Job Results:
Ligand
Structure
Job ID
43b2a7cfabc12f2b0137afb38036989c
Job name
NA
Time
2025-03-05 09:37:27
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Histamine H3 receptor (H3R) | 7F61 | 4.03 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -5.49 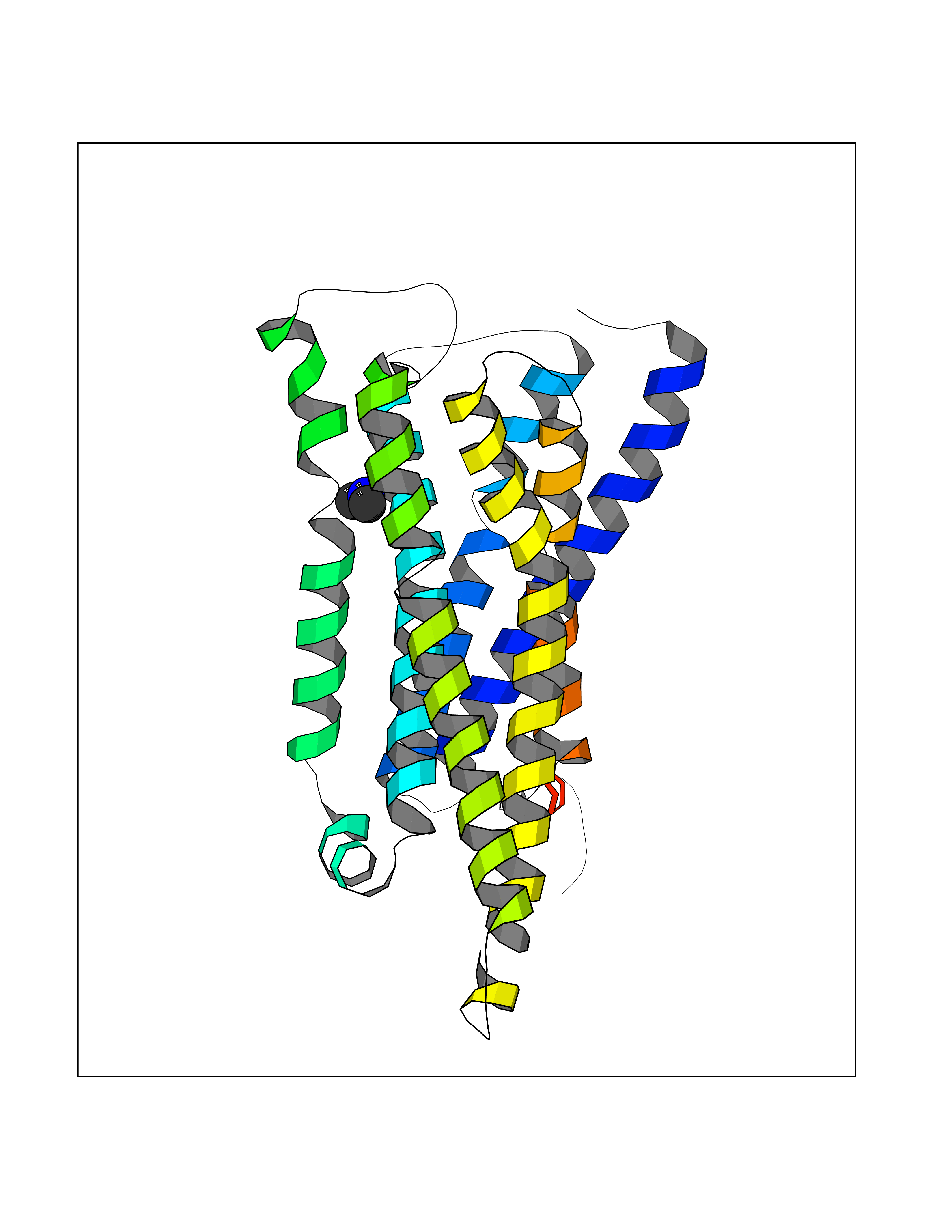 Molscript Map 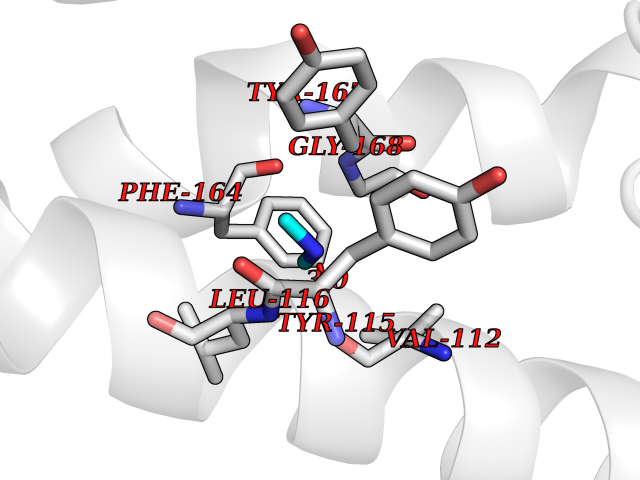 Pymol Map 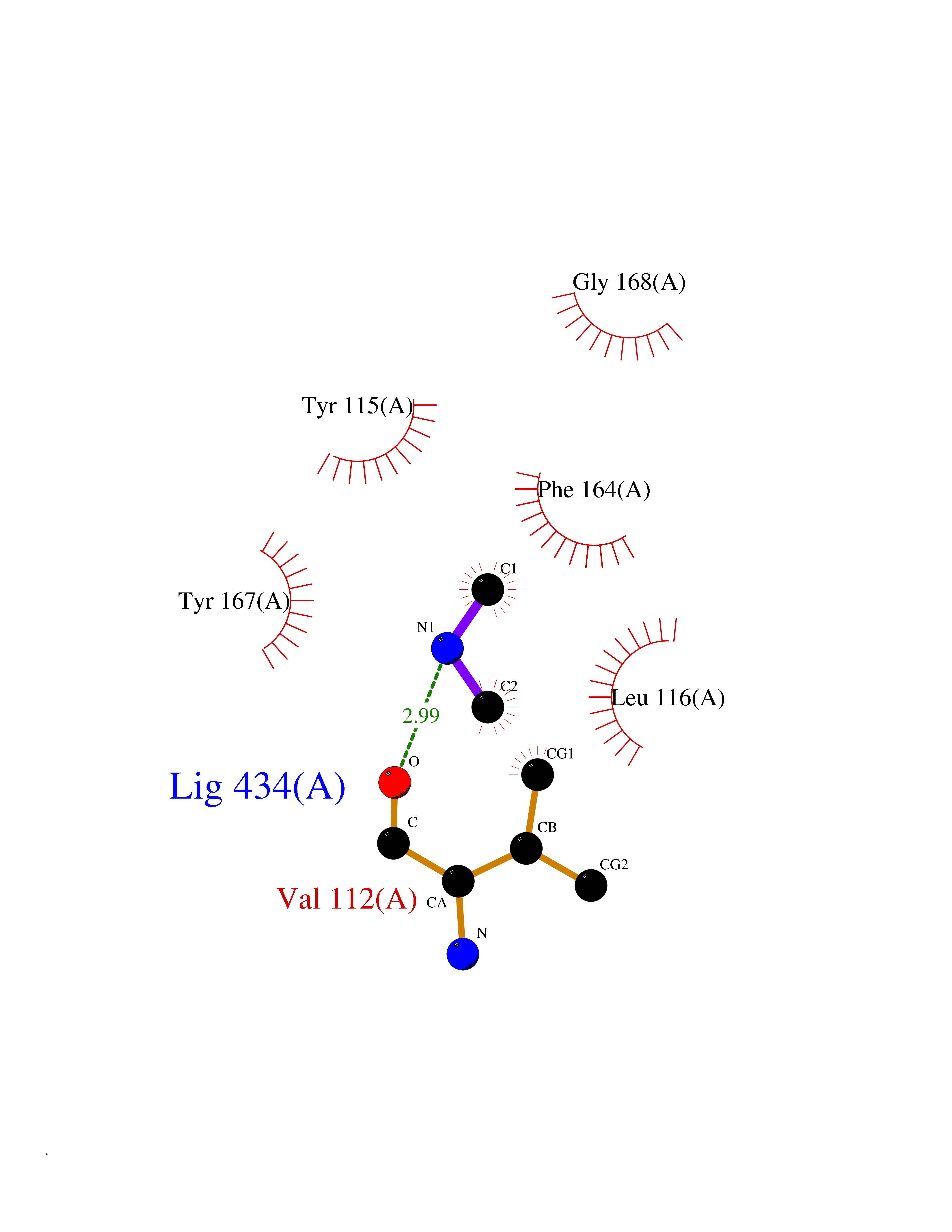 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Deubiquitinating enzyme 1 (USP1) | 7ZH4 | 4.03 | |
Target general information Gen name USP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hUBP; Ubiquitin-specific-processing protease 1; Ubiquitin thioesterase 1; Ubiquitin carboxyl-terminal hydrolase 1 Protein family Peptidase C19 family Biochemical class Peptidase Function Involved in PCNA-mediated translesion synthesis (TLS) by deubiquitinating monoubiquitinated PCNA. Has almost no deubiquitinating activity by itself and requires the interaction with WDR48 to have a high activity. Negative regulator of DNA damage repair which specifically deubiquitinates monoubiquitinated FANCD2. Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8TAF3; Q8TAF3-1 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Autocatalytic cleavage; DNA damage; DNA repair; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID D Molecular weight (Da) 32426 Length 285 Aromaticity 0.1 Instability index 50.73 Isoelectric point 5.85 Charge (pH=7) -4.67 2D Binding mode Binding energy (Kcal/mol) -5.49 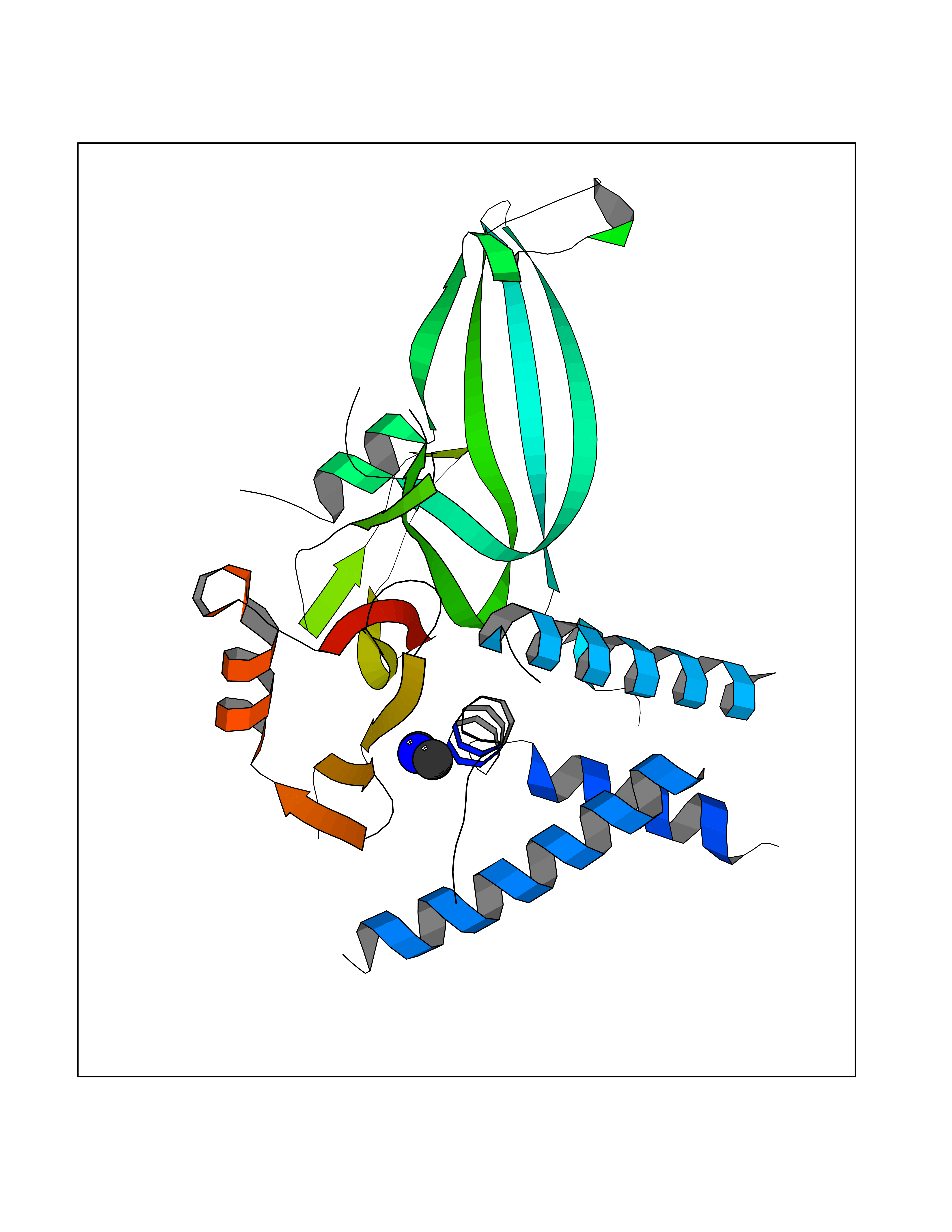 Molscript Map 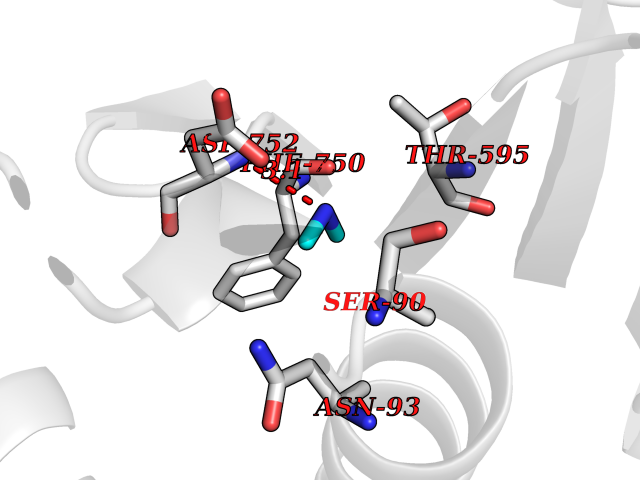 Pymol Map 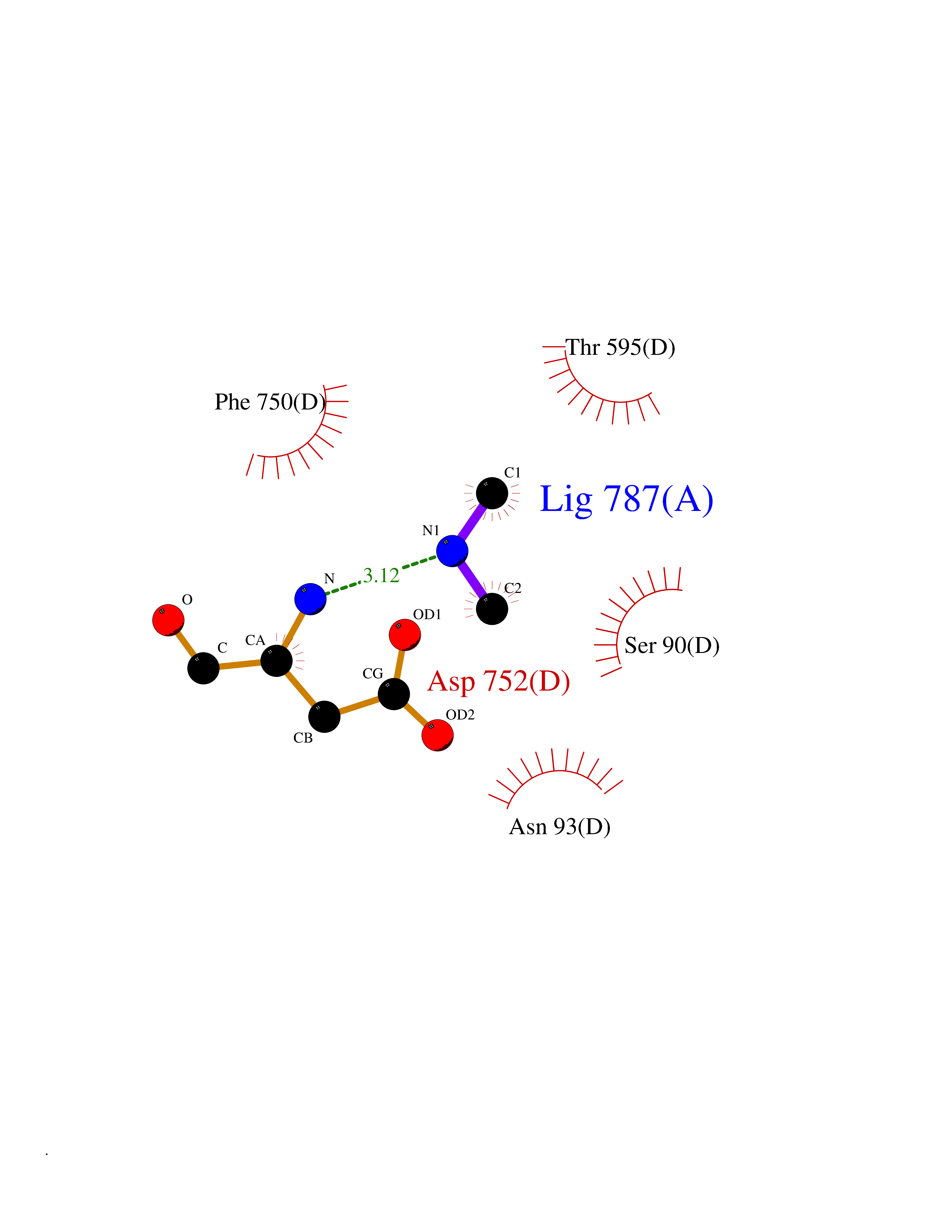 Ligplot Map 3D Binding mode Sequence GLNNLGNTSYLNSILQVLYFCPGFKSGVKHLFNIISRKKYELICSLQSLIISVEQLQASFLLNPLQHDAQEVLQCILGNIQETCQLLKKGFELVEKLFQGQLVLRTRCLECESLTERREDFQDISVPVQEDMKTLRWAISQFASVERIVGEDKYFCENCHHYTEAERSLLFDKMPEVITIHLKCFAASGLSKINTPLLTPLKLSLEEWSTKPTNDSYGLFAVVMHSGITISSGHYTASVKVTYEGKWLLFDDSEVKVTEEKDFLNSLSPSTSPTSTPYLLFYKKL Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Urokinase-type plasminogen activator (PLAU) | 4JNI | 4.02 | |
Target general information Gen name PLAU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UPA; U-plasminogen activator Protein family Peptidase S1 family Biochemical class Peptidase Function Specifically cleaves the zymogen plasminogen to form the active enzyme plasmin. Related diseases Quebec platelet disorder (QPD) [MIM:601709]: An autosomal dominant bleeding disorder due to a gain-of-function defect in fibrinolysis. Although affected individuals do not exhibit systemic fibrinolysis, they show delayed onset bleeding after challenge, such as surgery. The hallmark of the disorder is markedly increased PLAU levels within platelets, which causes intraplatelet plasmin generation and secondary degradation of alpha-granule proteins. {ECO:0000269|PubMed:20007542}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07129; DB07122; DB01905; DB02287; DB03729; DB01725; DB08072; DB07625; DB07626; DB08697; DB03136; DB01977; DB07076; DB03082; DB02705; DB02473; DB02398; DB02551; DB03865; DB06855; DB06856; DB03046; DB04059; DB04172; DB00594; DB03127; DB02526; DB03159; DB05254; DB03782; DB06857; DB16701; DB03876; DB03476 Interacts with Q9UKQ2; P05067; Q03405-1; P05121; P55000 EC number EC 3.4.21.73 Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Direct protein sequencing; Disulfide bond; EGF-like domain; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Phosphoprotein; Plasminogen activation; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID U Molecular weight (Da) 25825.3 Length 229 Aromaticity 0.1 Instability index 47.36 Isoelectric point 8.65 Charge (pH=7) 5.38 2D Binding mode Binding energy (Kcal/mol) -5.48 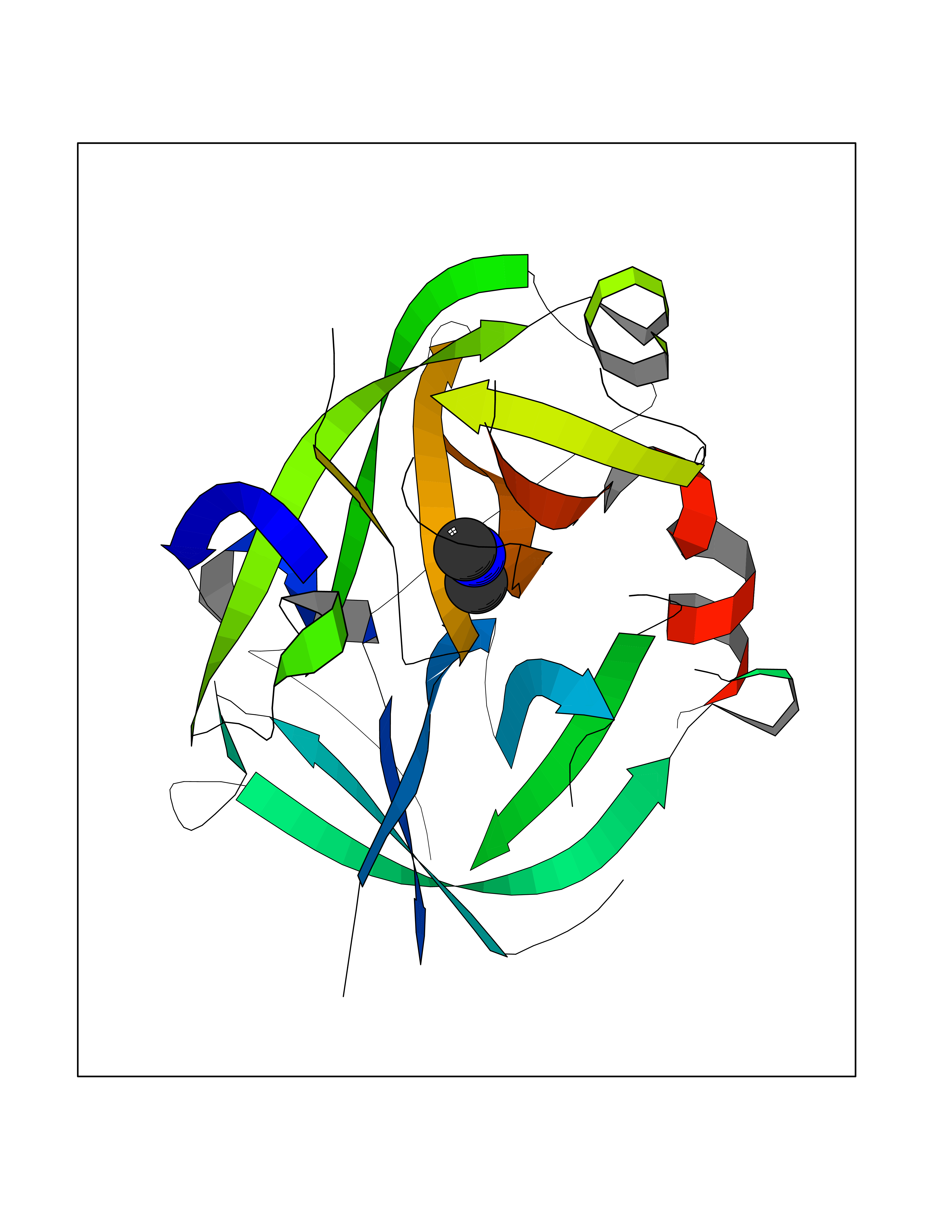 Molscript Map 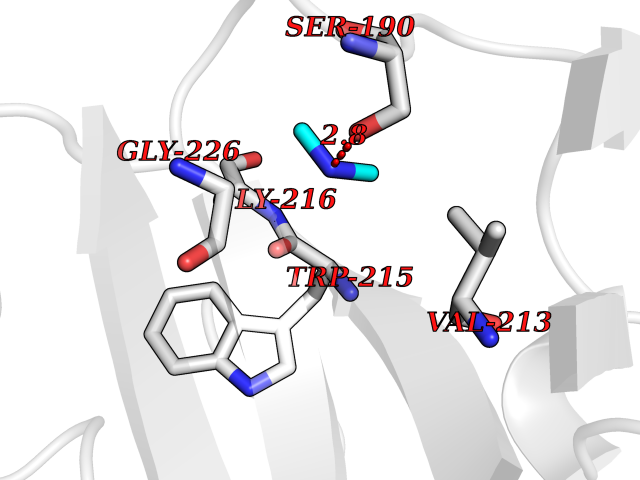 Pymol Map 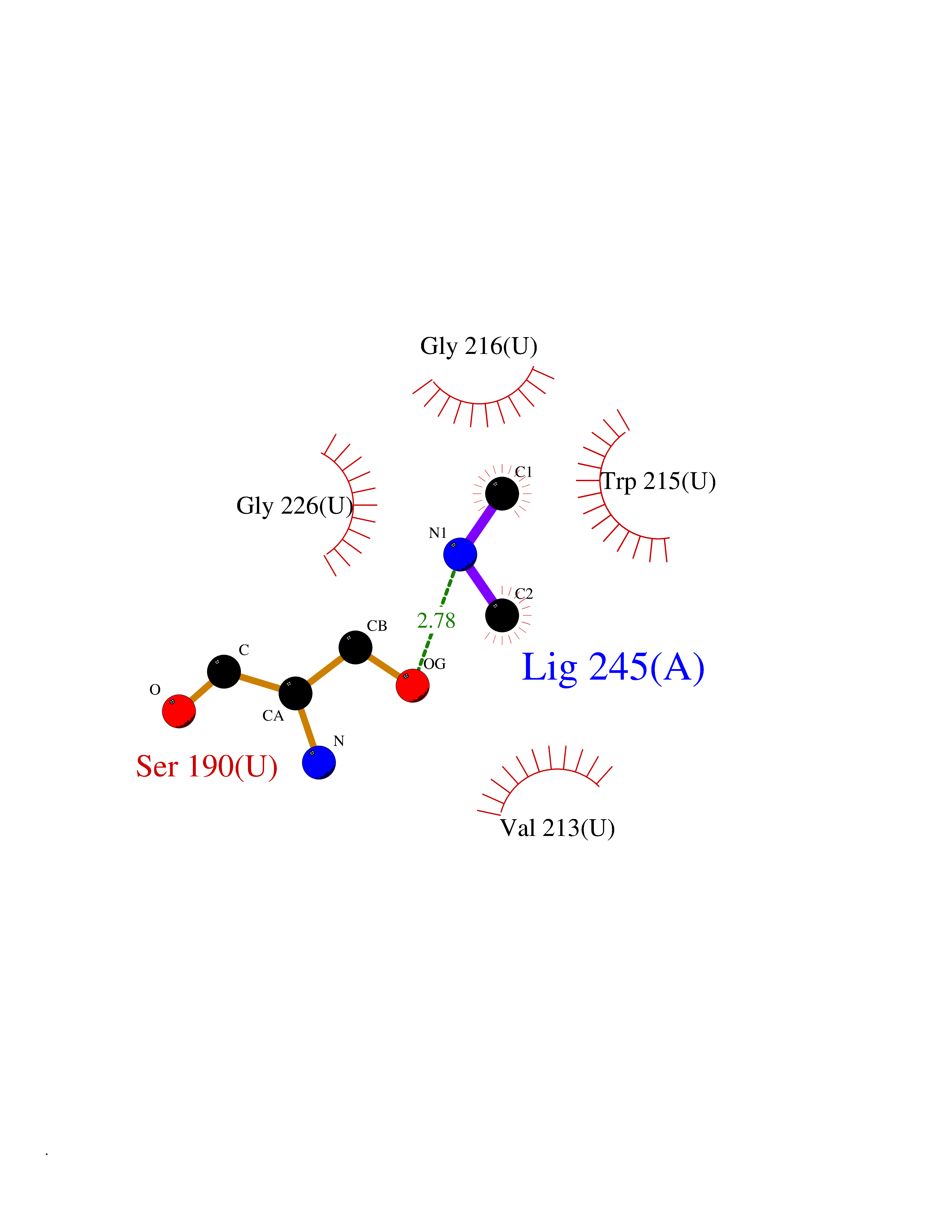 Ligplot Map 3D Binding mode Sequence IIGGEFTTIENQPWFAAIYRRSVTYVCGGSLISPCWVISATHCFPKKEDYIVYLGRSRLNSNTQGEMKFEVENLILHKDYSALAHHNDIALLKIRRCAQPSRTIQTIALPSMYNDPQFGTSCEITGFGKEQSTDYLYPEQLKMTVVKLISHRECQQHYYGSEVTTKMLCAAQWKTDSCQGDSGGPLVCSLQGRMTLTGIVSWGRGCALDKPGVYTRVSHFLPWIRSHTK Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | DNA topoisomerase 4 subunit A | 1ZVT | 4.02 | |
Target general information Gen name parC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3019;JW2987 Protein family Type II topoisomerase GyrA/ParC subunit family, ParC type 1 subfamily Biochemical class Isomerase Function ATP binding.DNA binding.DNA topoisomerase type II (ATP-hydrolyzing) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11943; DB12924; DB00817 Interacts with P22523; P0A7K2 EC number 5.6.2.2 Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; DNA-binding; Isomerase; Membrane; Reference proteome; Topoisomerase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 26490.3 Length 246 Aromaticity 0.04 Instability index 46.03 Isoelectric point 8.94 Charge (pH=7) 2.83 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map 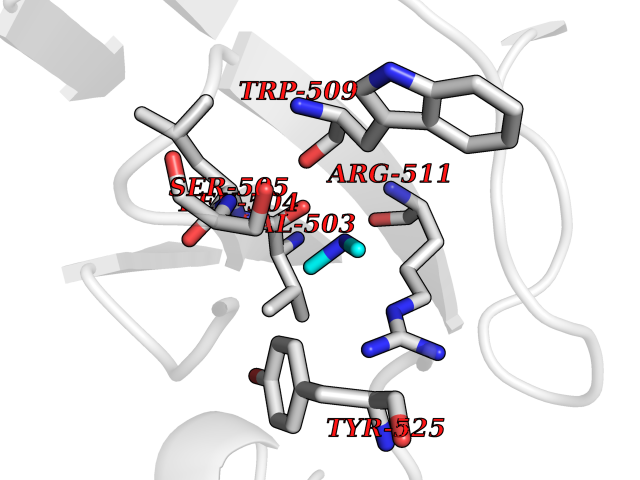 Pymol Map 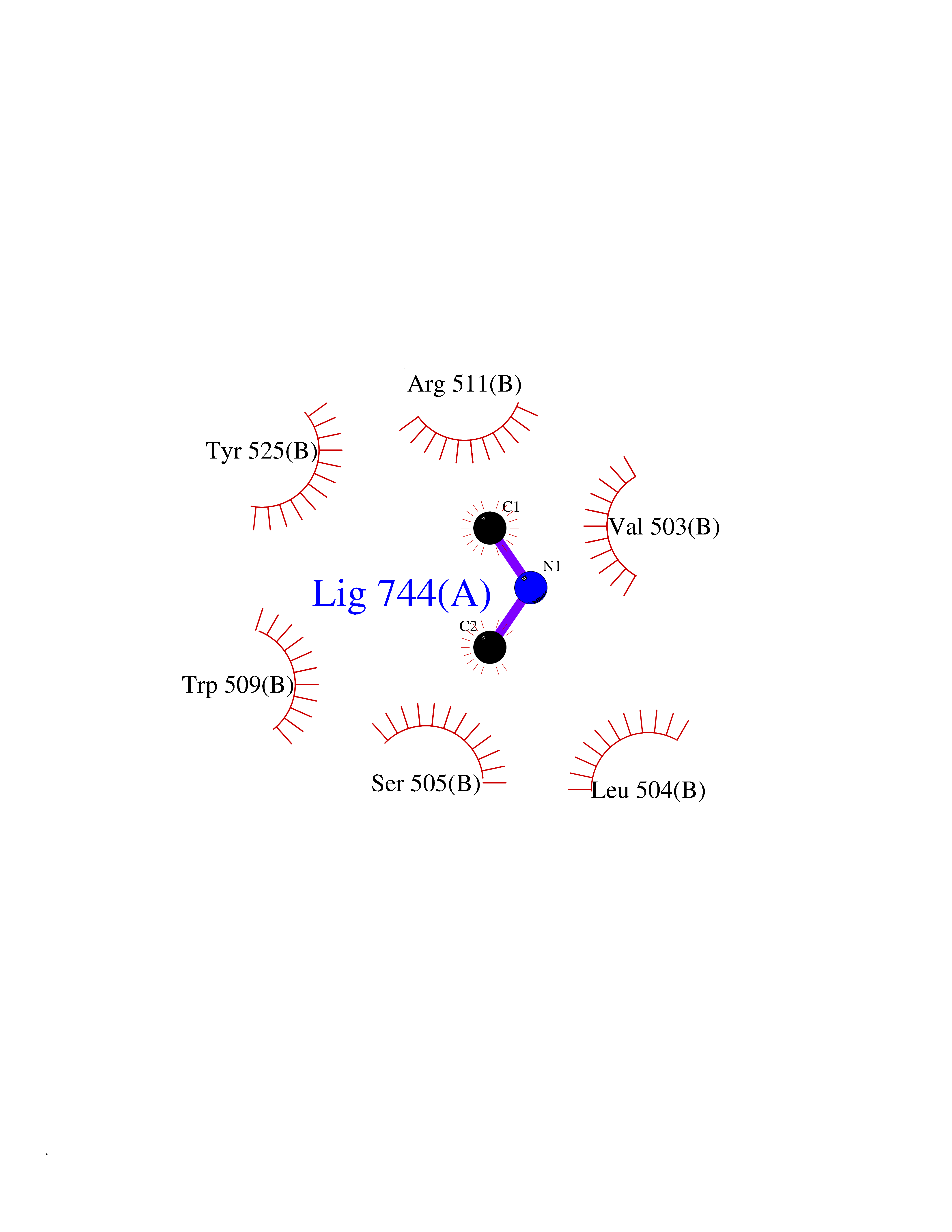 Ligplot Map 3D Binding mode Sequence SEPVTIVLSQMGWVRSAKGHDIDAPGLNYKAGDSFKAAVKGKSNQPVVFVDSTGRSYAIDPITLPSARGQGEPLTGKLTLPPGATVDHMLMESDDQKLLMASDAGYGFVCTFNDLVARNRAGKALITLPENAHVMPPVVIEDASDMLLAITQAGRMLMFPVSDLPQLSKGKGNKIINIPSAEAARGEDGLAQLYVLPPQSTLTIHVGKRKIKLRPEELQKVTGERGRRGTLMRGLQRIDRVEIDSP Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Bifunctional dihydrofolate reductase-thymidylate synthase | 1J3K | 4.02 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate K1 / Thailand) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydrofolate reductase family; Thymidylate synthase family Biochemical class Oxidoreductase Function Dihydrofolate reductase activity.Thymidylate synthase activity. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01131; DB00205; DB01299 Interacts with NA EC number 1.5.1.3; 2.1.1.45 Uniprot keywords 3D-structure; Methyltransferase; Multifunctional enzyme; NADP; Nucleotide biosynthesis; One-carbon metabolism; Oxidoreductase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61720.6 Length 525 Aromaticity 0.13 Instability index 33.9 Isoelectric point 8.79 Charge (pH=7) 11.61 2D Binding mode Binding energy (Kcal/mol) -5.49 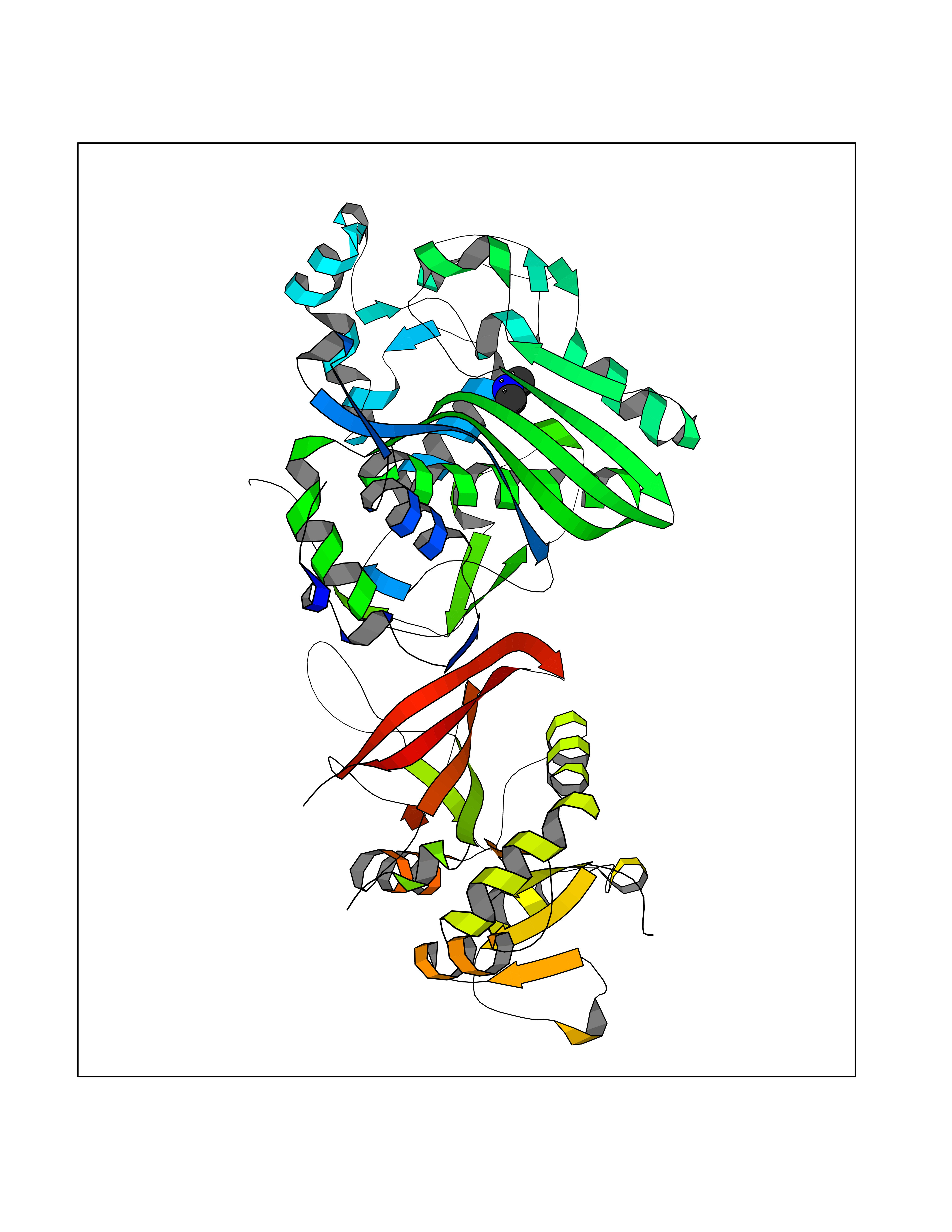 Molscript Map 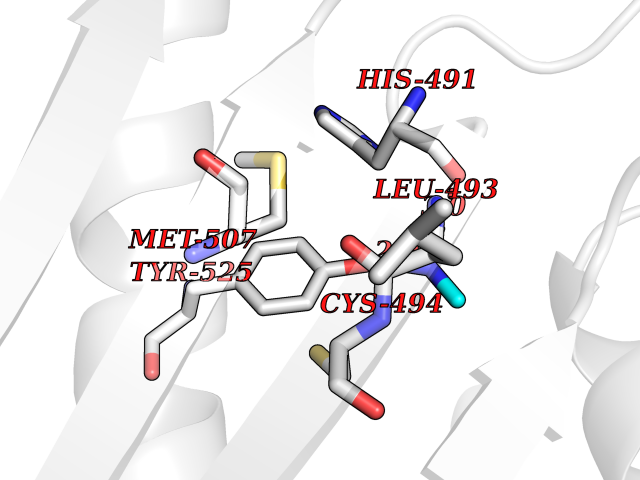 Pymol Map 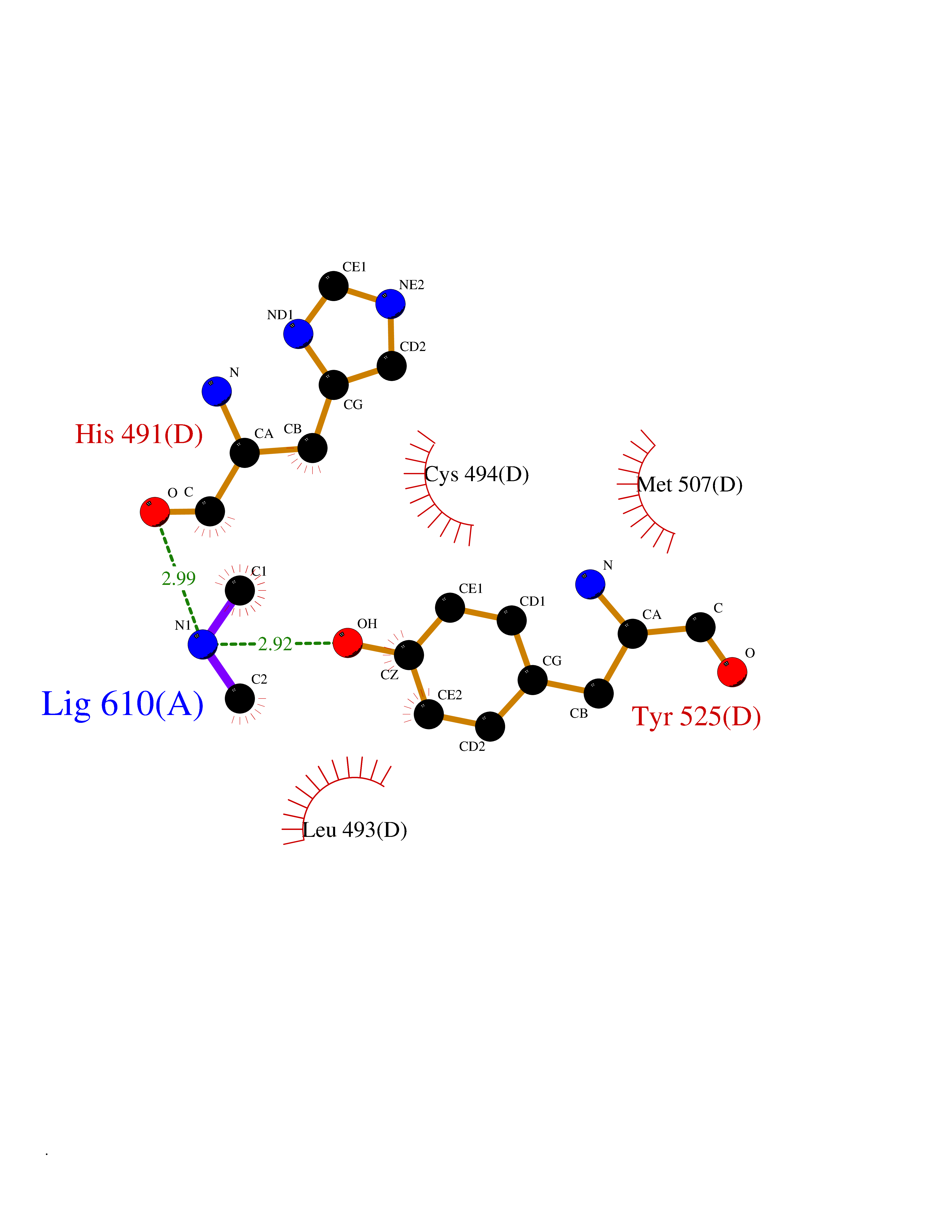 Ligplot Map 3D Binding mode Sequence NSIHPNDFQIYNSLKYKYHPEYQYLNIIYDIMMNGNKQSDRTGVGVLSKFGYIMKFDLSQYFPLLTTKKLFLRGIIEELLWFIRGETNGNTLLNKNVRIWEANGTREFLDNRKLFHREVNDLGPIYGFQWRHFGAEYTNMYDNYENKGVDQLKNIINLIKNDPTSRRILLCAWNVKDLDQMALPPCHILCQFYVFDGKLSCIMYQRSCDLGLGVPFNIASYSIFTHMIAQVCNLQPAQFIHVLGNAHVYNNHIDSLKIQLNRIPYPFPTLKLNPDIKNIEDFTISDFTIQNYVHHEKISMDMAAMMEQVCDVFDIYAICACCKVESKNEGKKNEVFNNYTFRGLGNKGVLPWKCISLDMKYFRAVTTYVNESKYEKLKYKRCKYLPNSKKLQNVVVMGRTNWESIPKKFKPLSNRINVILSRTLKKEDFDEDVYIINKVEDLIVLLGKLNYYKCFILGGSVVYQEFLEKKLIKKIYFTRINSTYECDVFFPEINENEYQIISVSDVYTSNNTTLDFIIYKKTNNK Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Flavodoxin/ferredoxin--NADP reductase | 1FDR | 4.02 | |
Target general information Gen name fpr Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms mvrA;b3924;JW3895 Protein family Ferredoxin--NADP reductase type 1 family Biochemical class Flavoprotein Function FAD binding.Ferredoxin-NADP+ reductase activity.Oxidoreductase activity. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.18.1.2; 1.19.1.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; NADP; Nucleotide-binding; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 27346.2 Length 244 Aromaticity 0.08 Instability index 30.68 Isoelectric point 7.25 Charge (pH=7) 0.42 2D Binding mode Binding energy (Kcal/mol) -5.48 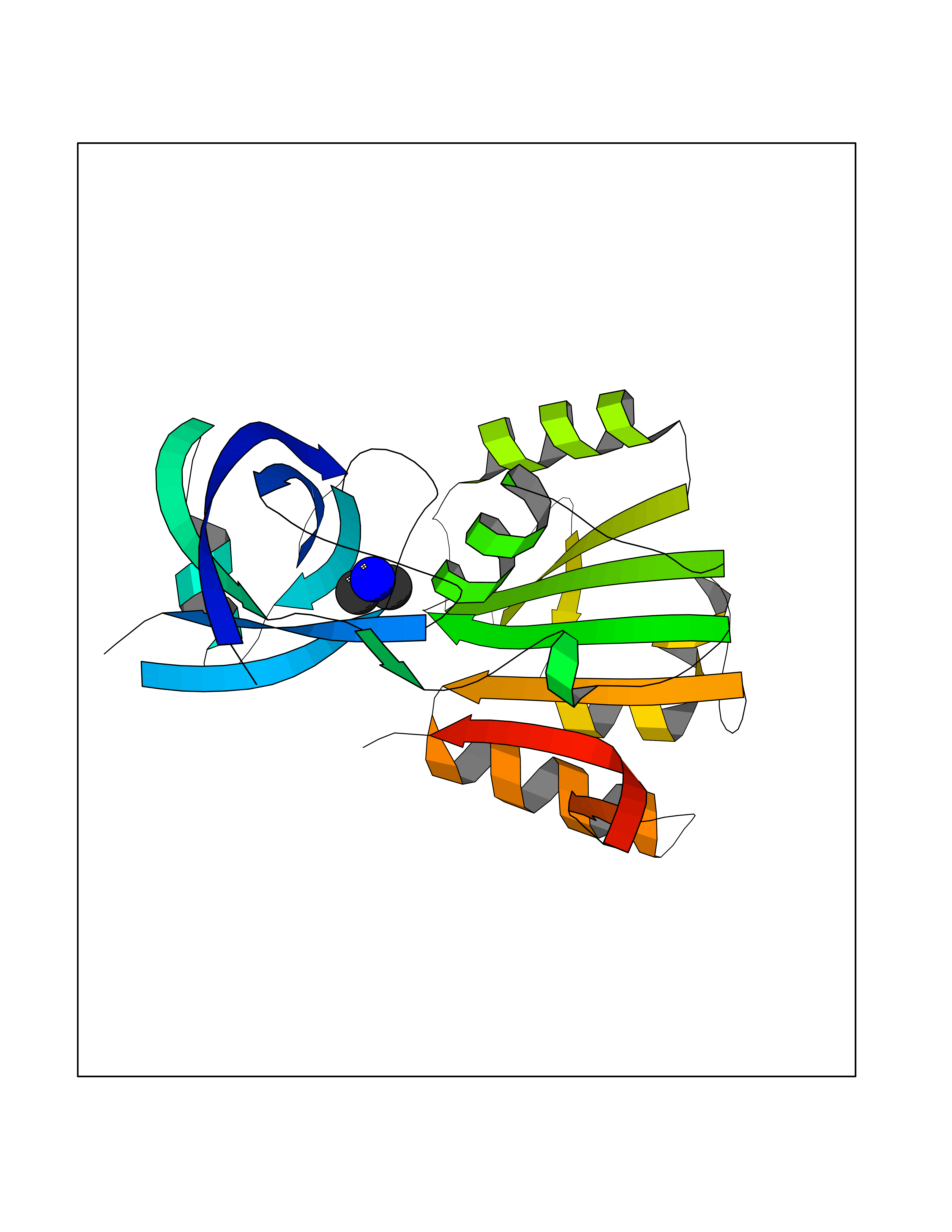 Molscript Map 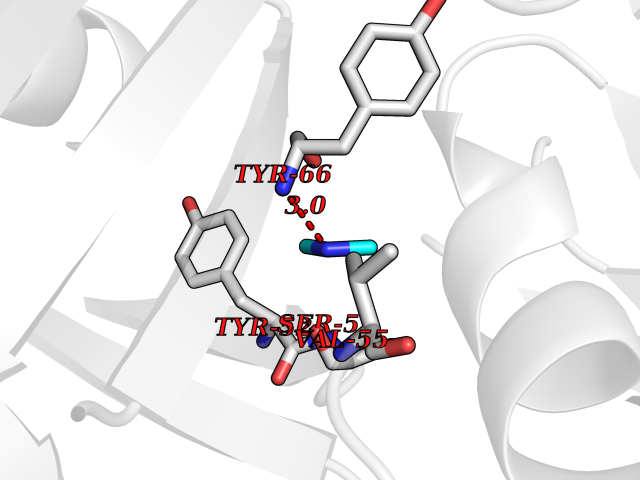 Pymol Map 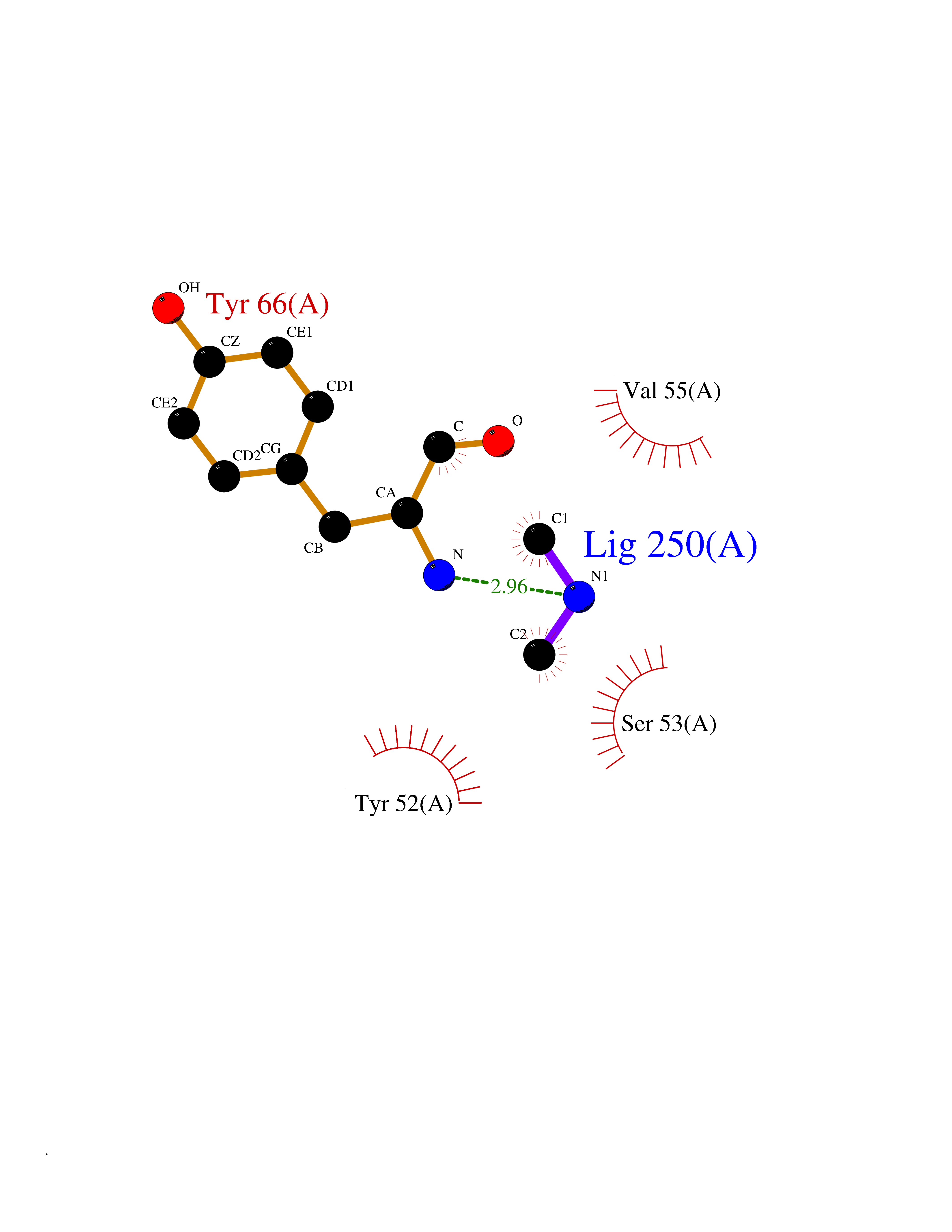 Ligplot Map 3D Binding mode Sequence ADWVTGKVTKVQNWTDALFSLTVHAPVLPFTAGQFTKLGLEIRVQRAYSYVNSPDNPDLEFYLVTVPDGKLSPRLAALKPGDEVQVVSEAAGFFVLDEVPHCETLWMLATGTAIGPYLSILRLGKDLDRFKNLVLVHAARYAADLSYLPLMQELEKRYEGKLRIQTVVSRETAAGSLTGRIPALIESGELESTIGLPMNKETSHVMLCGNPQMVRDTQQLLKETRQMTKHLRRRPGHMTAEHYW Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | S-adenosylmethionine synthase isoform type-1 | 2OBV | 4.02 | |
Target general information Gen name MAT1A Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MATA1;AMS1 Protein family AdoMet synthase family Biochemical class Transferase Function ATP binding.Identical protein binding.Metal ion binding.Methionine adenosyltransferase activity.Selenomethionine adenosyltransferase activity. Related diseases Methionine adenosyltransferase deficiency (MATD) [MIM:250850]: An inborn error of metabolism resulting in isolated hypermethioninemia. Most patients have no clinical abnormalities, although some neurologic symptoms may be present in rare cases with severe loss of methionine adenosyltransferase activity. {ECO:0000269|PubMed:10677294, ECO:0000269|PubMed:7560086, ECO:0000269|PubMed:8770875, ECO:0000269|PubMed:9042912}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03191; DB00118; DB03611; DB00134 Interacts with P05067; P42858; Q00266; P31153 EC number 2.5.1.6 Uniprot keywords 3D-structure; ATP-binding; Disease variant; Disulfide bond; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Potassium; Proteomics identification; Reference proteome; S-nitrosylation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42222.9 Length 381 Aromaticity 0.08 Instability index 41.95 Isoelectric point 6.14 Charge (pH=7) -4.58 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map 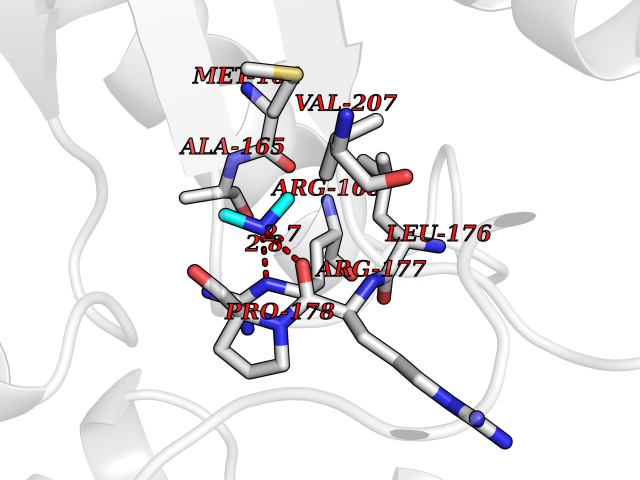 Pymol Map 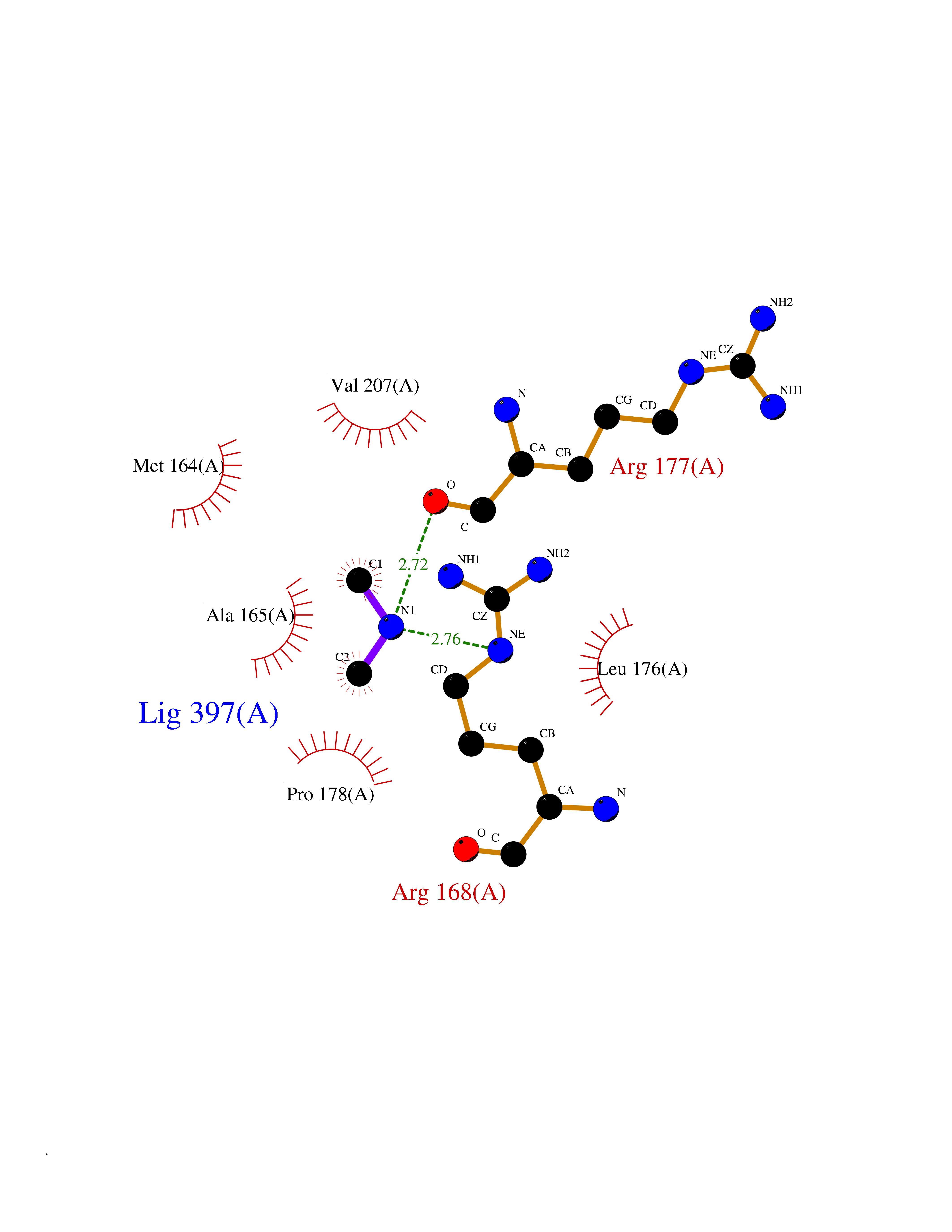 Ligplot Map 3D Binding mode Sequence MGVFMFTSESVGEGHPDKICDQISDAVLDAHLKQDPNAKVACETVCKTGMVLLCGEITSMAMVDYQRVVRDTIKHIGYDDSAKGFDFKTCNVLVALEQQSPDIAQCVHLDRNEEDVGAGDQGLMFGYATDETEECMPLTIILAHKLNARMADLRRSGLLPWLRPDSKTQVTVQYMQDNGAVIPVRIHTIVISVQHNEDITLEEMRRALKEQVIRAVVPAKYLDEDTVYHLQPSGRFVIGGPQGDAGVTGRKIIVDTYGGWGAHGGGAFSGKDYTKVDRSAAYAARWVAKSLVKAGLCRRVLVQVSYAIGVAEPLSISIFTYGTSQKTERELLDVVHKNFDLRPGVIVRDLDLKKPIYQKTACYGHFGRSEFPWEVPRKLVF Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Beta-xylanase | 2D22 | 4.02 | |
Target general information Gen name N/A Organism Streptomyces olivaceoviridis (Streptomyces corchorusii) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 10 (cellulase F) family Biochemical class Hydrolase Function Endo-1,4-beta-xylanase activity. Related diseases Mitochondrial DNA depletion syndrome 8A (MTDPS8A) [MIM:612075]: A disorder due to mitochondrial dysfunction characterized by various combinations of neonatal hypotonia, neurological deterioration, respiratory distress, lactic acidosis, and renal tubulopathy. {ECO:0000269|PubMed:17486094, ECO:0000269|PubMed:18504129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 8B (MTDPS8B) [MIM:612075]: A disease due to mitochondrial dysfunction and characterized by ophthalmoplegia, ptosis, gastrointestinal dysmotility, cachexia, peripheral neuropathy. {ECO:0000269|PubMed:19667227}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal dominant, 5 (PEOA5) [MIM:613077]: A disorder characterized by progressive weakness of ocular muscles and levator muscle of the upper eyelid. In a minority of cases, it is associated with skeletal myopathy, which predominantly involves axial or proximal muscles and which causes abnormal fatigability and even permanent muscle weakness. Ragged-red fibers and atrophy are found on muscle biopsy. A large proportion of chronic ophthalmoplegias are associated with other symptoms, leading to a multisystemic pattern of this disease. Additional symptoms are variable, and may include cataracts, hearing loss, sensory axonal neuropathy, ataxia, depression, hypogonadism, and parkinsonism. {ECO:0000269|PubMed:19664747}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy, sensorineural deafness, and Fanconi-type renal dysfunction (RCDFRD) [MIM:268315]: An autosomal recessive disease characterized by visual impairment due to rod-cone dystrophy, sensorineural hearing loss, and Fanconi-type renal dysfunction resulting in rickets-like skeletal changes. Death may occur in childhood or young adulthood due to renal failure. Disease onset is before age 5 years. {ECO:0000269|PubMed:32827185}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03389; DB02379; DB04465 Interacts with NA EC number 3.2.1.8 Uniprot keywords 3D-structure; Carbohydrate metabolism; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32170.2 Length 295 Aromaticity 0.11 Instability index 29.09 Isoelectric point 6.66 Charge (pH=7) -0.7 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map 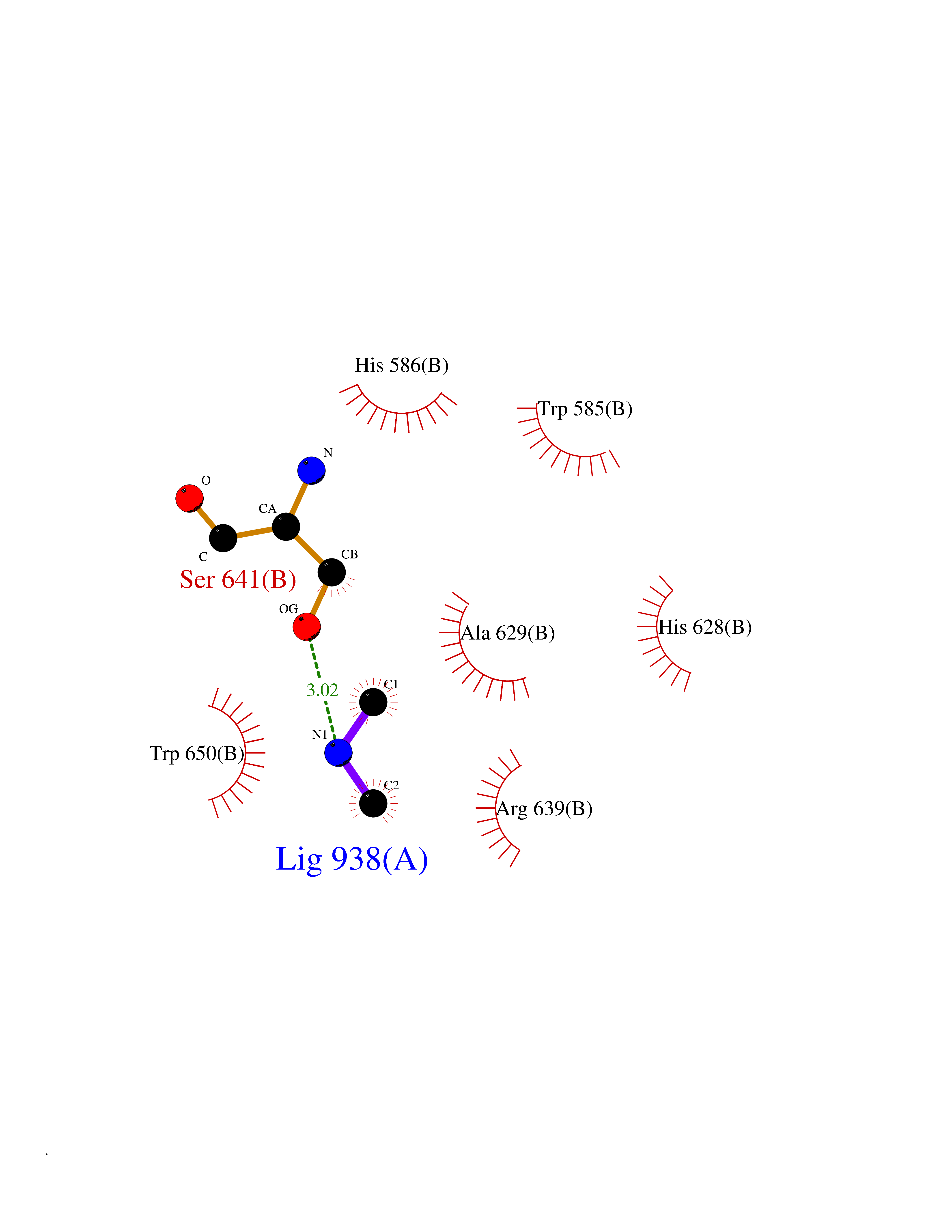 Ligplot Map 3D Binding mode Sequence AESTLGAAAAQSGRYFGTAIASGKLGDSAYTTIASREFNMVTAENEMKIDATEPQRGQFNFSAGDRVYNWAVQNGKQVRGHTLAWHSQQPGWMQSLSGSTLRQAMIDHINGVMGHYKGKIAQWDVVSHAFSDDGSGGRRDSNLQRTGNDWIEVAFRTARAADPAAKLCYNDYNIENWTWAKTQGVYNMVRDFKQRGVPIDCVGFQSHFNSGSPYNSNFRTTLQNFAALGVDVAITELDIQGASSSTYAAVTNDCLAVSRCLGITVWGVRDTDSWRSGDTPLLFNGDGSKKAAYTA Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Histone deacetylase 2 (HDAC2) | 4LY1 | 4.02 | |
Target general information Gen name HDAC2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HD2 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Forms transcriptional repressor complexes by associating with MAD, SIN3, YY1 and N-COR. Interacts in the late S-phase of DNA-replication with DNMT1 in the other transcriptional repressor complex composed of DNMT1, DMAP1, PCNA, CAF1. Deacetylates TSHZ3 and regulates its transcriptional repressor activity. Component of a RCOR/GFI/KDM1A/HDAC complex that suppresses, via histone deacetylase (HDAC) recruitment, a number of genes implicated in multilineage blood cell development. May be involved in the transcriptional repression of circadian target genes, such as PER1, mediated by CRY1 through histone deacetylation. Involved in MTA1-mediated transcriptional corepression of TFF1 and CDKN1A. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Ventricular tachycardia, catecholaminergic polymorphic, 1, with or without atrial dysfunction and/or dilated cardiomyopathy (CPVT1) [MIM:604772]: An arrhythmogenic disorder characterized by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and cause sudden death. Patients present with recurrent syncope, seizures, or sudden death after physical activity or emotional stress. CPVT1 inheritance is autosomal dominant. {ECO:0000269|PubMed:11157710, ECO:0000269|PubMed:11159936, ECO:0000269|PubMed:11208676, ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:12106942, ECO:0000269|PubMed:14571276, ECO:0000269|PubMed:15046072, ECO:0000269|PubMed:15046073, ECO:0000269|PubMed:15466642, ECO:0000269|PubMed:15544015, ECO:0000269|PubMed:16188589, ECO:0000269|PubMed:24793461, ECO:0000269|PubMed:25372681, ECO:0000269|PubMed:27733687}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ventricular arrhythmias due to cardiac ryanodine receptor calcium release deficiency syndrome (VACRDS) [MIM:115000]: An autosomal dominant arrhythmogenic disorder characterized by syncope, cardiac arrest and/or sudden unexpected death, often in association with physical exertion or acute emotional stress. Patients who survive manifest polymorphic ventricular tachycardia and ventricular fibrillation. Unlike typical catecholaminergic ventricular tachycardia, arrhythmias are not reproducible on exercise stress testing or adrenaline challenge. {ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:17984046, ECO:0000269|PubMed:33536282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB01223; DB01076; DB05015; DB01262; DB11841; DB01095; DB12645; DB00227; DB11830; DB01303; DB06603; DB06819; DB05223; DB00175; DB03766; DB12847; DB06176; DB00641; DB00277; DB09091; DB00313; DB02546 Interacts with Q9C0K0; Q9HCU9; P68400; Q9UER7; P51610; Q13547; Q9UIS9; Q13330; P01106; P06748; P48382; Q96ST3; O95863; Q9HD15; O43463; Q9H3M7; Q92618; Q17R98; Q2HR82; PRO_0000449623 [P0DTD1] EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Cytoplasm; Hydrolase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 42020.5 Length 366 Aromaticity 0.13 Instability index 29.52 Isoelectric point 6.52 Charge (pH=7) -2.16 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KKKVCYYYDGDIGNYYYGQGHPMKPHRIRMTHNLLLNYGLYRKMEIYRPHKATAEEMTKYHSDEYIKFLRSIRPDNMSEYSKQMQRFNVGEDCPVFDGLFEFCQLSTGGSVAGAVKLNRQQTDMAVNWAGGLHHAKKSEASGFCYVNDIVLAILELLKYHQRVLYIDIDIHHGDGVEEAFYTTDRVMTVSFHKYGEYFPGTGDLRDIGAGKGKYYAVNFPMRDGIDDESYGQIFKPIISKVMEMYQPSAVVLQCGADSLSGDRLGCFNLTVKGHAKCVEVVKTFNLPLLMLGGGGYTIRNVARCWTYETAVALDCEIPNELPYNDYFEYFGPDFKLHISPSNMTNQNTPEYMEKIKQRLFENLRML Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Leucine carboxyl methyltransferase 1 | 3IEI | 4.02 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Fumarate reductase flavoprotein subunit | 1QO8 | 4.02 | |
Target general information Gen name ifcA Organism Shewanella frigidimarina (strain NCIMB 400) Uniprot ID TTD ID NA Synonyms Sfri_2586 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Fumarate reductase (menaquinone).Metal ion binding.Succinate dehydrogenase activity. Related diseases Myopathy with lactic acidosis and sideroblastic anemia 2 (MLASA2) [MIM:613561]: A rare oxidative phosphorylation disorder specific to skeletal muscle and bone marrow. Affected individuals manifest sideroblastic anemia, progressive lethargy, muscle weakness, and exercise intolerance associated with persistent lactic acidemia. {ECO:0000269|PubMed:20598274, ECO:0000269|PubMed:22504945}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.3.2.- Uniprot keywords 3D-structure; Direct protein sequencing; Electron transport; FAD; Flavoprotein; Heme; Iron; Metal-binding; Oxidoreductase; Periplasm; Reference proteome; Signal; Transport Protein physicochemical properties Chain ID A,D Molecular weight (Da) 34344.3 Length 328 Aromaticity 0.05 Instability index 25.18 Isoelectric point 6.27 Charge (pH=7) -3.25 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence WDDGWDQDKIQKAIAAGPSETTQVLVVGAGSAGFNASLAAKKAGANVILVDKAPFSGGNSMISAGGMNAVGTKQQTAHGVEDKVEWFIEDAMKGGRQQNDIKLVTILAEQSADGVQWLESLGANLDDLKRSGGARVDRTHRPHGGKSSGPEIIDTLRKAAKEQGIDTRLNSRVVKLVVNDDHSVVGAVVHGKHTGYYMIGAKSVVLATGGYGMNKEMIAYYRPTMKDMTSSNNITATGDGVLMAKEIGASMTDIDWVQAHIHHTMGGVAINTTASVLDLQSKPIDGLFAAGEVTGGVHGYNRLGGNAIADTVVFGRIAGDNAAKHALD Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | N-acetylgalactosamine 6 sulfatase (GALNS) | 4FDJ | 4.02 | |
Target general information Gen name GALNS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms N-acetylgalactosamine-6-sulfate sulfatase; N-acetylgalactosamine-6-sulfatase; Galactose-6-sulfate sulfatase; GalNAc6S sulfatase; GalN6S; Chondroitinsulfatase; Chondroitinase Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Catalyzes the chemical reaction of cleaving off the 6-sulfate groups of the N-acetyl-D-galactosamine 6-sulfate units of the macromolecule chondroitin sulfate and, similarly, of the D-galactose 6-sulfate units of the macromolecule keratan sulfate. Related diseases Mucopolysaccharidosis 4A (MPS4A) [MIM:253000]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:1522213, ECO:0000269|PubMed:16287098, ECO:0000269|PubMed:24726177, ECO:0000269|PubMed:7581409, ECO:0000269|PubMed:7633425, ECO:0000269|PubMed:7668283, ECO:0000269|PubMed:7795586, ECO:0000269|PubMed:8651279, ECO:0000269|PubMed:8826435, ECO:0000269|PubMed:9298823, ECO:0000269|PubMed:9375852, ECO:0000269|PubMed:9521421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09301 Interacts with NA EC number EC 3.1.6.4 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Glycoprotein; Hydrolase; Lysosome; Metal-binding; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 55013.8 Length 493 Aromaticity 0.11 Instability index 35.46 Isoelectric point 6.14 Charge (pH=7) -6.48 2D Binding mode Binding energy (Kcal/mol) -5.48 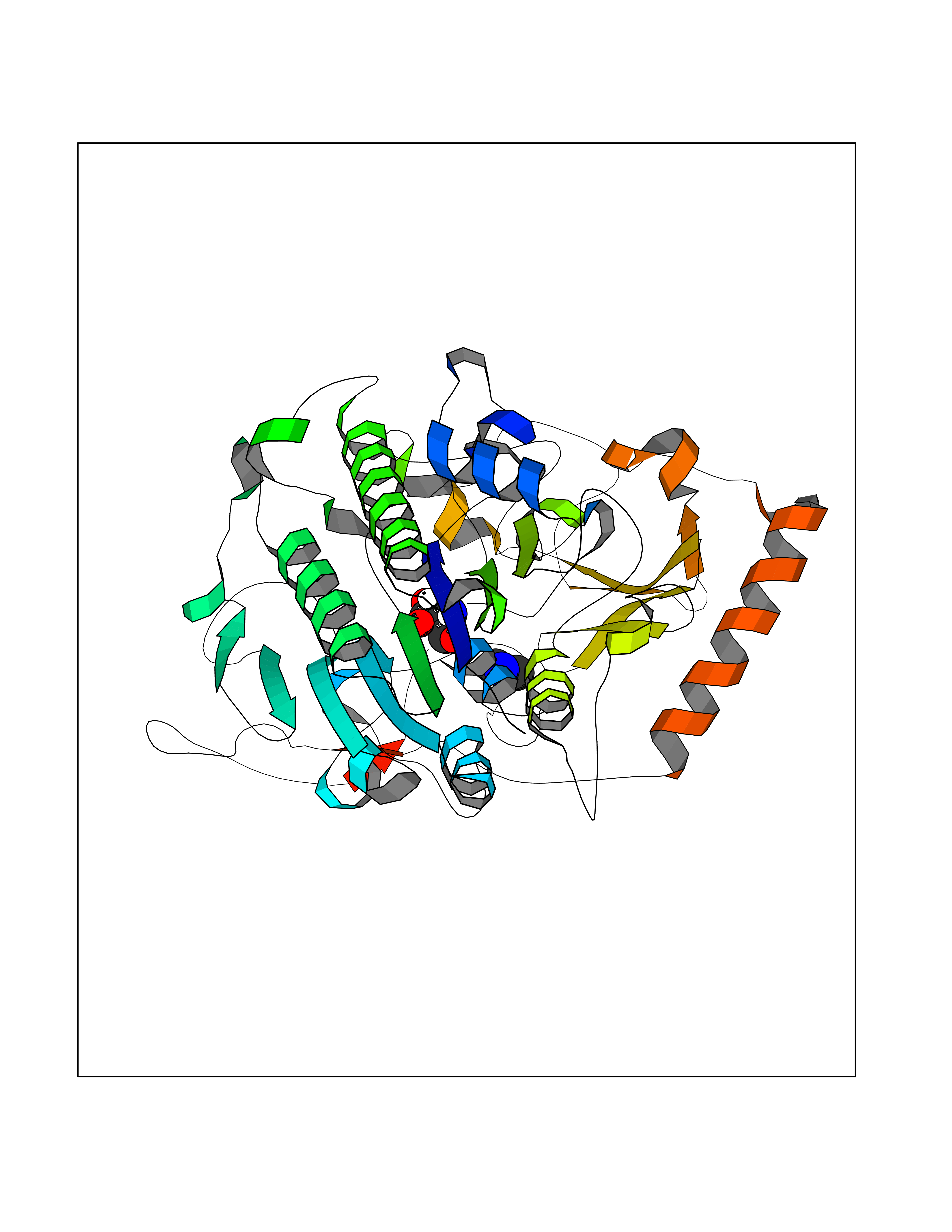 Molscript Map 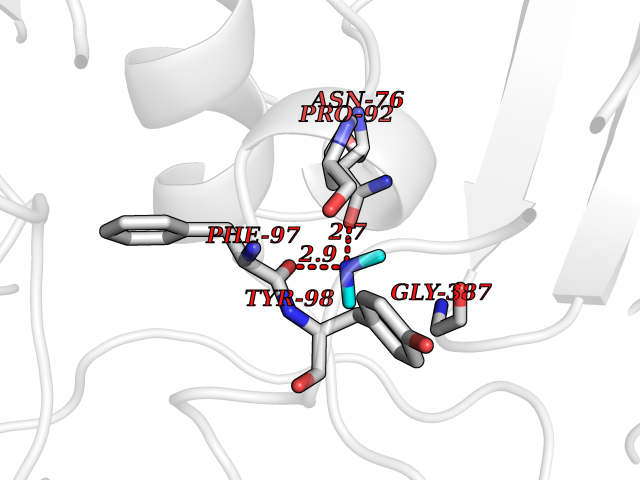 Pymol Map 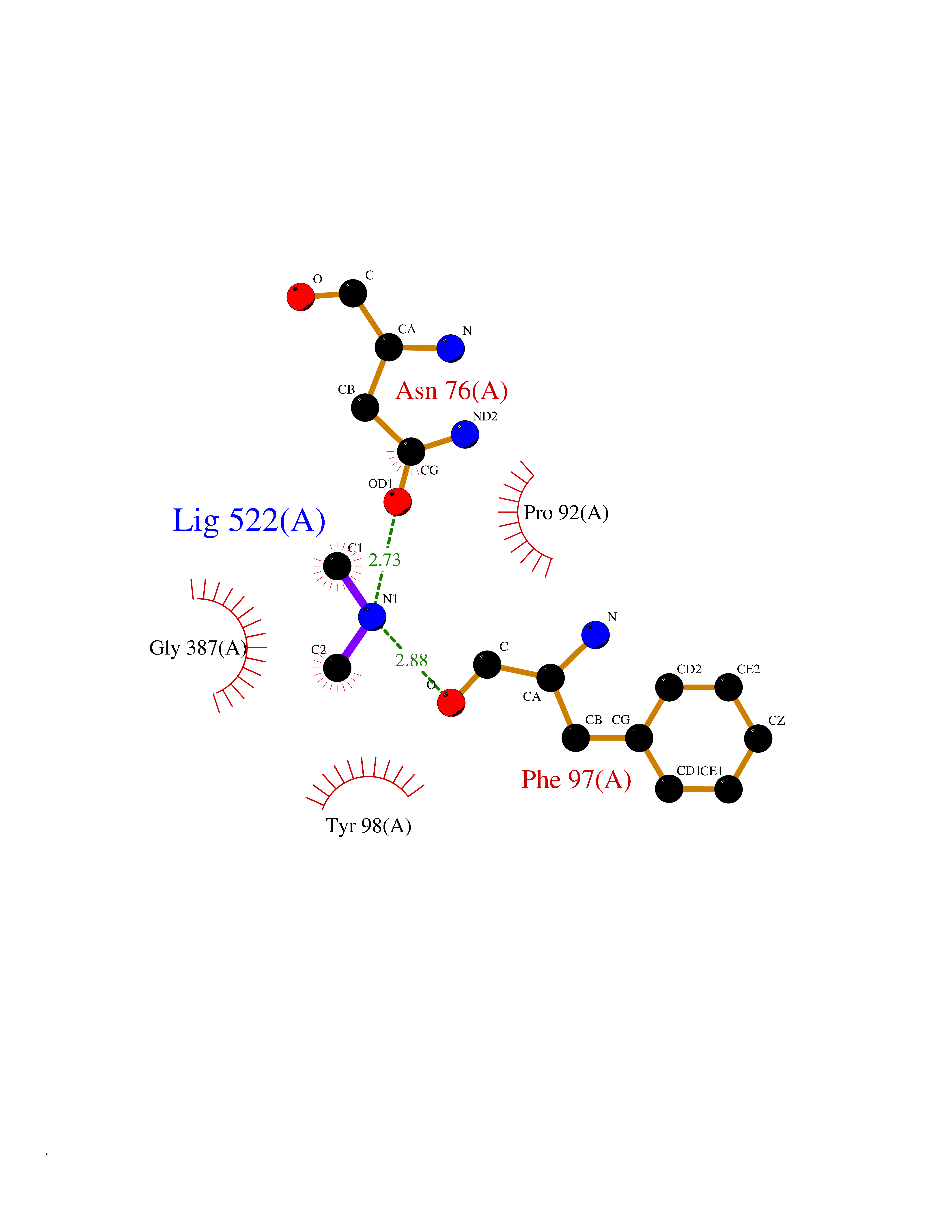 Ligplot Map 3D Binding mode Sequence PQPPNILLLLMDDMGWGDLGVYGEPSRETPNLDRMAAEGLLFPNFYSANPLXSPSRAALLTGRLPIRNGFYTTNAHARNAYTPQEIVGGIPDSEQLLPELLKKAGYVSKIVGKWHLGHRPQFHPLKHGFDEWFGSPNCHFGPYDNKARPNIPVYRDWEMVGRYYEEFPINLKTGEANLTQIYLQEALDFIKRQARHHPFFLYWAVDATHAPVYASKPFLGTSQRGRYGDAVREIDDSIGKILELLQDLHVADNTFVFFTSDNGAALISAPEQGGSNGPFLCGKQTTFEGGMREPALAWWPGHVTAGQVSHQLGSIMDLFTTSLALAGLTPPSDRAIDGLNLLPTLLQGRLMDRPIFYYRGDTLMAATLGQHKAHFWTWTNSWENFRQGIDFCPGQNVSGVTTHNLEDHTKLPLIFHLGRDPGERFPLSFASAEYQEALSRITSVVQQHQEALVPAQPQLNVCNWAVMNWAPPGCEKLGKCLTPPESIPKKCLW Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Tankyrase-2 (TNKS-2) | 3U9H | 4.02 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -5.48 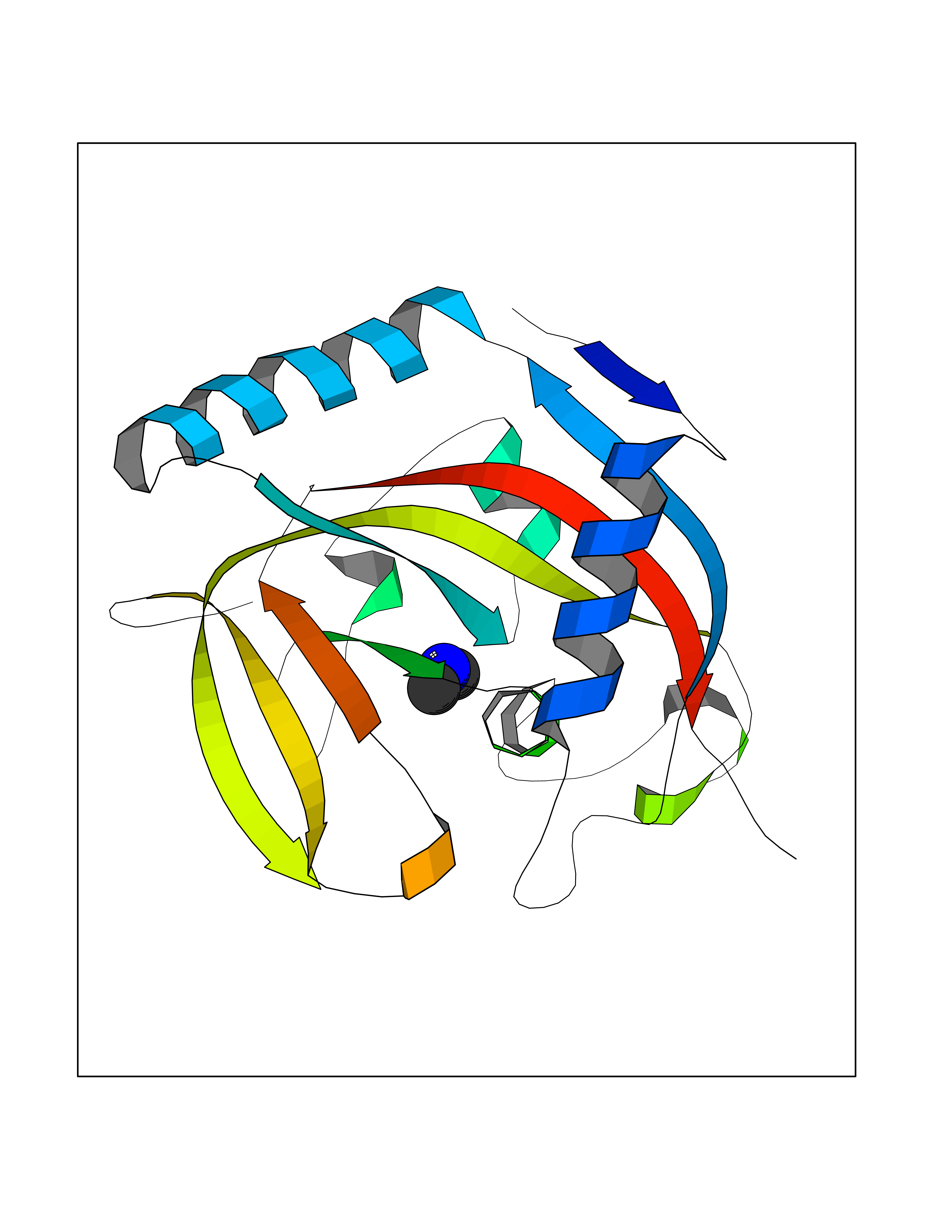 Molscript Map 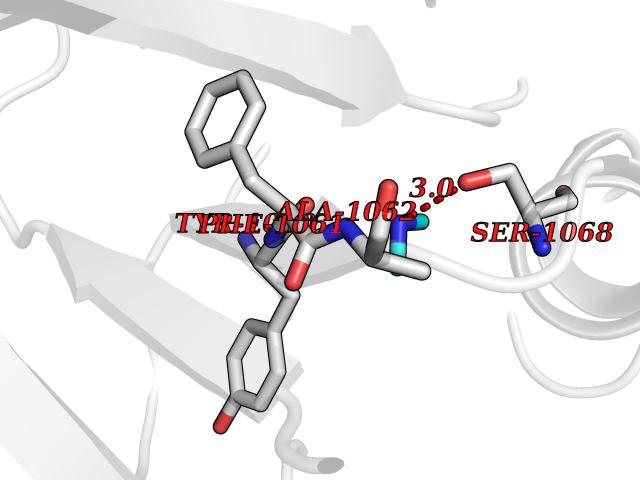 Pymol Map 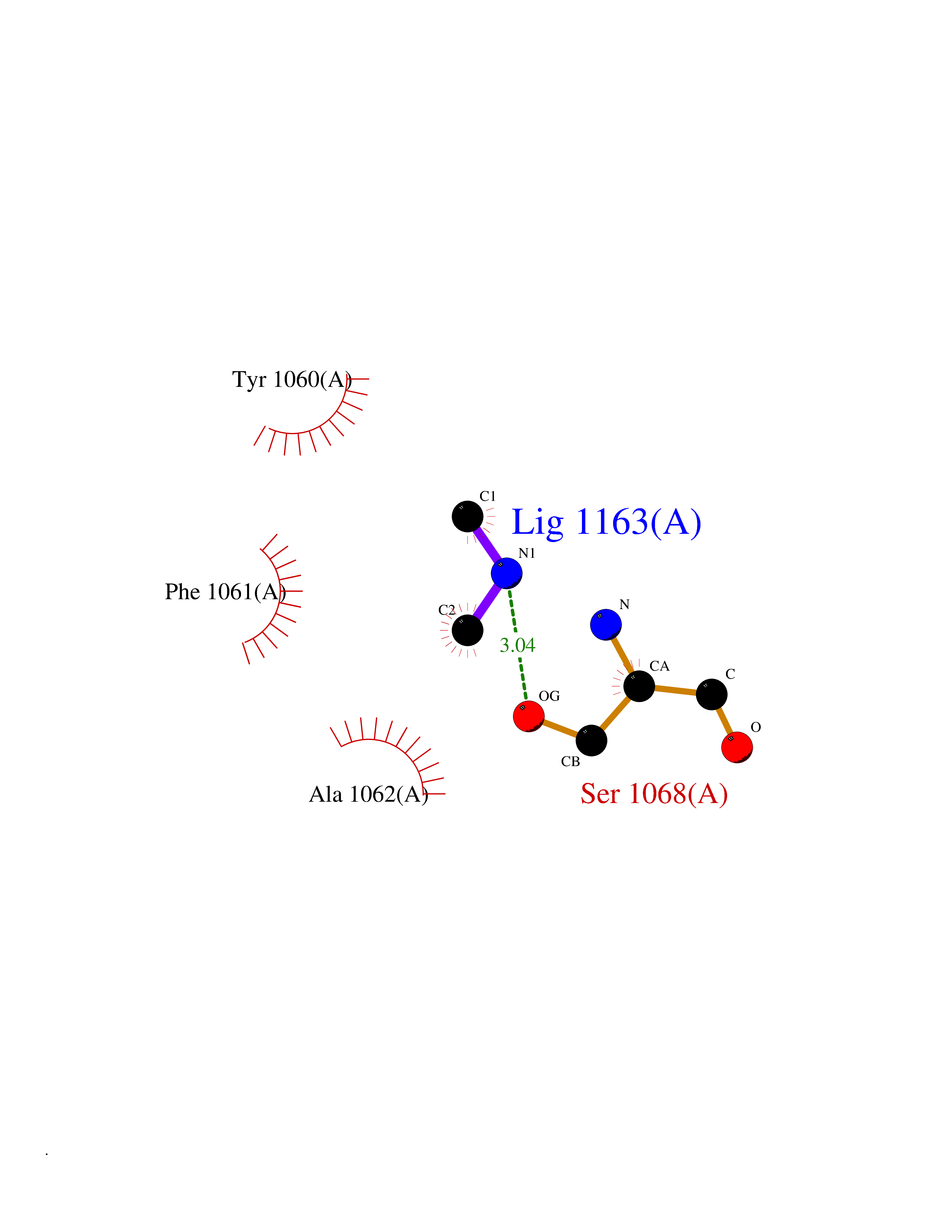 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Complement C1s component (C1S) | 1ELV | 4.02 | |
Target general information Gen name C1S Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complement component 1 subcomponent s; Complement C1s subcomponent; C1-esterase; C1 esterase Protein family Peptidase S1 family Biochemical class Peptidase Function C1r activates C1s so that it can, in turn, activate C2 and C4. C1s B chain is a serine protease that combines with C1q and C1r to form C1, the first component of the classical pathway of the complement system. Related diseases Complement component C1s deficiency (C1SD) [MIM:613783]: A rare defect resulting in C1 deficiency and impaired activation of the complement classical pathway. C1 deficiency generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. {ECO:0000269|PubMed:11390518}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ehlers-Danlos syndrome, periodontal type, 2 (EDSPD2) [MIM:617174]: A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSPD2 is characterized by the association of typical features of Ehlers-Danlos syndrome with gingival recession and severe early-onset periodontal disease, leading to premature loss of permanent teeth. EDSPD2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:27745832}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02371; DB09228; DB09130; DB12831; DB06404; DB14996; DB01593; DB14487; DB14533; DB14548 Interacts with P00736; P09871; P06681; O43889-2; Q9H6H4; P05155 EC number EC 3.4.21.42 Uniprot keywords 3D-structure; Calcium; Complement pathway; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Ehlers-Danlos syndrome; Glycoprotein; Hydrolase; Hydroxylation; Immunity; Innate immunity; Metal-binding; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal; Sushi Protein physicochemical properties Chain ID A Molecular weight (Da) 33278.6 Length 303 Aromaticity 0.1 Instability index 33.69 Isoelectric point 5.16 Charge (pH=7) -7.95 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 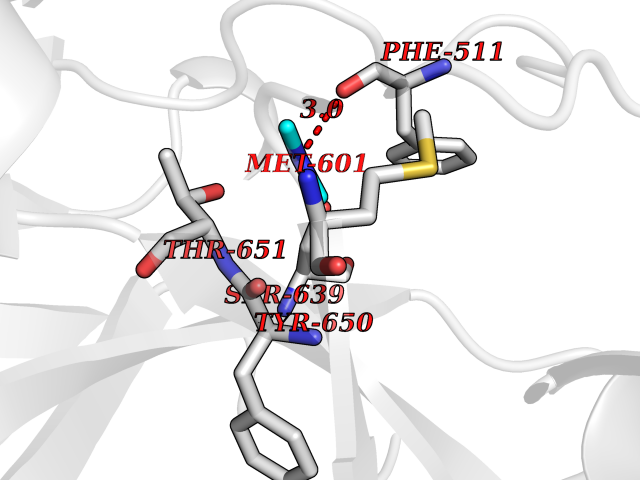 Pymol Map 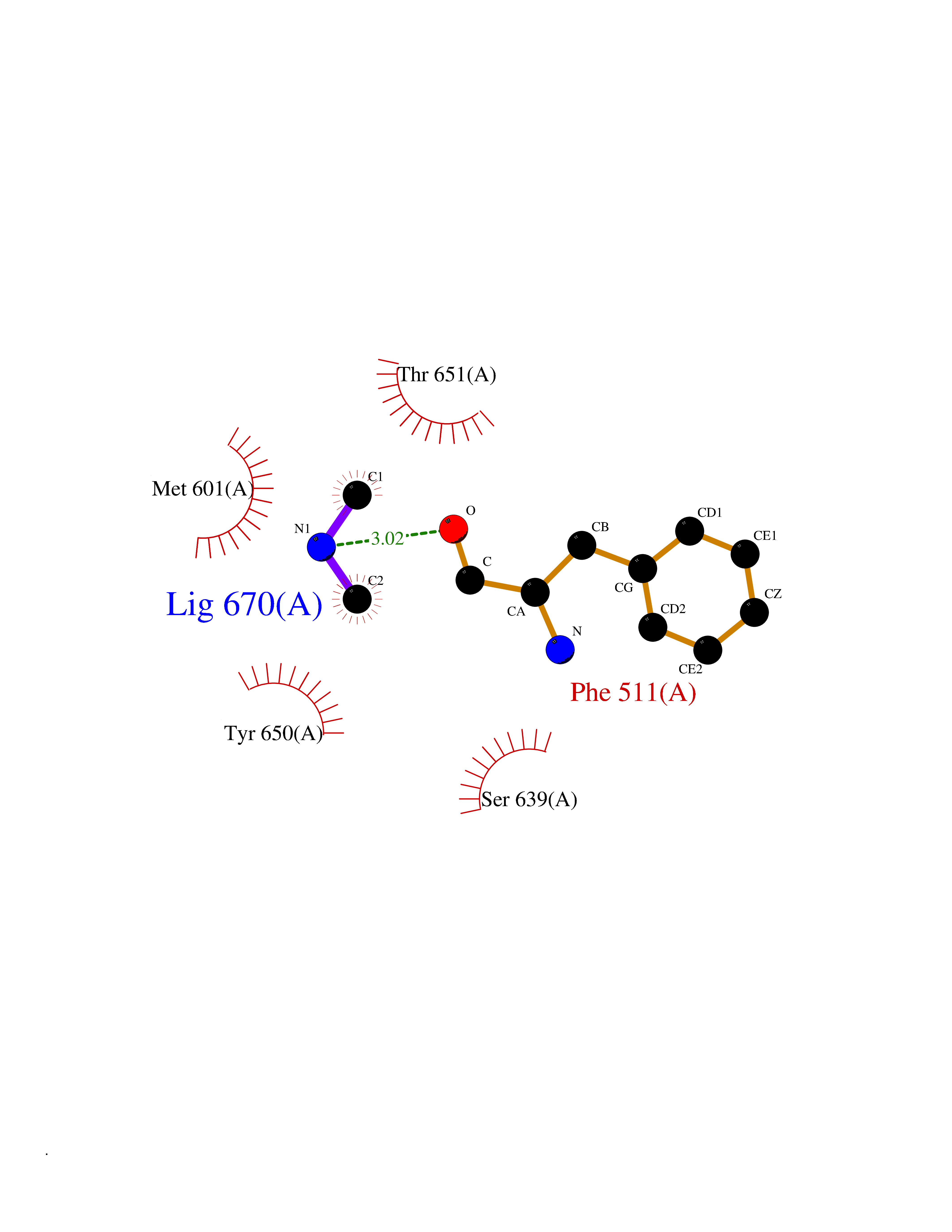 Ligplot Map 3D Binding mode Sequence LDCGIPESIENGKVEDPESTLFGSVIRYTCEEPYYYMEGGGEYHCAGNGSWVNEVLGPELPKCVPVCGVPREPFIIGGSDADIKNFPWQVFFDNPWAGGALINEYWVLTAAHVVEGNREPTMYVGSTSVQKMLTPEHVFIHPGWKLLAVPEGRTNFDNDIALVRLKDPVKMGPTVSPICLPGTSSDYNLMDGDLGLISGWGRTEKRDRAVRLKAARLPVAPLRKCKEVAYVFTPNMICAGGEKGMDSCKGDSGGAFAVQDPNDKTKFYAAGLVSWGPQCGTYGLYTRVKNYVDWIMKTMQENS Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Cathepsin G (CTSG) | 1KYN | 4.02 | |
Target general information Gen name CTSG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CG Protein family Peptidase S1 family Biochemical class Peptidase Function Cleaves complement C3. Has antibacterial activity against the Gram-negative bacterium P. aeruginosa, antibacterial activity is inhibited by LPS from P. aeruginosa, Z-Gly-Leu-Phe-CH2Cl and phenylmethylsulfonyl fluoride. Serine protease with trypsin- and chymotrypsin-like specificity. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04016; DB02360 Interacts with A8MQ03; Q15323; Q7Z3S9; Q9NRD5 EC number EC 3.4.21.20 Uniprot keywords 3D-structure; Antibiotic; Antimicrobial; Cell membrane; Chemotaxis; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Membrane; Nucleus; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 25353.8 Length 223 Aromaticity 0.06 Instability index 66.1 Isoelectric point 11.51 Charge (pH=7) 23.24 2D Binding mode Binding energy (Kcal/mol) -5.48 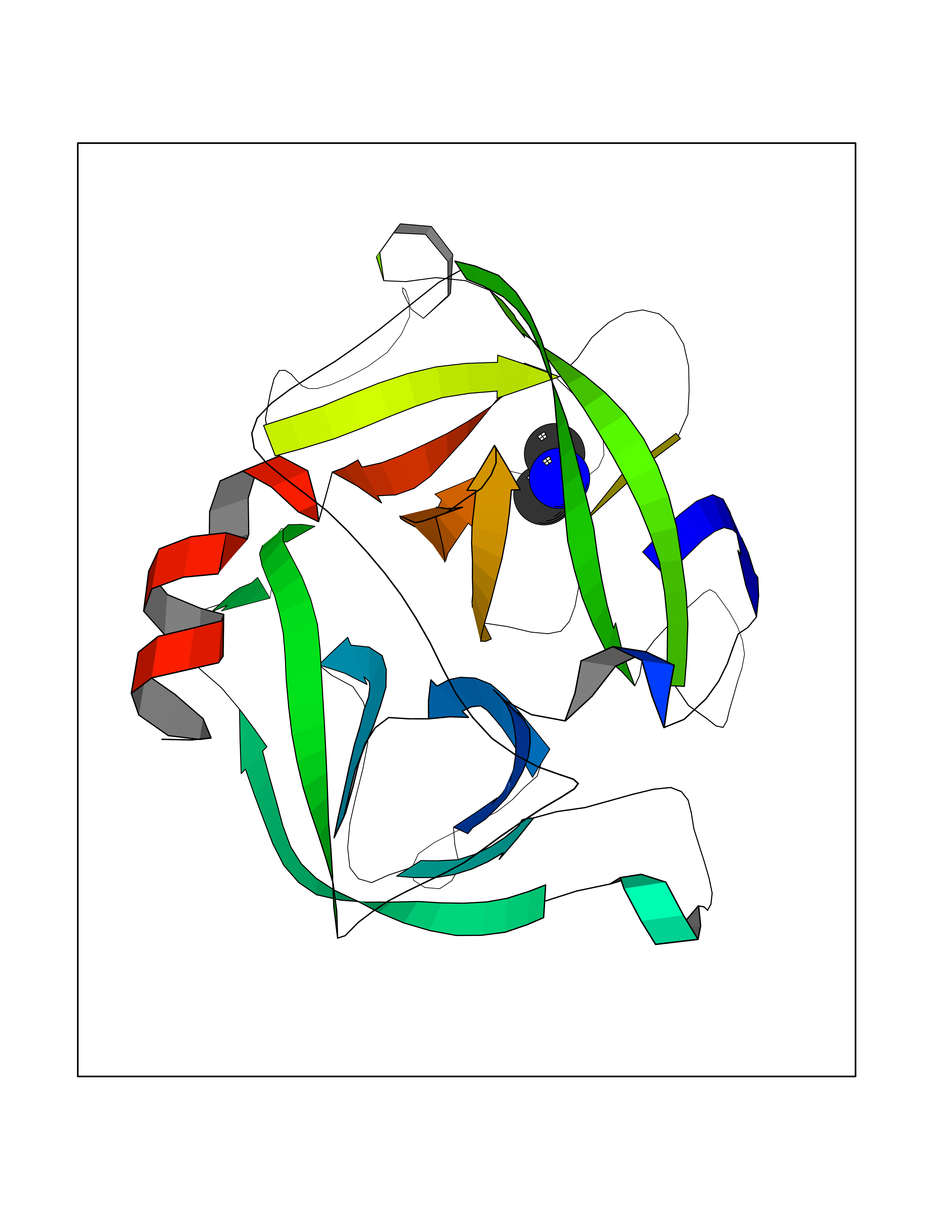 Molscript Map 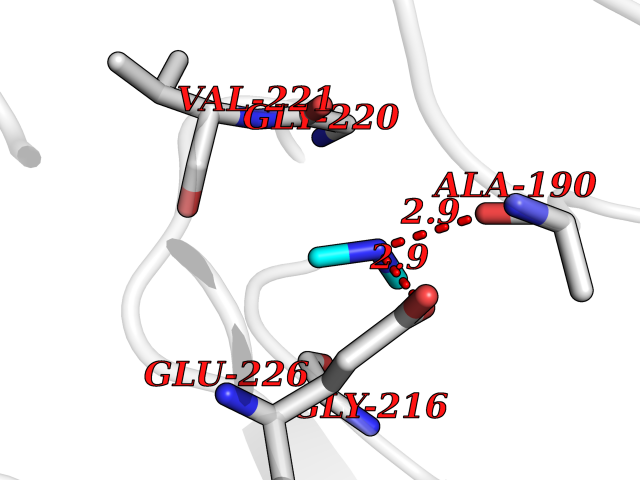 Pymol Map 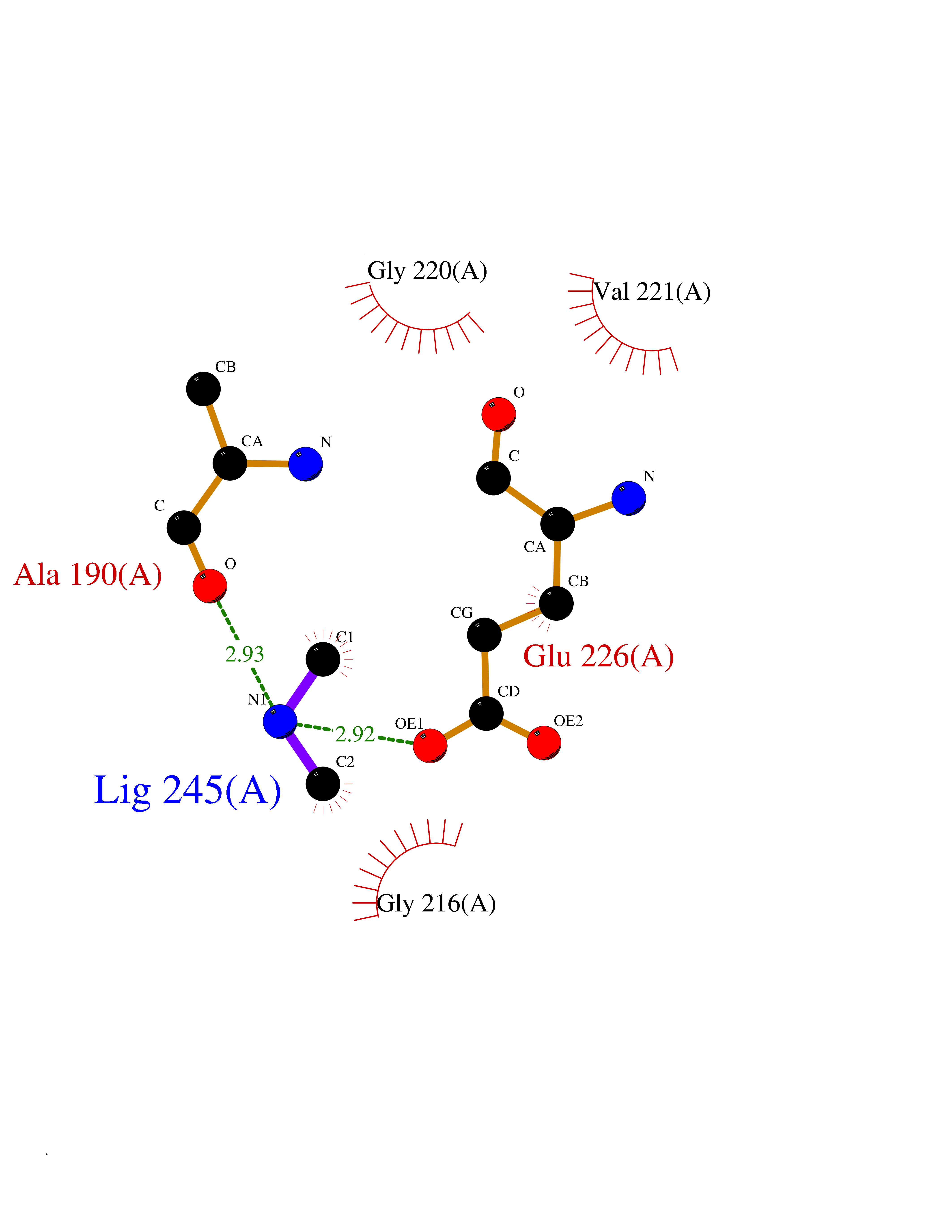 Ligplot Map 3D Binding mode Sequence IIGGRESRPHSRPYMAYLQIQSPAGQSRCGGFLVREDFVLTAAHCWGSNINVTLGAHNIQRRENTQQHITARRAIRHPQYNQRTIQNDIMLLQLSRRVRRNRNVNPVALPRAQEGLRPGTLCTVAGWGRVSMRRGTDTLREVQLRVQRDRQCLRIFGSYDPRRQICVGDRRERKAAFKGDSGGPLLCNNVAHGIVSYGKSSGVPPEVFTRVSSFLPWIRTTMR Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Kynureninase (KYNU) | 3E9K | 4.01 | |
Target general information Gen name KYNU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms L-kynurenine hydrolase; KYNU Protein family Kynureninase family Biochemical class Carbon-carbon bonds hydrolase Function Catalyzes the cleavage of L-kynurenine (L-Kyn) and L-3- hydroxykynurenine (L-3OHKyn) into anthranilic acid (AA) and 3- hydroxyanthranilic acid (3-OHAA), respectively. Has a preference for the L-3-hydroxy form. Also has cysteine-conjugate-beta-lyase activity. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160; DB07069; DB00114 Interacts with Q8WUE5; P56545-3; Q9NVL1-2; P61968; P59942; Q8TDC0; P78356-2; Q86WH2 EC number EC 3.7.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Proteomics identification; Pyridine nucleotide biosynthesis; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 50204.5 Length 446 Aromaticity 0.09 Instability index 39.91 Isoelectric point 6.36 Charge (pH=7) -4.59 2D Binding mode Binding energy (Kcal/mol) -5.47 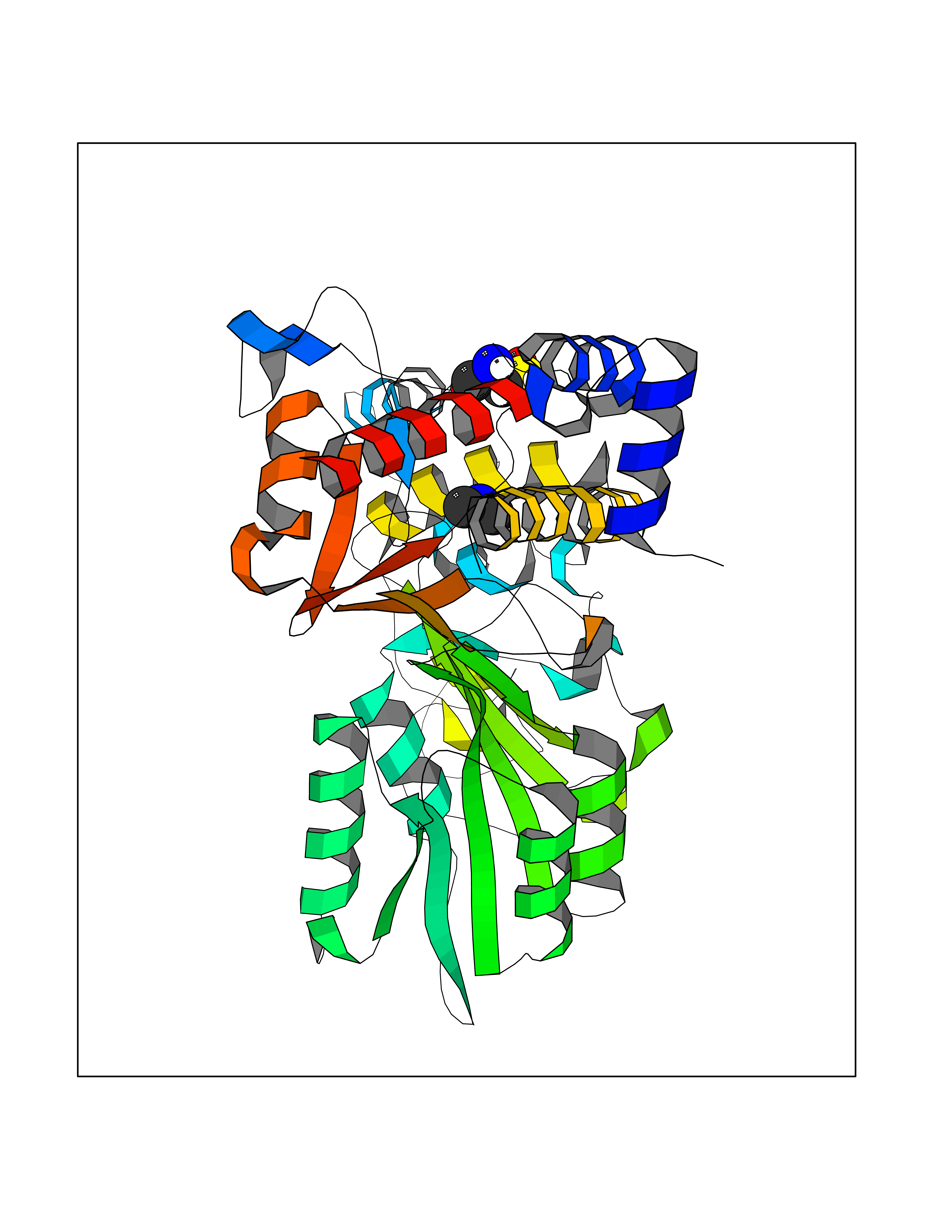 Molscript Map 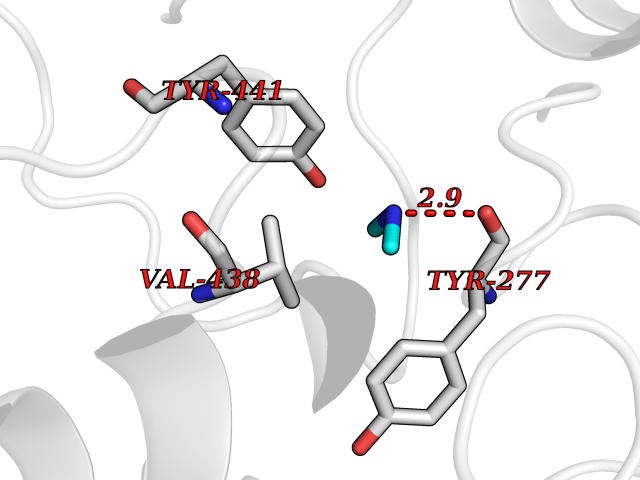 Pymol Map 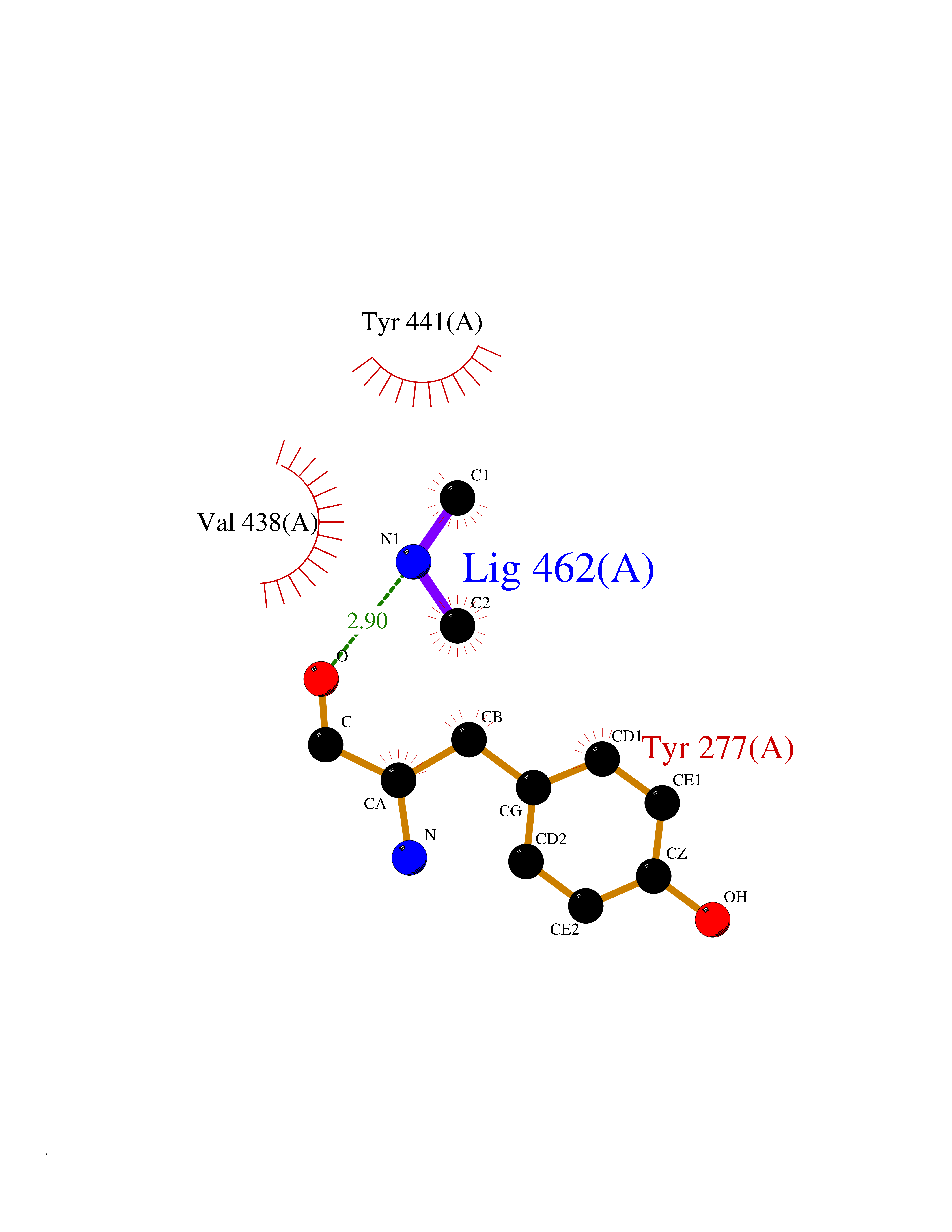 Ligplot Map 3D Binding mode Sequence LELPADTVQRIAAELKCHPTDERVALHLDEEDKLRHFREXFYIPKIQDLPPVDLSLVNKDENAIYFLGNSLGLQPKMVKTYLEEELDKWAKIAAYGHEVGKRPWITGDESIVGLMKDIVGANEKEIALMNALTVNLHLLMLSFFKPTPKRYKILLEAKAFPSDHYAIESQLQLHGLNIEESMRMIKPREGEETLRIEDILEVIEKEGDSIAVILFSGVHFYTGQHFNIPAITKAGQAKGCYVGFDLAHAVGNVELYLHDWGVDFACWCSYKYLNAGAGGIAGAFIHEKHAHTIKPALVGWFGHELSTRFKMDNKLQLIPGVCGFRISNPPILLVCSLHASLEIFKQATMKALRKKSVLLTGYLEYLIKHNYGVVNIITPSHVEERGCQLTITFSVPNKDVFQELEKRGVVCDKRNPNGIRVAPVPLYNSFHDVYKFTNLLTSILDS Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Lanosterol 14-alpha-demethylase (EC 1.14.13.70) | 4G3J | 4.01 | |
Target general information Gen name Tb11.02.4080 Organism Trypanosoma brucei brucei (strain 927/4 GUTat10.1) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class NA Function NA Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with NA EC number 1.14.13.70 Uniprot keywords 3D-structure; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID D Molecular weight (Da) 50691.4 Length 448 Aromaticity 0.08 Instability index 54.09 Isoelectric point 6.99 Charge (pH=7) -0.03 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 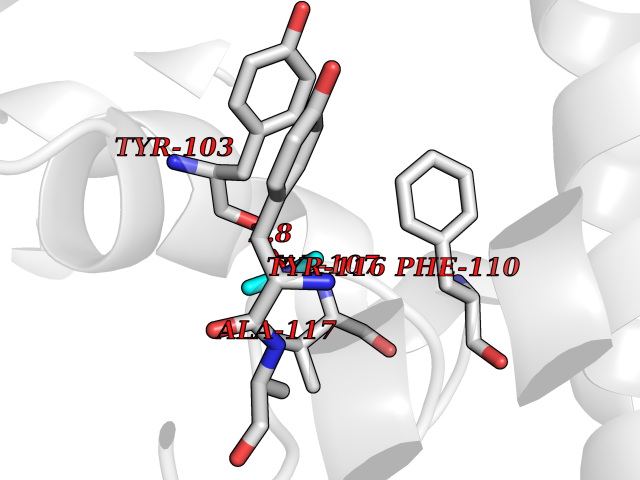 Pymol Map 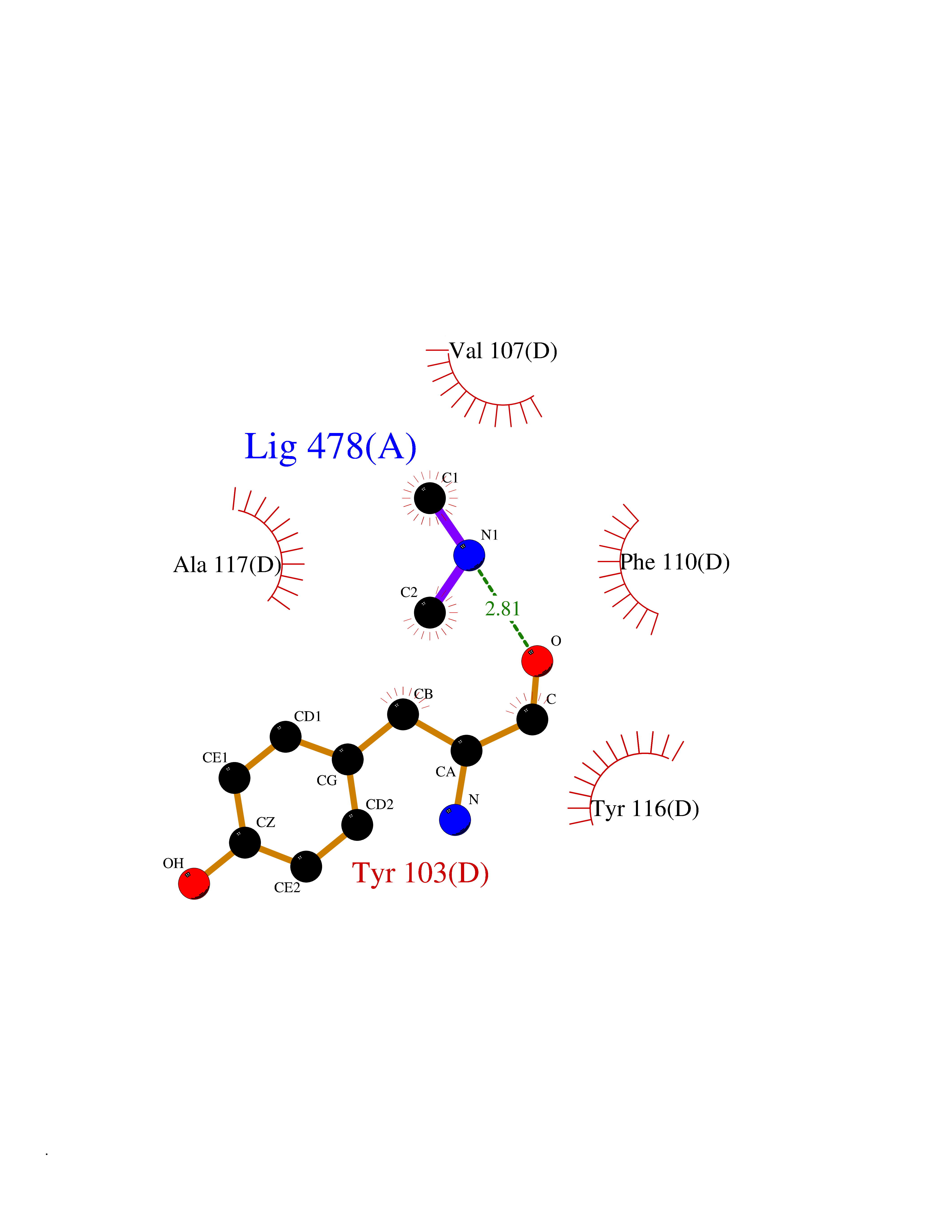 Ligplot Map 3D Binding mode Sequence GKLPPVYPVTVPILGHIIQFGKSPLGFMQECKRQLKSGIFTINIVGKRVTIVGDPHEHSRFFLPRNEVLSPREVYSFMVPVFGEGVAYAAPYPRMREQLNFLAEELTIAKFQNFVPAIQHEVRKFMAANWDKDEGEINLLEDCSTMIINTACQCLFGEDLRKRLDARRFAQLLAKMESSLIPAAVFLPILLKLPLPQSARCHEARTELQKILSEIIIARKEEEVNKDSSTSDLLSGLLSAVYRDGTPMSLHEVCGMIVAAMFAGQHTSSITTTWSMLHLMHPANVKHLEALRKEIEEFPAQLNYNNVMDEMPFAERCARESIRRDPPLLMLMRKVMADVKVGSYVVPKGDIIACSPLLSHHDEEAFPEPRRWDPERDEKVEGAFIGFGAGVHKCIGQKFGLLQVKTILATAFRSYDFQLLRDEVPDPDYHTMVVGPTASQCRVKYIRR Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Protein cereblon (CRBN) | 5FQD | 4.01 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 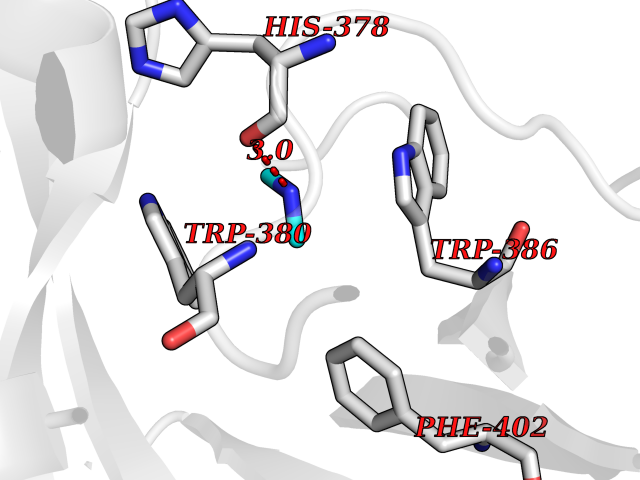 Pymol Map 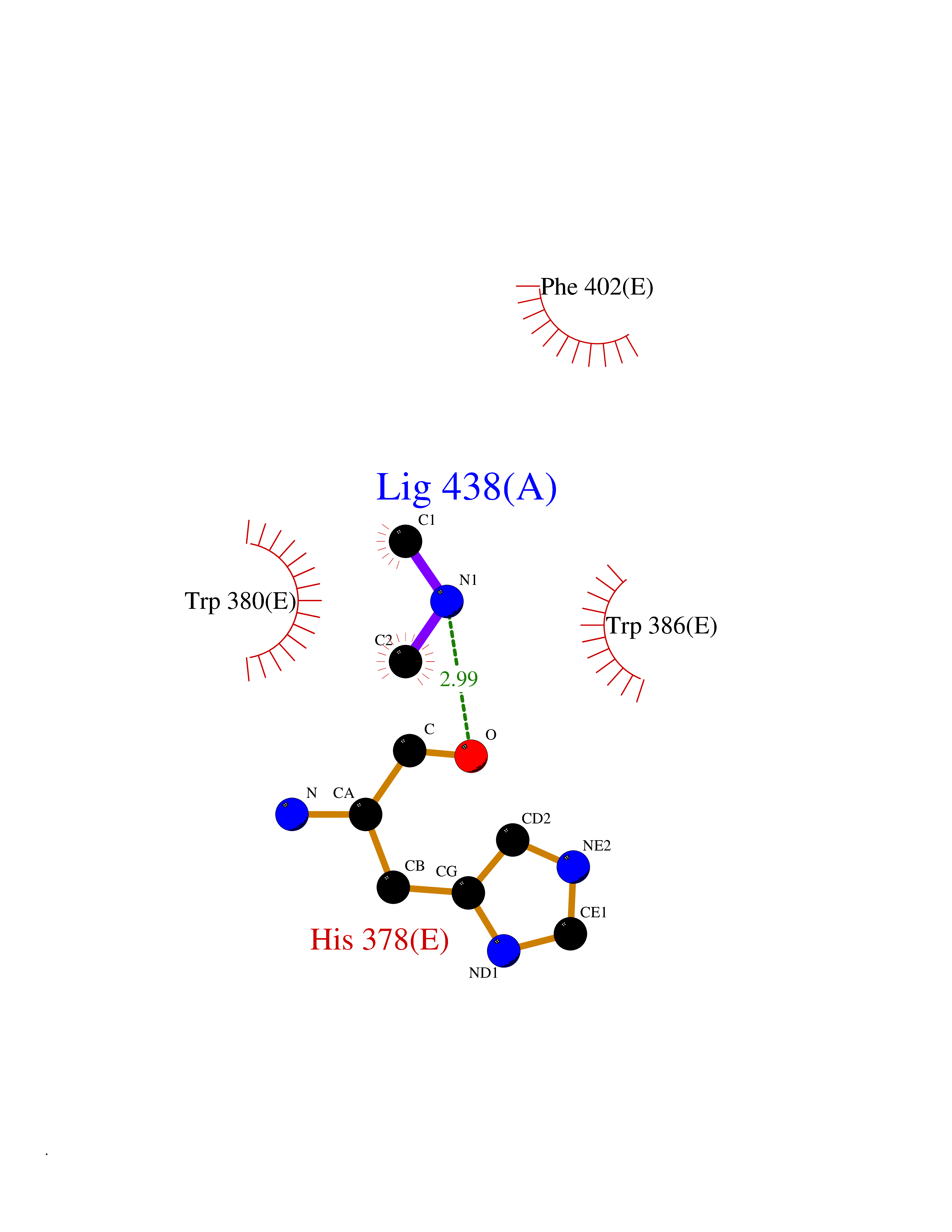 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Peroxisomal trans-2-enoyl-CoA reductase | 1YXM | 4.01 | |
Target general information Gen name PECR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PRO1004;SDR29C1 Protein family Short-chain dehydrogenases/reductases (SDR) family Biochemical class Oxidoreductase Function Receptor binding.Trans-2-enoyl-CoA reductase (NADPH) activity. Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00173 Interacts with NA EC number 1.3.1.38 Uniprot keywords 3D-structure; Alternative splicing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 30349.5 Length 283 Aromaticity 0.08 Instability index 38.34 Isoelectric point 8.89 Charge (pH=7) 5.26 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RSYLAPGLLQGQVAIVTGGATGIGKAIVKELLELGSNVVIASRKLERLKSAADELQANLPPTKQARVIPIQCNIRNEEEVNNLVKSTLDTFGKINFLVNNGGGQFLSPAEHISSKGWHAVLETNLTGTFYMCKAVYSSWMKEHGGSIVNIIVPTKAGFPLAVHSGAARAGVYNLTKSLALEWACSGIRINCVAPGVIYSQTAQSFFEGSFQKIPAKRIGVPEEVSSVVCFLLSPAASFITGQSVDVDGGRSLYTHSYEVPDHDNWPKGAGDLSVVKKMKETFK Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Haemophilus influenzae NadR protein (Hae-influ nadR) | 1LW7 | 4.01 | |
Target general information Gen name Hae-influ nadR Organism Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Uniprot ID TTD ID Synonyms nadR; Transcriptional regulator nadR Protein family Bacterial NMN adenylyltransferase family; Bacterial RNK family Biochemical class Nicotinamide ribonucleoside uptake permease Function This enzyme has twoactivities: nicotinamide mononucleotide (NMN) adenylyltransferase and ribosylnicotinamide (RN) kinase. The RN kinase activity catalyzes the phosphorylation of RN to form nicotinamide ribonucleotide. The NMN adenylyltransferase activity catalyzes the transfer of the AMP moiety of ATP to nicotinamide ribonucleotide to form NAD(+). Related diseases Involved in the epigenetic regulation of ESR1 expression in breast cancer in a TFAP2C, IFI16 and HDAC4/5/6-dependent manner. {ECO:0000269|PubMed:24413532}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell membrane; Cytoplasm; Kinase; Membrane; Multifunctional enzyme; NAD; Nucleotide-binding; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39581.5 Length 344 Aromaticity 0.14 Instability index 41.39 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 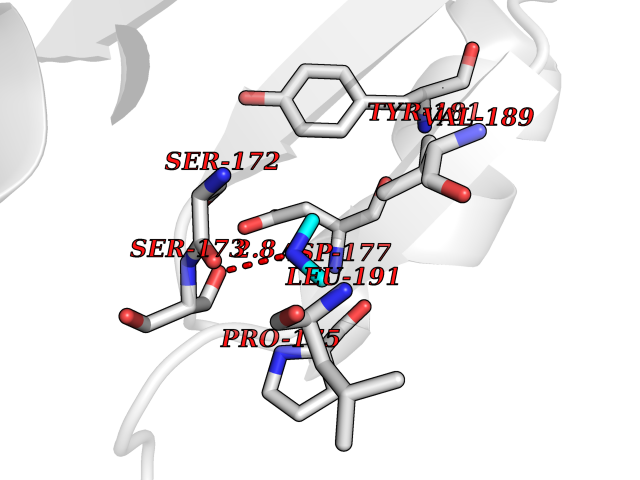 Pymol Map 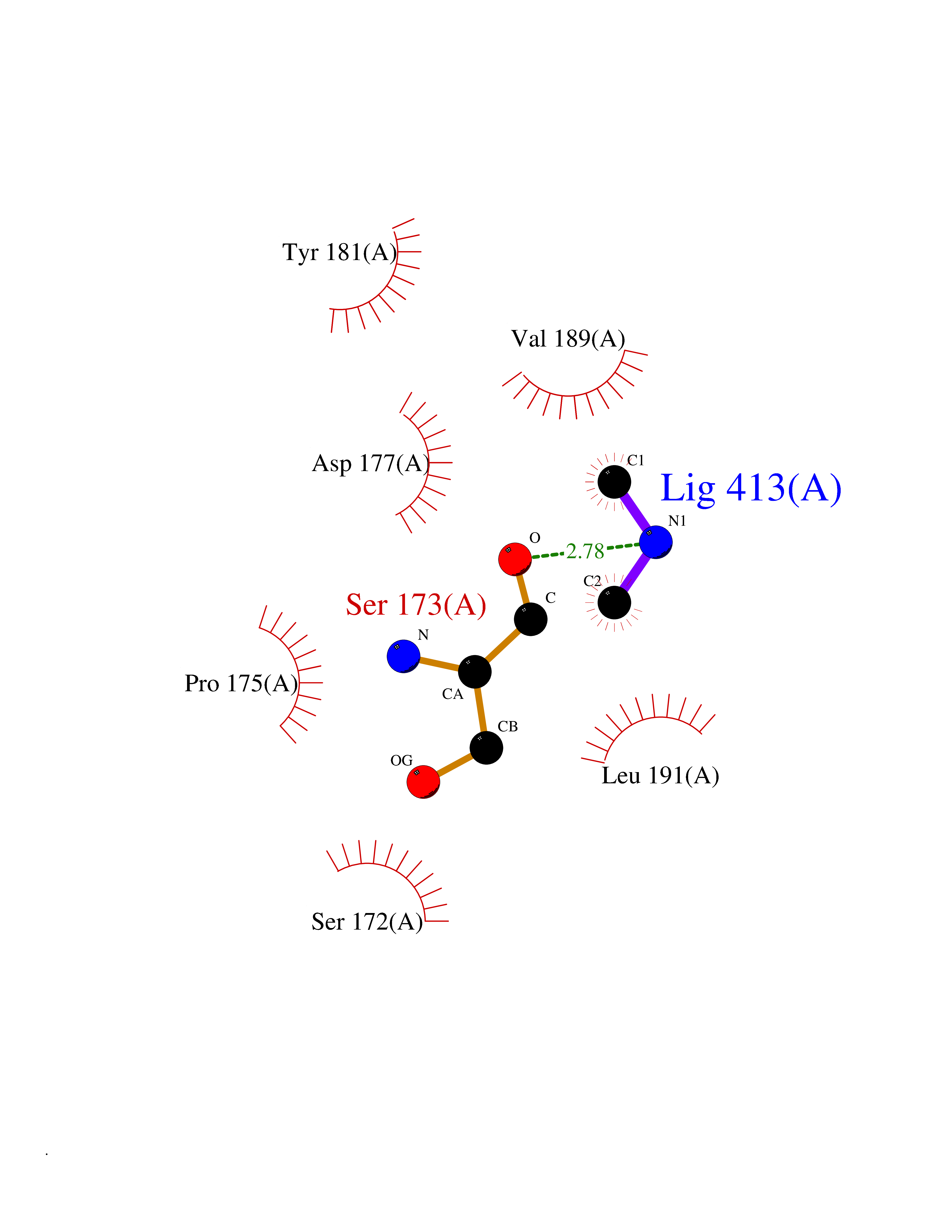 Ligplot Map 3D Binding mode Sequence EKKVGVIFGKFYPVHTGHINXIYEAFSKVDELHVIVCSDTVRDLKLFYDSKXKRXPTVQDRLRWXQQIFKYQKNQIFIHHLVEDGIPSYPNGWQSWSEAVKTLFHEKHFEPSIVFSSEPQDKAPYEKYLGLEVSLVDPDRTFFNVSATKIRTTPFQYWKFIPKEARPFFAKTVAILGGESSGKSVLVNKLAAVFNTTSAWEYGREFVFEKLGGDEQAMQYSDYPQXALGHQRYIDYAVRHSHKIAFIDTDFITTQAFCIQYEGKAHPFLDSXIKEYPFDVTILLKNNTEQKQRQQFQQLLKKLLDKYKVPYIEIESPSYLDRYNQVKAVIEKVLNEEEISELQN Hydrogen bonds contact Hydrophobic contact | ||||