Job Results:
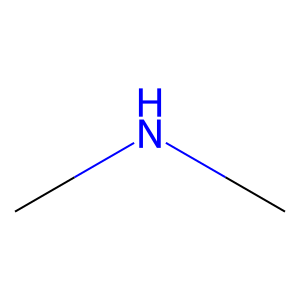
Ligand
Structure
Job ID
369cc27f15dbe7b3a0e0dfe22b2eb6d9
Job name
NA
Time
2025-03-05 09:37:27
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Protoporphyrinogen oxidase (PPOX) | 3NKS | 4.03 | |
Target general information Gen name PPOX Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PPO Protein family Protoporphyrinogen/coproporphyrinogen oxidase family, Protoporphyrinogen oxidase subfamily Biochemical class NA Function Catalyzes the 6-electron oxidation of protoporphyrinogen-IX to form protoporphyrin-IX. Related diseases Variegate porphyria (VP) [MIM:176200]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. Variegate porphyria is an acute hepatic form characterized by partial reduction of protoporphyrinogen oxidase activity, increased photosensitivity, skin blistering and scarring of sun-exposed areas, skin hyperpigmentation, abdominal pain, and neuropsychiatric symptoms. High fecal levels of protoporphyrin and coproporphyrin, increased urine uroporphyrins and iron overload are typical markers of the disease. Inheritance is autosomal dominant with incomplete penetrance. {ECO:0000269|PubMed:10486317, ECO:0000269|PubMed:11074242, ECO:0000269|PubMed:11102990, ECO:0000269|PubMed:11348478, ECO:0000269|PubMed:11350188, ECO:0000269|PubMed:11474578, ECO:0000269|PubMed:12380696, ECO:0000269|PubMed:12655566, ECO:0000269|PubMed:12859407, ECO:0000269|PubMed:12922165, ECO:0000269|PubMed:14669009, ECO:0000269|PubMed:16433813, ECO:0000269|PubMed:16621625, ECO:0000269|PubMed:16922948, ECO:0000269|PubMed:16947091, ECO:0000269|PubMed:18350656, ECO:0000269|PubMed:18570668, ECO:0000269|PubMed:19320019, ECO:0000269|PubMed:21048046, ECO:0000269|PubMed:23430901, ECO:0000269|PubMed:23467411, ECO:0000269|PubMed:24073655, ECO:0000269|PubMed:8817334, ECO:0000269|PubMed:8852667, ECO:0000269|PubMed:9763307}. The disease is caused by variants affecting the gene represented in this entry. Mutations leading to severe PPOX deficiency cause the rare homozygous variant form of VP. Missense mutations that preserve 10%-25% of wild-type activity may not cause clinically overt VP in heterozygotes (PubMed:9811936). Mutations with intermediate effect on catalytic activity may cause VP, but with a low clinical penetrance (PubMed:10486317). {ECO:0000269|PubMed:10486317, ECO:0000269|PubMed:9811936}.; DISEASE: Variegate porphyria, childhood-onset (VPCO) [MIM:620483]: An autosomal recessive form of variegate porphyria, a disorder of heme biosynthesis that results from diminished activity of protoporphyrinogen oxidase. VPCO is characterized by severe protoporphyrinogen oxidase deficiency, onset of photosensitization by porphyrins in early childhood, skin scarring and hyperpigmentation, and skeletal abnormalities of the hand. Additional variable features are short stature, impaired intellectual development, and seizures. VPCO patients rarely experience acute neuropsychiatric or abdominal attacks. {ECO:0000269|PubMed:10870850, ECO:0000269|PubMed:11286631, ECO:0000269|PubMed:33159949, ECO:0000269|PubMed:8673113, ECO:0000269|PubMed:9541112, ECO:0000269|PubMed:9811936}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.3.3.4 Uniprot keywords 3D-structure; Disease variant; FAD; Flavoprotein; Heme biosynthesis; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Porphyrin biosynthesis; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 49470.2 Length 465 Aromaticity 0.05 Instability index 48.23 Isoelectric point 7.8 Charge (pH=7) 1.98 2D Binding mode Binding energy (Kcal/mol) -5.5 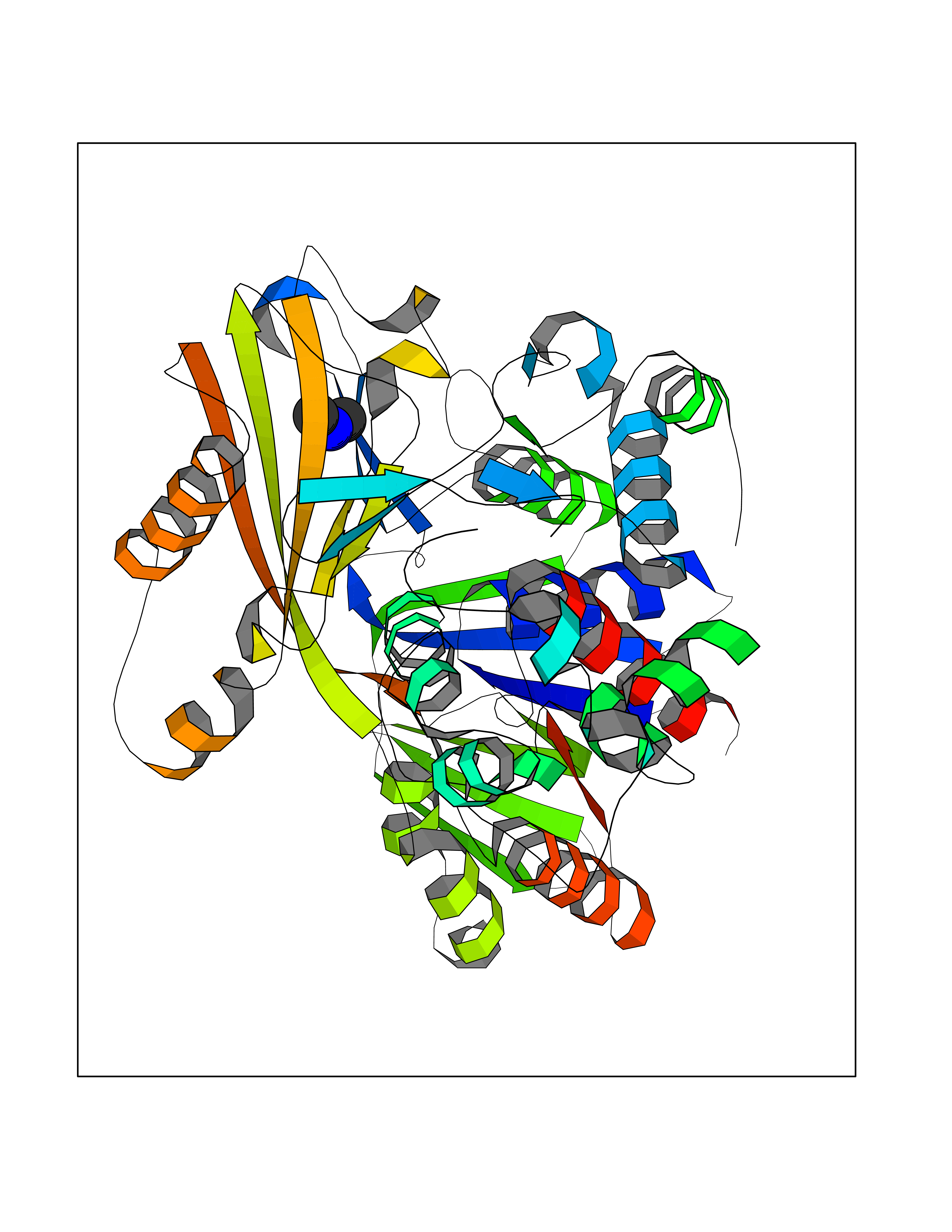 Molscript Map 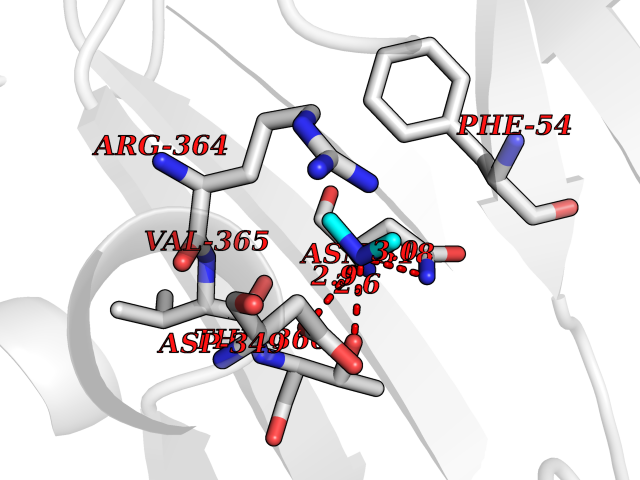 Pymol Map 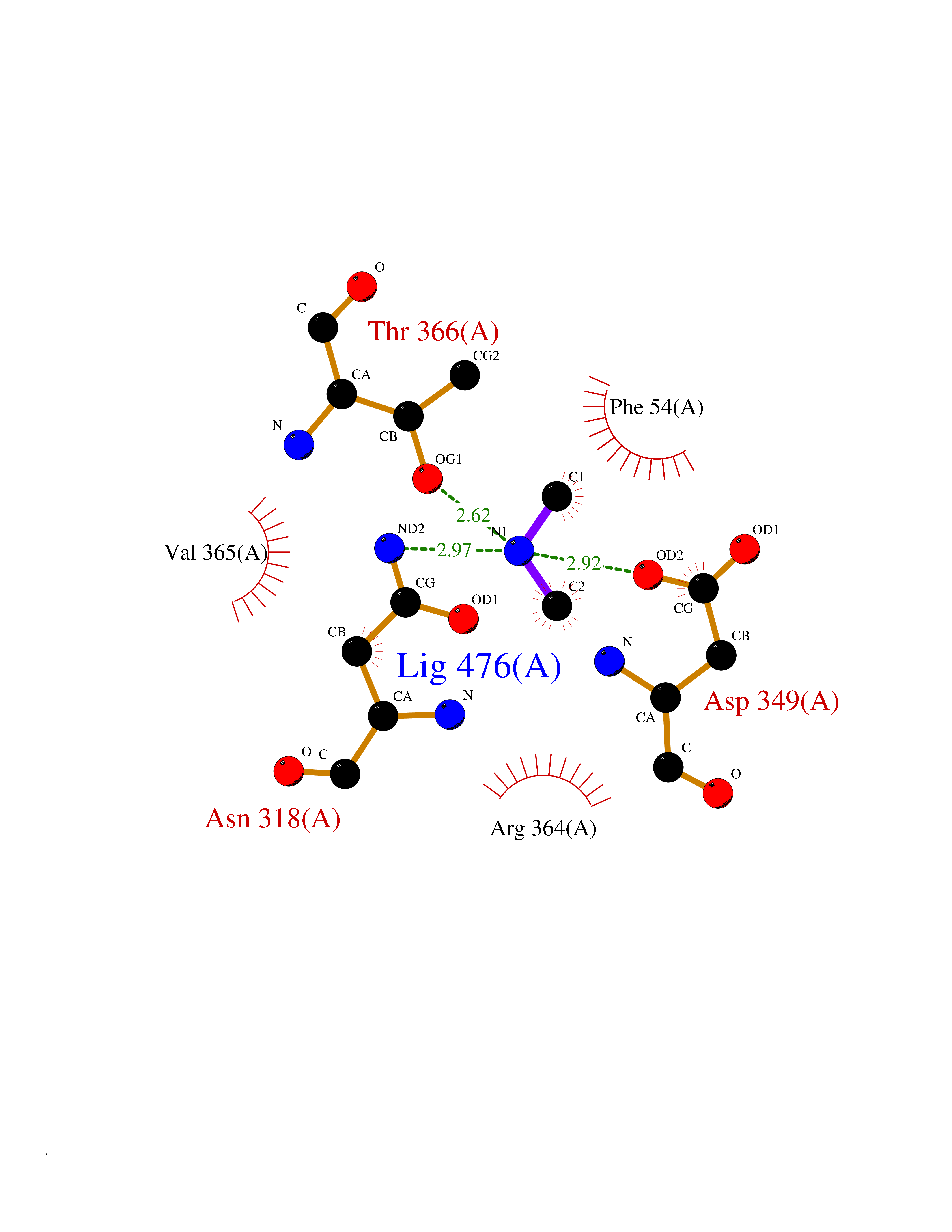 Ligplot Map 3D Binding mode Sequence GRTVVVLGGGISGLAASYHLSRAPCPPKVVLVESSERLGGWIRSVRGPNGAIFELGPRGIRPAGALGARTLLLVSELGLDSEVLPVRGDHPAAQNRFLYVGGALHALPTGLRGPSPPFSKPLFWAGLRELTKPRGKEPDETVHSFAQRRLGPEVASLAMDSLCRGVFAGNSRELSIRSCFPSLFQAEQTHRSILLGLLLGQPDSALIRQALAERWSQWSLRGGLEMLPQALETHLTSRGVSVLRGQPVCGLSLQAEGRWKVSLRDSSLEADHVISAIPASVLSELLPAEAAPLARALSAITAVSVAVVNLQYQGAHLPVQGFGHLVPSSEDPGVLGIVYDSVAFPEQDGSPPGLRVTVMLGGSWLQTLEASGCVLSQELFQQRAQEAAATQLGLKEMPSHCLVHLHKNCIPQYTLGHWQKLESARQFLTAHRLPLTLAGASYEGVAVNDCIESGRQAAVSVLGTE Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Plasma kallikrein (KLKB1) | 6T7P | 4.03 | |
Target general information Gen name KLKB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Plasma prekallikrein; Plasma kallikrein light chain; Plasma kallikrein heavy chain; PKK; Kininogenin; KLK3; Fletcher factor Protein family Peptidase S1 family, Plasma kallikrein subfamily Biochemical class Peptidase Function It activates, in a reciprocal reaction, factor XII after its binding to a negatively charged surface. It also releases bradykinin from HMW kininogen and may also play a role in the renin-angiotensin system by converting prorenin into renin. The enzyme cleaves Lys-Arg and Arg-Ser bonds. Related diseases Prekallikrein deficiency (PKKD) [MIM:612423]: An autosomal recessive condition characterized by a clotting defect due to prolongation of activated partial thromboplastin time. Affected individuals are clinically asymptomatic. {ECO:0000269|PubMed:14652634, ECO:0000269|PubMed:17598838, ECO:0000269|PubMed:34847617}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15982; DB09228; DB05311; DB12831; DB06404; DB14597; DB01593; DB14487; DB14533; DB14548 Interacts with Q9UI10; O00746; C9J082; O14744; Q8TAS3; O00233; Q8IYM2; Q9UMY4; O43493-5; Q8NFB2; Q8N0U8 EC number EC 3.4.21.34 Uniprot keywords 3D-structure; Blood coagulation; Direct protein sequencing; Disease variant; Disulfide bond; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Inflammatory response; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 26696.2 Length 237 Aromaticity 0.1 Instability index 34.33 Isoelectric point 8.07 Charge (pH=7) 2.21 2D Binding mode Binding energy (Kcal/mol) -5.5  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence IVGGTNSSWGEWPWQVSLQVKLTAQRHLCGGSLIGHQWVLTAAHCFDGLPLQDVWRIYSGILNLSDITKDTPFSQIKEIIIHQNYKVSEGNHDIALIKLQAPLNYTEFQKPICLPSKGDTSTIYTNCWVTGWGFSKEKGEIQNILQKVNIPLVTNEECQKRYQDYKITQRMVCAGYKEGGKDACKGDSGGPLVCKHNGMWRLVGITSWGEGCARREQPGVYTKVAEYMDWILEKTQS Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | DNA topoisomerase 4 subunit A | 1ZVT | 4.02 | |
Target general information Gen name parC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3019;JW2987 Protein family Type II topoisomerase GyrA/ParC subunit family, ParC type 1 subfamily Biochemical class Isomerase Function ATP binding.DNA binding.DNA topoisomerase type II (ATP-hydrolyzing) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11943; DB12924; DB00817 Interacts with P22523; P0A7K2 EC number 5.6.2.2 Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; DNA-binding; Isomerase; Membrane; Reference proteome; Topoisomerase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 26490.3 Length 246 Aromaticity 0.04 Instability index 46.03 Isoelectric point 8.94 Charge (pH=7) 2.83 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map  Pymol Map 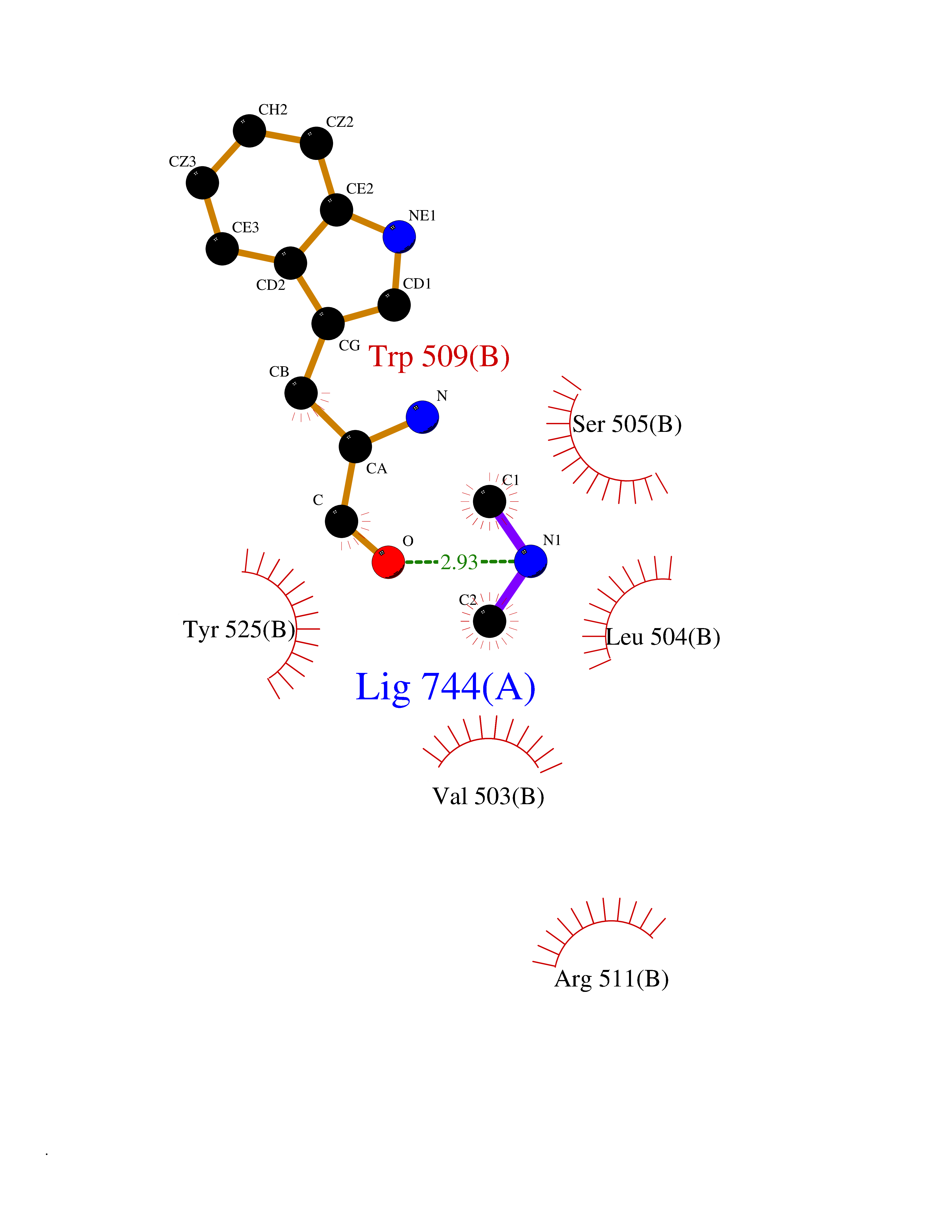 Ligplot Map 3D Binding mode Sequence SEPVTIVLSQMGWVRSAKGHDIDAPGLNYKAGDSFKAAVKGKSNQPVVFVDSTGRSYAIDPITLPSARGQGEPLTGKLTLPPGATVDHMLMESDDQKLLMASDAGYGFVCTFNDLVARNRAGKALITLPENAHVMPPVVIEDASDMLLAITQAGRMLMFPVSDLPQLSKGKGNKIINIPSAEAARGEDGLAQLYVLPPQSTLTIHVGKRKIKLRPEELQKVTGERGRRGTLMRGLQRIDRVEIDSP Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Tartrate-resistant acid phosphatase type 5 | 2BQ8 | 4.02 | |
Target general information Gen name ACP5 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Metallophosphoesterase superfamily, Purple acid phosphatase family Biochemical class Hydrolase Function Acid phosphatase activity.Ferric iron binding.Ferrous iron binding. Related diseases Spondyloenchondrodysplasia with immune dysregulation (SPENCDI) [MIM:607944]: A disease characterized by vertebral and metaphyseal dysplasia, spasticity with cerebral calcifications, and strong predisposition to autoimmune diseases. The skeletal dysplasia is characterized by radiolucent and irregular spondylar and metaphyseal lesions that represent islands of chondroid tissue within bone. {ECO:0000269|PubMed:21217752, ECO:0000269|PubMed:21217755}. The disease is caused by variants affecting the gene represented in this entry. ACP5 inactivating mutations result in a functional excess of phosphorylated osteopontin causing deregulation of osteopontin signaling and consequential autoimmune disease. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.3.2 Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Iron; Lysosome; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID X Molecular weight (Da) 34330.6 Length 304 Aromaticity 0.12 Instability index 42.3 Isoelectric point 9.11 Charge (pH=7) 6.75 2D Binding mode Binding energy (Kcal/mol) -5.48 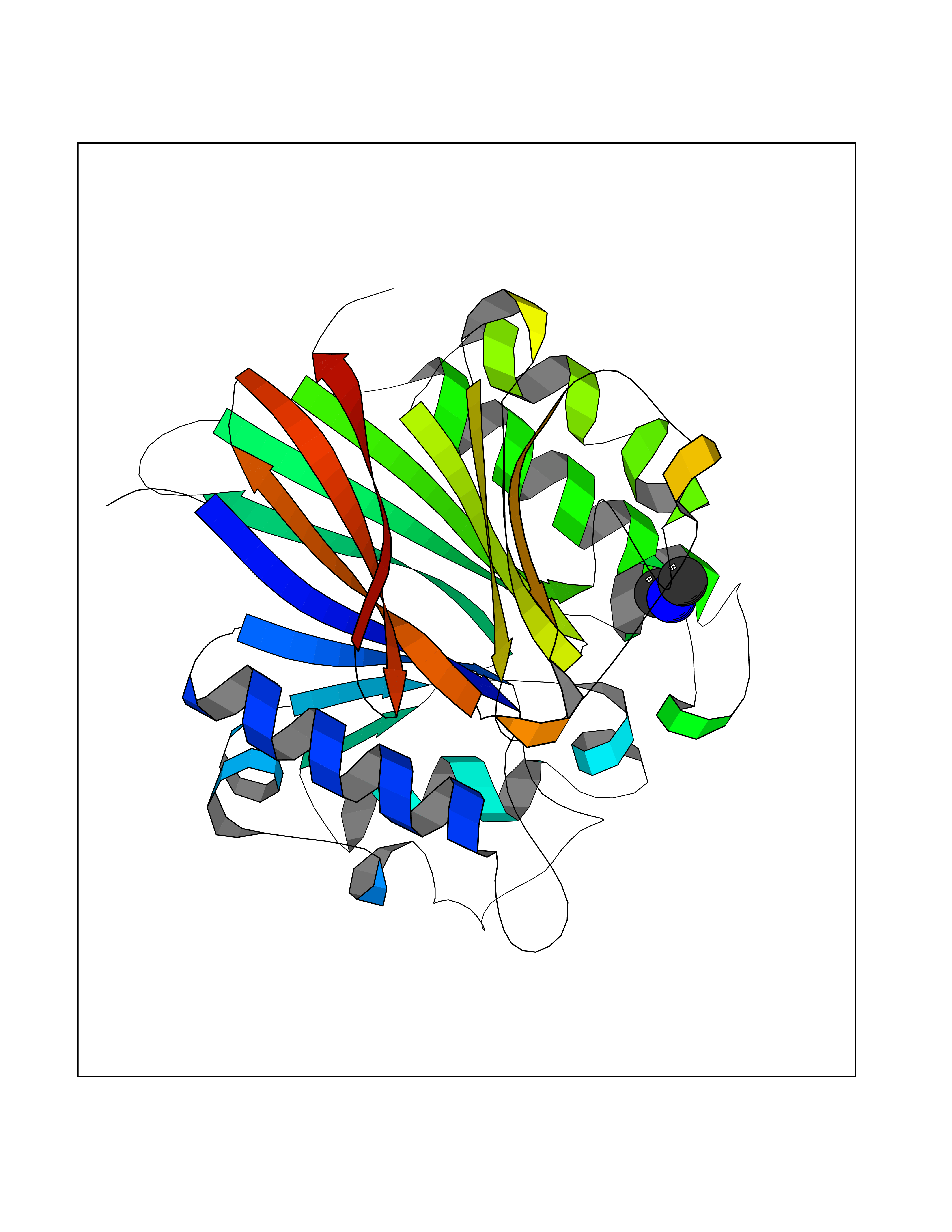 Molscript Map 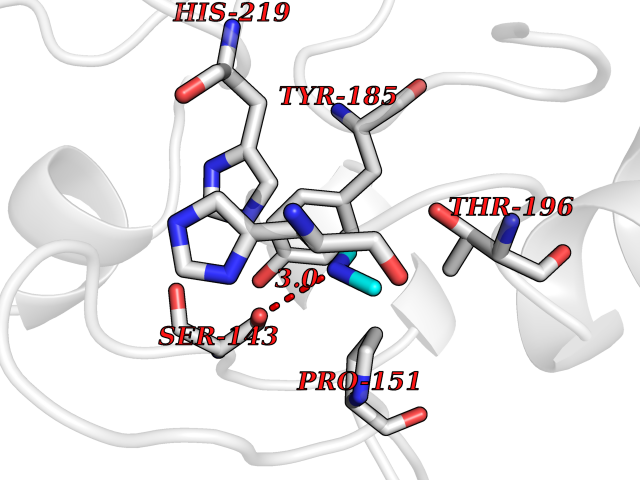 Pymol Map 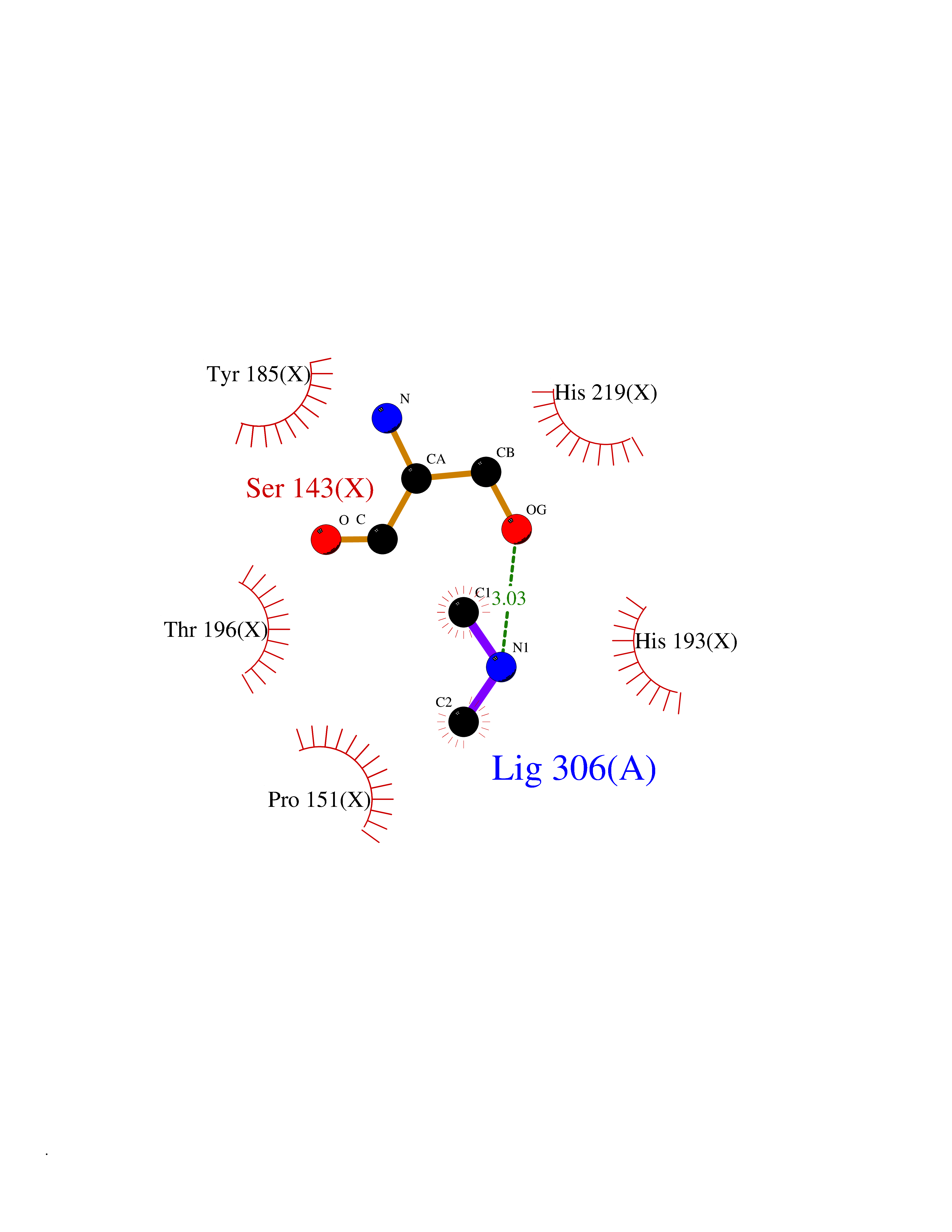 Ligplot Map 3D Binding mode Sequence ATPALRFVAVGDWGGVPNAPFHTAREMANAKEIARTVQILGADFILSLGDNFYFTGVQDINDKRFQETFEDVFSDRSLRKVPWYVLAGNHDHLGNVSAQIAYSKISKRWNFPSPFYRLHFKIPQTNVSVAIFMLDTVTLCGNSDDFLSQQPERPRDVKLARTQLSWLKKQLAAAREDYVLVAGHYPVWSIAEHGPTHCLVKQLRPLLATYGVTAYLCGHDHNLQYLQDENGVGYVLSGAGNFMDPSKRHQRKVPNGYLRFHYGTEDSLGGFAYVEISSKEMTVTYIEASGKSLFKTRLPRRARP Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | 3-oxoacyl-[acyl-carrier-protein] synthase 1 | 2VBA | 4.02 | |
Target general information Gen name fabB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2320;fabC;b2323 Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Transferase Function 3-oxoacyl-[acyl-carrier-protein] synthase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08627; DB08628; DB08359; DB04302; DB03600; DB04519; DB01034; DB03017 Interacts with NA EC number 2.3.1.41 Uniprot keywords 3D-structure; Acyltransferase; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40198.1 Length 383 Aromaticity 0.06 Instability index 38.17 Isoelectric point 5.17 Charge (pH=7) -13.62 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MKRAVITGLGIVSSIGNNQQEVLASLREGRSGITFSQELKDSGMRSHVWGNVKLDTTGLIDRKVVRFMSDASIYAFLSMEQAIADAGLSPEAYQNNPRVGLIAGSGGGSPRFVTKAMASGVSACLATPFKIHGVNYSISSACATSAHCIGNAVEQIQLGKQDIVFAGGGEELCWEMACEFDAMGALSTKYNDTPEKASRTYDAHRDGFVIAGGGGMVVVEELEHALARGAHIYAEIVGYGATSDGADMVAPSGEGAVRCMKMAMHGVDTPIDYLNSHGTSTPVGDVKELAAIREVFGDKSPAISATKAMTGHSLGAAGVQEAIYSLLMLEHGFIAPSINIEELDEQAAGLNIVTETTDRELTTVMSNSFGFGGTNATLVMRKL Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Phosphoinositide dependent protein kinase-1 (PDPK1) | 5LVO | 4.02 | |
Target general information Gen name PDPK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PDK1; HPDK1; 3-phosphoinositide-dependent protein kinase 1; 3-Phosphoinositide-dependent kinase-1; 3'-phosphoinositide dependent kinase 1 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, PDPK1 subfamily Biochemical class Kinase Function Its targets include: protein kinase B (PKB/AKT1, PKB/AKT2, PKB/AKT3), p70 ribosomal protein S6 kinase (RPS6KB1), p90 ribosomal protein S6 kinase (RPS6KA1, RPS6KA2 and RPS6KA3), cyclic AMP-dependent protein kinase (PRKACA), protein kinase C (PRKCD and PRKCZ), serum and glucocorticoid-inducible kinase (SGK1, SGK2 and SGK3), p21-activated kinase-1 (PAK1), protein kinase PKN (PKN1 and PKN2). Plays a central role in the transduction of signals from insulin by providing the activating phosphorylation to PKB/AKT1, thus propagating the signal to downstream targets controlling cell proliferation and survival, as well as glucose and amino acid uptake and storage. Negatively regulates the TGF-beta-induced signaling by: modulating the association of SMAD3 and SMAD7 with TGF-beta receptor, phosphorylating SMAD2, SMAD3, SMAD4 and SMAD7, preventing the nuclear translocation of SMAD3 and SMAD4 and the translocation of SMAD7 from the nucleus to the cytoplasm in response to TGF-beta. Activates PPARG transcriptional activity and promotes adipocyte differentiation. Activates the NF-kappa-B pathway via phosphorylation of IKKB. The tyrosine phosphorylated form is crucial for the regulation of focal adhesions by angiotensin II. Controls proliferation, survival, and growth of developing pancreatic cells. Participates in the regulation of Ca(2+) entry and Ca(2+)-activated K(+) channels of mast cells. Essential for the motility of vascular endothelial cells (ECs) and is involved in the regulation of their chemotaxis. Plays a critical role in cardiac homeostasis by serving as a dual effector for cell survival and beta-adrenergic response. Plays an important role during thymocyte development by regulating the expression of key nutrient receptors on the surface of pre-T cells and mediating Notch-induced cell growth and proliferative responses. Provides negative feedback inhibition to toll-like receptor-mediated NF-kappa-B activation in macrophages. Isoform 3 is catalytically inactive. Serine/threonine kinase which acts as a master kinase, phosphorylating and activating a subgroup of the AGC family of protein kinases. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB07132; DB06932; DB07300; DB07456; DB07457; DB07033; DB01933; DB03777; DB01946; DB00482; DB04522; DB12010; DB01863; DB02010 Interacts with P31749; Q00005; Q9Y4P3; O75385; P54252; P42858; Q8WXH2; Q8IUH5 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; ATP-binding; Cell junction; Cell membrane; Cytoplasm; Direct protein sequencing; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32750.5 Length 286 Aromaticity 0.12 Instability index 40.9 Isoelectric point 8.55 Charge (pH=7) 3.58 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 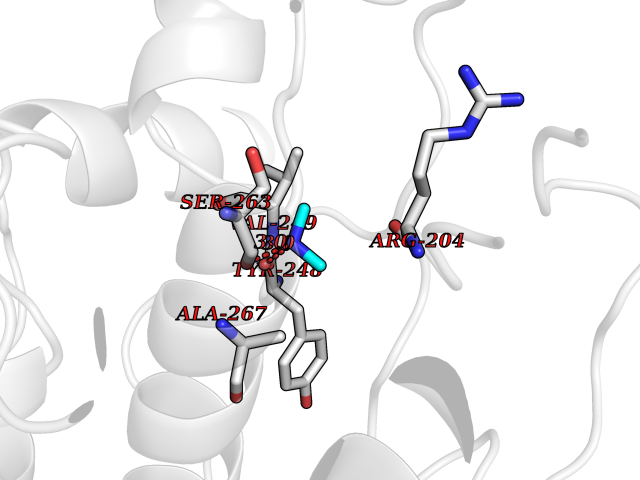 Pymol Map 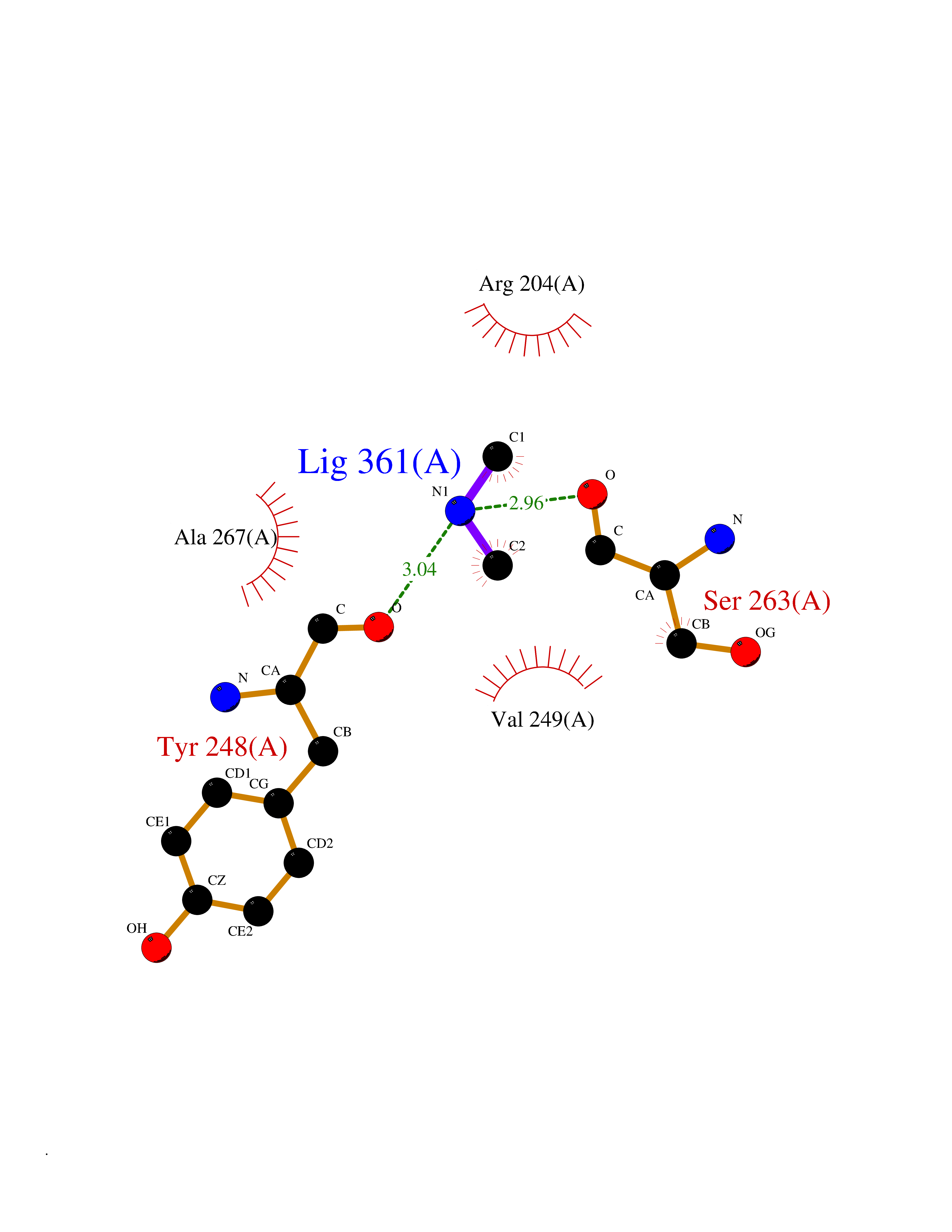 Ligplot Map 3D Binding mode Sequence PRKKRPEDFKFGKILGEGSFSTVVLARELATSREYAIKILEKRHIIKENKVPYVTRERDVMSRLDHPFFVKLYFTFQDDEKLYFGLSYAKNGELLKYIRKIGSFDETCTRFYTAEIVSALEYLHGKGIIHRDLKPENILLNEDMHIQITDFGTAKVLSPESKQARANXFVGTAQYVSPELLTEKSACKSSDLWALGCIIYQLVAGLPPFRAGNEGLIFAKIIKLEYDFPEKFFPKARDLVEKLLVLDATKRLGCEEMEGYGPLKAHPFFESVTWENLHQQTPPKLT Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Urokinase-type plasminogen activator (PLAU) | 4JNI | 4.02 | |
Target general information Gen name PLAU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UPA; U-plasminogen activator Protein family Peptidase S1 family Biochemical class Peptidase Function Specifically cleaves the zymogen plasminogen to form the active enzyme plasmin. Related diseases Quebec platelet disorder (QPD) [MIM:601709]: An autosomal dominant bleeding disorder due to a gain-of-function defect in fibrinolysis. Although affected individuals do not exhibit systemic fibrinolysis, they show delayed onset bleeding after challenge, such as surgery. The hallmark of the disorder is markedly increased PLAU levels within platelets, which causes intraplatelet plasmin generation and secondary degradation of alpha-granule proteins. {ECO:0000269|PubMed:20007542}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07129; DB07122; DB01905; DB02287; DB03729; DB01725; DB08072; DB07625; DB07626; DB08697; DB03136; DB01977; DB07076; DB03082; DB02705; DB02473; DB02398; DB02551; DB03865; DB06855; DB06856; DB03046; DB04059; DB04172; DB00594; DB03127; DB02526; DB03159; DB05254; DB03782; DB06857; DB16701; DB03876; DB03476 Interacts with Q9UKQ2; P05067; Q03405-1; P05121; P55000 EC number EC 3.4.21.73 Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Direct protein sequencing; Disulfide bond; EGF-like domain; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Phosphoprotein; Plasminogen activation; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID U Molecular weight (Da) 25825.3 Length 229 Aromaticity 0.1 Instability index 47.36 Isoelectric point 8.65 Charge (pH=7) 5.38 2D Binding mode Binding energy (Kcal/mol) -5.48 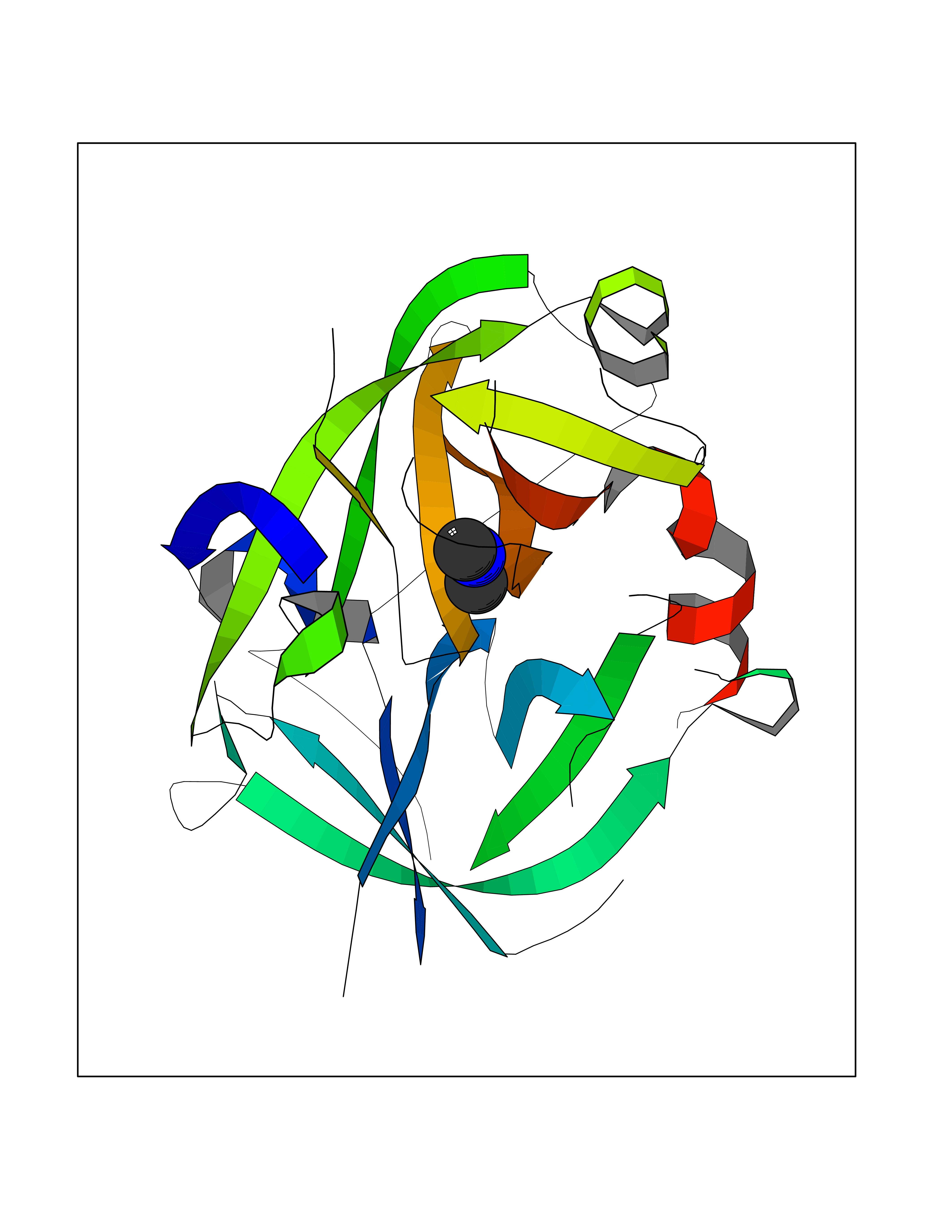 Molscript Map 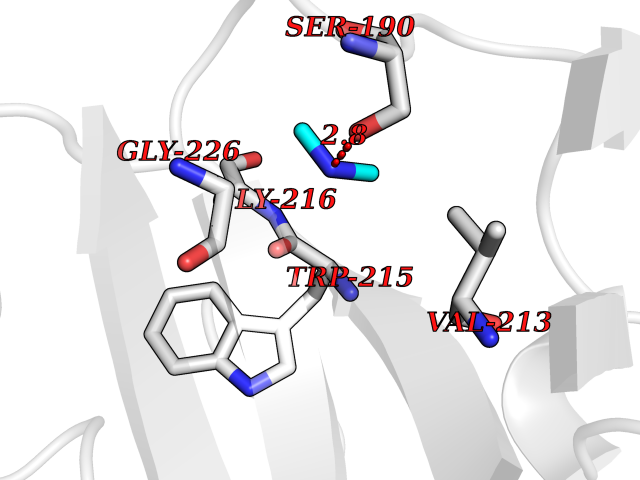 Pymol Map 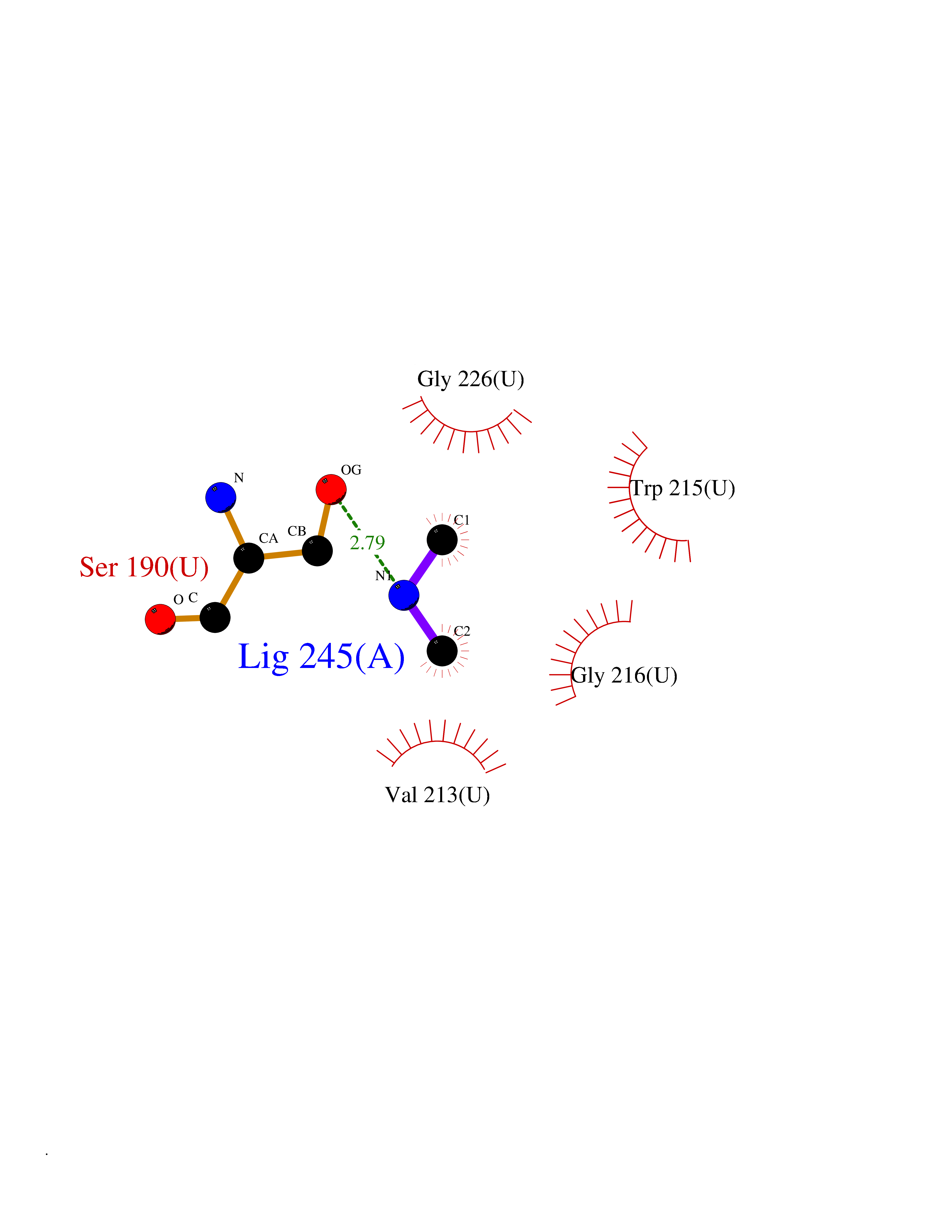 Ligplot Map 3D Binding mode Sequence IIGGEFTTIENQPWFAAIYRRSVTYVCGGSLISPCWVISATHCFPKKEDYIVYLGRSRLNSNTQGEMKFEVENLILHKDYSALAHHNDIALLKIRRCAQPSRTIQTIALPSMYNDPQFGTSCEITGFGKEQSTDYLYPEQLKMTVVKLISHRECQQHYYGSEVTTKMLCAAQWKTDSCQGDSGGPLVCSLQGRMTLTGIVSWGRGCALDKPGVYTRVSHFLPWIRSHTK Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Aromatic-L-amino-acid decarboxylase (DDC) | 3RCH | 4.02 | |
Target general information Gen name DDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DOPA decarboxylase; AADC Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the decarboxylation of L-3,4-dihydroxyphenylalanine (DOPA) to dopamine, L-5-hydroxytryptophan to serotonin and L-tryptophan to tryptamine. Related diseases Aromatic L-amino-acid decarboxylase deficiency (AADCD) [MIM:608643]: An inborn error in neurotransmitter metabolism that leads to combined serotonin and catecholamine deficiency. It causes developmental and psychomotor delay, poor feeding, lethargy, ptosis, intermittent hypothermia, gastrointestinal disturbances. The onset is early in infancy and inheritance is autosomal recessive. {ECO:0000269|PubMed:14991824, ECO:0000269|PubMed:15079002, ECO:0000269|Ref.12}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB12783; DB00190; DB00260; DB06262; DB13848; DB00875; DB01235; DB00968; DB00114; DB00150 Interacts with P10275 EC number EC 4.1.1.28 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Disease variant; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 41117.4 Length 369 Aromaticity 0.09 Instability index 37.52 Isoelectric point 8.26 Charge (pH=7) 3.76 2D Binding mode Binding energy (Kcal/mol) -5.48 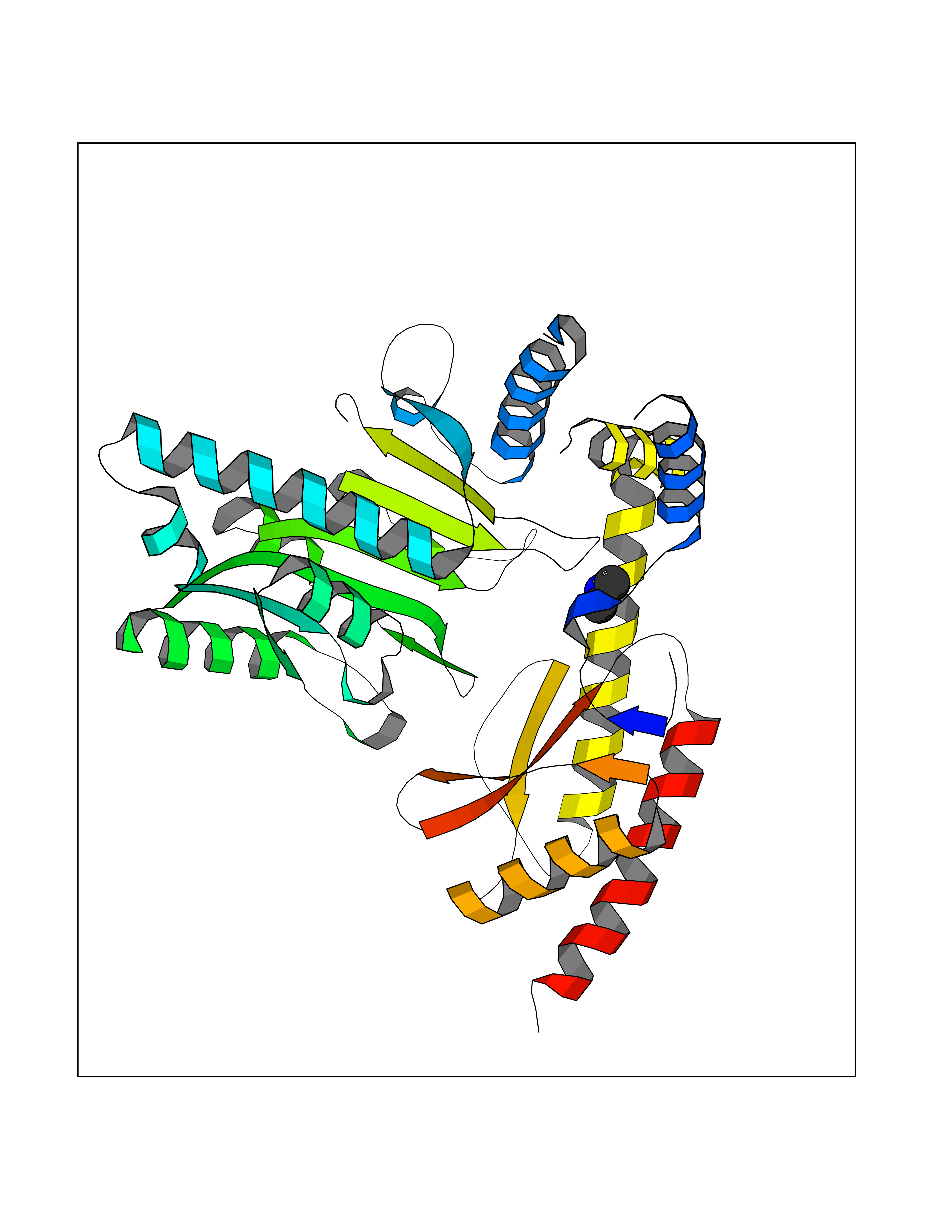 Molscript Map 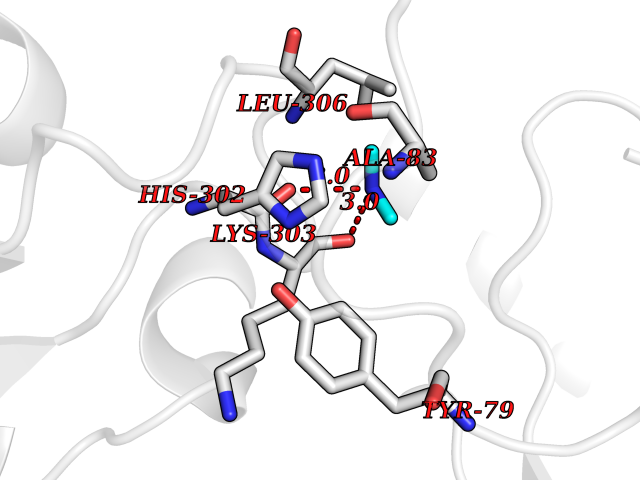 Pymol Map 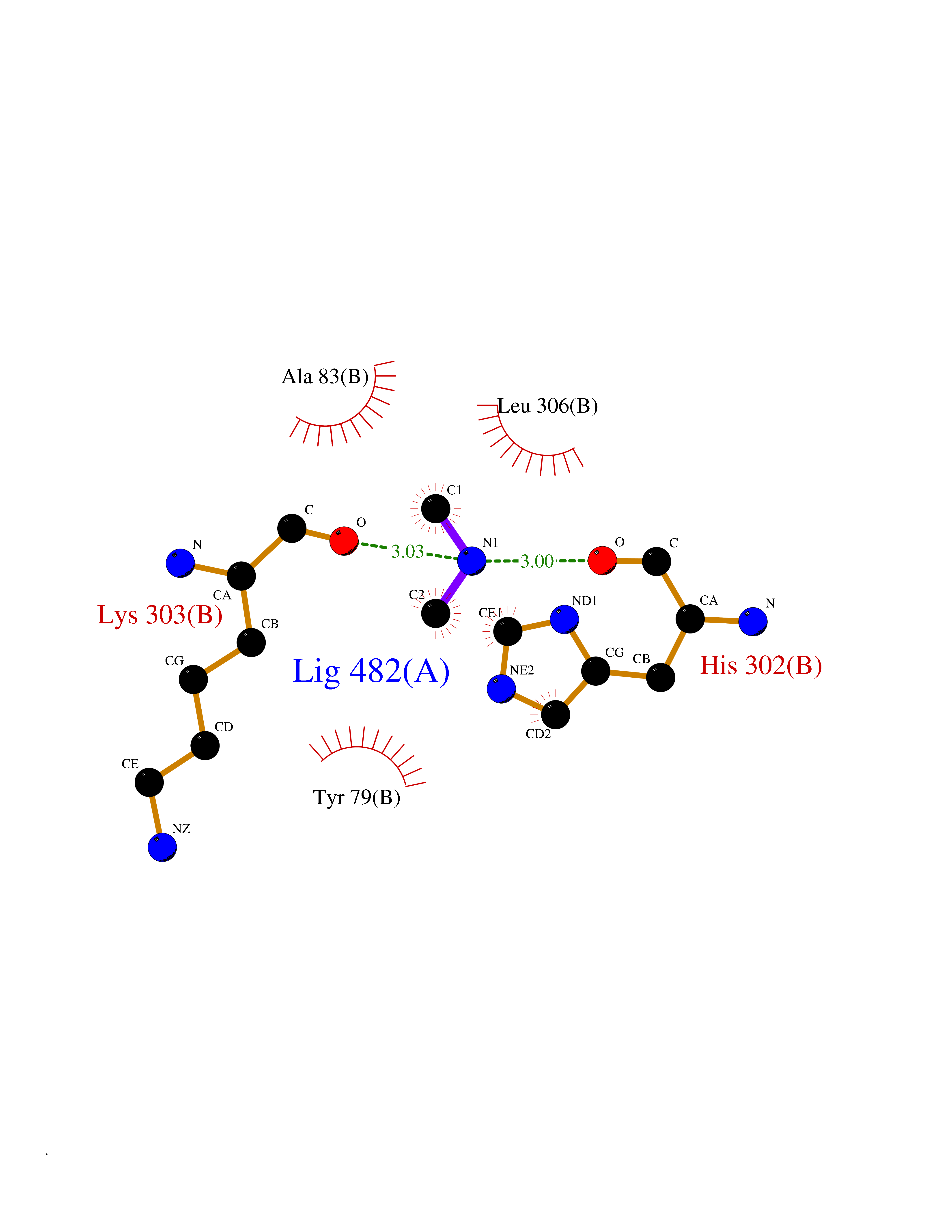 Ligplot Map 3D Binding mode Sequence HSPYFFAYFPTASSYPAMLADMLCGAIGASPACTELETVMMDWLGKMLELPKAFLNEKAGEGGGVIQGSASEATLVALLAARTKVIHRLQAASPELTQAAIMEKLVAYSSDQAHSSVERAGLIGGVKLKAIPSDGNFAMRASALQEALERDKAAGLIPFFMVATLGTTTCCSFDNLLEVGPICNKEDIWLHVDAAYAGSAFICPEFRHLLNGVEFADSFNFNPHKWLLVNFDCSAMWVKKRTDLRFRSLKMWFVFRMYGVKGLQAYIRKHVQLSHEFESLVRQDPRFEICVEVILGLVCFRLKGSNKVNEALLQRINSAKKIHLVPCHLRDKFVLRFAICSRTVESAHVQRAWEHIKELAADVLRAERE Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Aminoacylase-1 | 1Q7L | 4.02 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -5.49 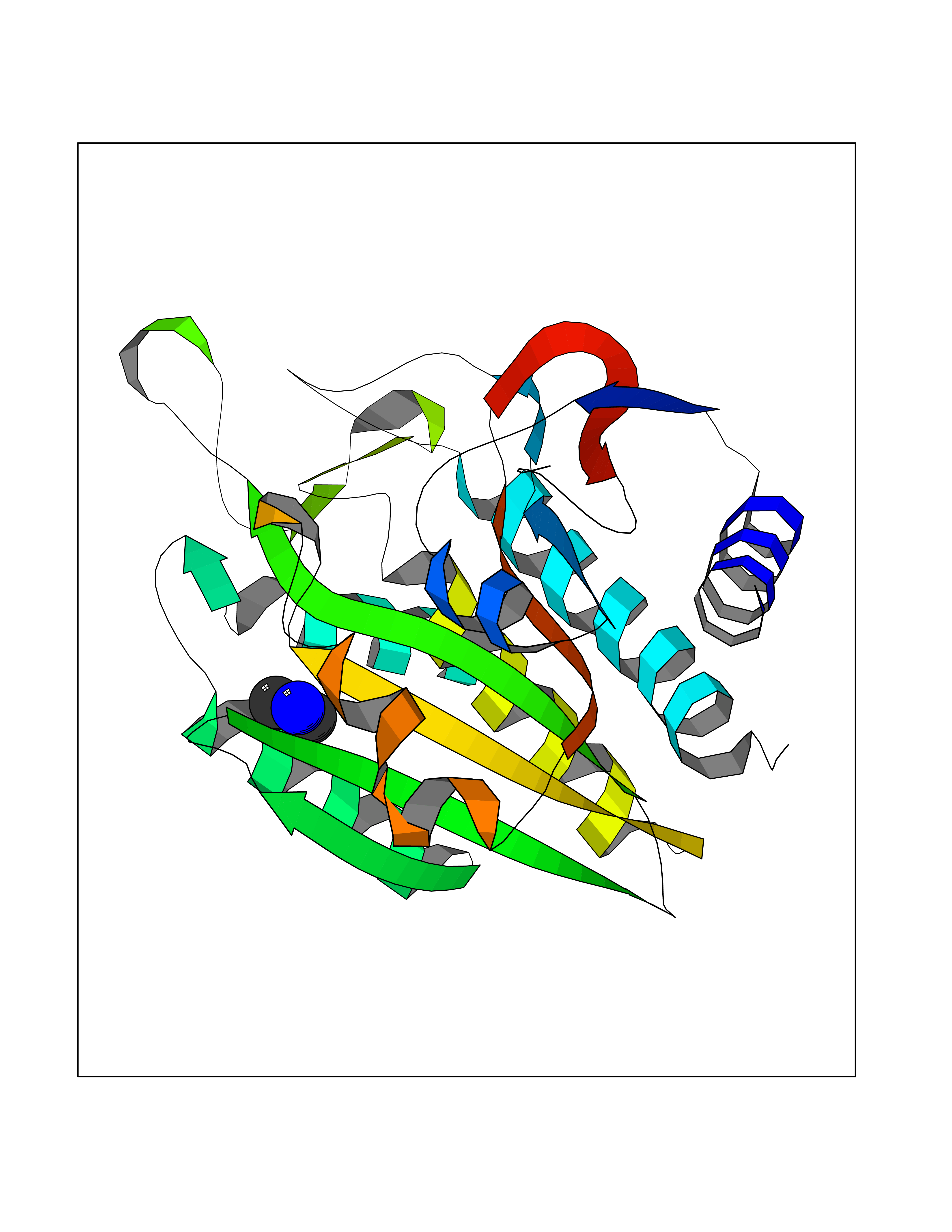 Molscript Map 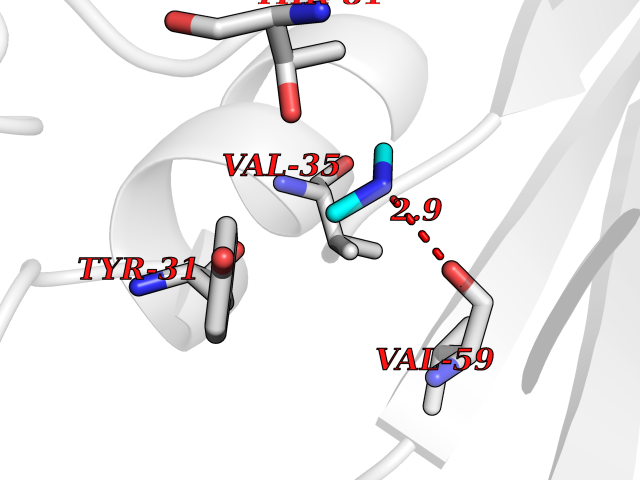 Pymol Map 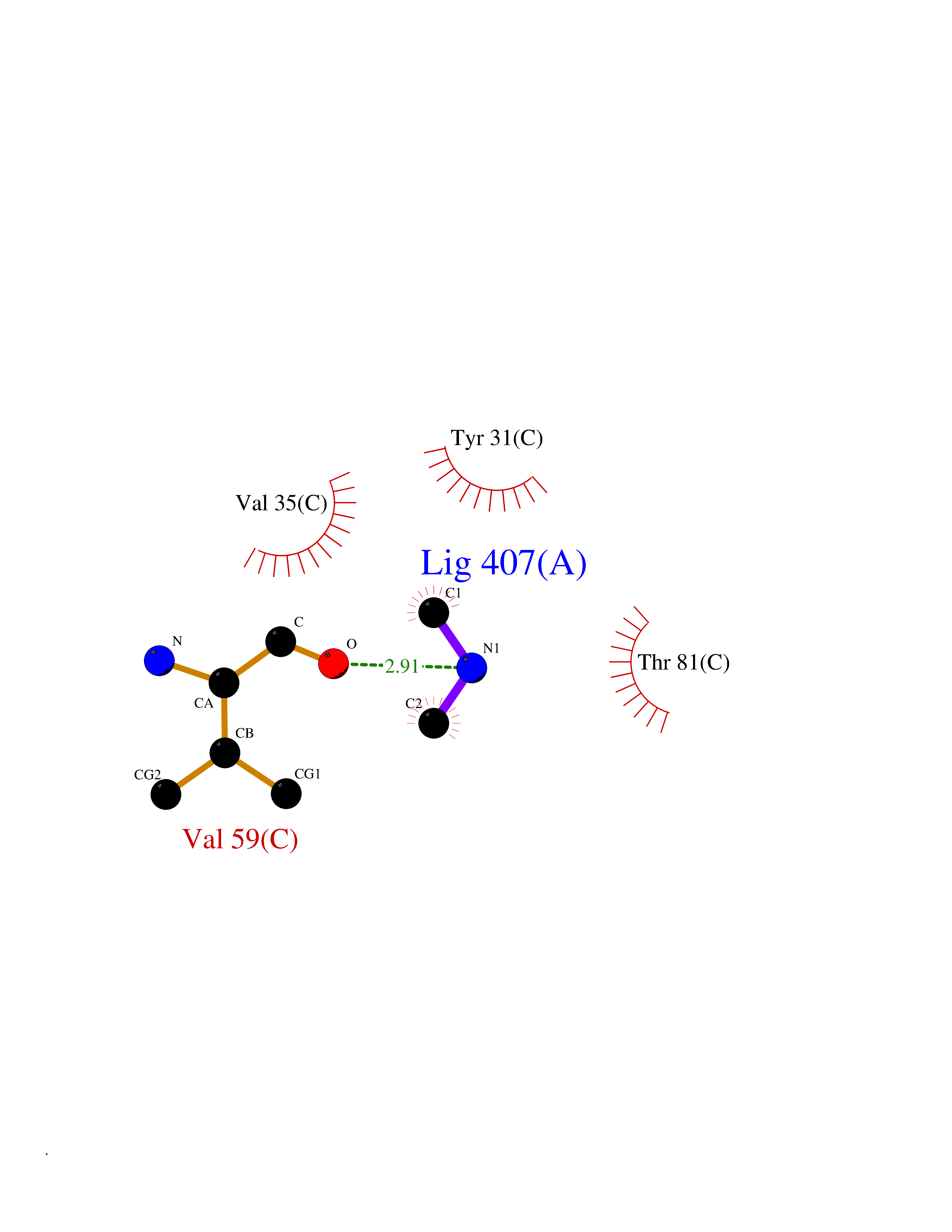 Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Carbapenem-hydrolyzing beta-lactamase KPC | 3RXW | 4.02 | |
Target general information Gen name bla Organism Klebsiella pneumoniae Uniprot ID TTD ID NA Synonyms kpc;kpc1 Protein family Class-A beta-lactamase family Biochemical class Hydrolase / hydrolase inhibitor Function Beta-lactamase activity. Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09060; DB12107 Interacts with NA EC number 3.5.2.6 Uniprot keywords 3D-structure; Antibiotic resistance; Hydrolase; Plasmid; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 27965.2 Length 264 Aromaticity 0.08 Instability index 26.27 Isoelectric point 6.14 Charge (pH=7) -1.42 2D Binding mode Binding energy (Kcal/mol) -5.48 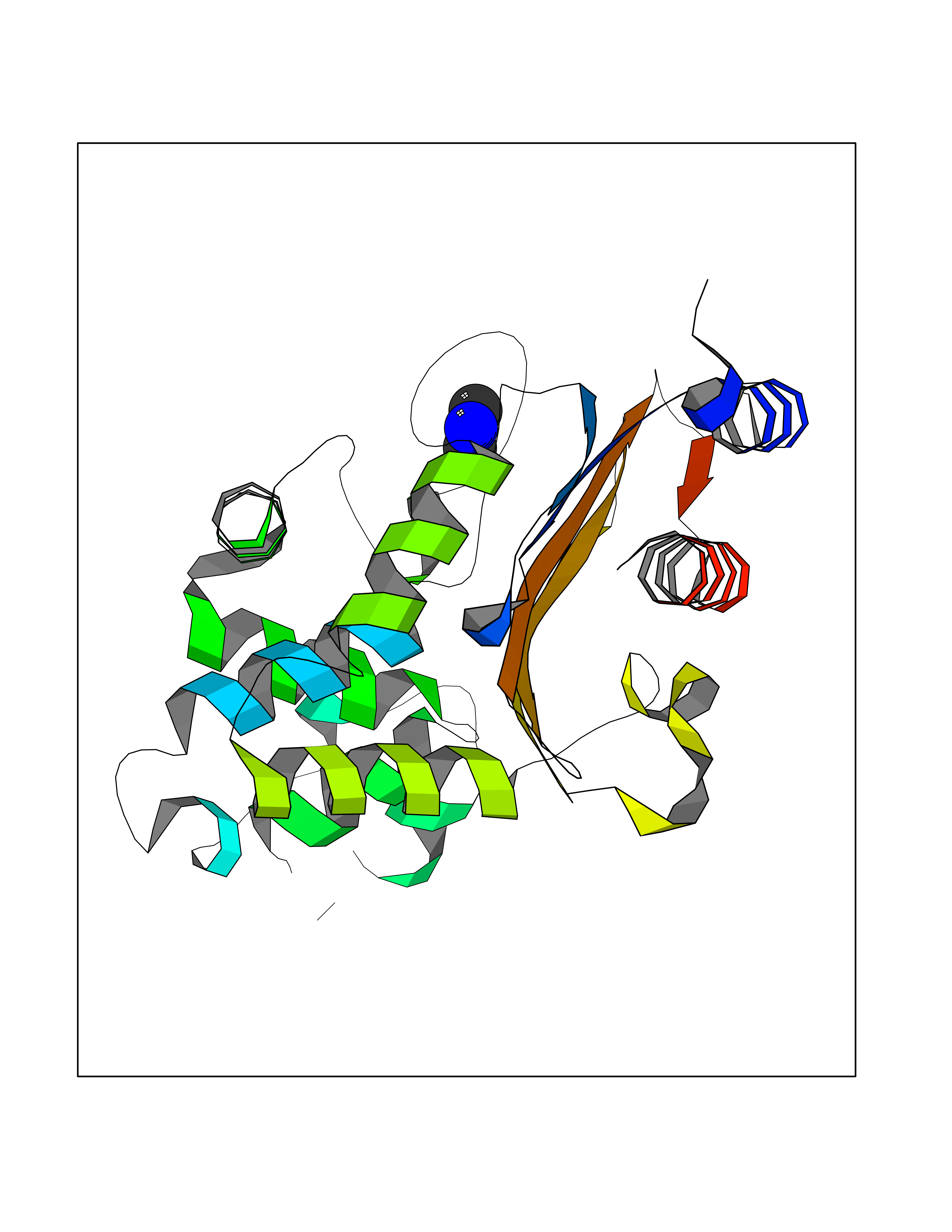 Molscript Map 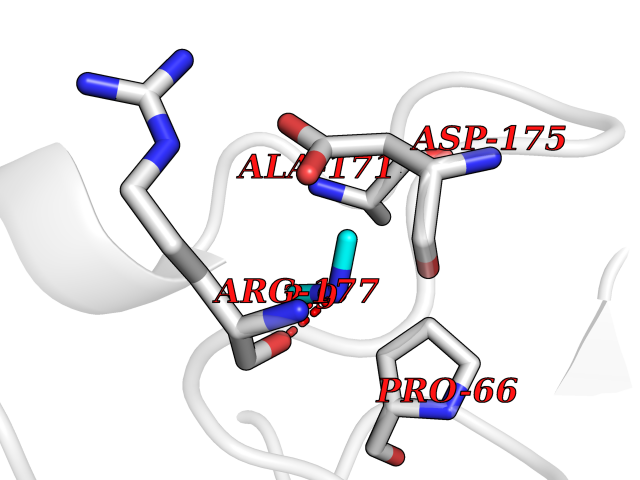 Pymol Map 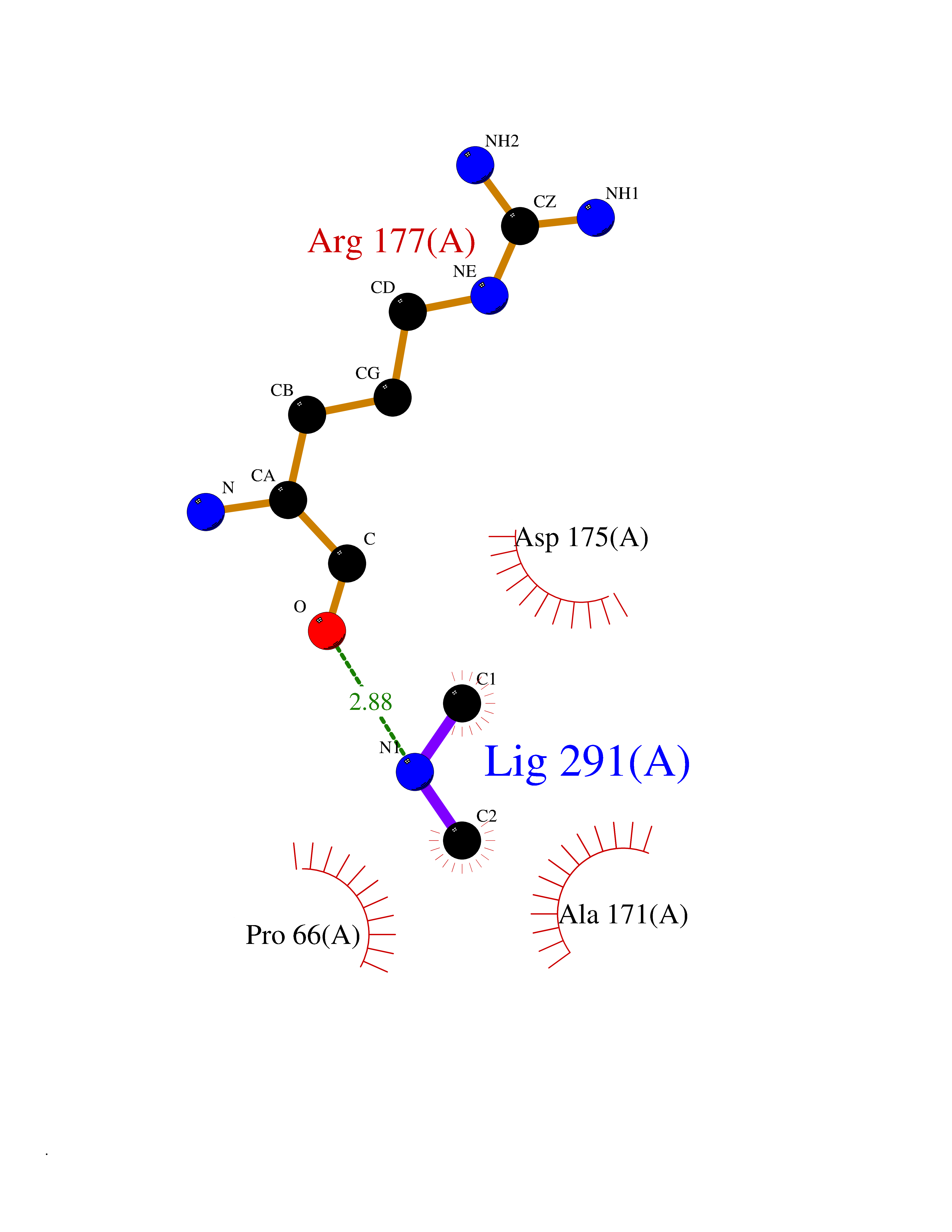 Ligplot Map 3D Binding mode Sequence TNLVAEPFAKLEQDFGGSIGVYAMDTGSGATVSYRAEERFPLCSSFKGFLAAAVLARSQQQAGLLDTPIRYGKNALVPWSPISEKYLTTGMTVAELSAAAVQYSDNAAANLLLKELGGPAGLTAFMRSIGDTTFRLDRWELELNSAIPGDARDTSSPRAVTESLQKLTLGSALAAPQRQQFVDWLKGNTTGNHRIRAAVPADWAVGDKTGTCGVYGTANDYAVVWPTGRAPIVLAVYTRAPNKDDKHSEAVIAAAARLALEGLG Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Pectate lyase | 1R76 | 4.02 | |
Target general information Gen name pelA Organism Niveispirillum irakense (Azospirillum irakense) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Lyase Function Lyase activity. Related diseases A chromosomal aberration involving ALK is found in a form of non-Hodgkin lymphoma. Translocation t(2;5)(p23;q35) with NPM1. The resulting chimeric NPM1-ALK protein homodimerize and the kinase becomes constitutively activated. The constitutively active fusion proteins are responsible for 5-10% of non-Hodgkin lymphomas. {ECO:0000269|PubMed:15938644}.; DISEASE: A chromosomal aberration involving ALK is associated with inflammatory myofibroblastic tumors (IMTs). Translocation t(2;11)(p23;p15) with CARS; translocation t(2;4)(p23;q21) with SEC31A. {ECO:0000269|PubMed:12112524, ECO:0000269|PubMed:16161041}.; DISEASE: A chromosomal aberration involving ALK is associated with anaplastic large-cell lymphoma (ALCL). Translocation t(2;17)(p23;q25) with ALO17. {ECO:0000269|PubMed:12112524}.; DISEASE: Neuroblastoma 3 (NBLST3) [MIM:613014]: A common neoplasm of early childhood arising from embryonic cells that form the primitive neural crest and give rise to the adrenal medulla and the sympathetic nervous system. {ECO:0000269|PubMed:18724359, ECO:0000269|PubMed:18923523, ECO:0000269|PubMed:18923525, ECO:0000269|PubMed:21242967, ECO:0000269|PubMed:22932897}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: The ALK signaling pathway plays an important role in glioblastoma, the most common malignant brain tumor of adults and one of the most lethal cancers. It regulates both glioblastoma migration and growth. {ECO:0000269|PubMed:15908427}.; DISEASE: A chromosomal aberration involving ALK is found in one subject with colorectal cancer. Translocation t(2;2)(p23.1;p23.3). A 5 million base pair tandem duplication generates an in-frame WDCP-ALK gene fusion. {ECO:0000269|PubMed:22327622}.; DISEASE: A chromosomal aberration involving ALK has been identified in a subset of patients with non-small-cell lung carcinoma. This aberration leads to the production of a fusion protein between the N-terminus of EML4 et the C-terminus of ALK. It is unclear whether the fusion protein is caused by a simple inversion within 2p (inv(2)(p21p23)) or whether the chromosome translocation involving 2p is more complex. When tested in a heterologous system, the fusion protein EML4-ALK possesses transforming activity that is dependent on ALK catalytic activity, possibly due to spontaneous dimerization mediated by the EML4 moiety, leading to ALK kinase activation. {ECO:0000269|PubMed:17625570}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Lyase; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41907.5 Length 384 Aromaticity 0.08 Instability index 43.72 Isoelectric point 6.11 Charge (pH=7) -3.46 2D Binding mode Binding energy (Kcal/mol) -5.48 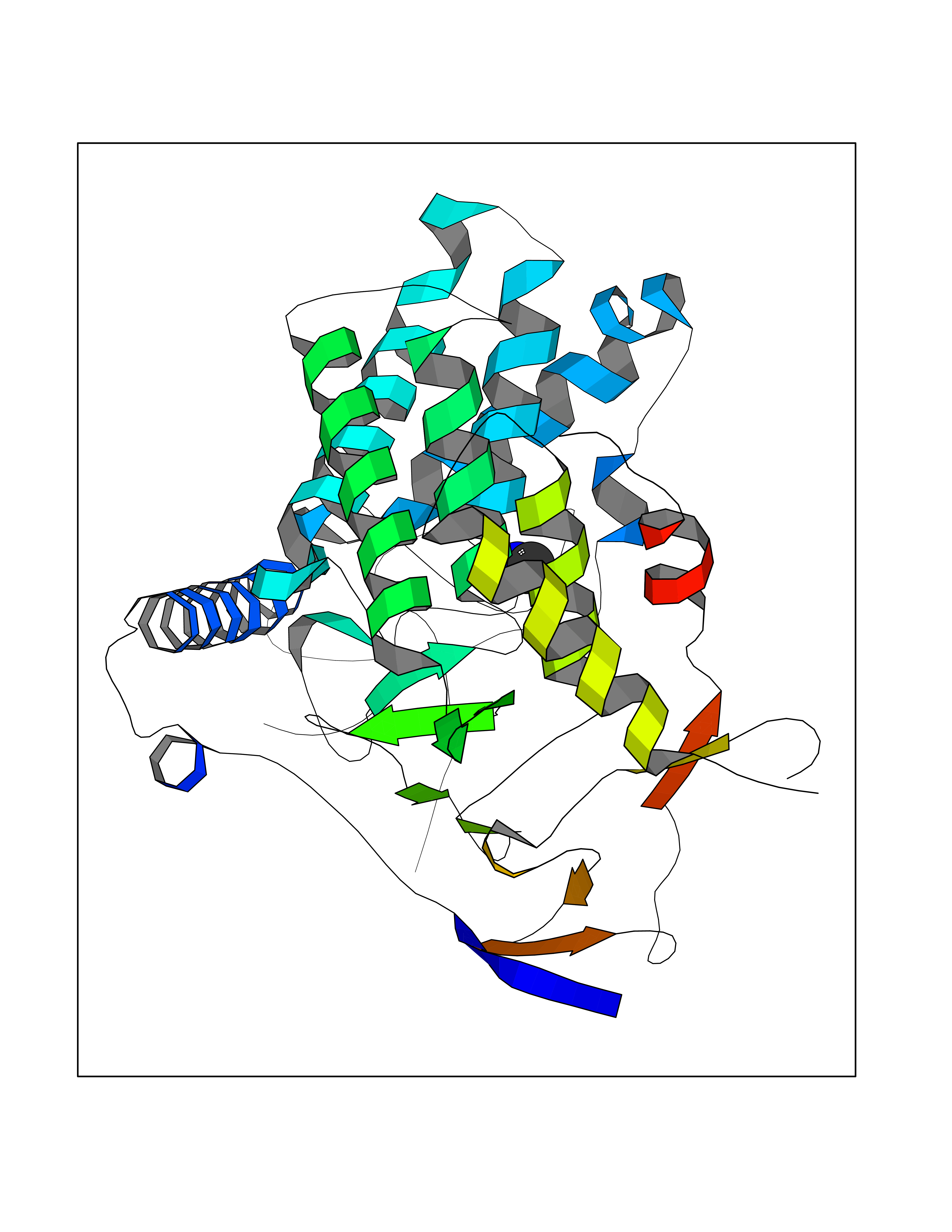 Molscript Map 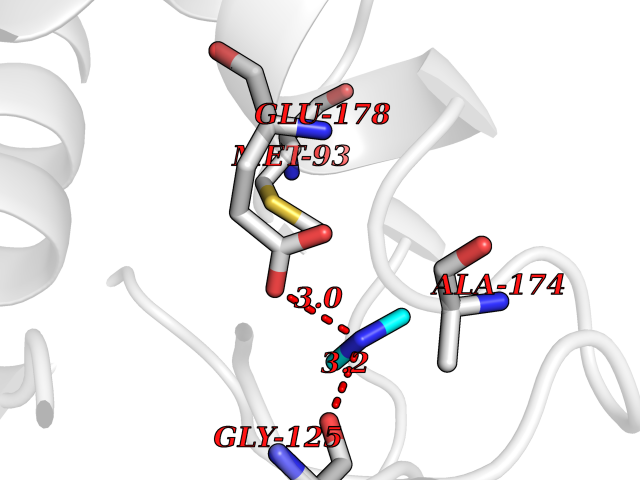 Pymol Map  Ligplot Map 3D Binding mode Sequence AVIGMNEAASALTPSRVSSLPDTQRAAWQEYLARSEAQLSRDKASLAAELAPGQPLPPPPAEGKGADTMPLDKPAAWYTSKAARHVADVIVSFQTPAGGWGKNQPRDGALRLPGQHYTGENVAKVKRDRDWHYVGTIDNDATVTEIRFLAQVVSQLAPEEAAPYRDAALKGIEYLLASQFPNGGWPQVWPLEGGYHDAITYNDDALVHVAELLSDIAAGRDGFGFVPPAIRTRALEATNAAIHCIVETQVVQDGKRLGWGQQHDALTLRPTSARNFEPAALSSTESARILLFLMEIEAPSDAVKQAIRGGVAWLNTSVIRDQGAKPLWSRFYSLDGNKPVFGDRDKTIHDDVMGISQERRTGYAWYTTSPQKALSAFTKWEKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | N-acetylgalactosamine 6 sulfatase (GALNS) | 4FDJ | 4.02 | |
Target general information Gen name GALNS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms N-acetylgalactosamine-6-sulfate sulfatase; N-acetylgalactosamine-6-sulfatase; Galactose-6-sulfate sulfatase; GalNAc6S sulfatase; GalN6S; Chondroitinsulfatase; Chondroitinase Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Catalyzes the chemical reaction of cleaving off the 6-sulfate groups of the N-acetyl-D-galactosamine 6-sulfate units of the macromolecule chondroitin sulfate and, similarly, of the D-galactose 6-sulfate units of the macromolecule keratan sulfate. Related diseases Mucopolysaccharidosis 4A (MPS4A) [MIM:253000]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:1522213, ECO:0000269|PubMed:16287098, ECO:0000269|PubMed:24726177, ECO:0000269|PubMed:7581409, ECO:0000269|PubMed:7633425, ECO:0000269|PubMed:7668283, ECO:0000269|PubMed:7795586, ECO:0000269|PubMed:8651279, ECO:0000269|PubMed:8826435, ECO:0000269|PubMed:9298823, ECO:0000269|PubMed:9375852, ECO:0000269|PubMed:9521421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09301 Interacts with NA EC number EC 3.1.6.4 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Glycoprotein; Hydrolase; Lysosome; Metal-binding; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 55013.8 Length 493 Aromaticity 0.11 Instability index 35.46 Isoelectric point 6.14 Charge (pH=7) -6.48 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PQPPNILLLLMDDMGWGDLGVYGEPSRETPNLDRMAAEGLLFPNFYSANPLXSPSRAALLTGRLPIRNGFYTTNAHARNAYTPQEIVGGIPDSEQLLPELLKKAGYVSKIVGKWHLGHRPQFHPLKHGFDEWFGSPNCHFGPYDNKARPNIPVYRDWEMVGRYYEEFPINLKTGEANLTQIYLQEALDFIKRQARHHPFFLYWAVDATHAPVYASKPFLGTSQRGRYGDAVREIDDSIGKILELLQDLHVADNTFVFFTSDNGAALISAPEQGGSNGPFLCGKQTTFEGGMREPALAWWPGHVTAGQVSHQLGSIMDLFTTSLALAGLTPPSDRAIDGLNLLPTLLQGRLMDRPIFYYRGDTLMAATLGQHKAHFWTWTNSWENFRQGIDFCPGQNVSGVTTHNLEDHTKLPLIFHLGRDPGERFPLSFASAEYQEALSRITSVVQQHQEALVPAQPQLNVCNWAVMNWAPPGCEKLGKCLTPPESIPKKCLW Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Trypanosoma Cruzipain (Trypano CYSP) | 1EWM | 4.02 | |
Target general information Gen name Trypano CYSP Organism Trypanosoma cruzi Uniprot ID TTD ID Synonyms Cruzaine; Major cysteine proteinase Protein family Peptidase C1 family Biochemical class Peptidase Function The cysteine protease may play an important role in the development and differentiation of the parasites at several stages of their life cycle. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02200; DB02051; DB01871; DB01810; DB02128; DB03536; DB04427; DB03691; DB04502; DB03573 Interacts with NA EC number EC 3.4.22.51 Uniprot keywords 3D-structure; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Signal; Thiol protease; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 22703 Length 215 Aromaticity 0.09 Instability index 28.98 Isoelectric point 4.37 Charge (pH=7) -13.9 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 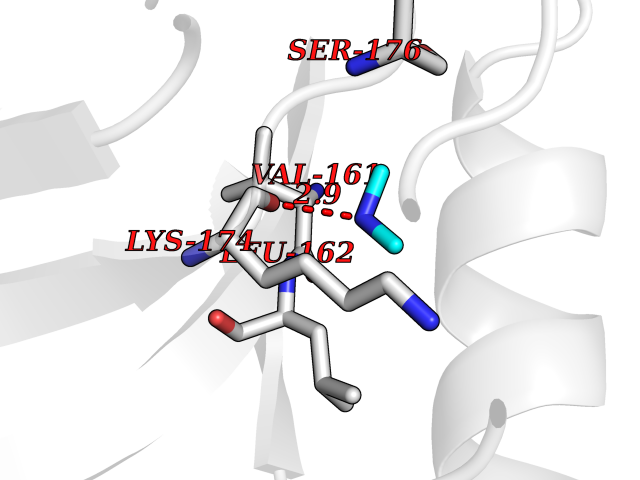 Pymol Map 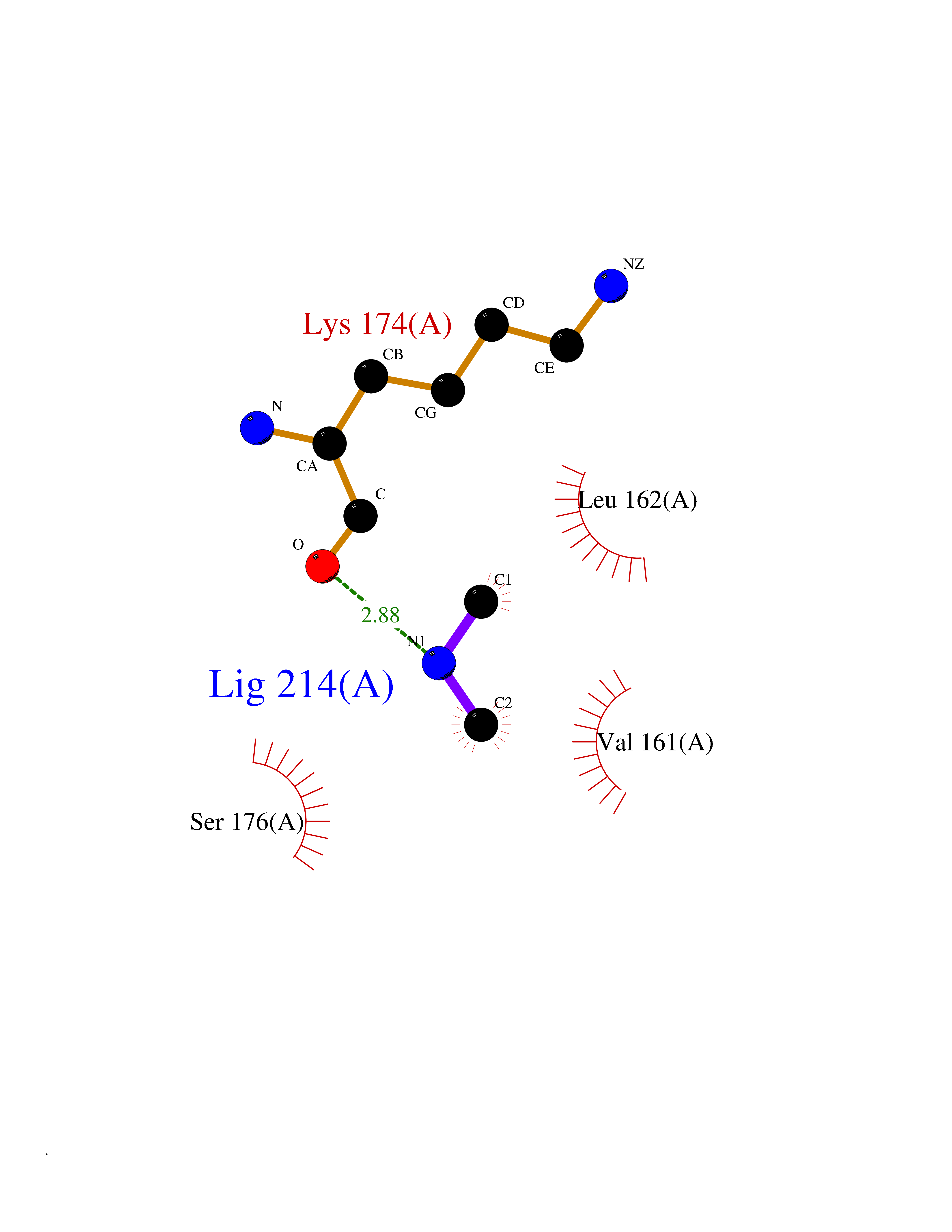 Ligplot Map 3D Binding mode Sequence APAAVDWRARGAVTAVKDQGQCGSCWAFSAIGNVECQWFLAGHPLTNLSEQMLVSCDKTDSGCSGGLMNNAFEWIVQENNGAVYTEDSYPYASGEGISPPCTTSGHTVGATITGHVELPQDEAQIAAWLAVNGPVAVAVDASSWMTYTGGVMTSCVSEQLDHGVLLVGYNDSAAVPYWIIKNSWTTQWGEEGYIRIAKGSNQCLVKEEASSAVVG Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Suppressor of tumorigenicity 14 protein (ST14) | 3P8G | 4.02 | |
Target general information Gen name ST14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor-associated differentially-expressed gene 15 protein; Tumor associated differentially-expressed gene-15 protein; TADG15; Serine protease TADG-15; Serine protease 14; SNC19; Prostamin; PRSS14; Mem Protein family Peptidase S1 family Biochemical class Peptidase Function Proposed to play a role in breast cancer invasion and metastasis. Exhibits trypsin-like activity as defined by cleavage of synthetic substrates with Arg or Lys as the P1 site. Involved in the terminal differentiation of keratinocytes through prostasin (PRSS8) activation and filaggrin (FLG) processing. Degrades extracellular matrix. Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03127; DB13729; DB00013 Interacts with NA EC number EC 3.4.21.109 Uniprot keywords 3D-structure; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Hypotrichosis; Ichthyosis; Membrane; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal-anchor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 26451.5 Length 241 Aromaticity 0.1 Instability index 30.45 Isoelectric point 5.6 Charge (pH=7) -5.69 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 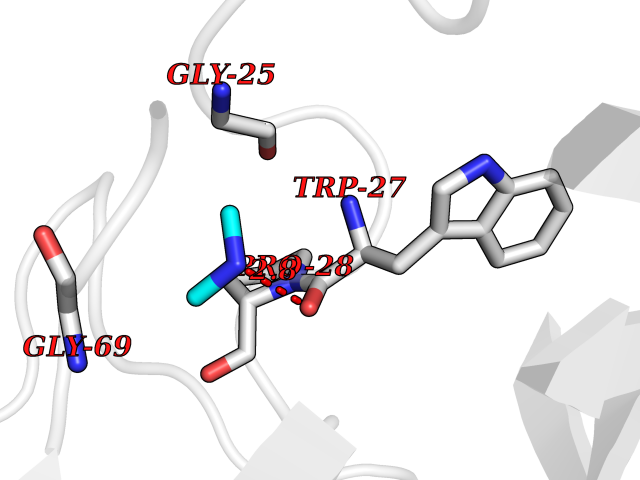 Pymol Map 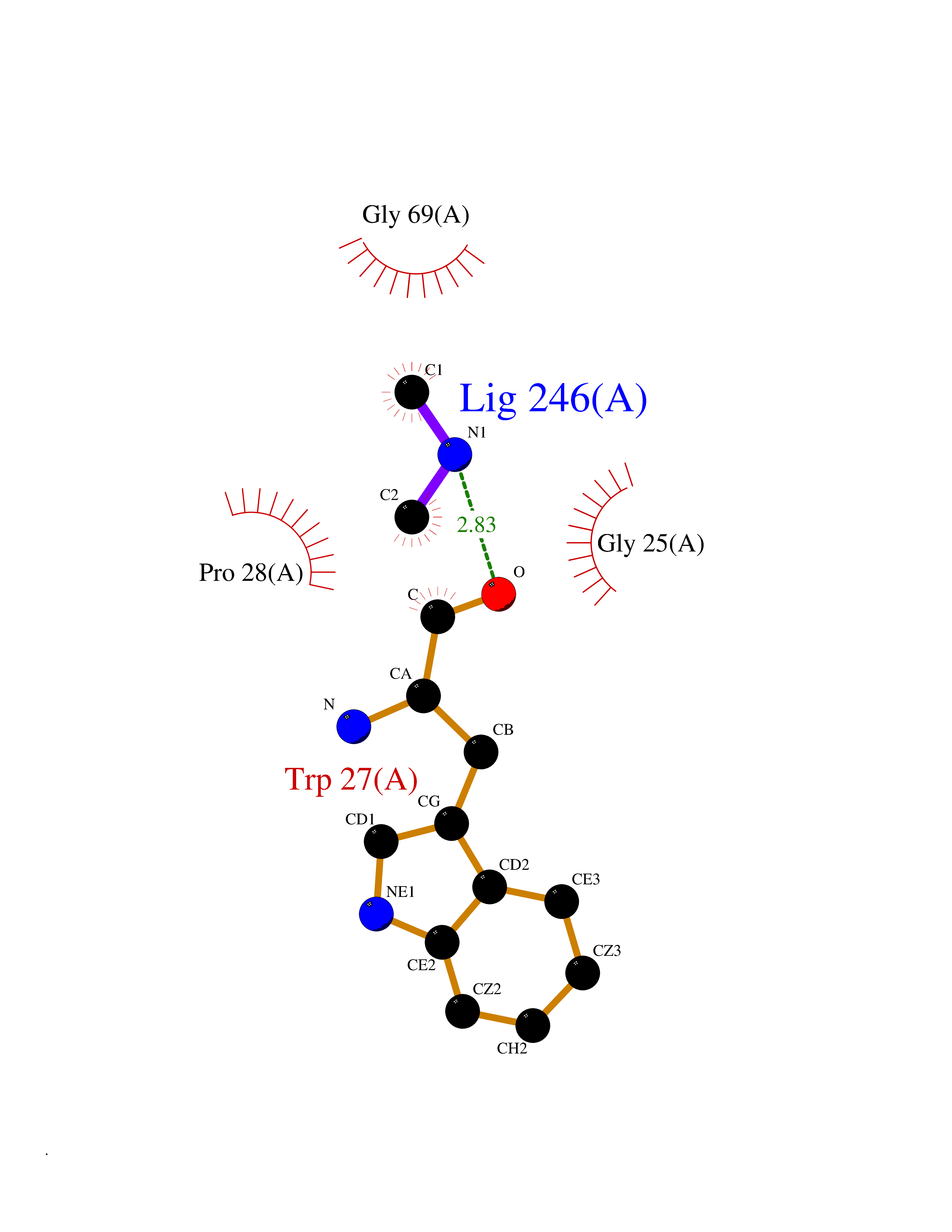 Ligplot Map 3D Binding mode Sequence VVGGTDADEGEWPWQVSLHALGQGHICGASLISPNWLVSAAHCYIDDRGFRYSDPTQWTAFLGLHDQSQRSAPGVQERRLKRIISHPFFNDFTFDYDIALLELEKPAEYSSMVRPICLPDASHVFPAGKAIWVTGWGHTQYGGTGALILQKGEIRVIQQTTCENLLPQQITPRMMCVGFLSGGVDSCQGDSGGPLSSVEADGRIFQAGVVSWGDGCAQRNKPGVYTRLPLFRDWIKENTGV Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Plasmodium DOXP reductoisomerase (Malaria DXR) | 3AU9 | 4.02 | |
Target general information Gen name Malaria DXR Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID Synonyms IspC; DXR; DXP reductoisomerase; DOXP reductoisomerase; 2-C-Methyl-d-erythritol 4-phosphate synthase; 1-deoxyxylulose-5-phosphate reductoisomerase Protein family DXR family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADP-dependent rearrangement and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol 4-phosphate (MEP). Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.1.267 Uniprot keywords 3D-structure; Apicoplast; Isoprene biosynthesis; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Plastid; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46644.4 Length 410 Aromaticity 0.09 Instability index 36.77 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 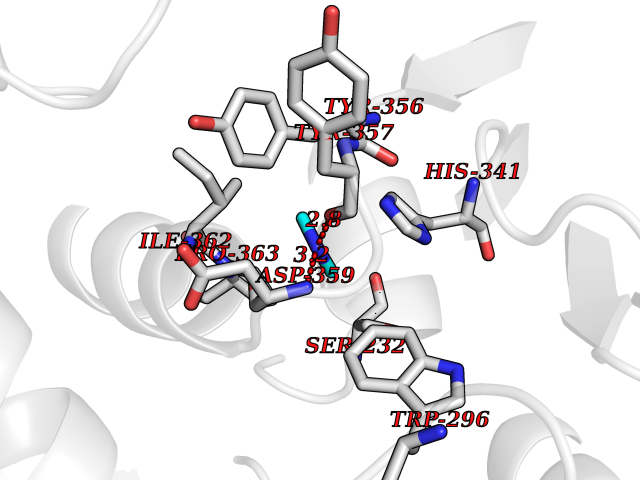 Pymol Map 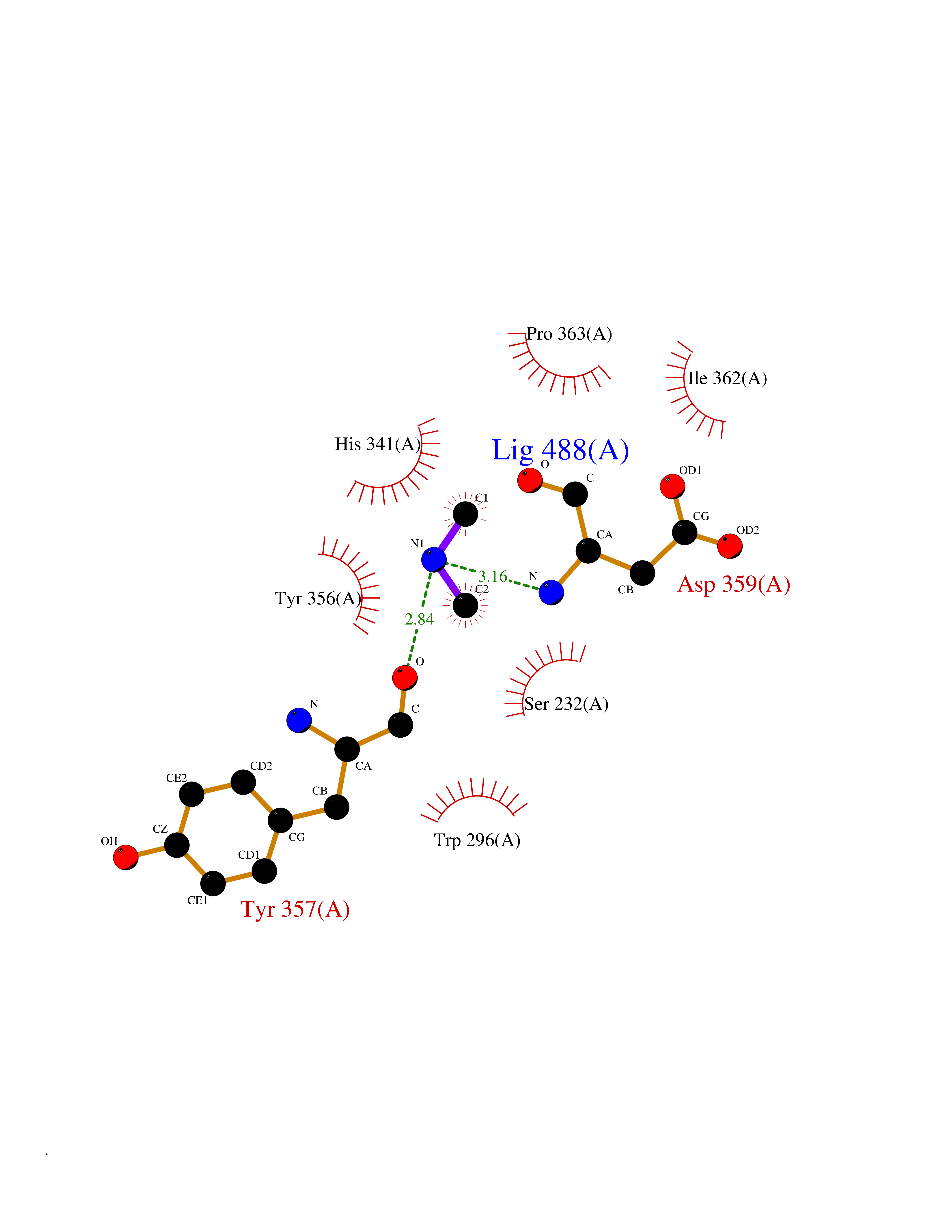 Ligplot Map 3D Binding mode Sequence PINVAIFGSTGSIGTNALNIIRECNKIENVFNVKALYVNKSVNELYEQAREFLPEYLCIHDKSVYEELKELVKNIKDYKPIILCGDEGMKEICSSNSIDKIVIGIDSFQGLYSTMYAIMNNKIVALANKESIVSAGFFLKKLLNIHKNAKIIPVDSEHSAIFQCLDNNKVLKTKCLQDNFSKINNINKIFLCSSGGPFQNLTMDELKNVTSENALKHPKWKMGKKITIDSATMMNKGLEVIETHFLFDVDYNDIEVIVHKECIIHSCVEFIDKSVISQMYYPDMQIPILYSLTWPDRIKTNLKPLDLAQVSTLTFHKPSLEHFPCIKLAYQAGIKGNFYPTVLNASNEIANNLFLNNKIKYFDISSIISQVLESFNSQKVSENSEDLMKQILQIHSWAKDKATDIYNKHN Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Tankyrase-2 (TNKS-2) | 3U9H | 4.02 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -5.48 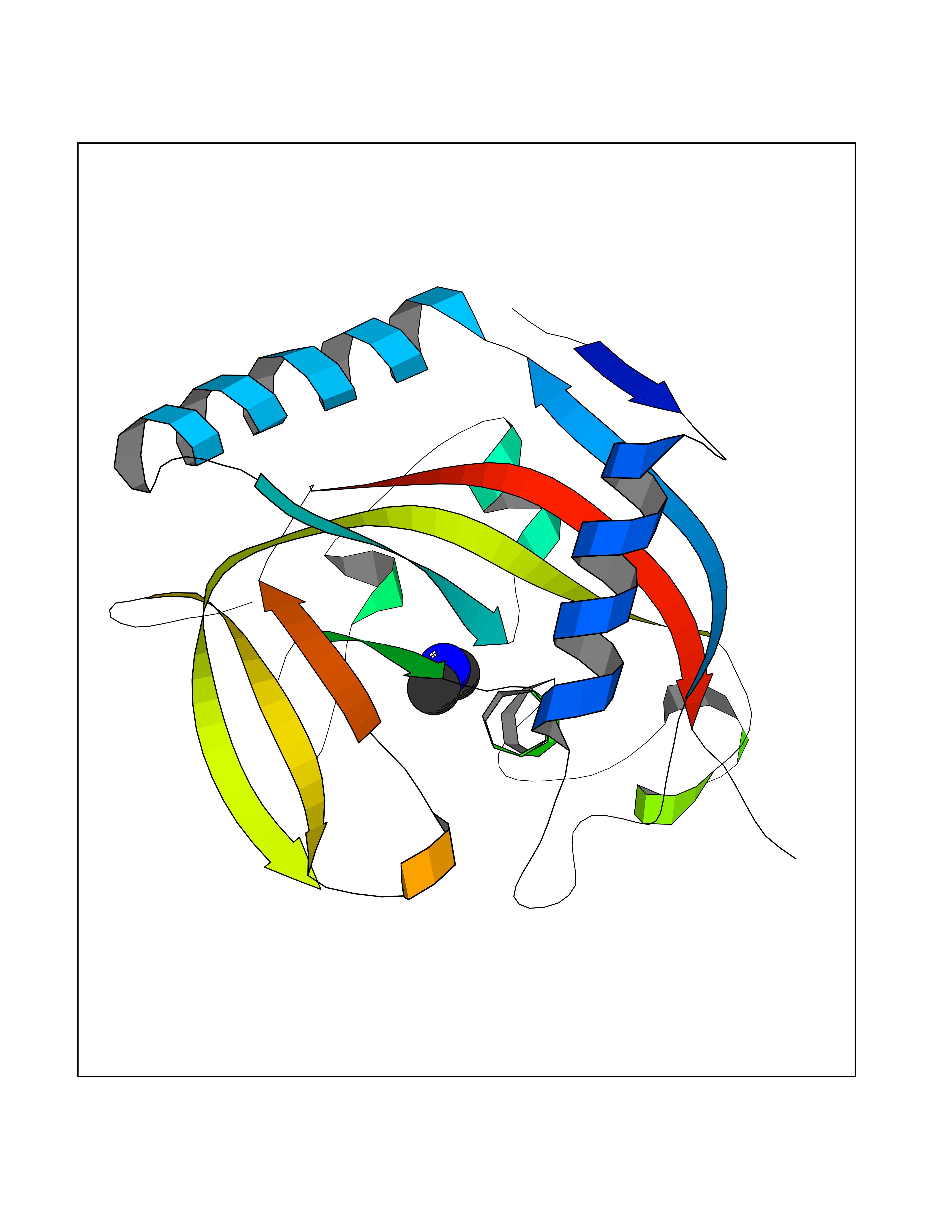 Molscript Map 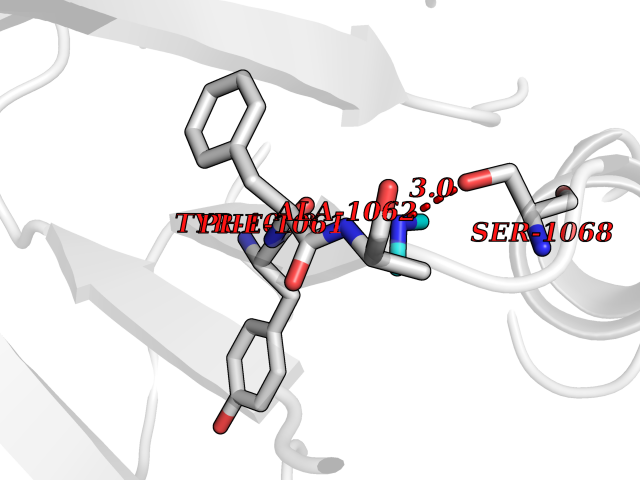 Pymol Map 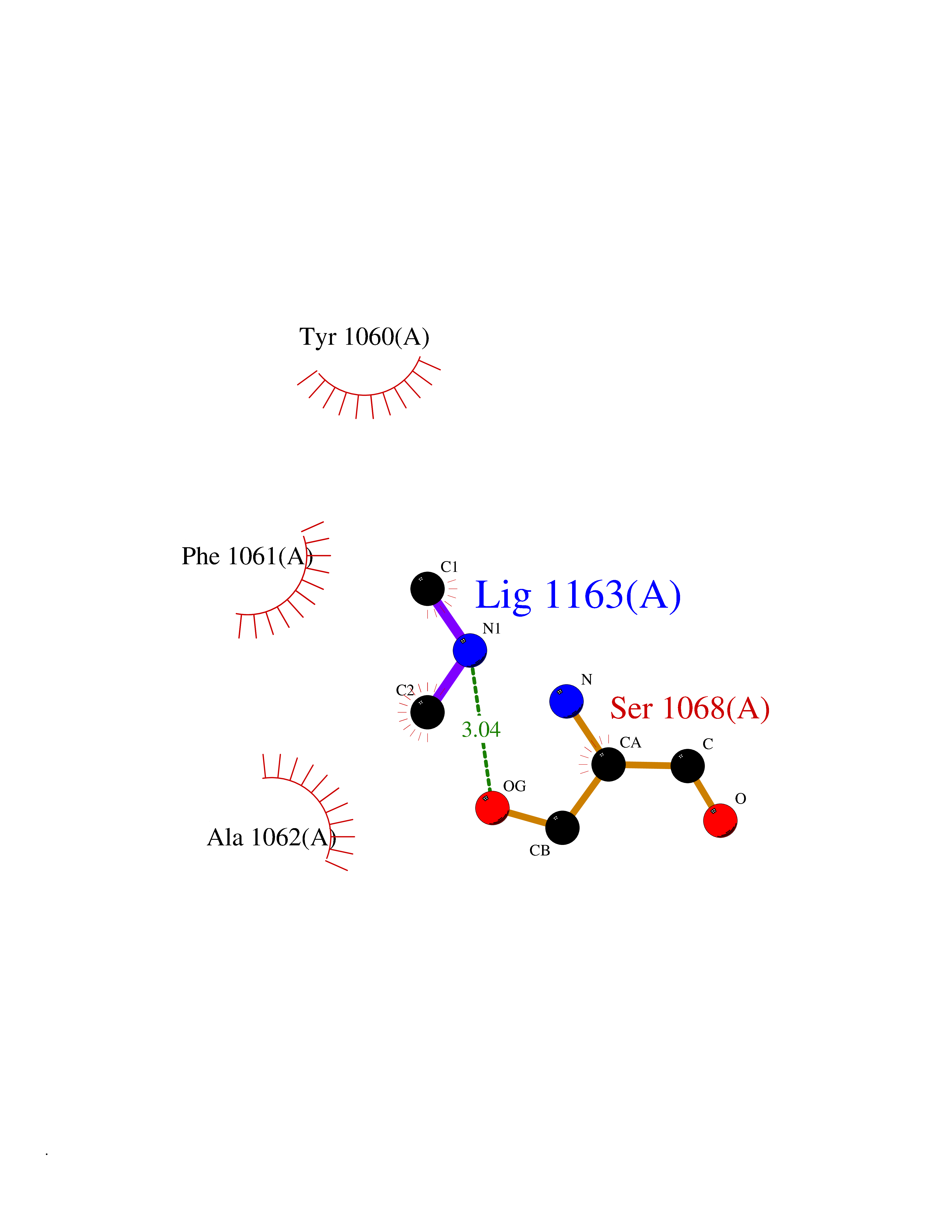 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Histamine H3 receptor (H3R) | 7F61 | 4.02 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -5.49 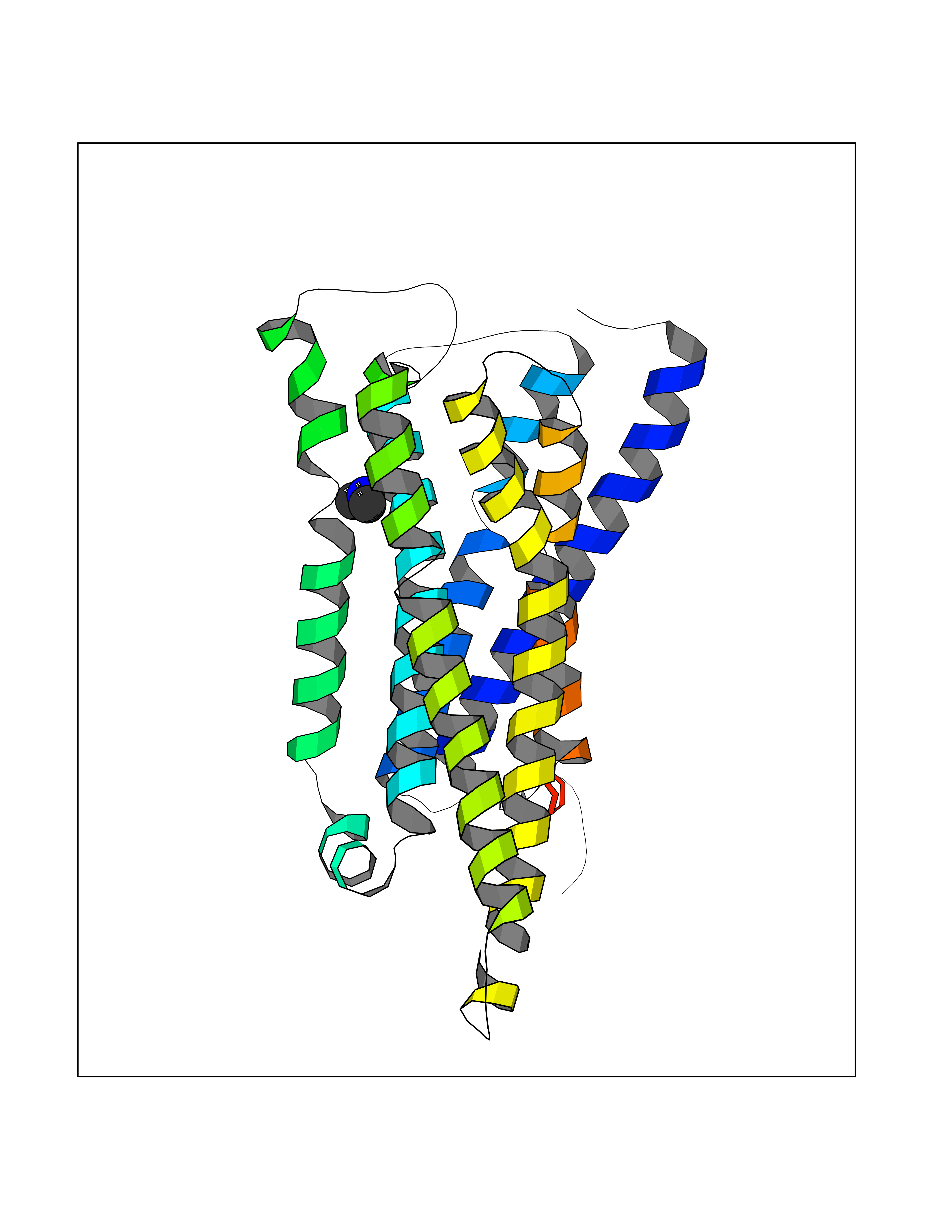 Molscript Map 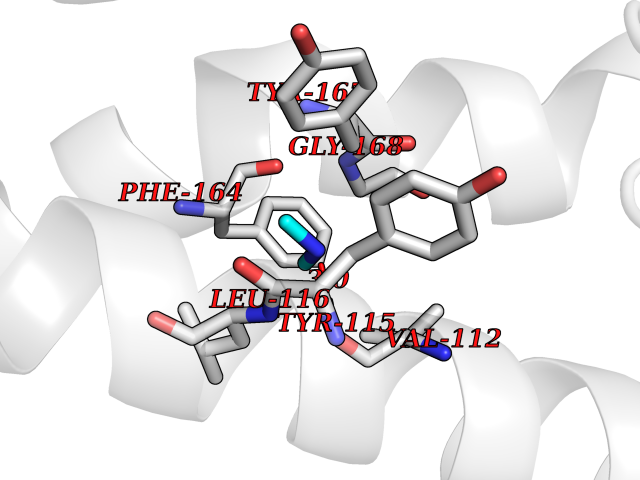 Pymol Map 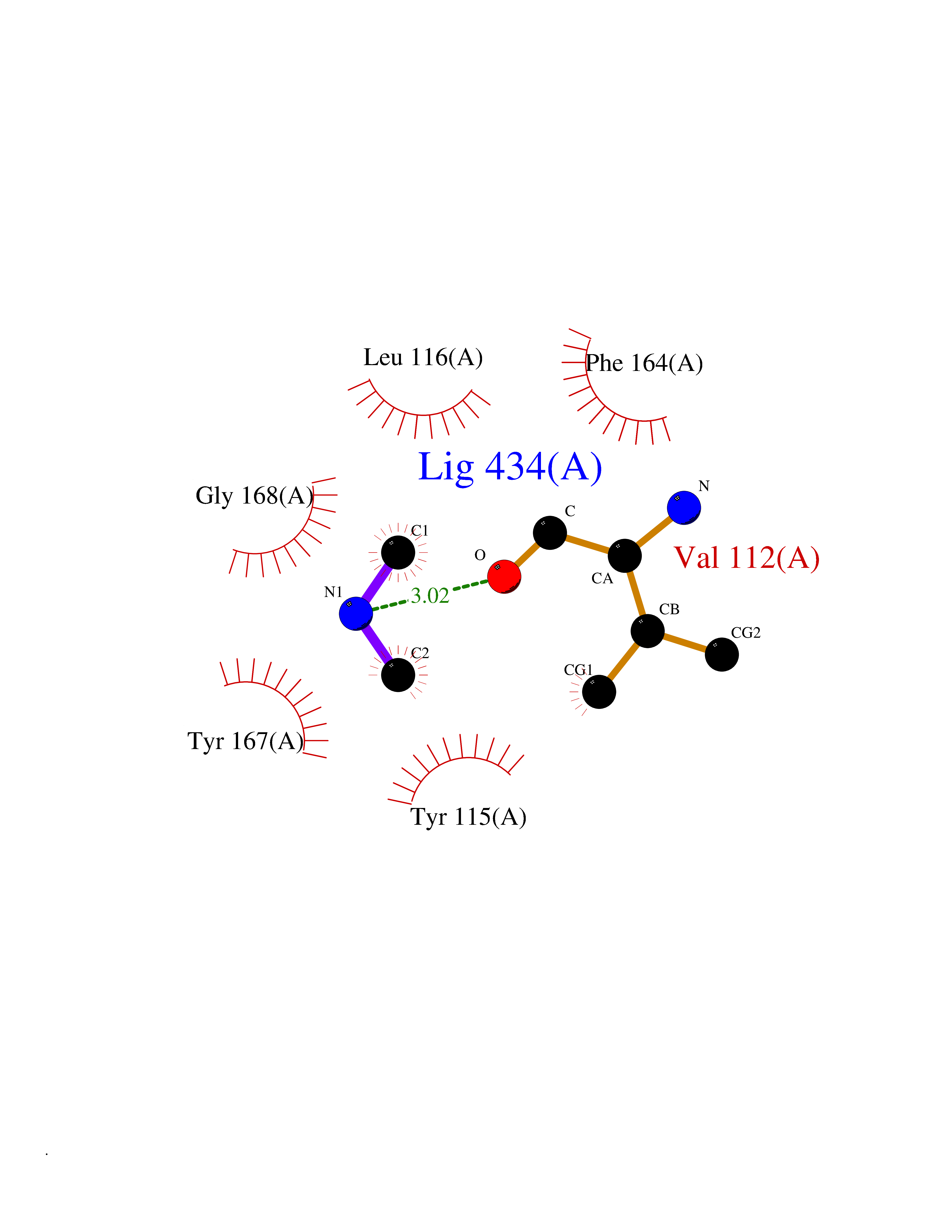 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Cathepsin D (CTSD) | 4OC6 | 4.02 | |
Target general information Gen name CTSD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CPSD; CD Protein family Peptidase A1 family Biochemical class Peptidase Function Plays a role in APP processing following cleavage and activation by ADAM30 which leads to APP degradation. Involved in the pathogenesis of several diseases such as breast cancer and possibly Alzheimer disease. Acid protease active in intracellular protein breakdown. Related diseases Ceroid lipofuscinosis, neuronal, 10 (CLN10) [MIM:610127]: A form of neuronal ceroid lipofuscinosis with onset at birth or early childhood. Neuronal ceroid lipofuscinoses are progressive neurodegenerative, lysosomal storage diseases characterized by intracellular accumulation of autofluorescent liposomal material, and clinically by seizures, dementia, visual loss, and/or cerebral atrophy. {ECO:0000269|PubMed:16670177, ECO:0000269|PubMed:16685649, ECO:0000269|PubMed:21990111}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03028; DB03096; DB07542; DB08740; DB02216 Interacts with P05067; Q9P1A6-3; I6L9I8; Q9H6S3; Q7Z602; P28799; PRO_0000012695 [P28799]; PRO_0000012696 [P28799]; PRO_0000012697 [P28799]; PRO_0000012698 [P28799]; PRO_0000012699 [P28799]; PRO_0000012700 [P28799]; PRO_0000012701 [P28799]; P68431; Q9Y6F6-3; Q12756; Q5TA79; Q86VF5-3; O15130-2; Q96LB9; P09565; Q9C004; Q8NBJ7; Q9BQG1; P28347-2; P45880; Q15007-2; O00308; Q5W0Z9-4; Q6ZNH5 EC number EC 3.4.23.5 Uniprot keywords 3D-structure; Alzheimer disease; Aspartyl protease; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Neurodegeneration; Neuronal ceroid lipofuscinosis; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37264.2 Length 341 Aromaticity 0.1 Instability index 32.32 Isoelectric point 5.6 Charge (pH=7) -4.86 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 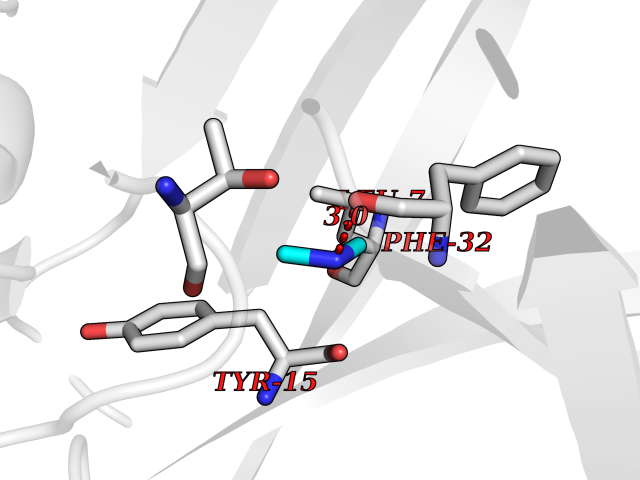 Pymol Map 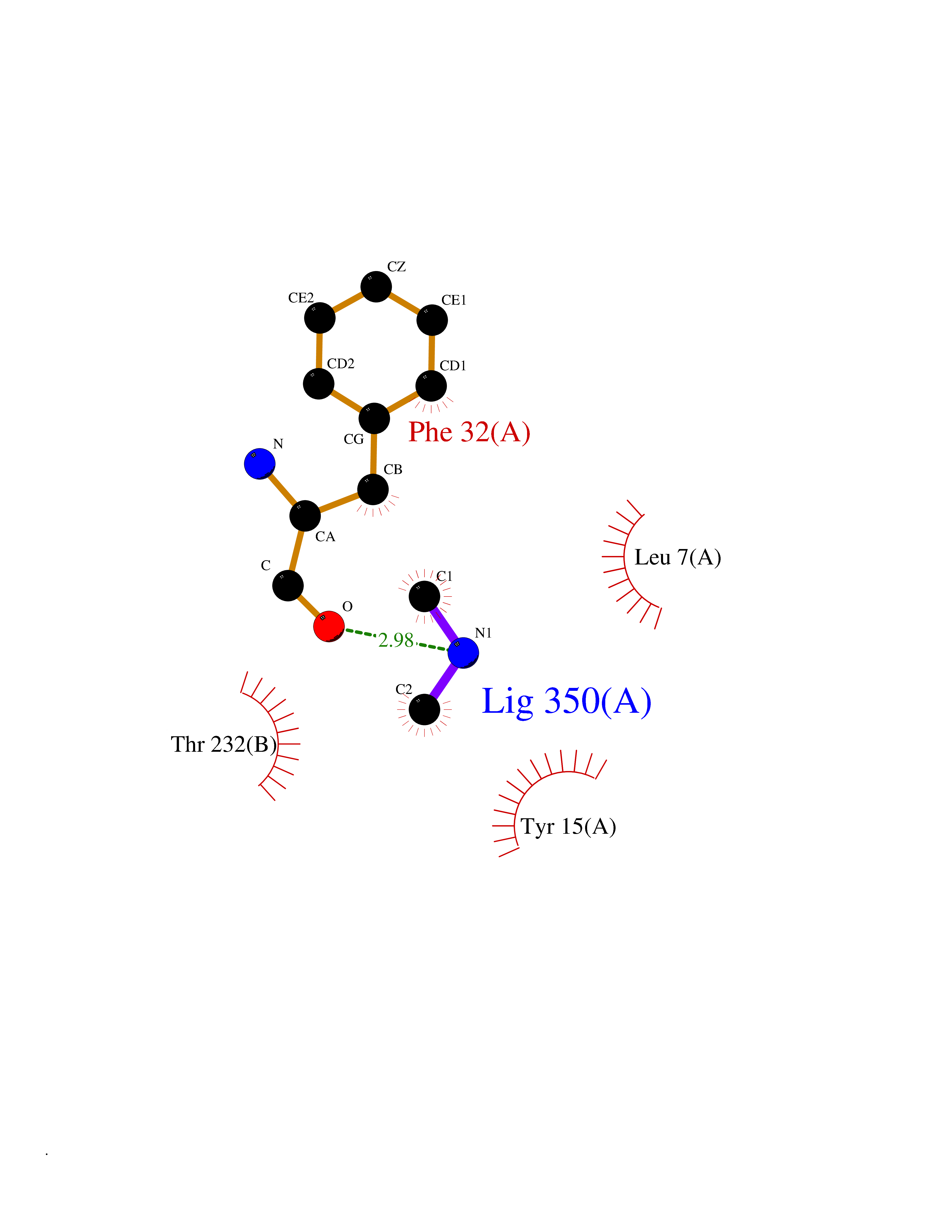 Ligplot Map 3D Binding mode Sequence GPIPEVLKNYMDAQYYGEIGIGTPPQCFTVVFDTGSSNLWVPSIHCKLLDIACWIHHKYNSDKSSTYVKNGTSFDIHYGSGSLSGYLSQDTVSVPCQSGGVKVERQVFGEATKQPGITFIAAKFDGILGMAYPRISVNNVLPVFDNLMQQKLVDQNIFSFYLSRDPDAQPGGELMLGGTDSKYYKGSLSYLNVTRKAYWQVHLDQVEVASGLTLCKEGCEAIVDTGTSLMVGPVDEVRELQKAIGAVPLIQGEYMIPCEKVSTLPAITLKLGGKGYKLSPEDYTLKVSQAGKTLCLSGFMGMDIPPPSGPLWILGDVFIGRYYTVFDRDNNRVGFAEAARL Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Complement C1s component (C1S) | 1ELV | 4.02 | |
Target general information Gen name C1S Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complement component 1 subcomponent s; Complement C1s subcomponent; C1-esterase; C1 esterase Protein family Peptidase S1 family Biochemical class Peptidase Function C1r activates C1s so that it can, in turn, activate C2 and C4. C1s B chain is a serine protease that combines with C1q and C1r to form C1, the first component of the classical pathway of the complement system. Related diseases Complement component C1s deficiency (C1SD) [MIM:613783]: A rare defect resulting in C1 deficiency and impaired activation of the complement classical pathway. C1 deficiency generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. {ECO:0000269|PubMed:11390518}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ehlers-Danlos syndrome, periodontal type, 2 (EDSPD2) [MIM:617174]: A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSPD2 is characterized by the association of typical features of Ehlers-Danlos syndrome with gingival recession and severe early-onset periodontal disease, leading to premature loss of permanent teeth. EDSPD2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:27745832}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02371; DB09228; DB09130; DB12831; DB06404; DB14996; DB01593; DB14487; DB14533; DB14548 Interacts with P00736; P09871; P06681; O43889-2; Q9H6H4; P05155 EC number EC 3.4.21.42 Uniprot keywords 3D-structure; Calcium; Complement pathway; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Ehlers-Danlos syndrome; Glycoprotein; Hydrolase; Hydroxylation; Immunity; Innate immunity; Metal-binding; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal; Sushi Protein physicochemical properties Chain ID A Molecular weight (Da) 33278.6 Length 303 Aromaticity 0.1 Instability index 33.69 Isoelectric point 5.16 Charge (pH=7) -7.95 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 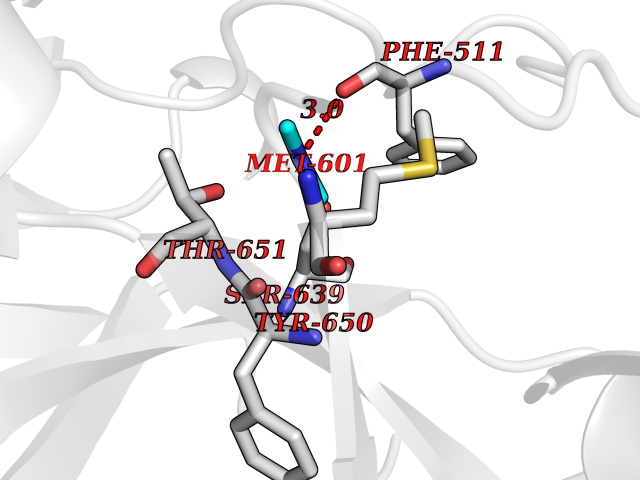 Pymol Map 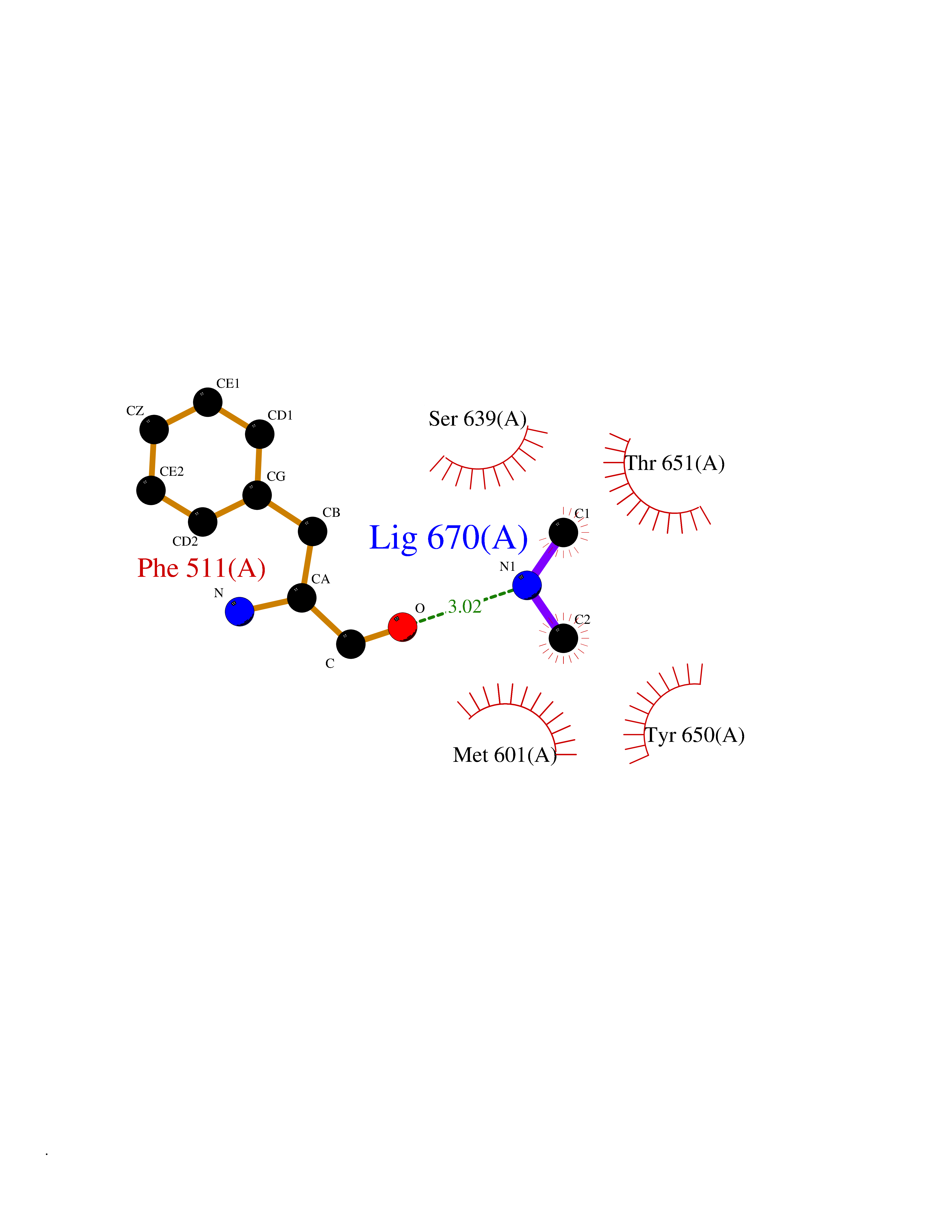 Ligplot Map 3D Binding mode Sequence LDCGIPESIENGKVEDPESTLFGSVIRYTCEEPYYYMEGGGEYHCAGNGSWVNEVLGPELPKCVPVCGVPREPFIIGGSDADIKNFPWQVFFDNPWAGGALINEYWVLTAAHVVEGNREPTMYVGSTSVQKMLTPEHVFIHPGWKLLAVPEGRTNFDNDIALVRLKDPVKMGPTVSPICLPGTSSDYNLMDGDLGLISGWGRTEKRDRAVRLKAARLPVAPLRKCKEVAYVFTPNMICAGGEKGMDSCKGDSGGAFAVQDPNDKTKFYAAGLVSWGPQCGTYGLYTRVKNYVDWIMKTMQENS Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Cathepsin G (CTSG) | 1KYN | 4.02 | |
Target general information Gen name CTSG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CG Protein family Peptidase S1 family Biochemical class Peptidase Function Cleaves complement C3. Has antibacterial activity against the Gram-negative bacterium P. aeruginosa, antibacterial activity is inhibited by LPS from P. aeruginosa, Z-Gly-Leu-Phe-CH2Cl and phenylmethylsulfonyl fluoride. Serine protease with trypsin- and chymotrypsin-like specificity. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04016; DB02360 Interacts with A8MQ03; Q15323; Q7Z3S9; Q9NRD5 EC number EC 3.4.21.20 Uniprot keywords 3D-structure; Antibiotic; Antimicrobial; Cell membrane; Chemotaxis; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Membrane; Nucleus; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 25353.8 Length 223 Aromaticity 0.06 Instability index 66.1 Isoelectric point 11.51 Charge (pH=7) 23.24 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 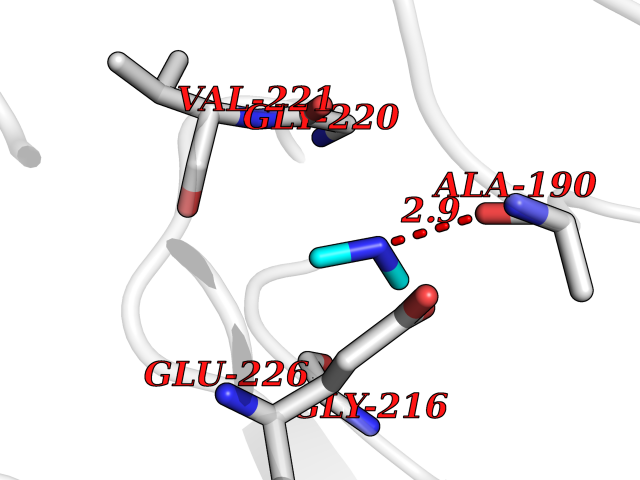 Pymol Map 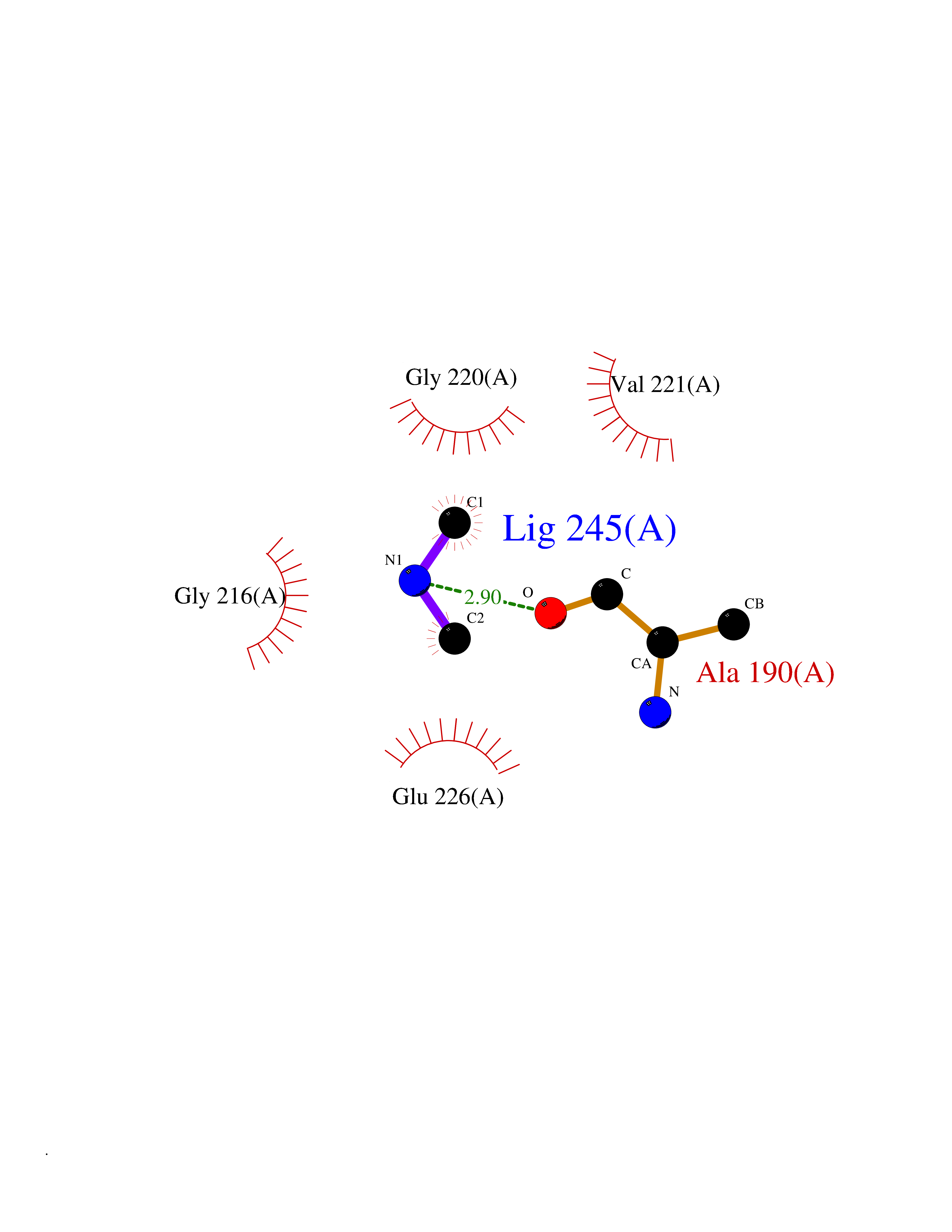 Ligplot Map 3D Binding mode Sequence IIGGRESRPHSRPYMAYLQIQSPAGQSRCGGFLVREDFVLTAAHCWGSNINVTLGAHNIQRRENTQQHITARRAIRHPQYNQRTIQNDIMLLQLSRRVRRNRNVNPVALPRAQEGLRPGTLCTVAGWGRVSMRRGTDTLREVQLRVQRDRQCLRIFGSYDPRRQICVGDRRERKAAFKGDSGGPLLCNNVAHGIVSYGKSSGVPPEVFTRVSSFLPWIRTTMR Hydrogen bonds contact Hydrophobic contact | ||||