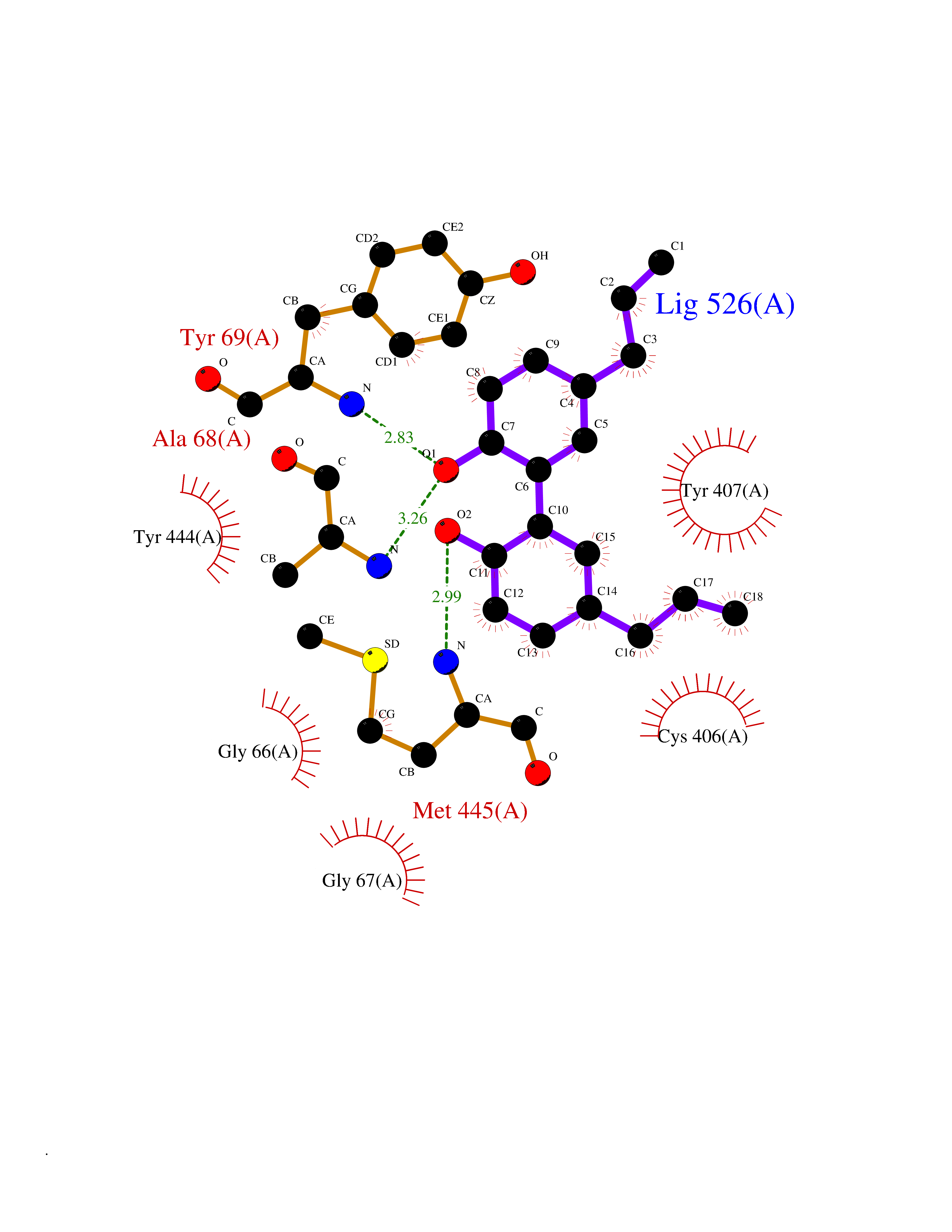 Ligplot Map 3D Binding mode Sequence HMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYVGPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRAAFPPVWNPIAYLDYNNLWRTIDNMGKEIPTDAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSVTNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAKIHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIMGFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYGRVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTFWERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLL Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Receptor-interacting protein 1 (RIPK1) | 5TX5 | 7.08 | |
Target general information Gen name RIPK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor-interacting serine/threonine-protein kinase 1; RIP1; RIP-1; RIP; Cell death protein RIP Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Upon activation of TNFR1 by the TNF-alpha family cytokines, TRADD and TRAF2 are recruited to the receptor. Phosphorylates DAB2IP at 'Ser-728' in a TNF-alpha-dependent manner, and thereby activates the MAP3K5-JNK apoptotic cascade. Ubiquitination by TRAF2 via 'Lys-63'-link chains acts as a critical enhancer of communication with downstream signal transducers in the mitogen-activated protein kinase pathway and the NF-kappa-B pathway, which in turn mediate downstream events including the activation of genes encoding inflammatory molecules. Polyubiquitinated protein binds to IKBKG/NEMO, the regulatory subunit of the IKK complex, a critical event for NF-kappa-B activation. Interaction with other cellular RHIM-containing adapters initiates gene activation and cell death. RIPK1 and RIPK3 association, in particular, forms a necrosis-inducing complex. Serine-threonine kinase which transduces inflammatory and cell-death signals (programmed necrosis) following death receptors ligation, activation of pathogen recognition receptors (PRRs), and DNA damage. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P04083; Q13490; Q13489; Q92851; Q14790; Q8IVM0; P48729; Q13158; Q9Y6K9; Q96AB6; Q9ULZ3; Q13546; Q9Y572; P19438; Q13077; Q12933; Q13114; Q13107; B7UI21; PRO_0000449629 [P0DTD1]; U5TQE9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Disease variant; Glycoprotein; Host-virus interaction; Inflammatory response; Isopeptide bond; Kinase; Membrane; Necrosis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29554.2 Length 259 Aromaticity 0.08 Instability index 48.26 Isoelectric point 6.29 Charge (pH=7) -2.52 2D Binding mode Binding energy (Kcal/mol) -9.66  Molscript Map 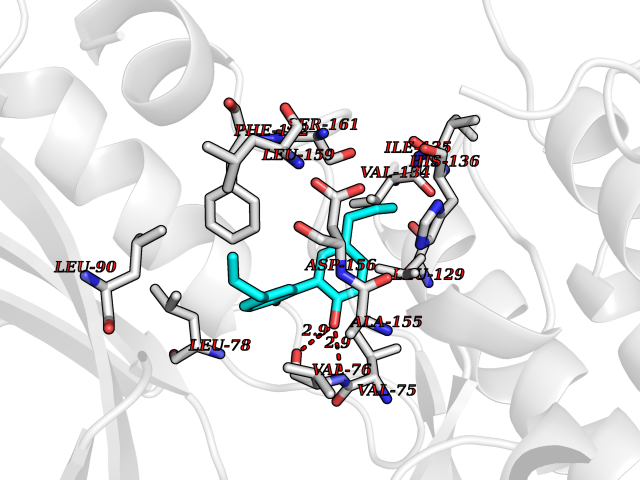 Pymol Map 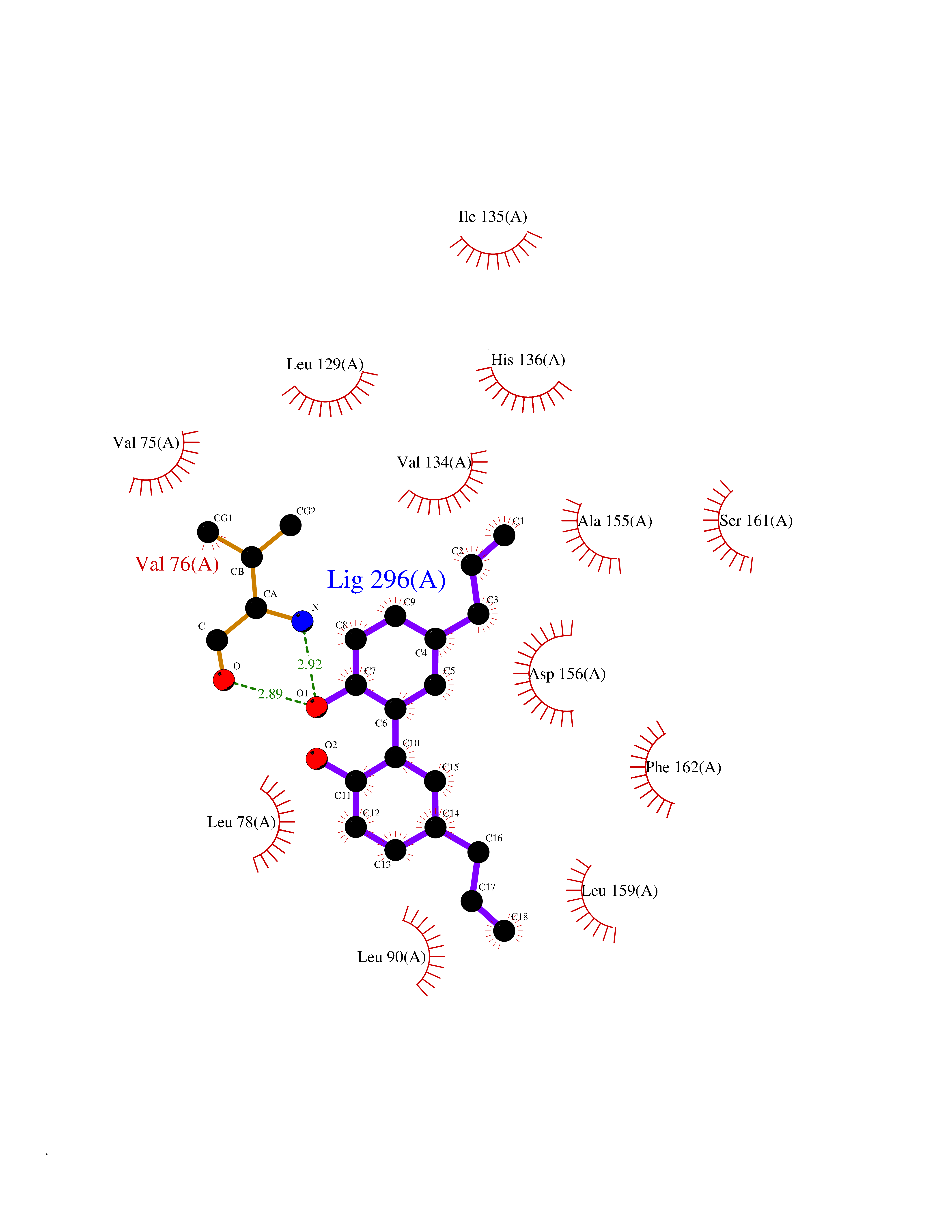 Ligplot Map 3D Binding mode Sequence IKMKSSDFLESAELDSGGKVSLAFHRTQGLMIMKTVYKGPNCIEHNEALLEEAKMMNRLRHSRVVKLLGVIIEEGKYSLVMEYMEKGNLMHVLKAEMSTPLSVKGRIILEIIEGMAYLHGKGVIHKDLKPENILVDNDFHIKIADLGLASFKMWSKLNGTLYYMAPEHLNDVNAKPTEKSDVYSFAVVLWAIFANKEPYQQLIMAIKSGNRPDVDDITEYCPREIISLMKLCWEANPEARPTFPGIEEKFRPFYLSQLE Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 7.07 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -9.65  Molscript Map  Pymol Map 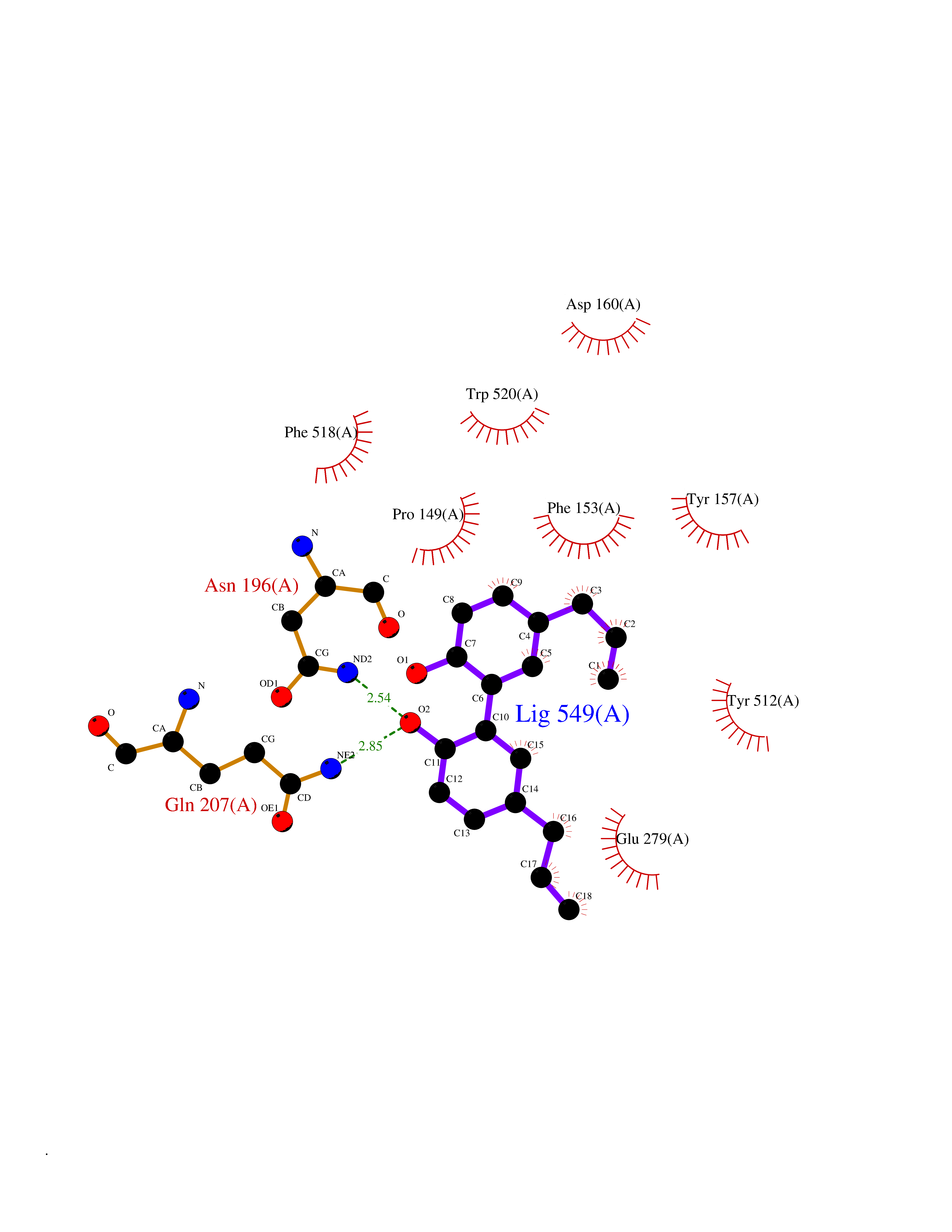 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 7.07 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -9.64 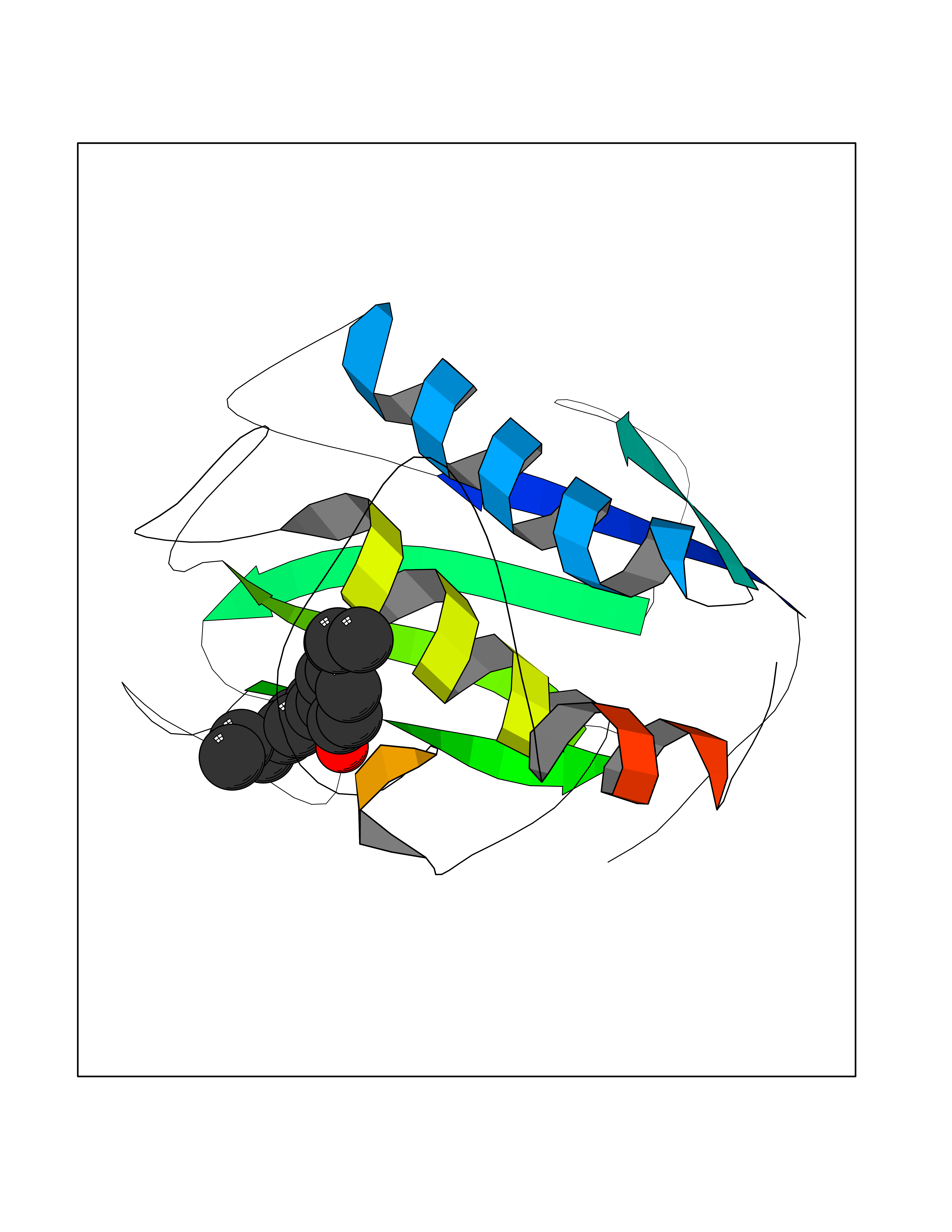 Molscript Map 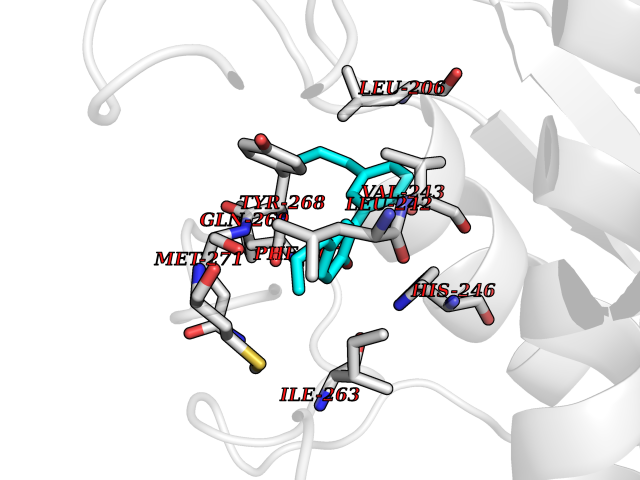 Pymol Map 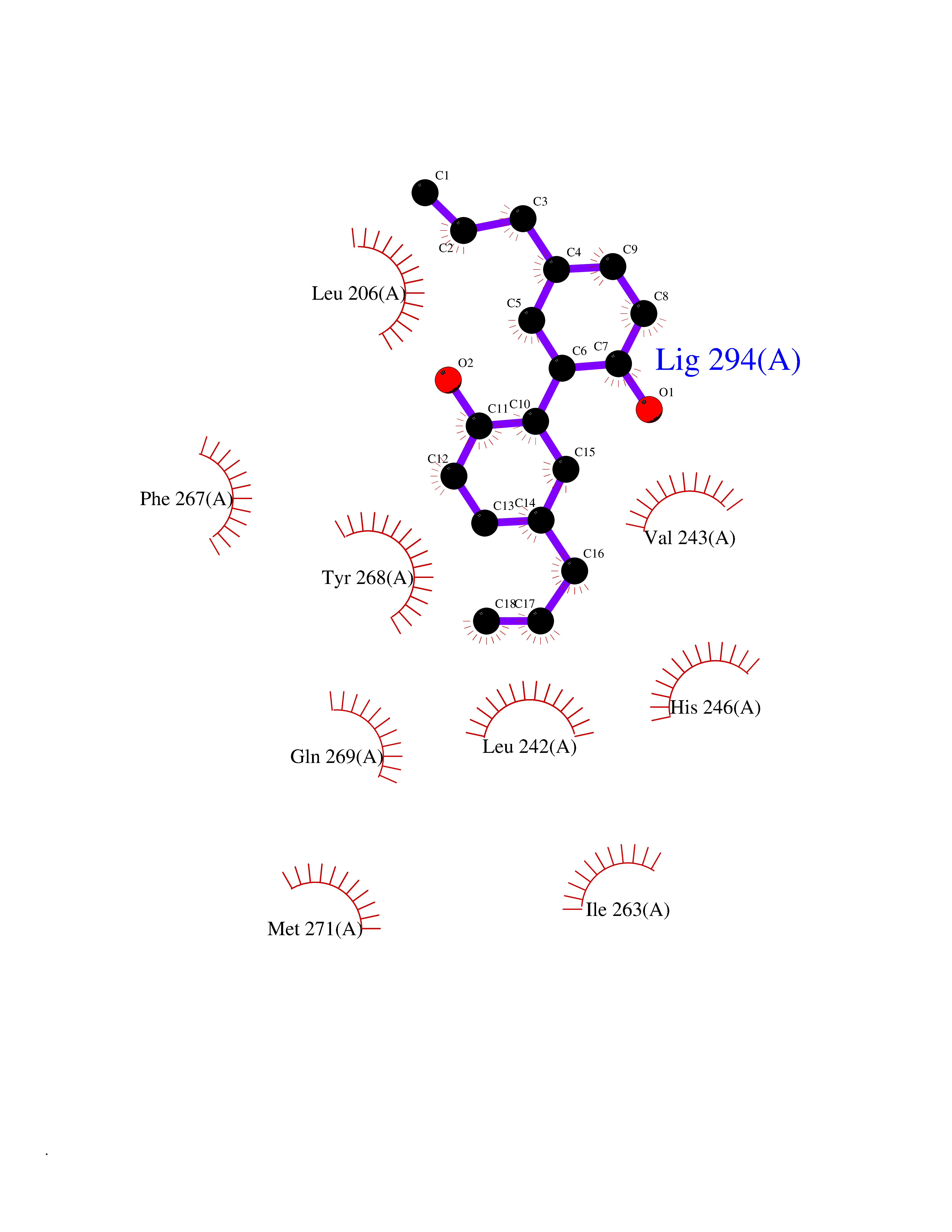 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 7.07 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -9.64 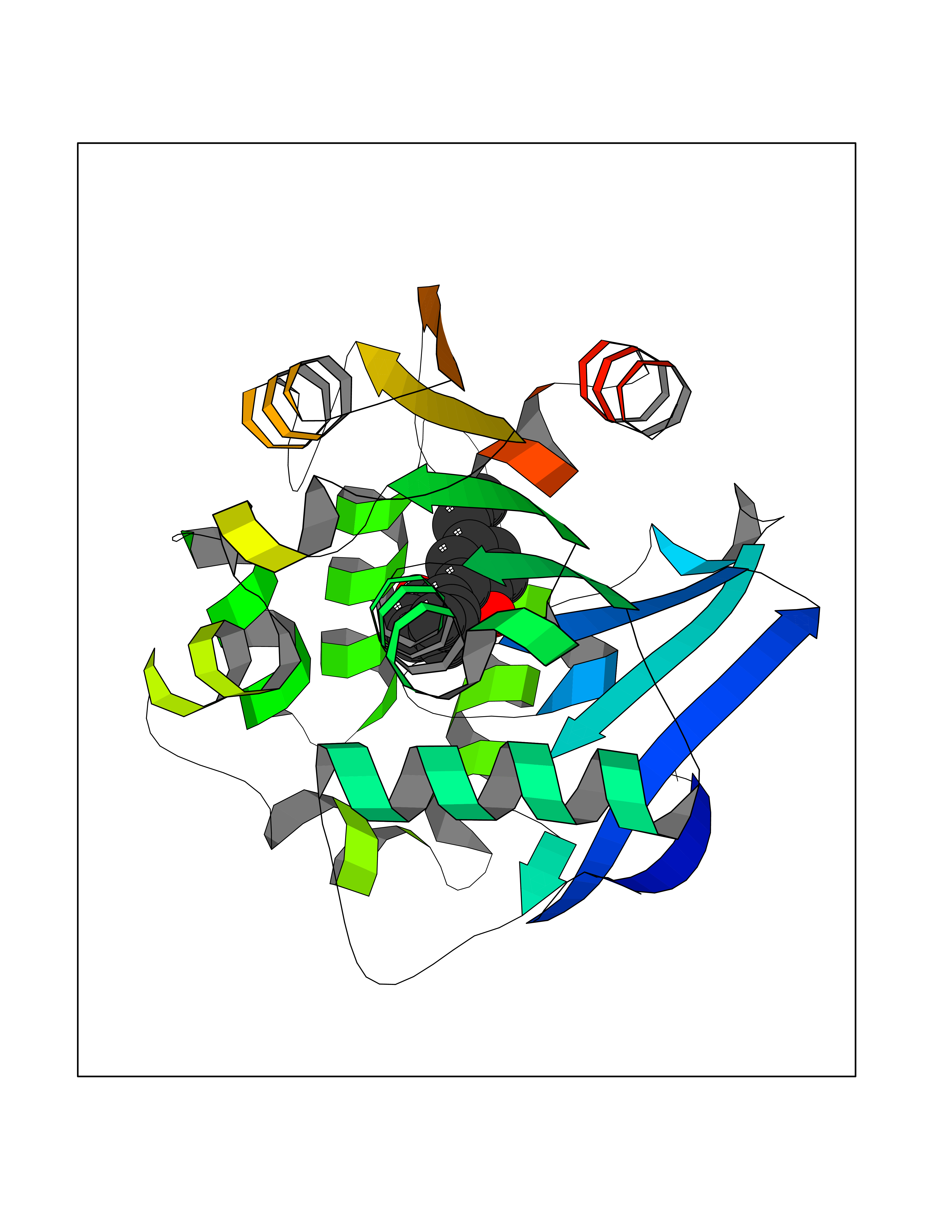 Molscript Map 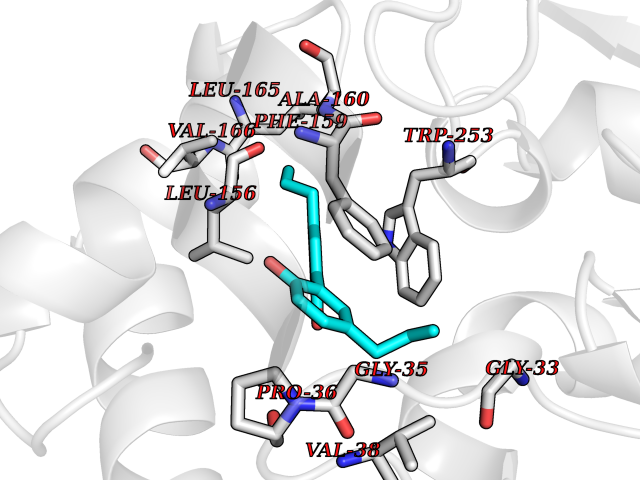 Pymol Map 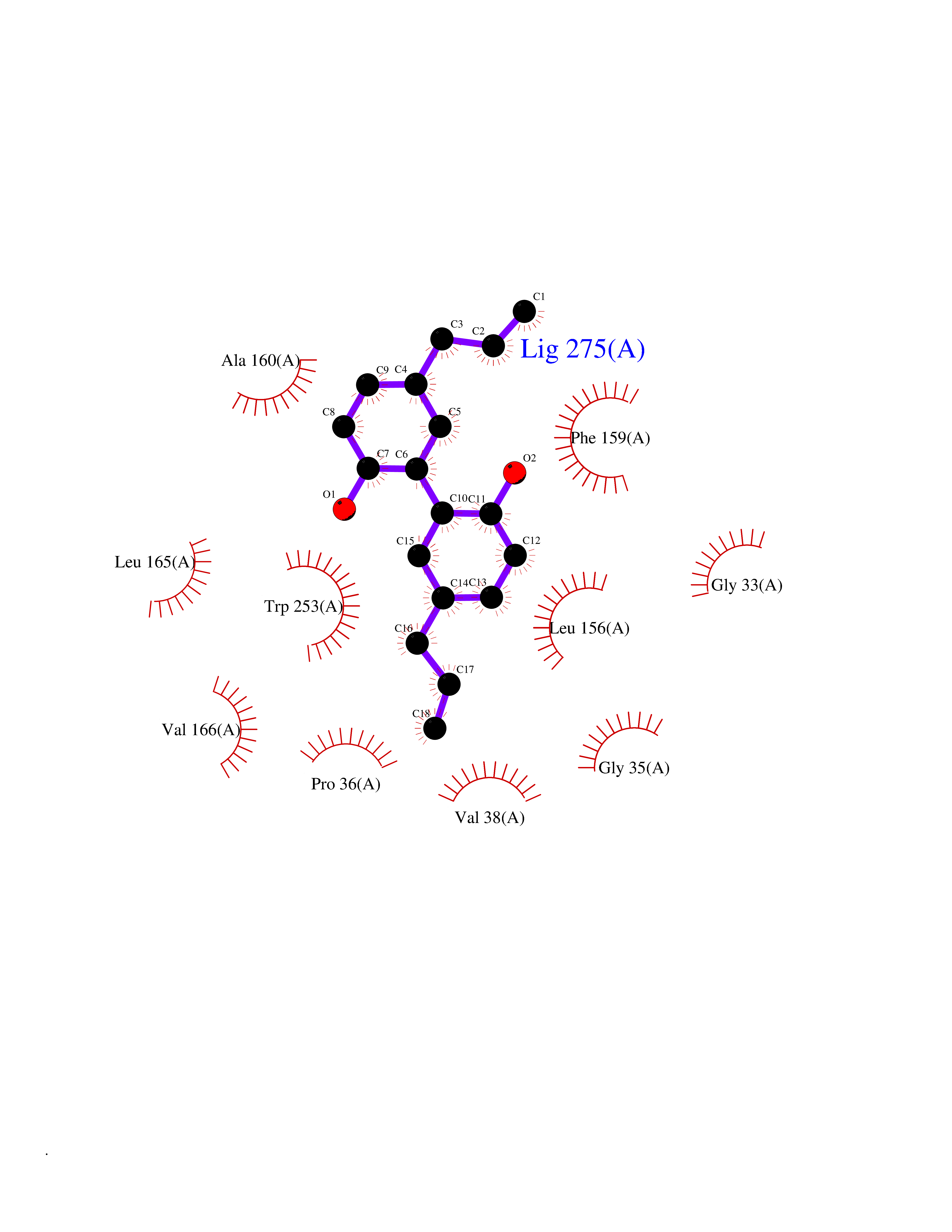 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 7.07 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -9.64 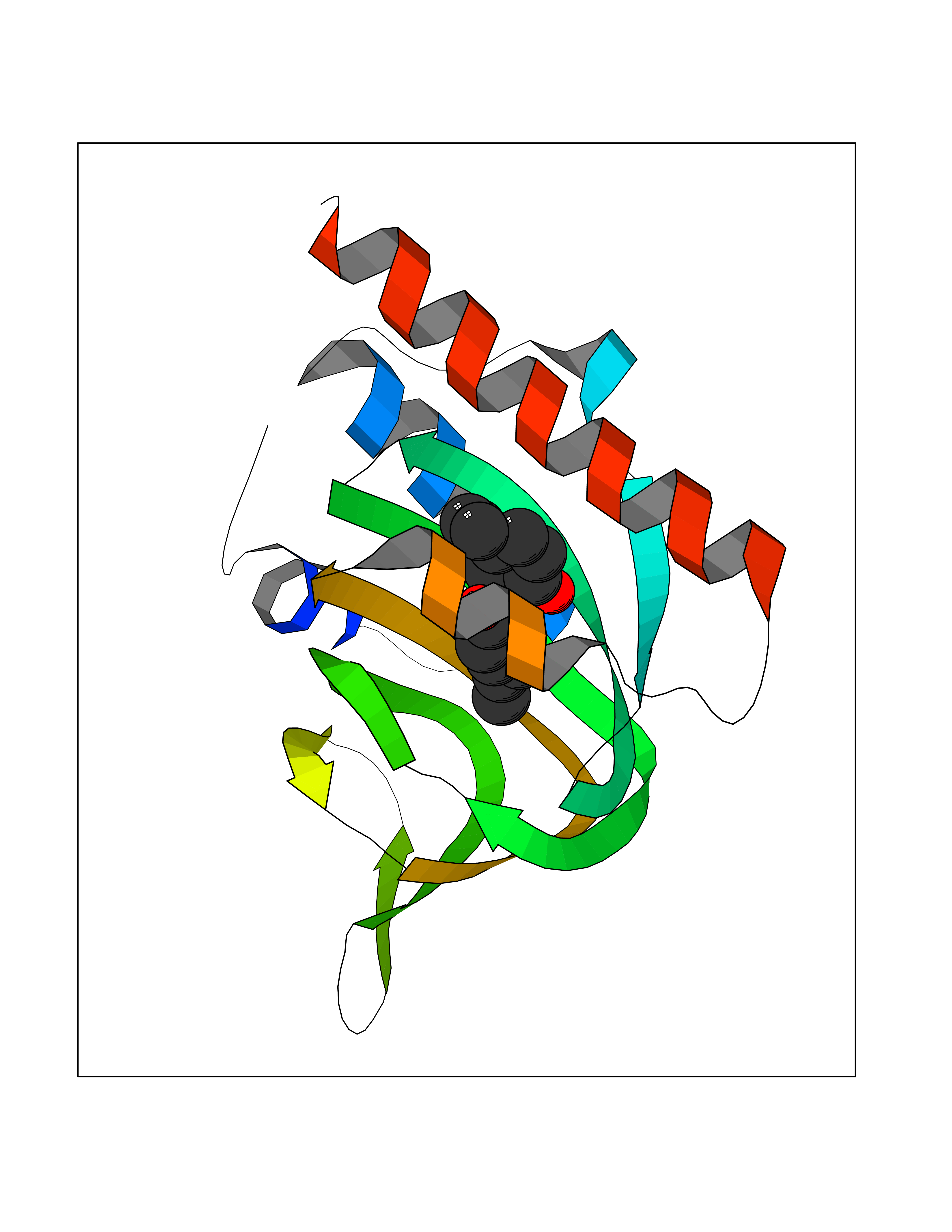 Molscript Map 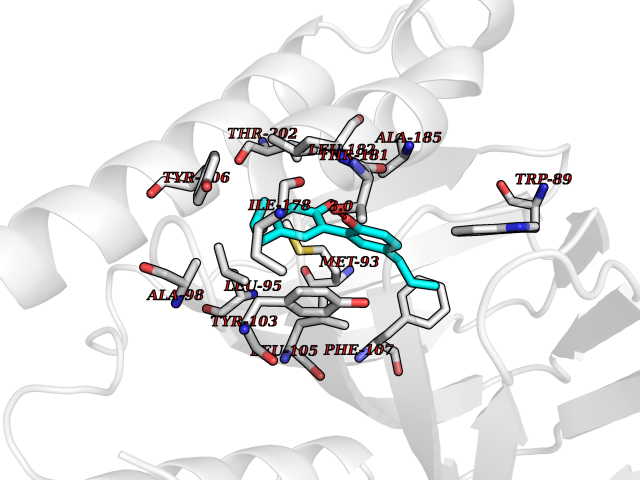 Pymol Map 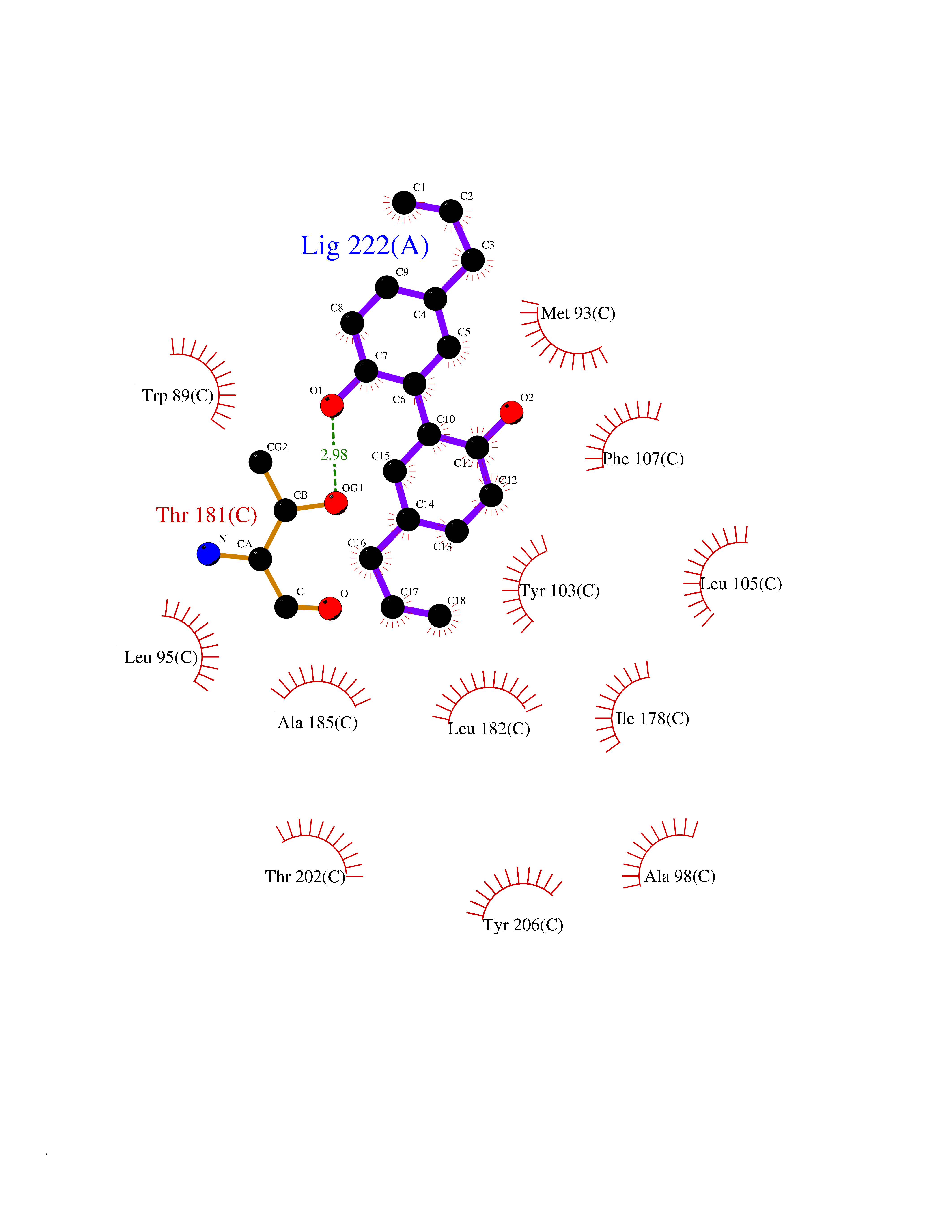 Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Cytochrome c oxidase subunit 2 | 3VRJ | 7.06 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -9.63 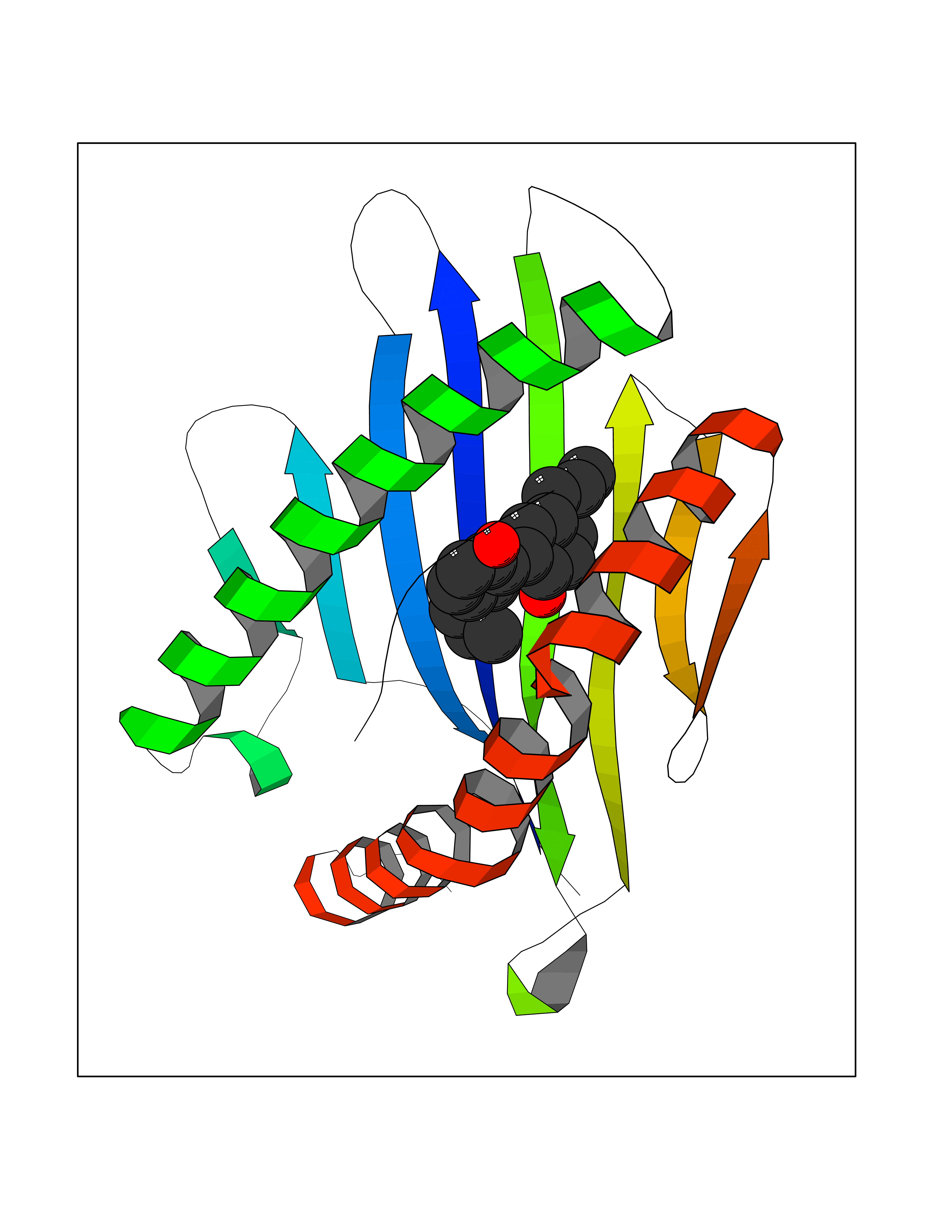 Molscript Map 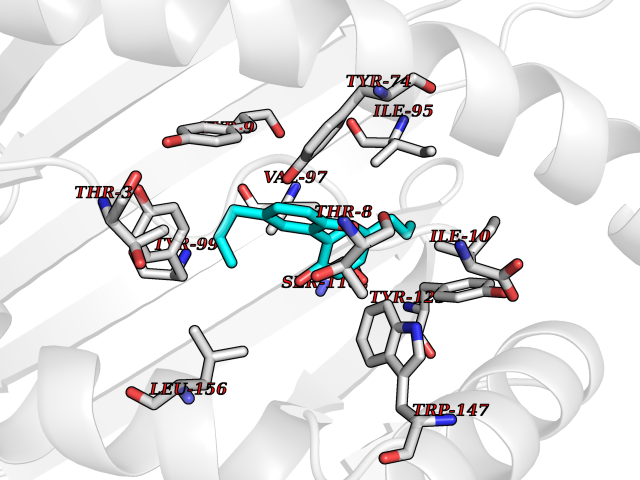 Pymol Map 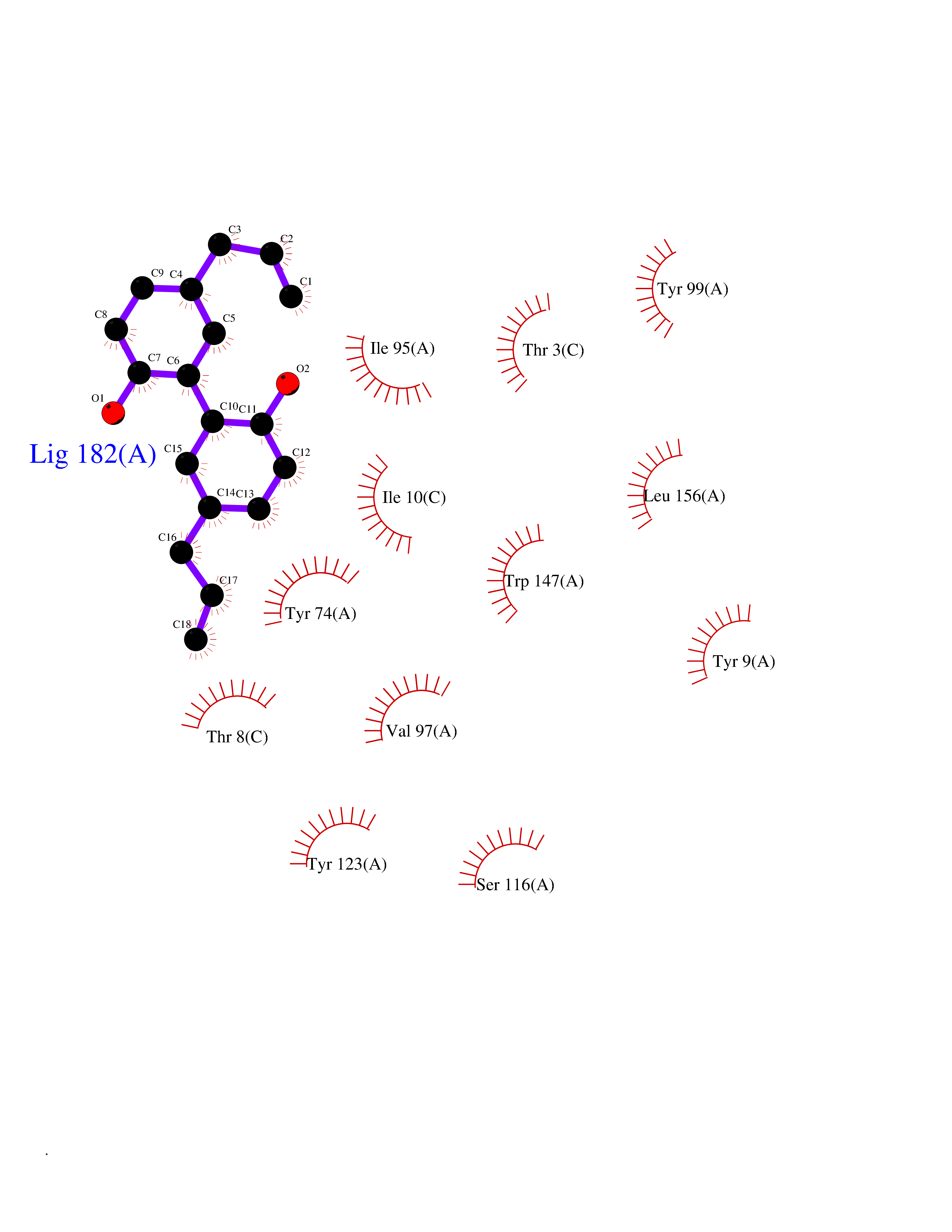 Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Aldose reductase (AKR1B1) | 1US0 | 7.06 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -9.62 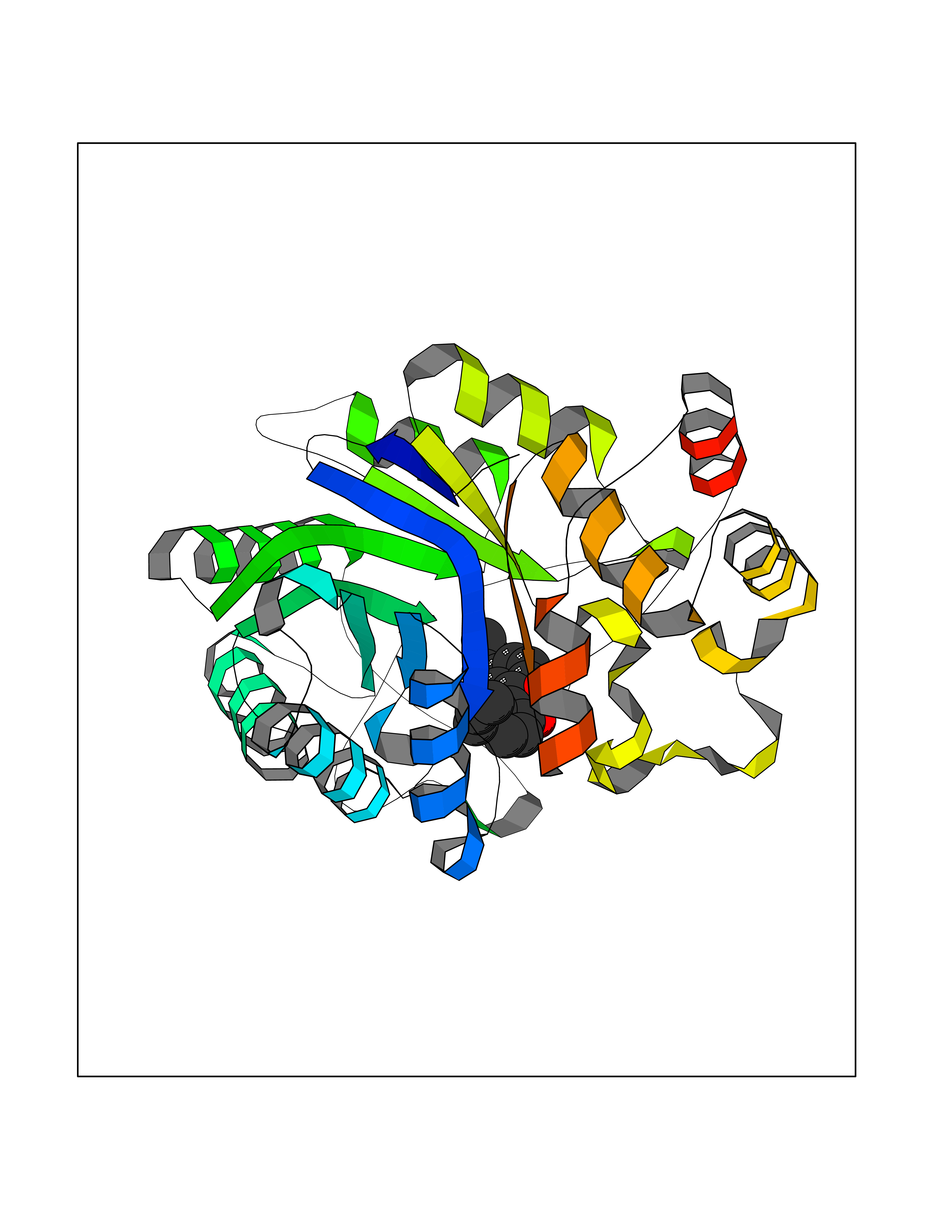 Molscript Map 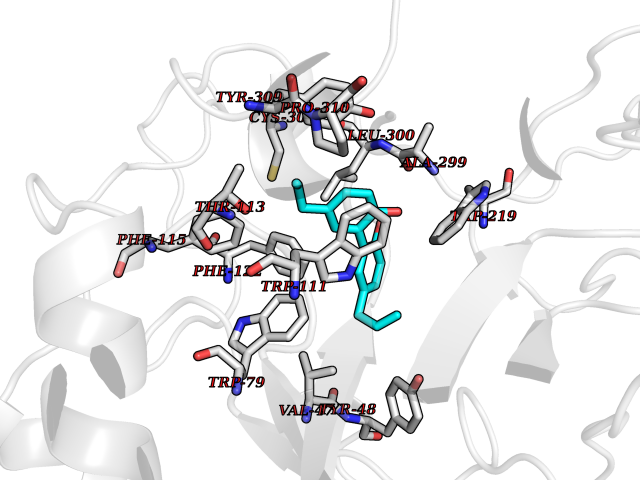 Pymol Map 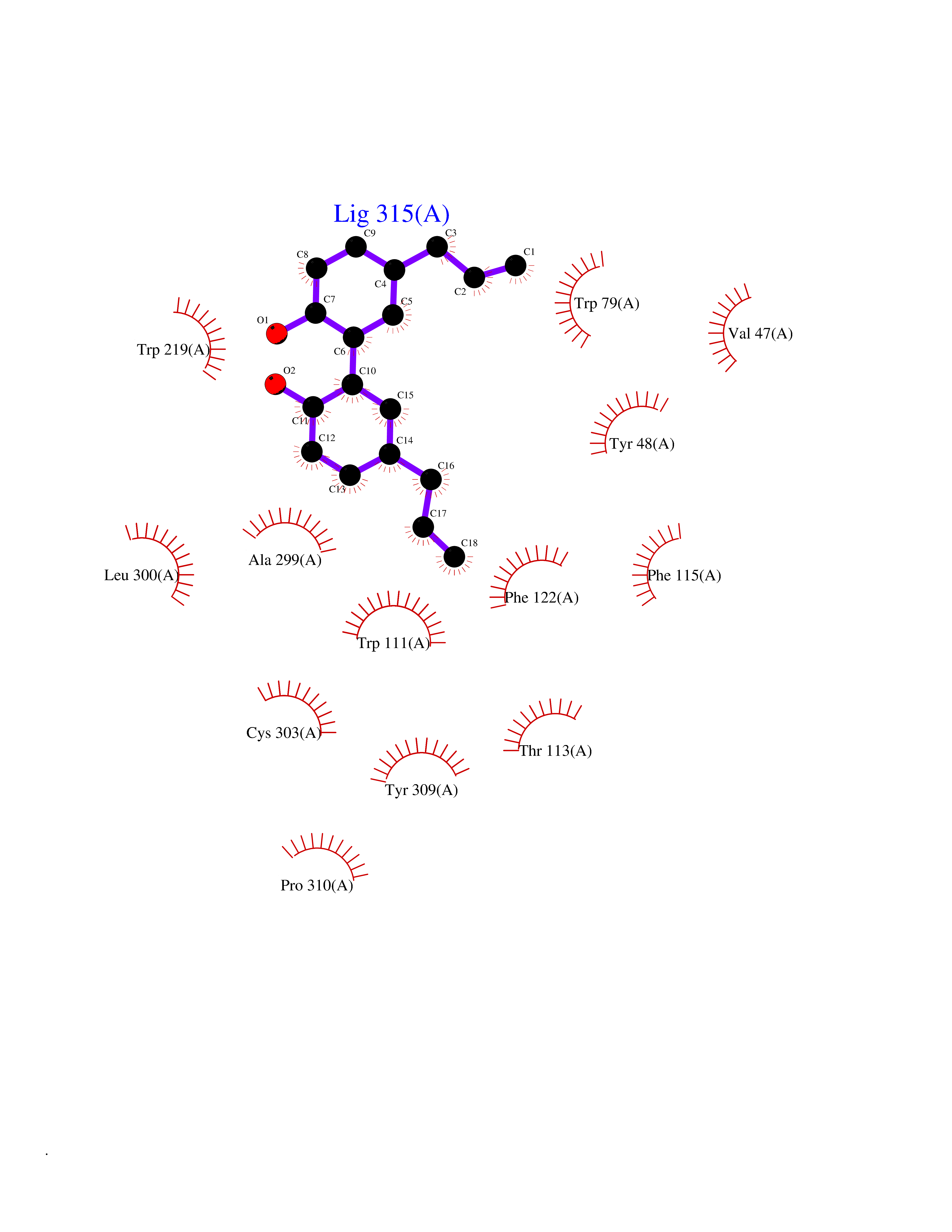 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Pyruvate dehydrogenase [ubiquinone] | 3EYA | 7.06 | |
Target general information Gen name poxB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0871;JW0855 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Identical protein binding.Lipid binding.Magnesium ion binding.Pyruvate dehydrogenase (quinone) activity.Thiamine pyrophosphate binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07003 EC number 1.2.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Lipid-binding; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate; Ubiquinone Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 113027 Length 1046 Aromaticity 0.07 Instability index 35.99 Isoelectric point 5.75 Charge (pH=7) -24.38 2D Binding mode Binding energy (Kcal/mol) -9.63  Molscript Map 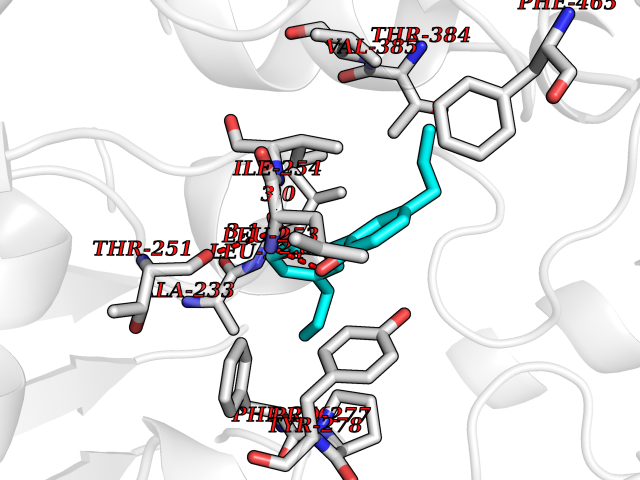 Pymol Map 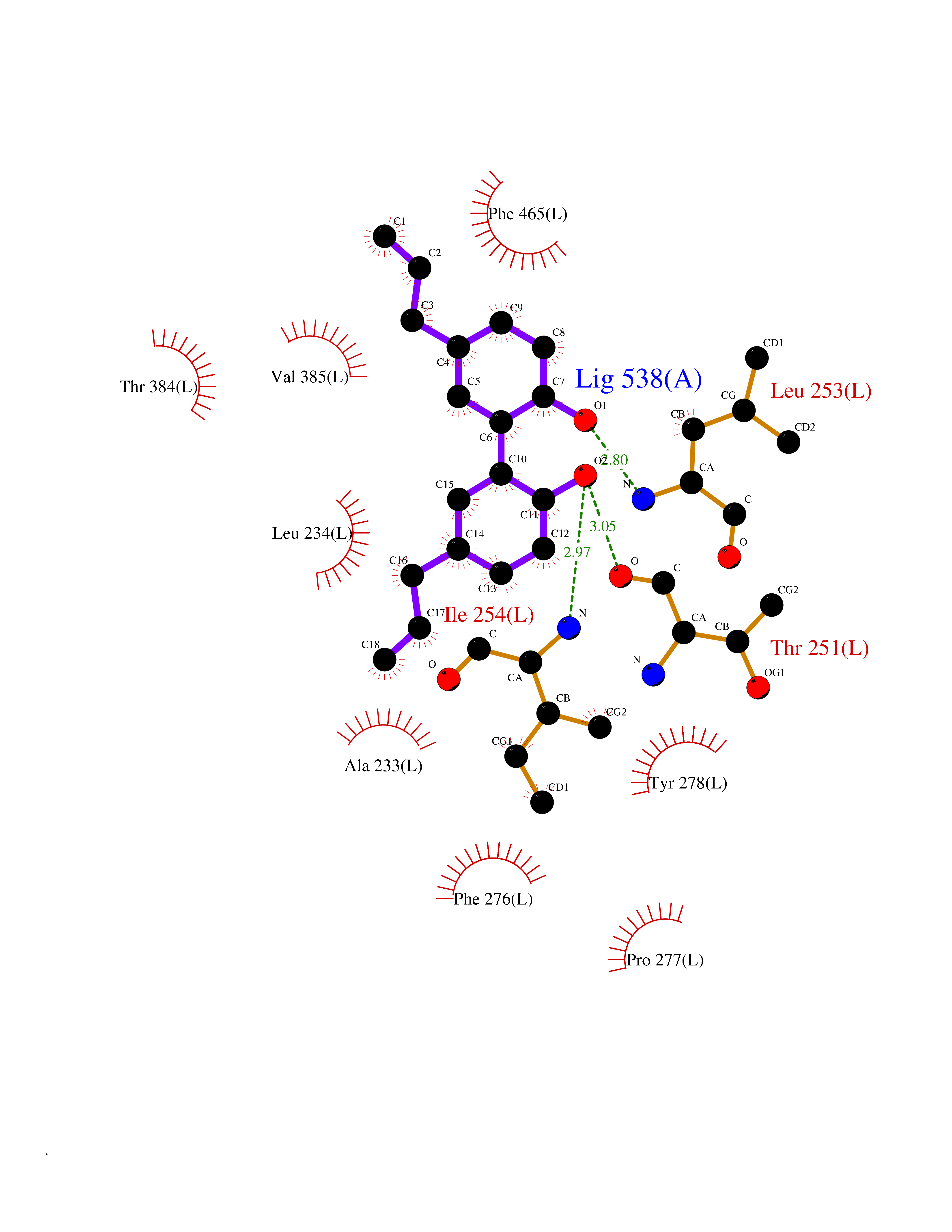 Ligplot Map 3D Binding mode Sequence MKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFDGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIPMKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFVGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIP Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Adrenergic receptor alpha-2A (ADRA2A) | 7EJ8 | 7.05 | |
Target general information Gen name ADRA2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Alpha-2AAR; Alpha-2A adrenoreceptor; Alpha-2A adrenoceptor; Alpha-2A adrenergic receptor; Alpha-2 adrenergic receptor subtype C10; ADRAR; ADRA2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRA2A sub-subfamily Biochemical class GPCR rhodopsin Function The rank order of potency for agonists of this receptor is oxymetazoline > clonidine > epinephrine > norepinephrine > phenylephrine > dopamine > p-synephrine > p-tyramine > serotonin = p-octopamine. For antagonists, the rank order is yohimbine > phentolamine = mianserine > chlorpromazine = spiperone = prazosin > propanolol > alprenolol = pindolol. Alpha-2 adrenergic receptors mediate the catecholamine-induced inhibition of adenylate cyclase through the action of G proteins. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00321; DB00543; DB00182; DB00714; DB00964; DB09229; DB01238; DB14185; DB06216; DB00865; DB00217; DB00484; DB01200; DB00248; DB01136; DB04846; DB00477; DB09202; DB00575; DB00363; DB01151; DB00633; DB01576; DB11273; DB13345; DB00320; DB00449; DB11278; DB09167; DB04855; DB06262; DB01363; DB05492; DB00751; DB00668; DB01049; DB00696; DB01175; DB06678; DB09194; DB00800; DB06623; DB00629; DB01018; DB00502; DB11577; DB00555; DB06707; DB00589; DB04948; DB09195; DB00408; DB08815; DB00934; DB01365; DB01577; DB01403; DB00968; DB06148; DB00370; DB09205; DB09242; DB06711; DB01149; DB00368; DB00540; DB06229; DB00935; DB01267; DB00715; DB01186; DB01608; DB00925; DB00692; DB00397; DB09286; DB09244; DB06153; DB00413; DB00457; DB00433; DB01069; DB00852; DB01224; DB11124; DB11738; DB00268; DB09304; DB06764; DB13025; DB00697; DB00797; DB00193; DB00656; DB00726; DB11477; DB06694; DB01392; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Methylation; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 30303.9 Length 263 Aromaticity 0.16 Instability index 35.08 Isoelectric point 9.66 Charge (pH=7) 16.82 2D Binding mode Binding energy (Kcal/mol) -9.61 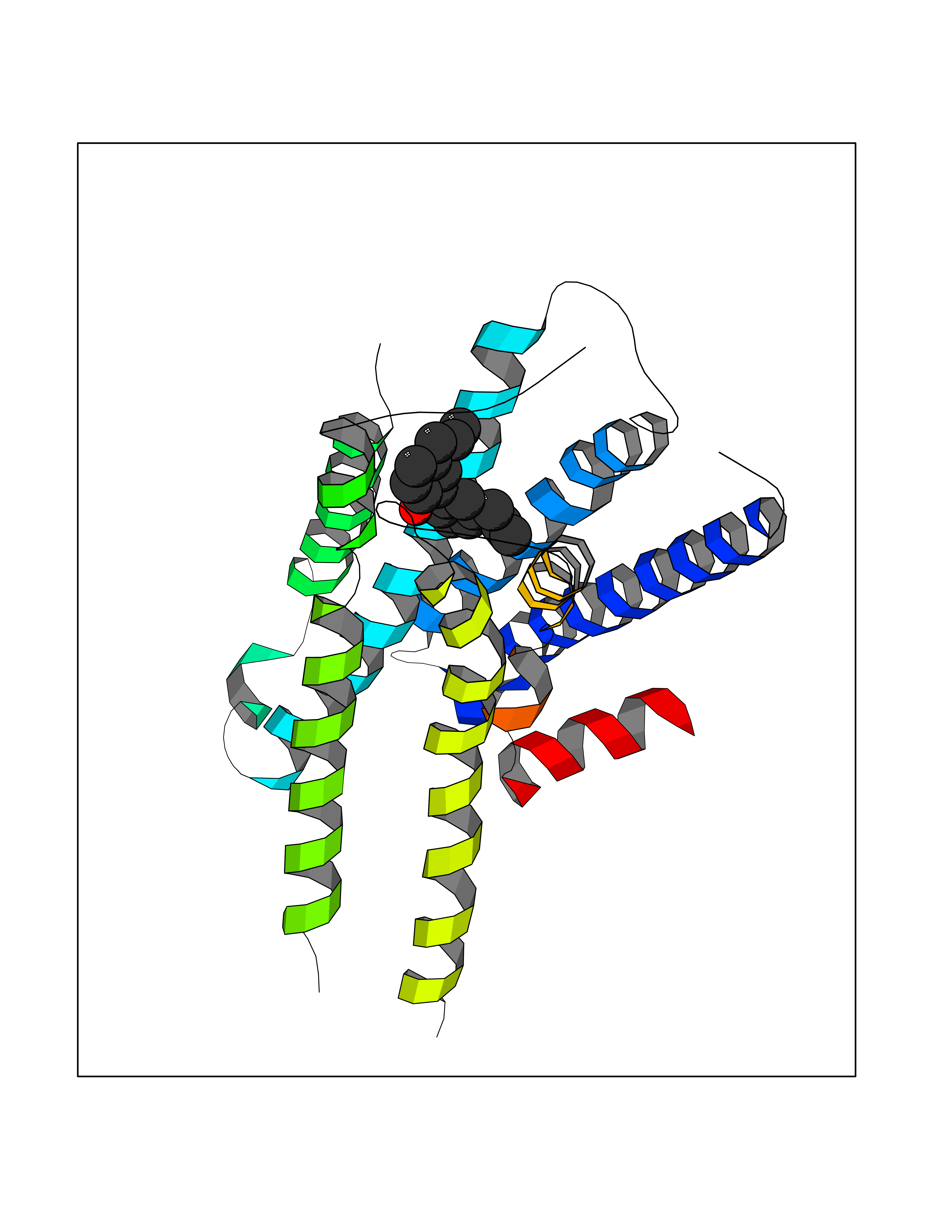 Molscript Map 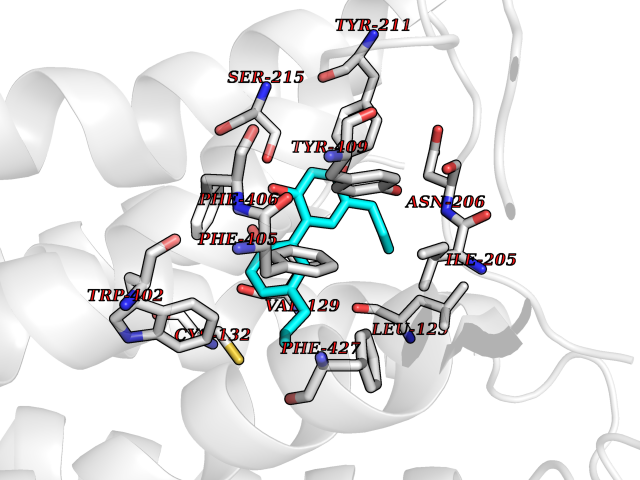 Pymol Map 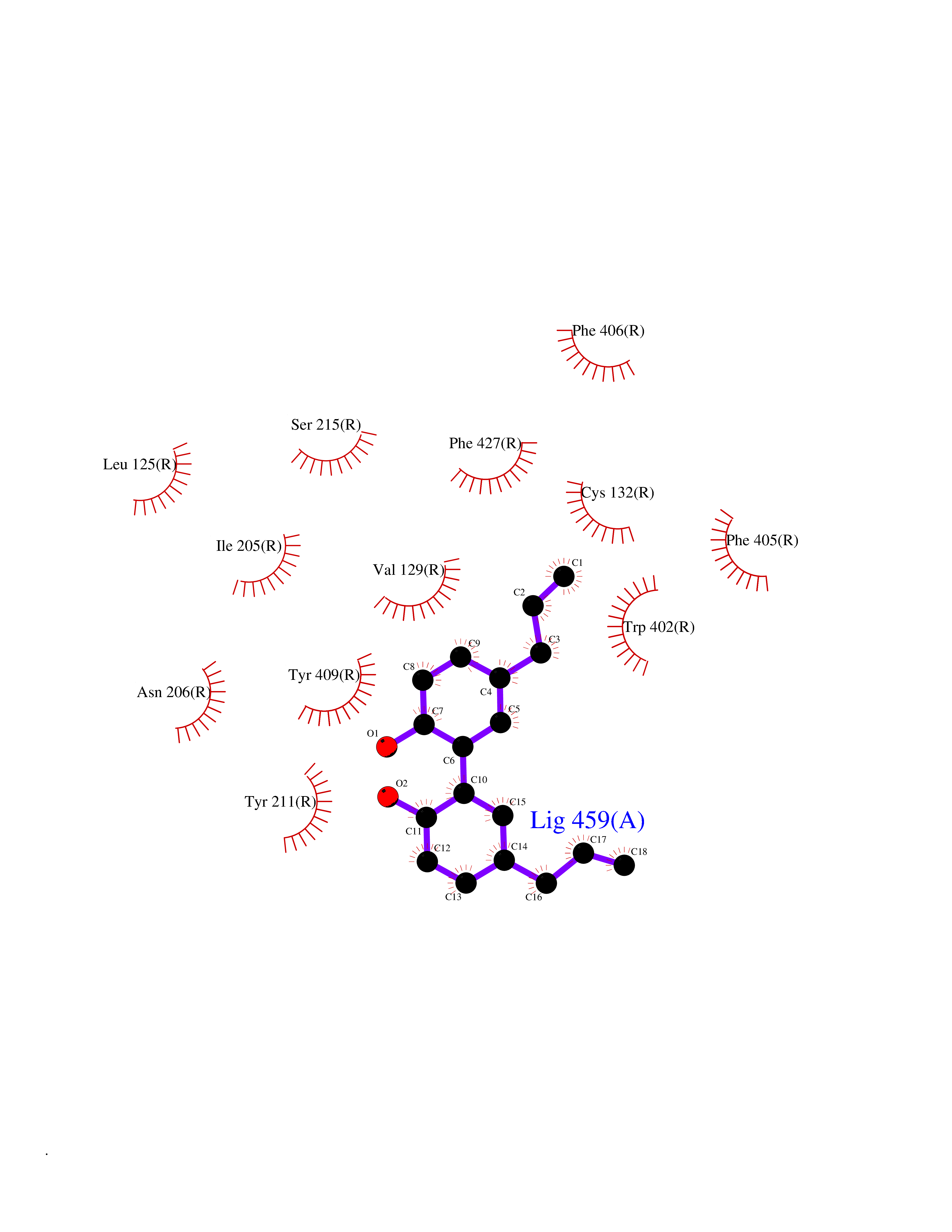 Ligplot Map 3D Binding mode Sequence YSLQVTLTLVCLAGLLMLLTVFGNVLVIIAVFTSRALKAPQNLFLVSLASADILVATLVIPFSLANEVMGYWYFGKAWCEIYLALDVLFCTSSIVHLCAISLDRYWSITQAIEYNLKRTPRRIKAIIITVWVISAVISFPPRCEINDQKWYVISSCIGSFFAPCLIMILVYVRIYQIAKRRTRRGRQNREKRFTFVLAVVIGVFVVCWFPFFFTYTLTAVGCSVPRTLFKFFFWFGYCNSSLNPVIYTIFNHDFRRAFKKILC Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 7.05 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -9.61 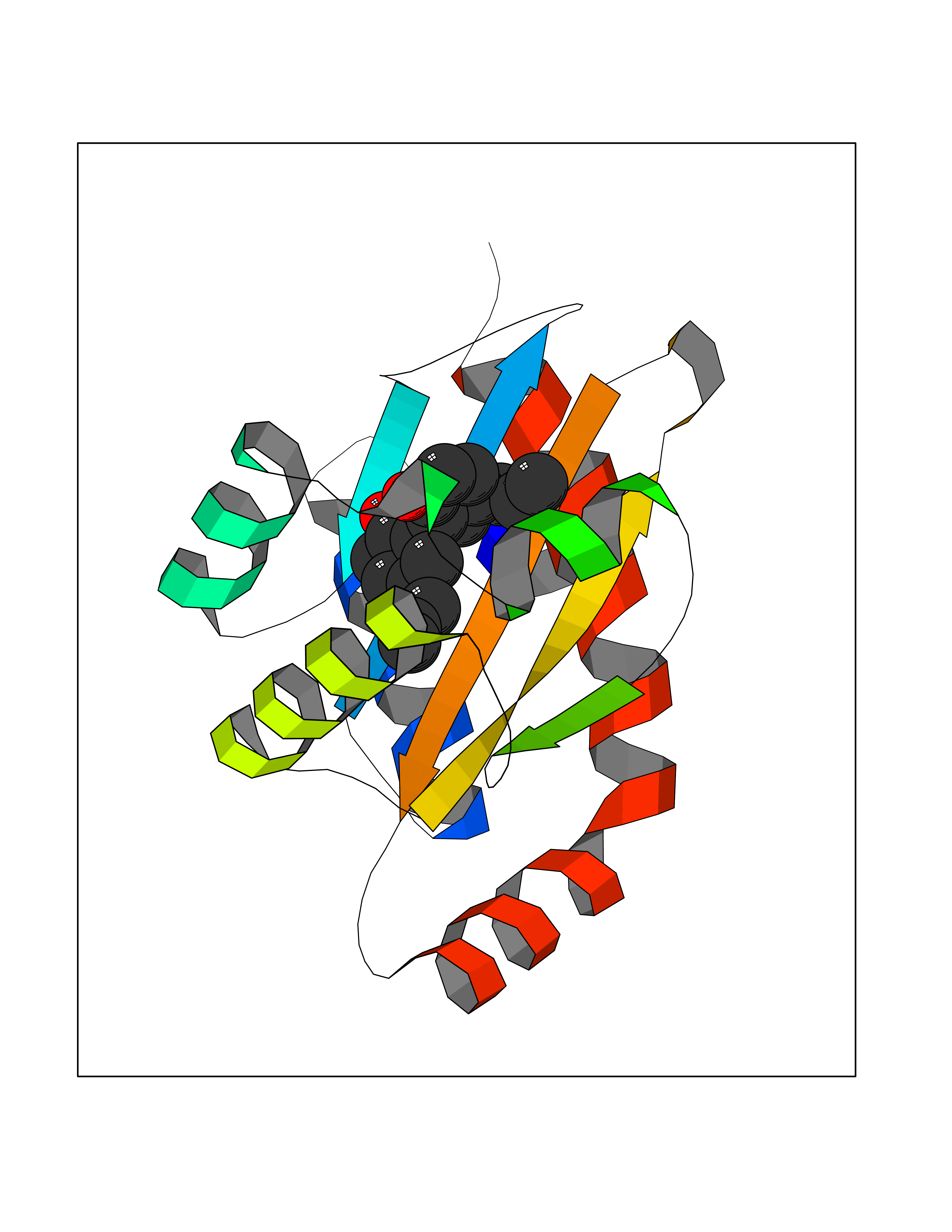 Molscript Map 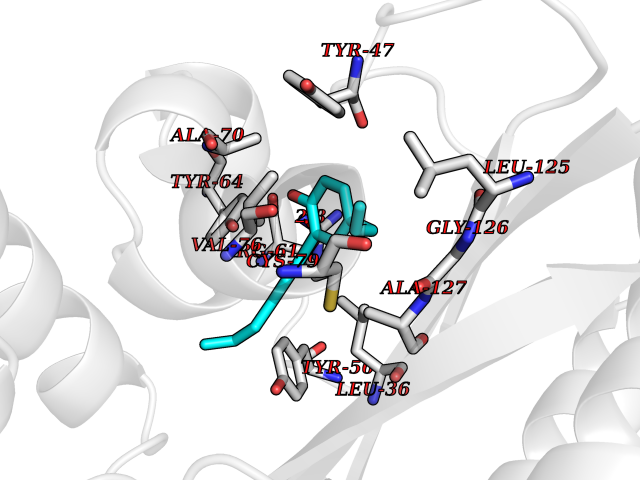 Pymol Map 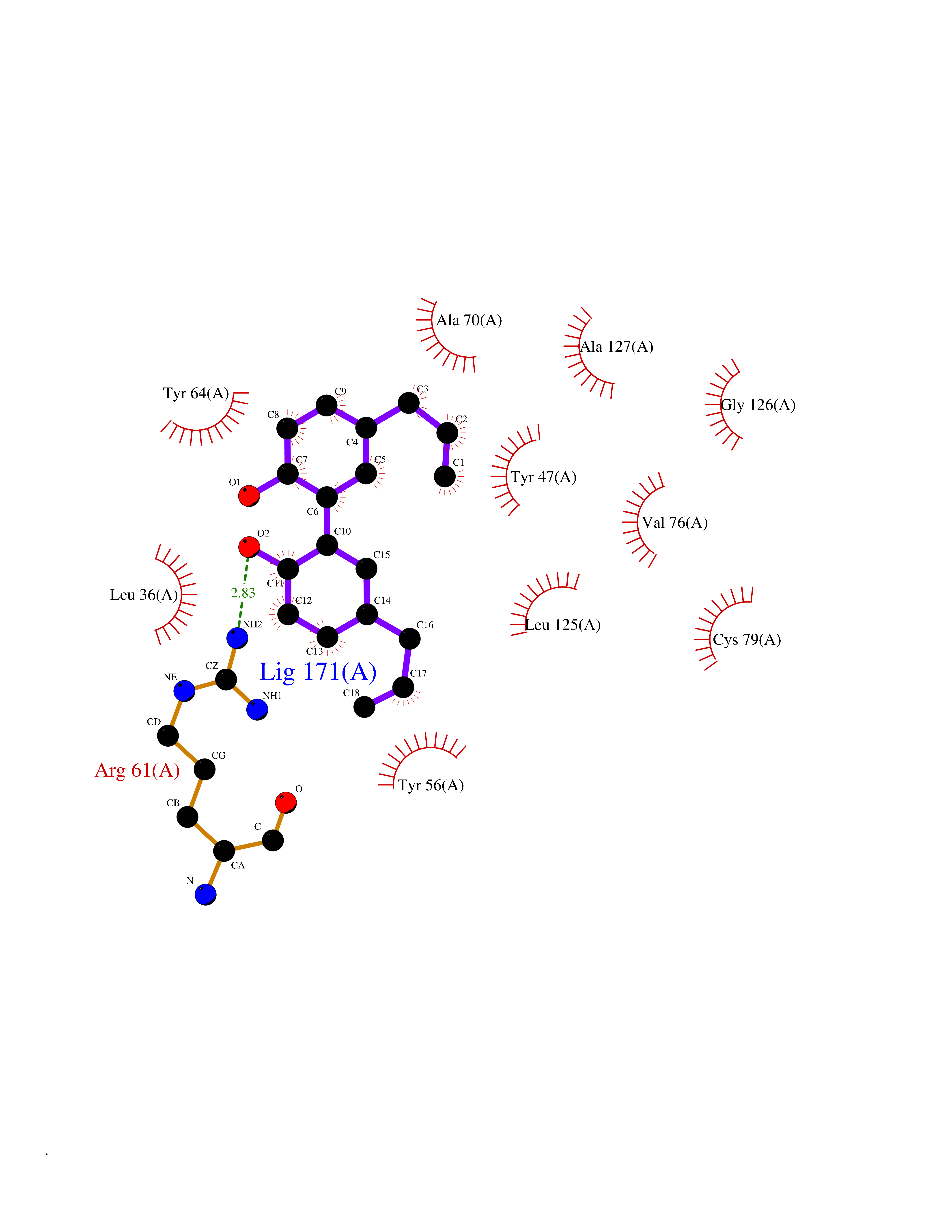 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | SEC14-like protein 2 | 4OMJ | 7.04 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -9.6 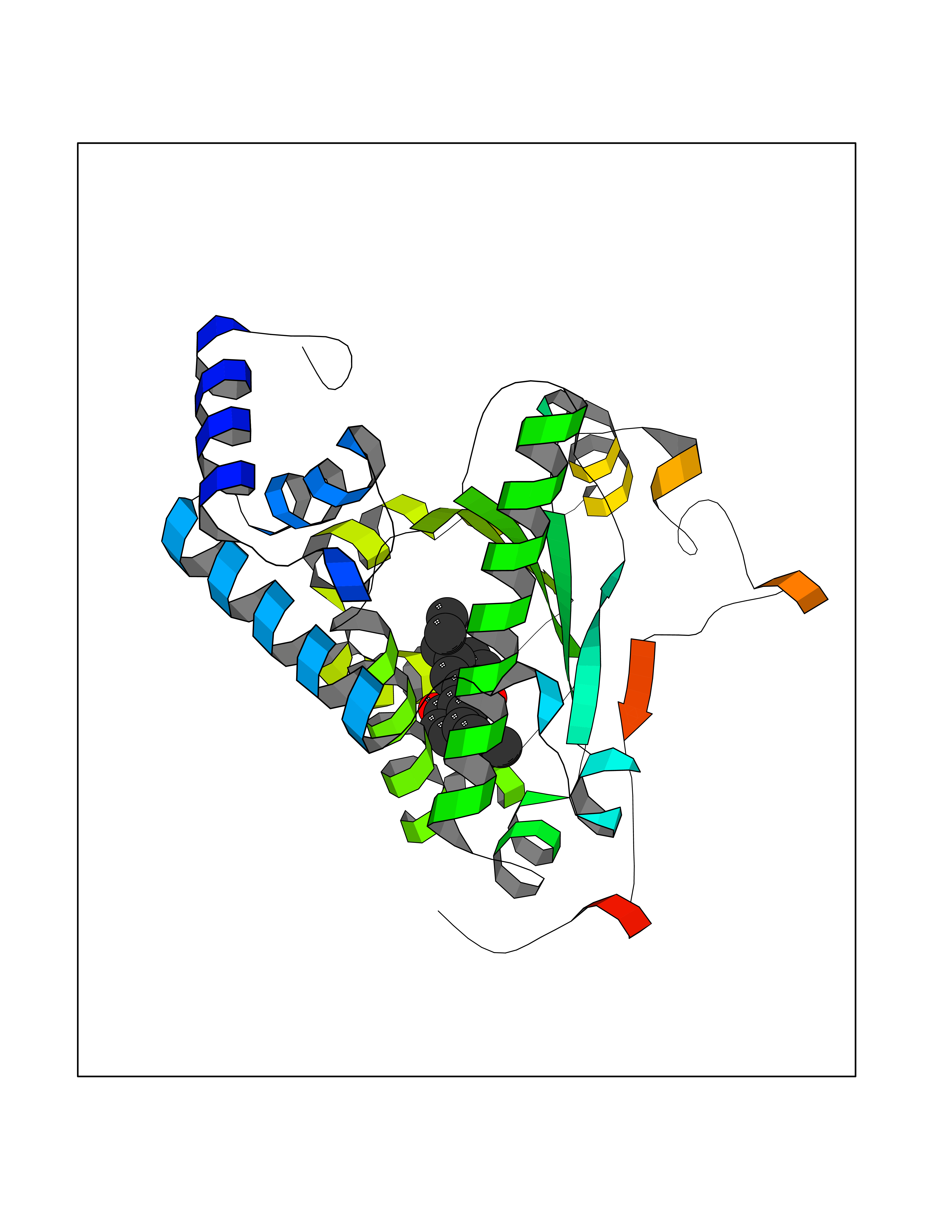 Molscript Map 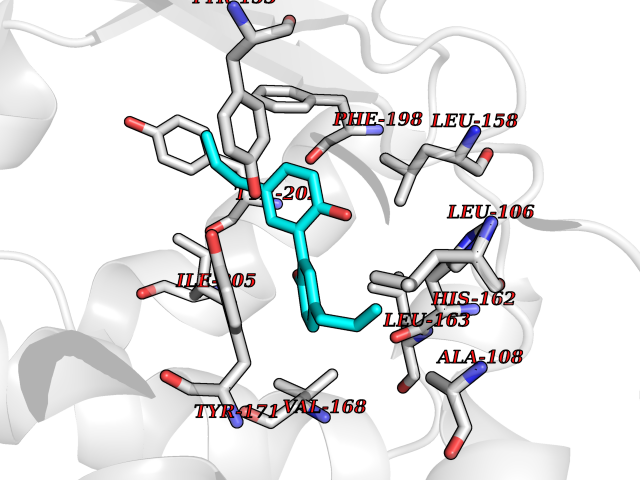 Pymol Map 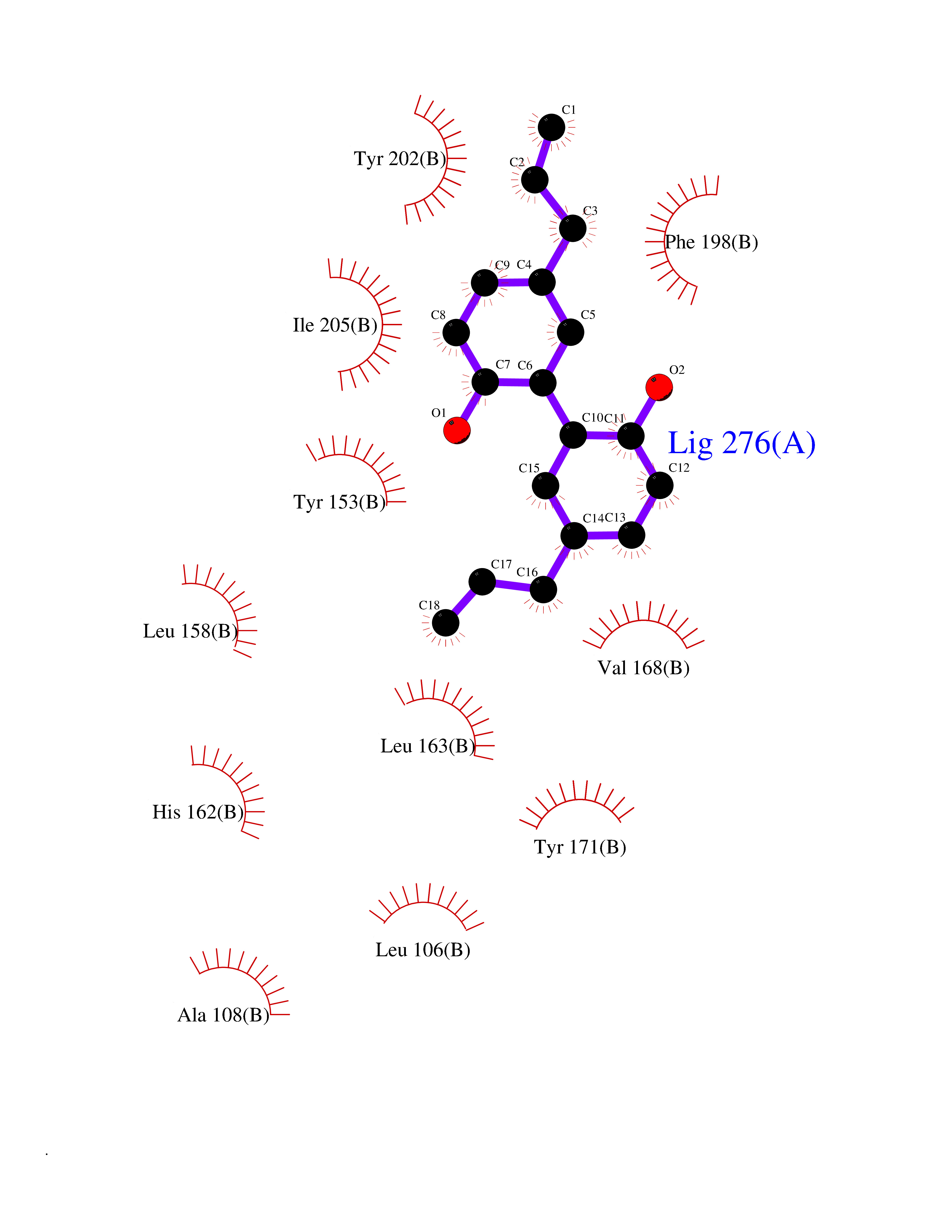 Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Monoglyceride lipase (MAGL) | 3PE6 | 7.04 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -9.6 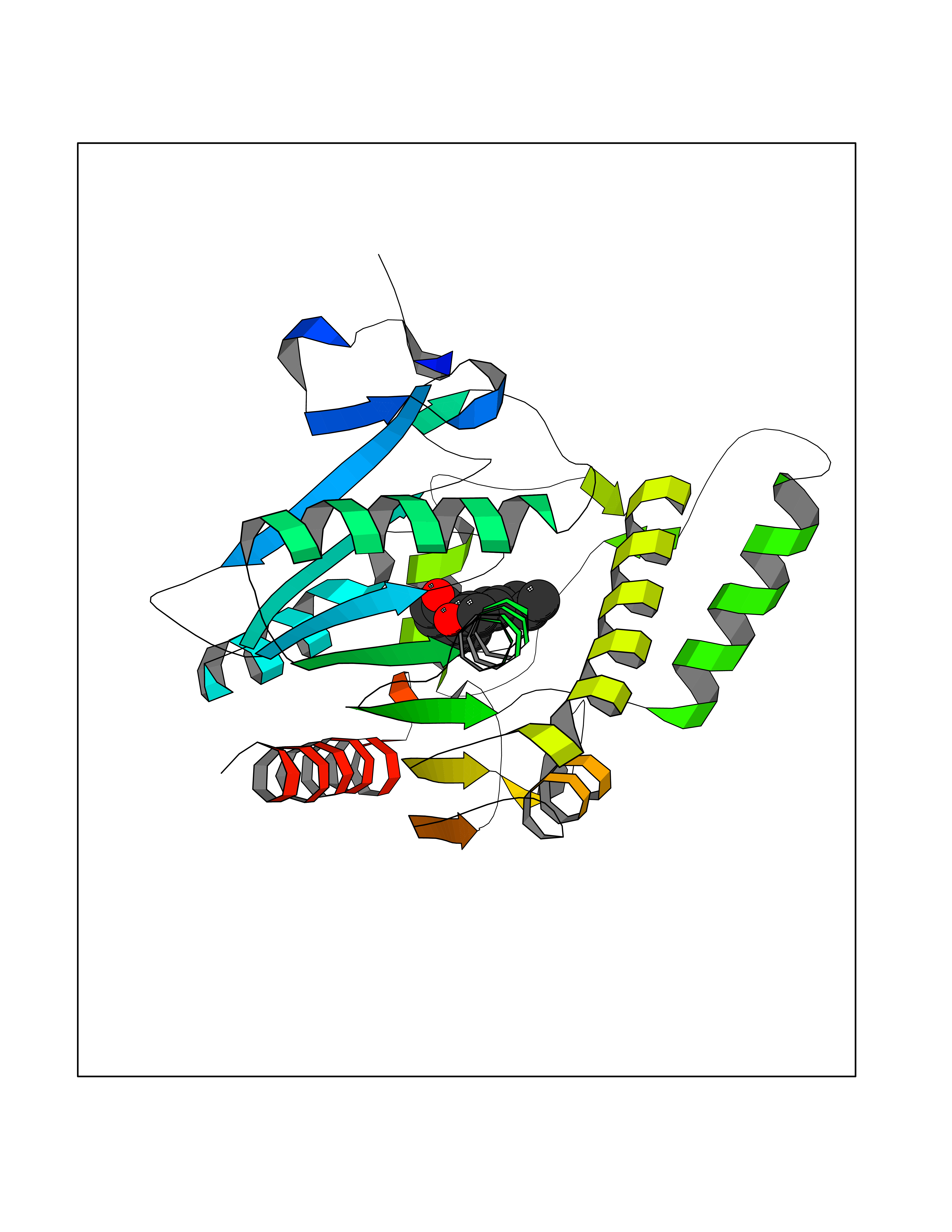 Molscript Map 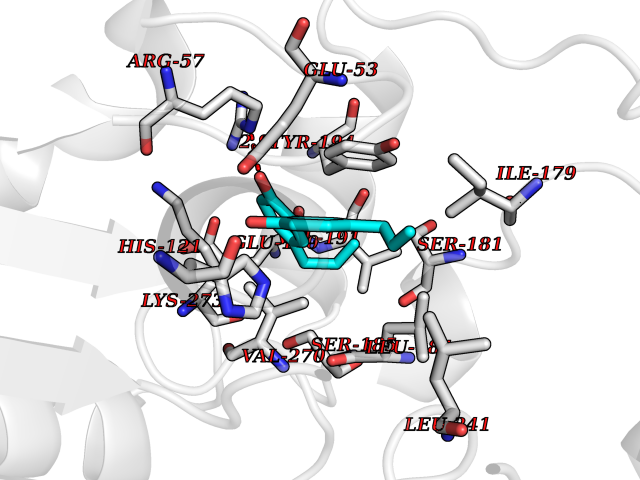 Pymol Map 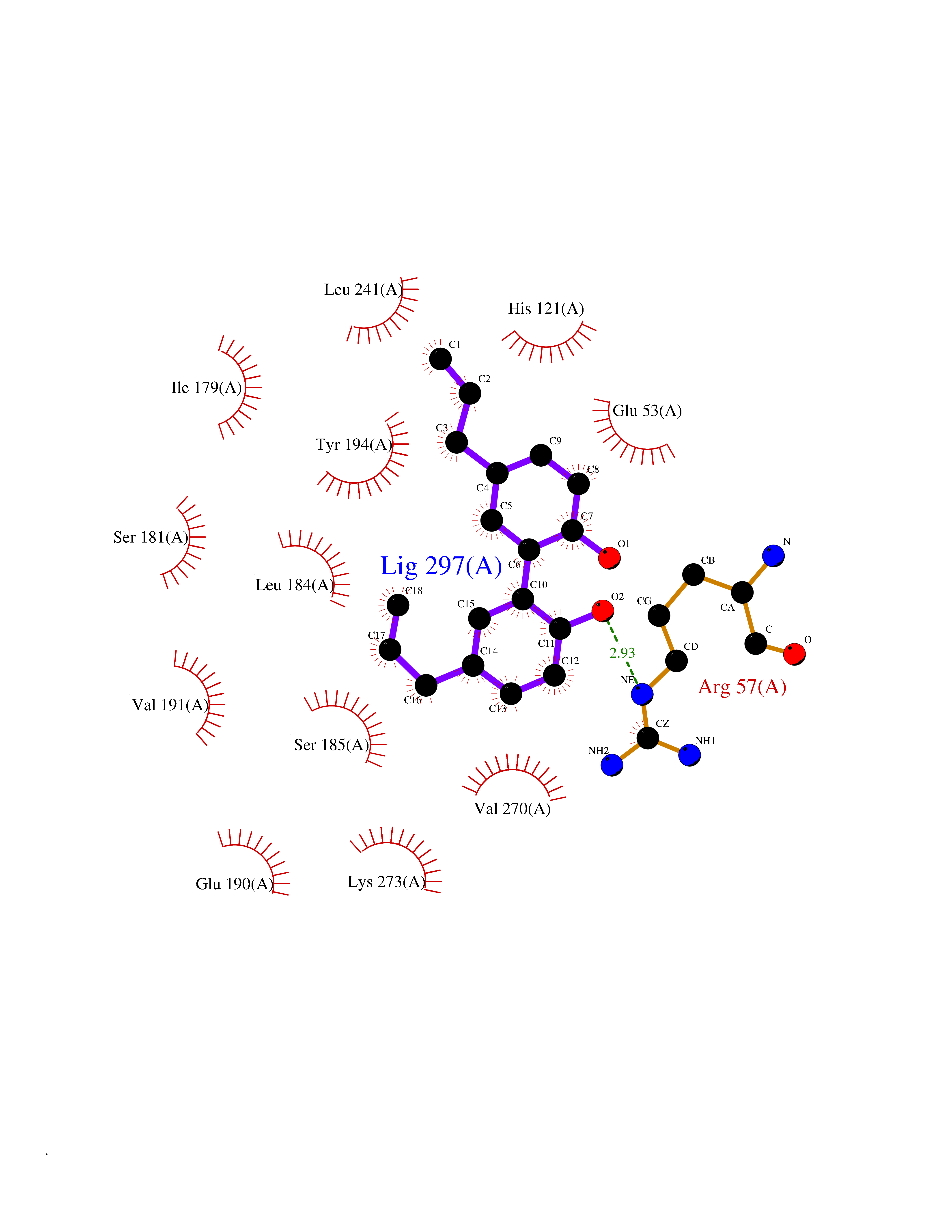 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Beta-arrestin-1 (ARRB1) | 6TKO | 7.04 | |
Target general information Gen name ARRB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Non-visual arrestin-2; Betaarrestin1; Arrestin beta1; Arrestin beta-1; ARR1 Protein family Arrestin family Biochemical class Arrestin protein Function During homologous desensitization, beta-arrestins bind to the GPRK-phosphorylated receptor and sterically preclude its coupling to the cognate G-protein; the binding appears to require additional receptor determinants exposed only in the active receptor conformation. The beta-arrestins target many receptors for internalization by acting as endocytic adapters (CLASPs, clathrin-associated sorting proteins) and recruiting the GPRCs to the adapter protein 2 complex 2 (AP-2) in clathrin-coated pits (CCPs). However, the extent of beta-arrestin involvement appears to vary significantly depending on the receptor, agonist and cell type. Internalized arrestin-receptor complexes traffic to intracellular endosomes, where they remain uncoupled from G-proteins. Two different modes of arrestin-mediated internalization occur. Class A receptors, like ADRB2, OPRM1, ENDRA, D1AR and ADRA1B dissociate from beta-arrestin at or near the plasma membrane and undergo rapid recycling. Class B receptors, like AVPR2, AGTR1, NTSR1, TRHR and TACR1 internalize as a complex with arrestin and traffic with it to endosomal vesicles, presumably as desensitized receptors, for extended periods of time. Receptor resensitization then requires that receptor-bound arrestin is removed so that the receptor can be dephosphorylated and returned to the plasma membrane. Involved in internalization of P2RY4 and UTP-stimulated internalization of P2RY2. Involved in phosphorylation-dependent internalization of OPRD1 ands subsequent recycling. Involved in the degradation of cAMP by recruiting cAMP phosphodiesterases to ligand-activated receptors. Beta-arrestins function as multivalent adapter proteins that can switch the GPCR from a G-protein signaling mode that transmits short-lived signals from the plasma membrane via small molecule second messengers and ion channels to a beta-arrestin signaling mode that transmits a distinct set of signals that are initiated as the receptor internalizes and transits the intracellular compartment. Acts as signaling scaffold for MAPK pathways such as MAPK1/3 (ERK1/2). ERK1/2 activated by the beta-arrestin scaffold is largely excluded from the nucleus and confined to cytoplasmic locations such as endocytic vesicles, also called beta-arrestin signalosomes. Recruits c-Src/SRC to ADRB2 resulting in ERK activation. GPCRs for which the beta-arrestin-mediated signaling relies on both ARRB1 and ARRB2 (codependent regulation) include ADRB2, F2RL1 and PTH1R. For some GPCRs the beta-arrestin-mediated signaling relies on either ARRB1 or ARRB2 and is inhibited by the other respective beta-arrestin form (reciprocal regulation). Inhibits ERK1/2 signaling in AGTR1- and AVPR2-mediated activation (reciprocal regulation). Is required for SP-stimulated endocytosis of NK1R and recruits c-Src/SRC to internalized NK1R resulting in ERK1/2 activation, which is required for the antiapoptotic effects of SP. Is involved in proteinase-activated F2RL1-mediated ERK activity. Acts as signaling scaffold for the AKT1 pathway. Is involved in alpha-thrombin-stimulated AKT1 signaling. Is involved in IGF1-stimulated AKT1 signaling leading to increased protection from apoptosis. Involved in activation of the p38 MAPK signaling pathway and in actin bundle formation. Involved in F2RL1-mediated cytoskeletal rearrangement and chemotaxis. Involved in AGTR1-mediated stress fiber formation by acting together with GNAQ to activate RHOA. Appears to function as signaling scaffold involved in regulation of MIP-1-beta-stimulated CCR5-dependent chemotaxis. Involved in attenuation of NF-kappa-B-dependent transcription in response to GPCR or cytokine stimulation by interacting with and stabilizing CHUK. May serve as nuclear messenger for GPCRs. Involved in OPRD1-stimulated transcriptional regulation by translocating to CDKN1B and FOS promoter regions and recruiting EP300 resulting in acetylation of histone H4. Involved in regulation of LEF1 transcriptional activity via interaction with DVL1 and/or DVL2 Also involved in regulation of receptors other than GPCRs. Involved in Toll-like receptor and IL-1 receptor signaling through the interaction with TRAF6 which prevents TRAF6 autoubiquitination and oligomerization required for activation of NF-kappa-B and JUN. Binds phosphoinositides. Binds inositolhexakisphosphate (InsP6). Involved in IL8-mediated granule release in neutrophils. Required for atypical chemokine receptor ACKR2-induced RAC1-LIMK1-PAK1-dependent phosphorylation of cofilin (CFL1) and for the up-regulation of ACKR2 from endosomal compartment to cell membrane, increasing its efficiency in chemokine uptake and degradation. Involved in the internalization of the atypical chemokine receptor ACKR3. Negatively regulates the NOTCH signaling pathway by mediating the ubiquitination and degradation of NOTCH1 by ITCH. Participates to the recruitment of the ubiquitin-protein ligase to the receptor. Functions in regulating agonist-mediated G-protein coupled receptor (GPCR) signaling by mediating both receptor desensitization and resensitization processes. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P63010-2; O15169; P0DP25; P20963; P25101; P50148; Q5JWF2; Q14749; P06396; Q16665; P11142; Q99683; P53779; P45984; Q00987; P19338; Q14978; P14618; P14859-6; P35813; O75688; Q13523; P06702; P12931; Q15208; Q13428; P04637; P27348; P25490; O43298; O95218; Q7DB77 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Coated pit; Cytoplasm; Cytoplasmic vesicle; Membrane; Nucleus; Phosphoprotein; Protein transport; Proteomics identification; Reference proteome; Signal transduction inhibitor; Transcription; Transcription regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32455.6 Length 293 Aromaticity 0.12 Instability index 28.99 Isoelectric point 9.12 Charge (pH=7) 9.86 2D Binding mode Binding energy (Kcal/mol) -9.6 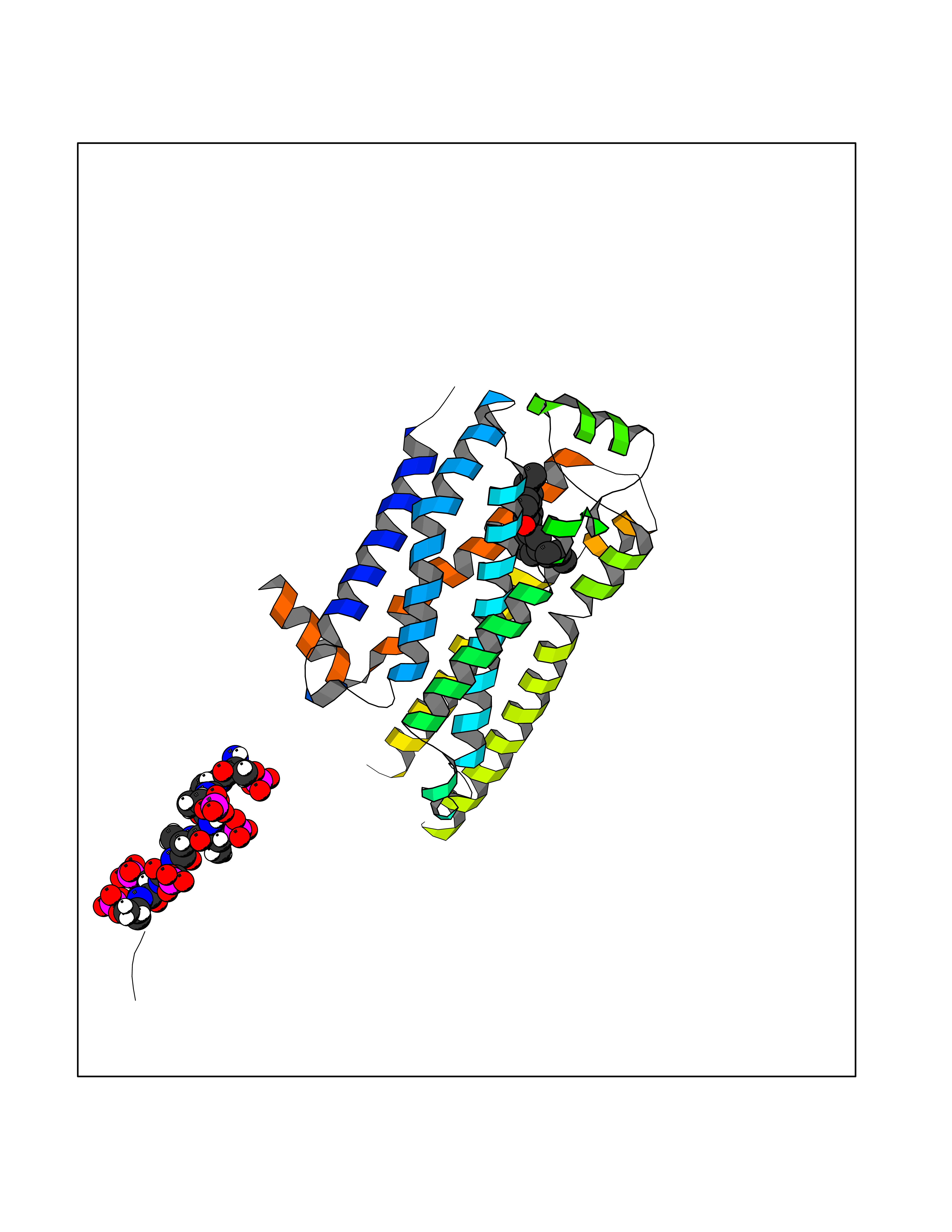 Molscript Map 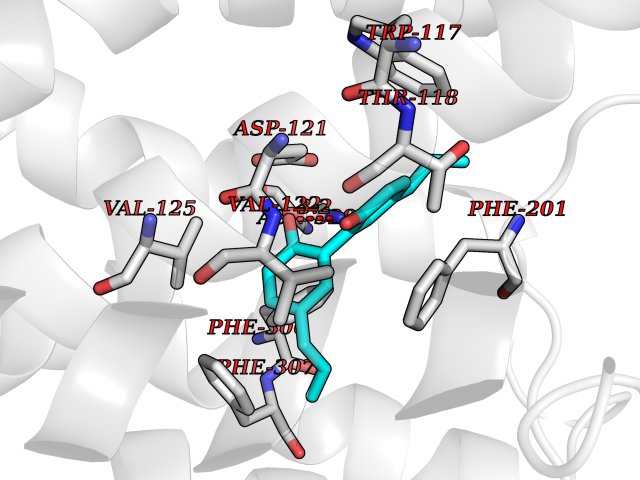 Pymol Map 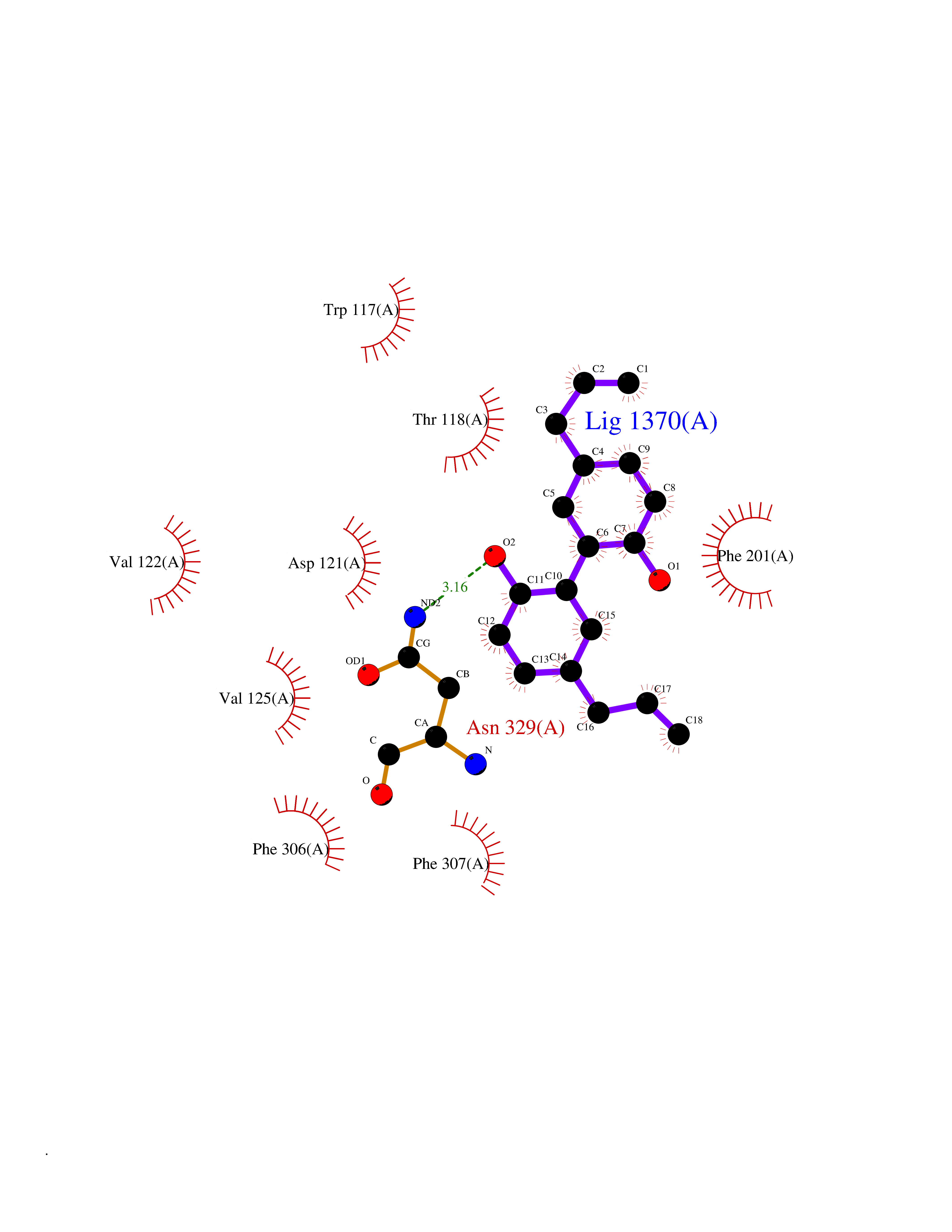 Ligplot Map 3D Binding mode Sequence AGCSLLMALVVLLIVAGNVLVIAAIGRTQRLQTLTNLFITSLACADLVVGLLVVPFGATLVCRGTWLWGSFLCELWTSLDVLCVTASIWTLCVIAIDRYLAITSPFRYQSLMTRARAKVIICTVWAISALVSFLPIMMHWWRDEDPQALKCYQDPGCCDFVTNRAYAIASSIISFYIPLLIMIFVYLRVYREAKEQIRKIDVMAMREHKALKTLGIIMGVFTLCWLPFFLVNIVNVFNRDLVPKWLFVAFNWLGYANSAMNPIIYCRSPDFRKAFKRLLAEXAXXAXXXLAKD Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Neuronal acetylcholine receptor beta-4 (CHRNB4) | 6PV7 | 7.04 | |
Target general information Gen name CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNB4; Beta-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-4/CHRNB4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00237; DB00565; DB00514; DB07720; DB00898; DB00472; DB01227; DB00184; DB01090; DB00202 Interacts with Q6FHY5 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -9.6 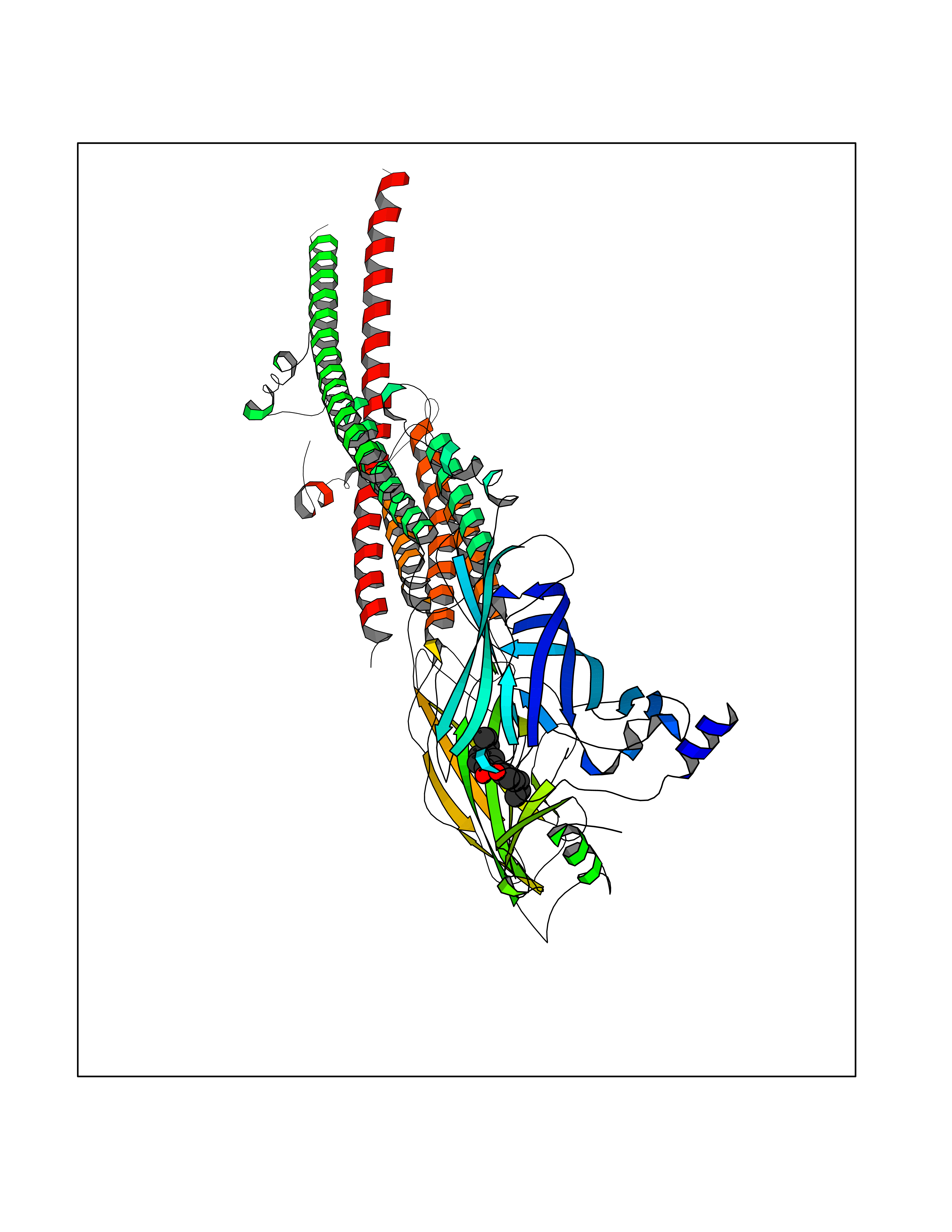 Molscript Map 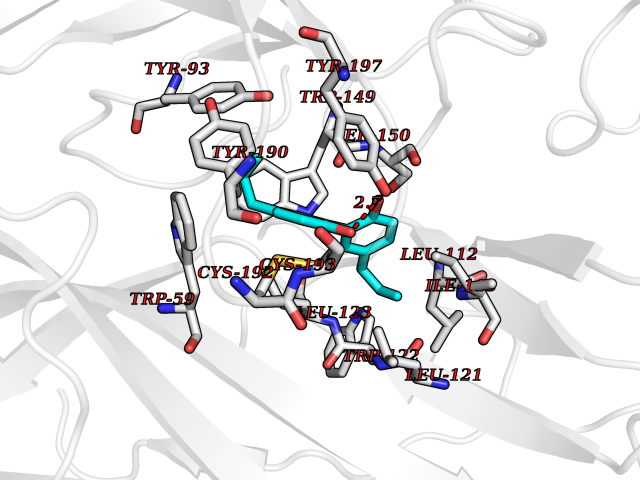 Pymol Map 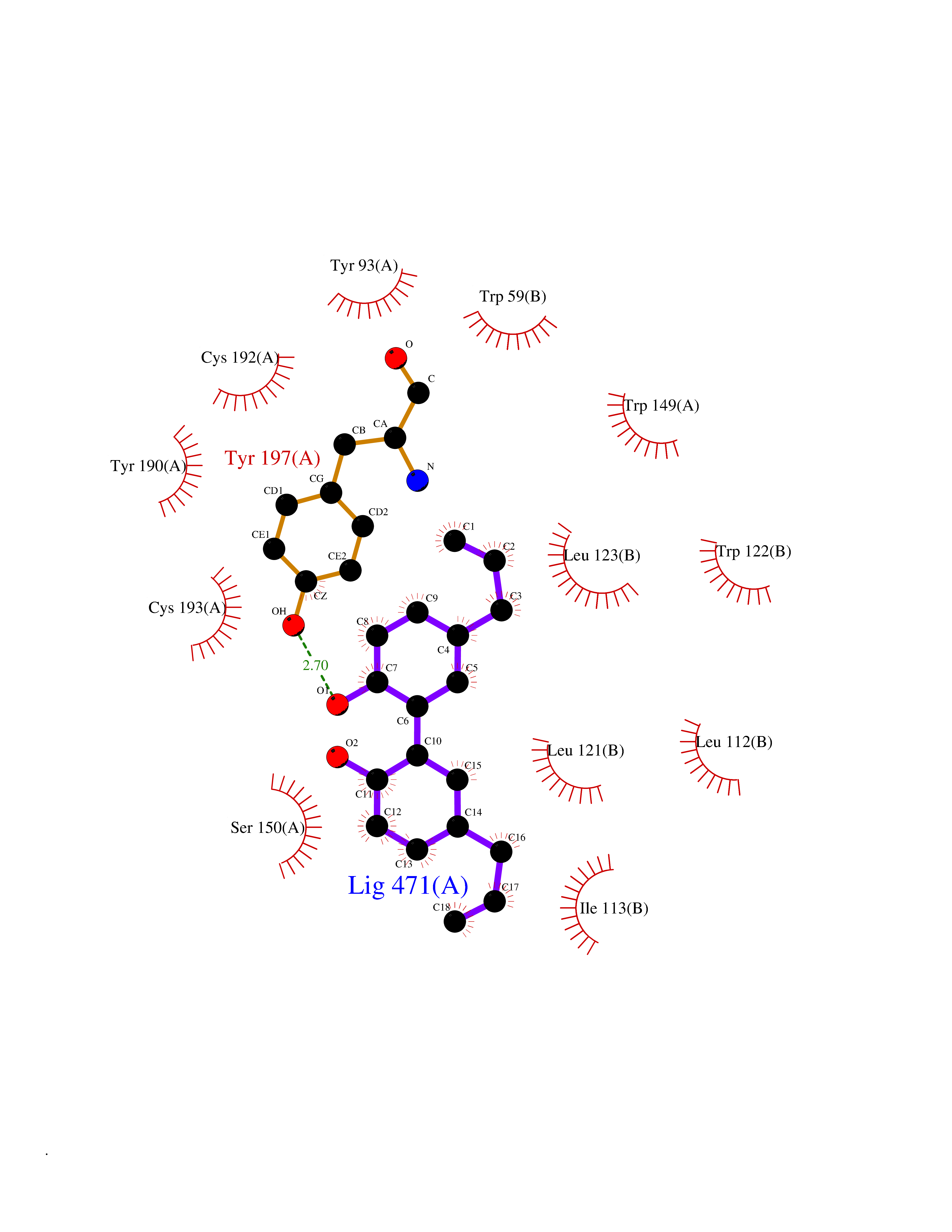 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||