Job Results:
Ligand
Structure
Job ID
4d96340bab84003cd843e60272b3611c
Job name
NA
Time
2025-02-13 15:23:33
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | D-alanyl-D-alanine carboxypeptidase DacA | 3MZF | 4.02 | |
Target general information Gen name dacA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0632;pfv;JW0627 Protein family Peptidase S11 family Biochemical class Hydrolase / antibiotic Function Beta-lactamase activity.Carboxypeptidase activity.Endopeptidase activity.Penicillin binding.Serine-type D-Ala-D-Ala carboxypeptidase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01602; DB04647; DB00578; DB09319; DB00671; DB00274; DB01329; DB01331; DB09050; DB01147; DB01000 Interacts with NA EC number 3.4.16.4; 3.5.2.6 Uniprot keywords 3D-structure; Carboxypeptidase; Cell inner membrane; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Direct protein sequencing; Hydrolase; Membrane; Peptidoglycan synthesis; Protease; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 38972 Length 355 Aromaticity 0.08 Instability index 21.23 Isoelectric point 7.1 Charge (pH=7) 0.14 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 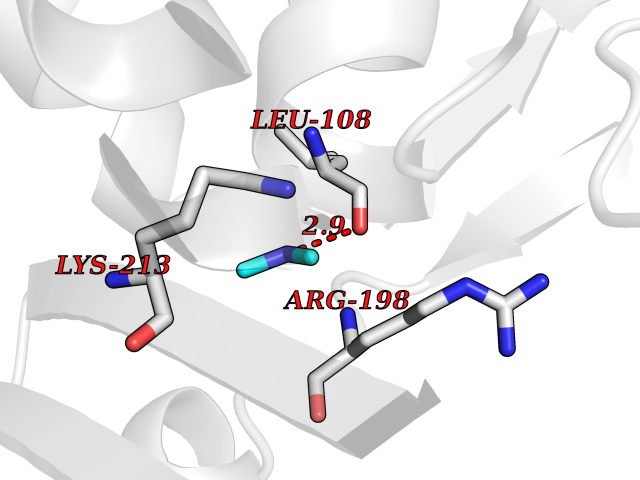 Pymol Map 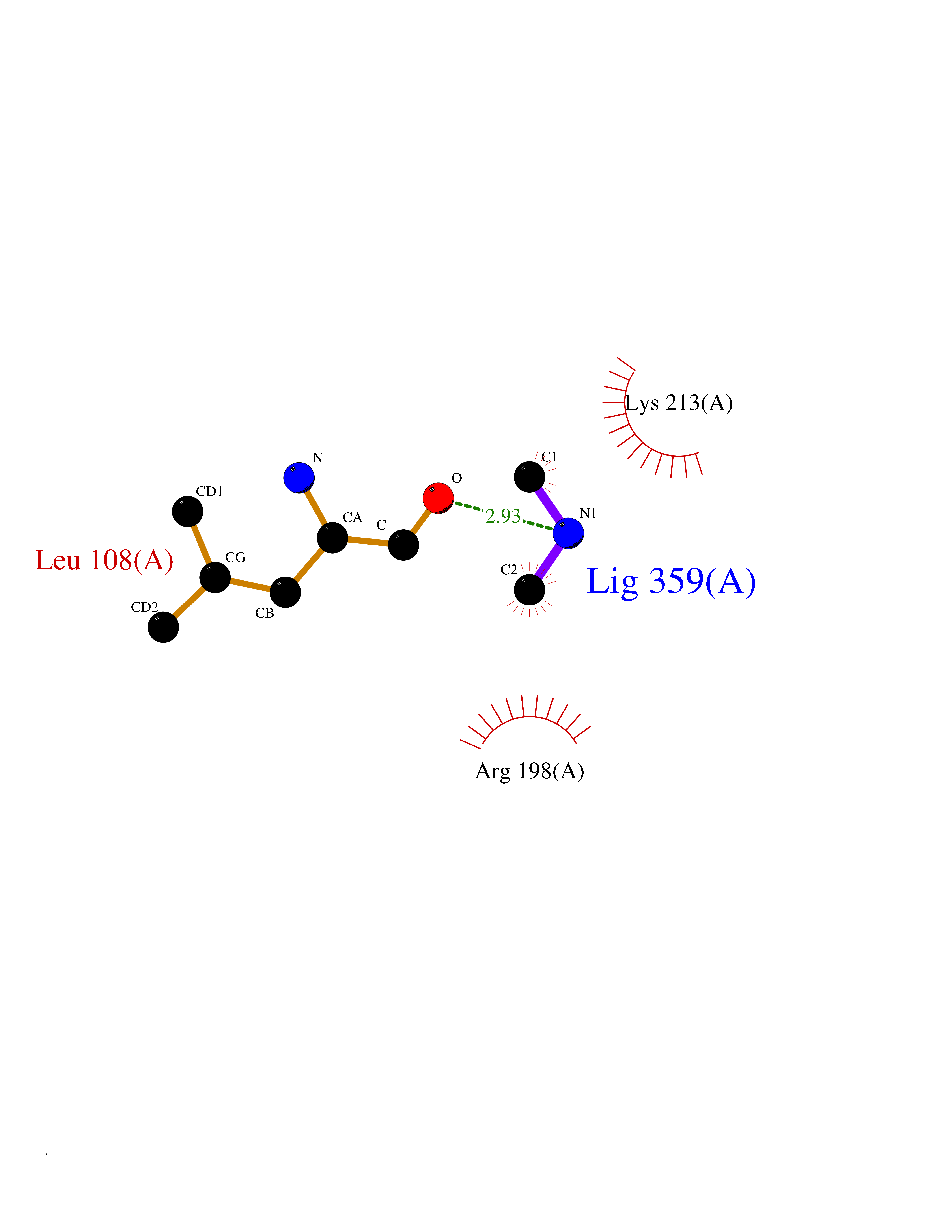 Ligplot Map 3D Binding mode Sequence LNIKTMIPGVPQIDAESYILIDYNSGKVLAEQNADVRRDPASLTKMMTSYVIGQAMKAGKFKETDLVTIGNDAWATGNPVFKGSSLMFLKPGMQVPVSQLIRGINLQSGNDACVAMADFAAGSQDAFVGLMNSYVNALGLKNTHFQTVHGLDADGQYSSARDMALIGQALIRDVPNEYSIYKEKEFTFNGIRQLNRNGLLWDNSLNVDGIKTGHTDKAGYNLVASATEGQMRLISAVMGGRTFKGREAESKKLLTWGFRFFETVNPLKVGKEFASEPVWFGDSDRASLGVDKDVYLTIPRGRMKDLKASYVLNSSELHAPLQKNQVVGTINFQLDGKTIEQRPLVVLQEIPEGNF Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Bifunctional dihydrofolate reductase-thymidylate synthase | 1J3K | 4.02 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate K1 / Thailand) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydrofolate reductase family; Thymidylate synthase family Biochemical class Oxidoreductase Function Dihydrofolate reductase activity.Thymidylate synthase activity. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01131; DB00205; DB01299 Interacts with NA EC number 1.5.1.3; 2.1.1.45 Uniprot keywords 3D-structure; Methyltransferase; Multifunctional enzyme; NADP; Nucleotide biosynthesis; One-carbon metabolism; Oxidoreductase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61720.6 Length 525 Aromaticity 0.13 Instability index 33.9 Isoelectric point 8.79 Charge (pH=7) 11.61 2D Binding mode Binding energy (Kcal/mol) -5.48 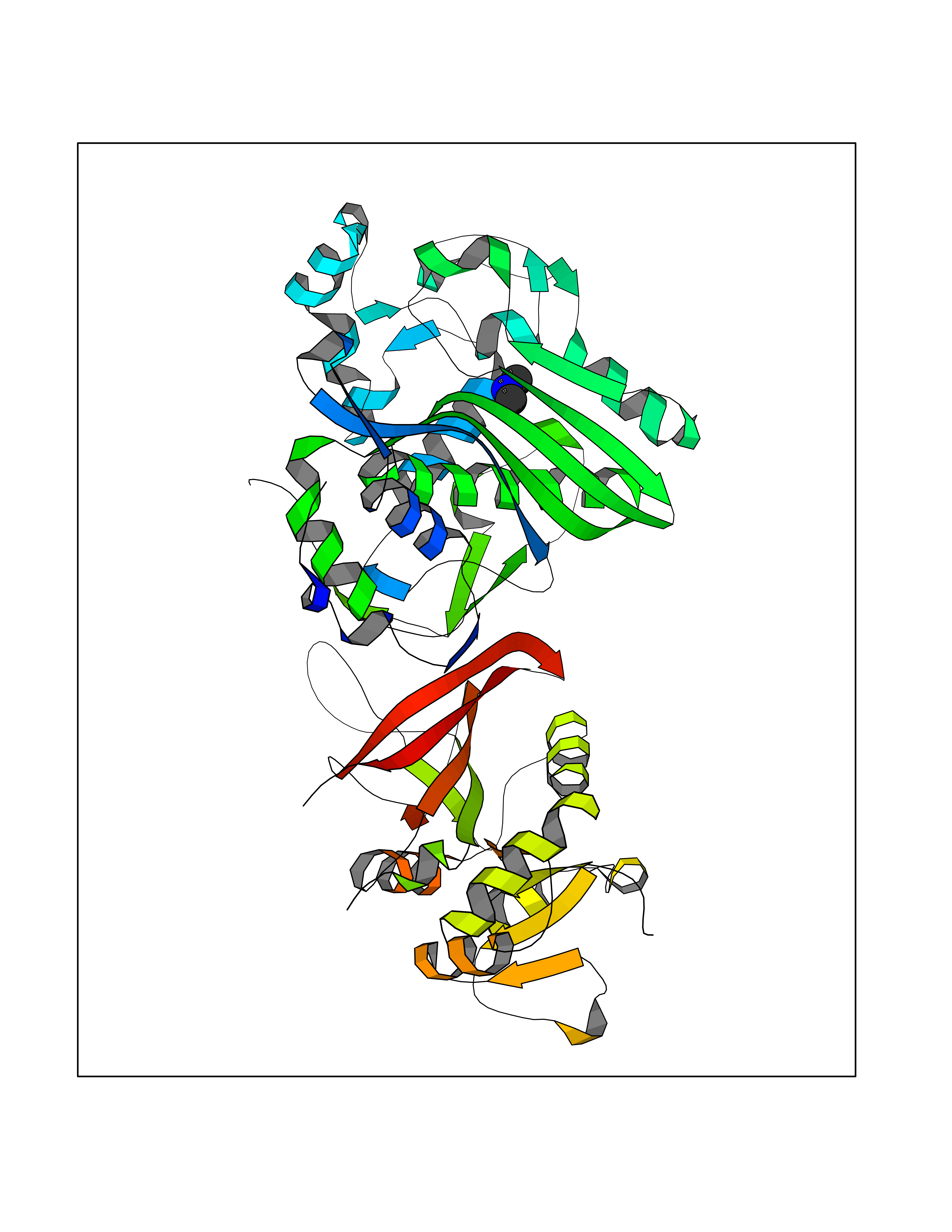 Molscript Map 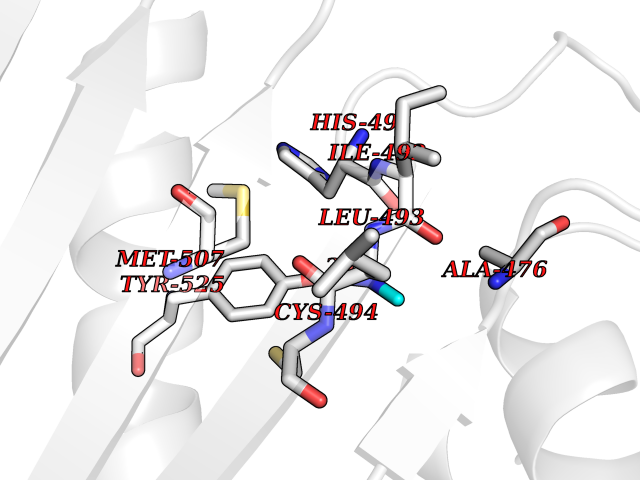 Pymol Map 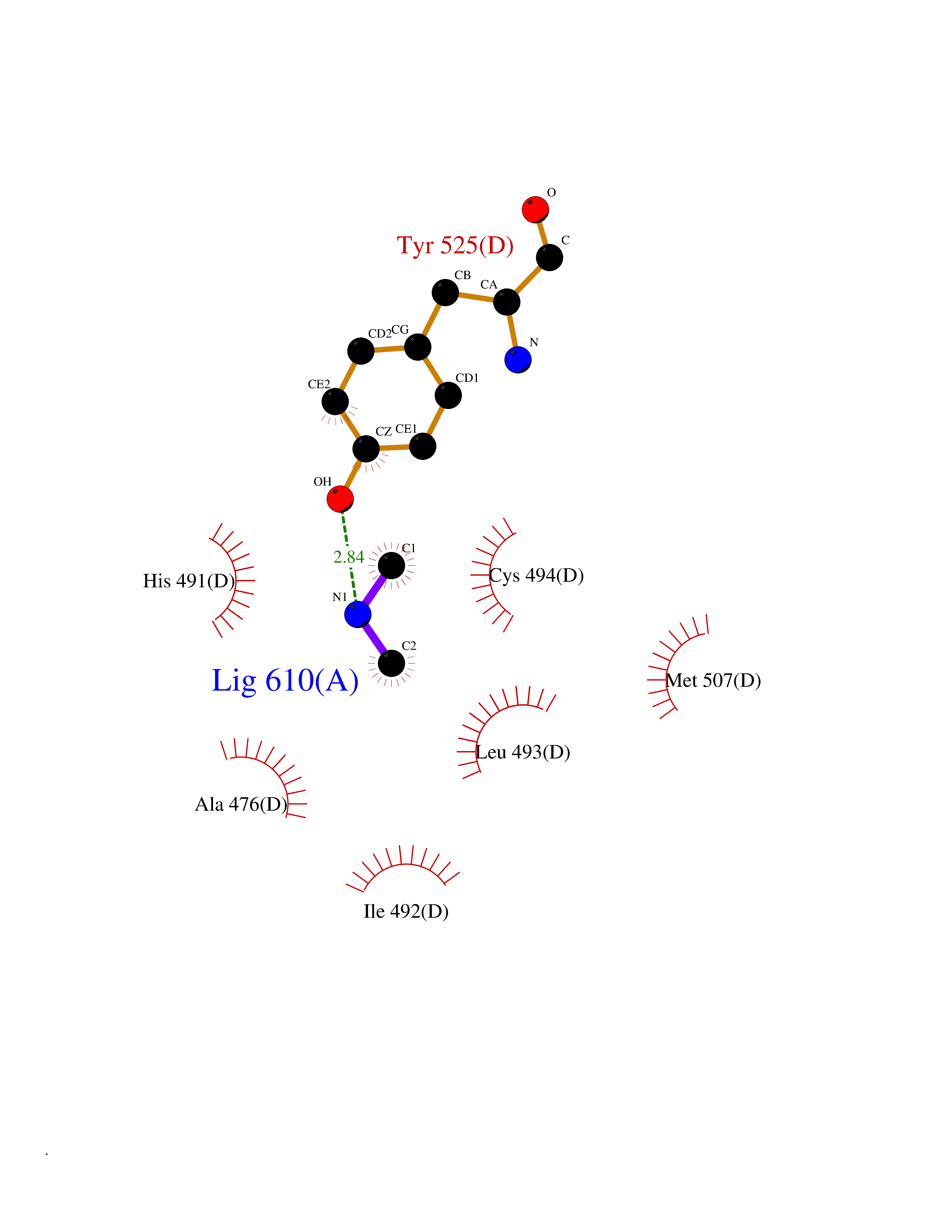 Ligplot Map 3D Binding mode Sequence NSIHPNDFQIYNSLKYKYHPEYQYLNIIYDIMMNGNKQSDRTGVGVLSKFGYIMKFDLSQYFPLLTTKKLFLRGIIEELLWFIRGETNGNTLLNKNVRIWEANGTREFLDNRKLFHREVNDLGPIYGFQWRHFGAEYTNMYDNYENKGVDQLKNIINLIKNDPTSRRILLCAWNVKDLDQMALPPCHILCQFYVFDGKLSCIMYQRSCDLGLGVPFNIASYSIFTHMIAQVCNLQPAQFIHVLGNAHVYNNHIDSLKIQLNRIPYPFPTLKLNPDIKNIEDFTISDFTIQNYVHHEKISMDMAAMMEQVCDVFDIYAICACCKVESKNEGKKNEVFNNYTFRGLGNKGVLPWKCISLDMKYFRAVTTYVNESKYEKLKYKRCKYLPNSKKLQNVVVMGRTNWESIPKKFKPLSNRINVILSRTLKKEDFDEDVYIINKVEDLIVLLGKLNYYKCFILGGSVVYQEFLEKKLIKKIYFTRINSTYECDVFFPEINENEYQIISVSDVYTSNNTTLDFIIYKKTNNK Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Tartrate-resistant acid phosphatase type 5 | 2BQ8 | 4.02 | |
Target general information Gen name ACP5 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Metallophosphoesterase superfamily, Purple acid phosphatase family Biochemical class Hydrolase Function Acid phosphatase activity.Ferric iron binding.Ferrous iron binding. Related diseases Spondyloenchondrodysplasia with immune dysregulation (SPENCDI) [MIM:607944]: A disease characterized by vertebral and metaphyseal dysplasia, spasticity with cerebral calcifications, and strong predisposition to autoimmune diseases. The skeletal dysplasia is characterized by radiolucent and irregular spondylar and metaphyseal lesions that represent islands of chondroid tissue within bone. {ECO:0000269|PubMed:21217752, ECO:0000269|PubMed:21217755}. The disease is caused by variants affecting the gene represented in this entry. ACP5 inactivating mutations result in a functional excess of phosphorylated osteopontin causing deregulation of osteopontin signaling and consequential autoimmune disease. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.3.2 Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Iron; Lysosome; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID X Molecular weight (Da) 34330.6 Length 304 Aromaticity 0.12 Instability index 42.3 Isoelectric point 9.11 Charge (pH=7) 6.75 2D Binding mode Binding energy (Kcal/mol) -5.48 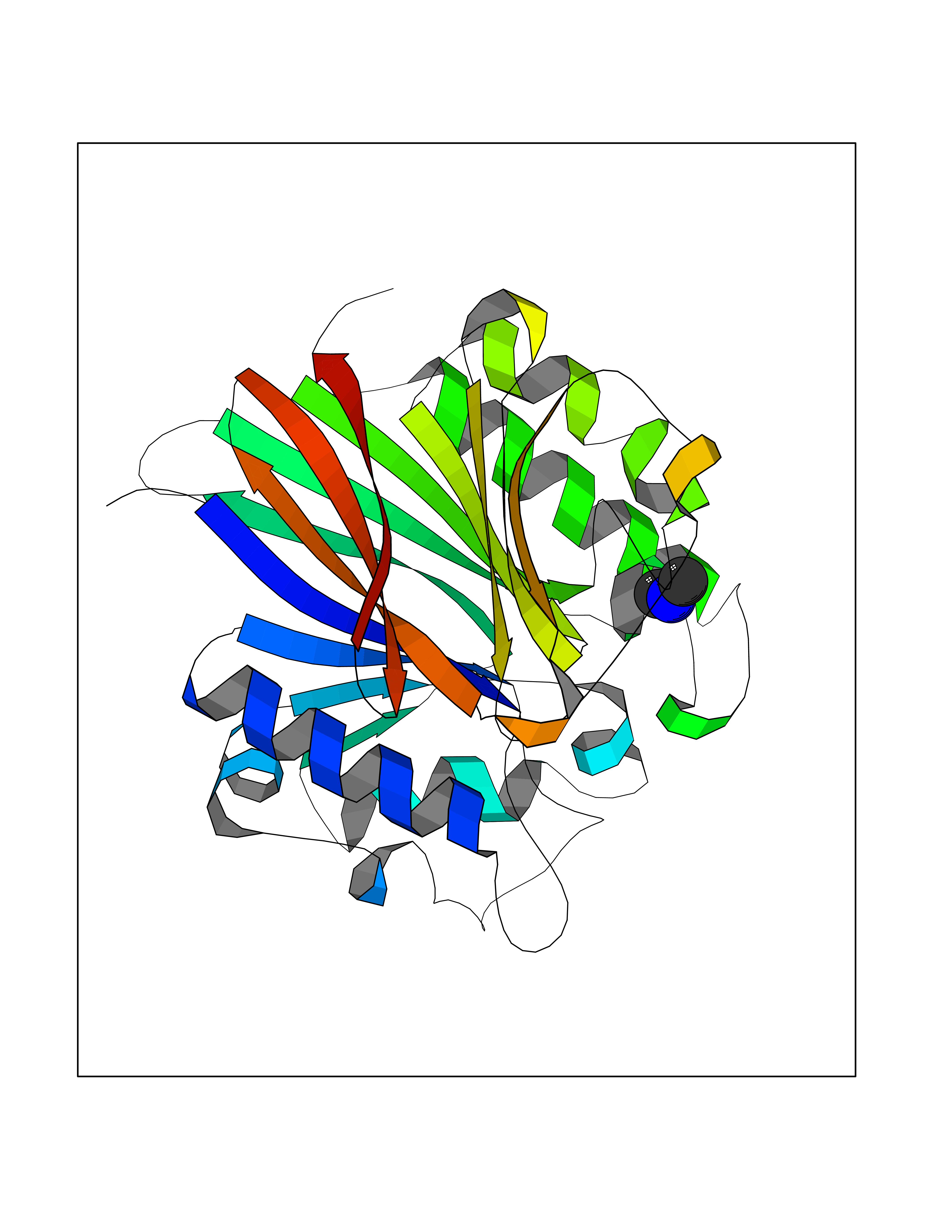 Molscript Map 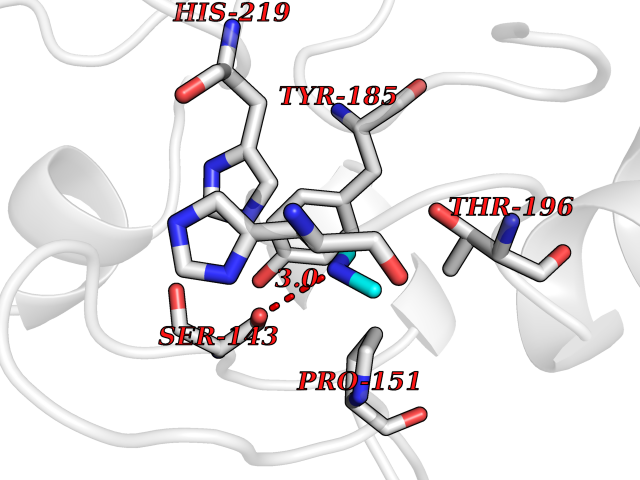 Pymol Map 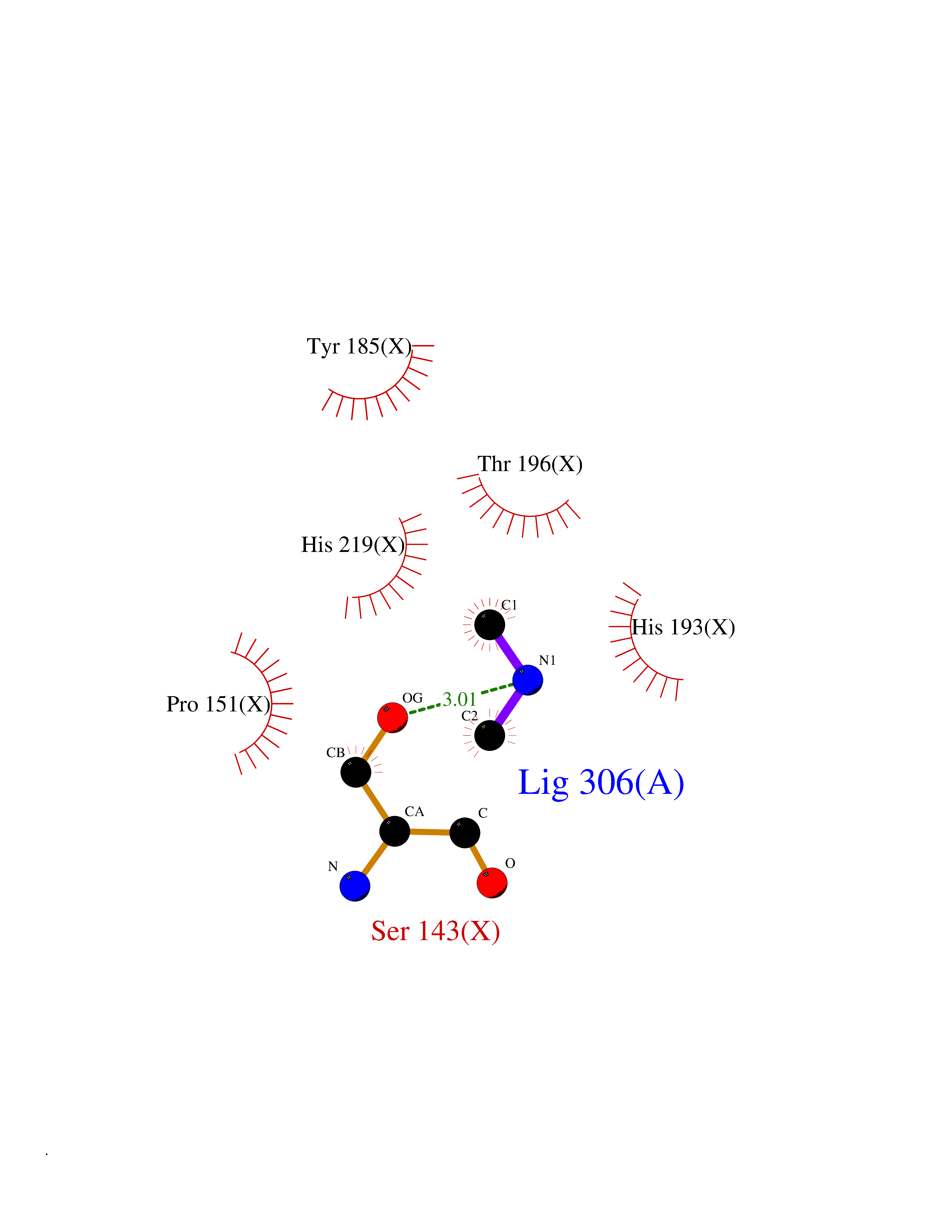 Ligplot Map 3D Binding mode Sequence ATPALRFVAVGDWGGVPNAPFHTAREMANAKEIARTVQILGADFILSLGDNFYFTGVQDINDKRFQETFEDVFSDRSLRKVPWYVLAGNHDHLGNVSAQIAYSKISKRWNFPSPFYRLHFKIPQTNVSVAIFMLDTVTLCGNSDDFLSQQPERPRDVKLARTQLSWLKKQLAAAREDYVLVAGHYPVWSIAEHGPTHCLVKQLRPLLATYGVTAYLCGHDHNLQYLQDENGVGYVLSGAGNFMDPSKRHQRKVPNGYLRFHYGTEDSLGGFAYVEISSKEMTVTYIEASGKSLFKTRLPRRARP Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 4.02 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map 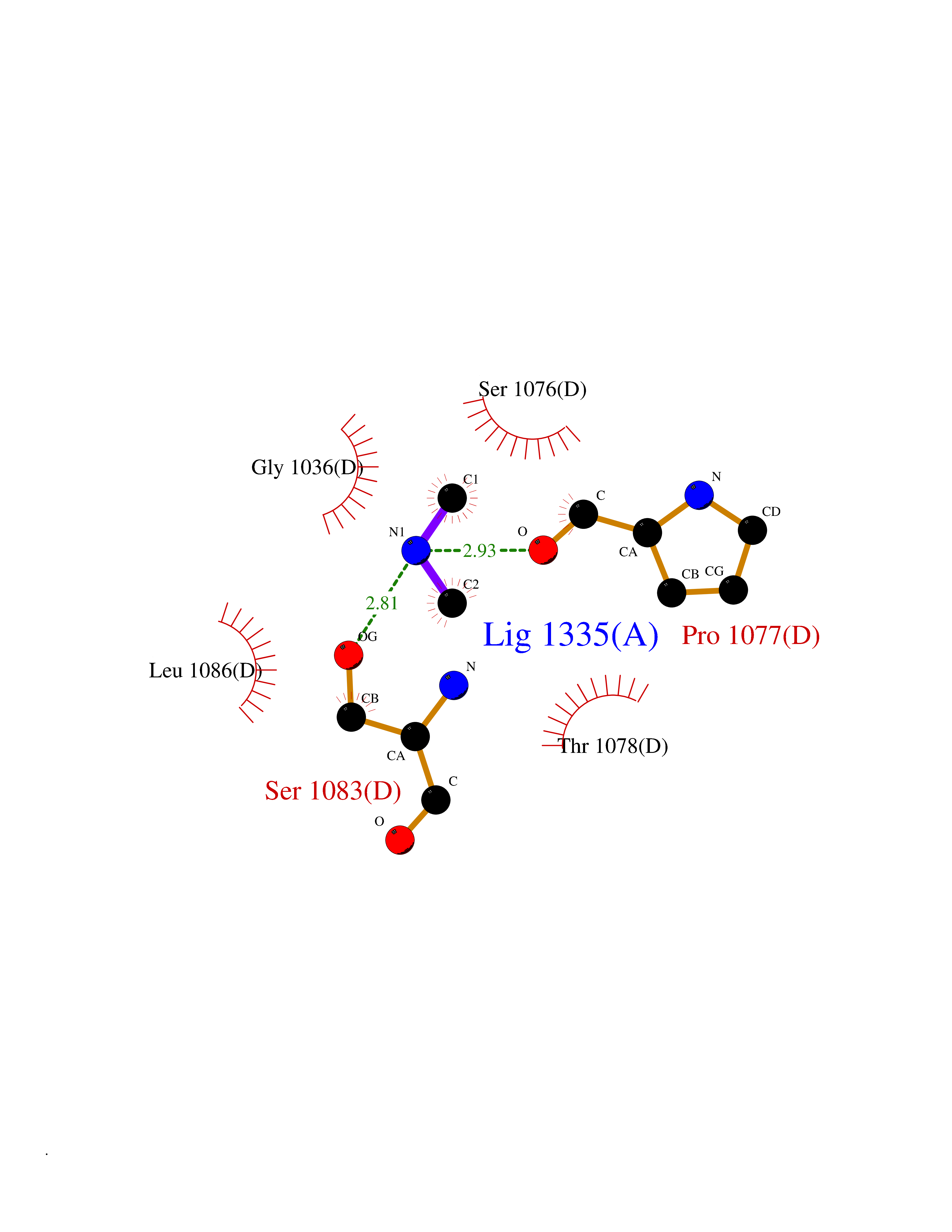 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Aminoacylase-1 | 1Q7L | 4.02 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -5.48 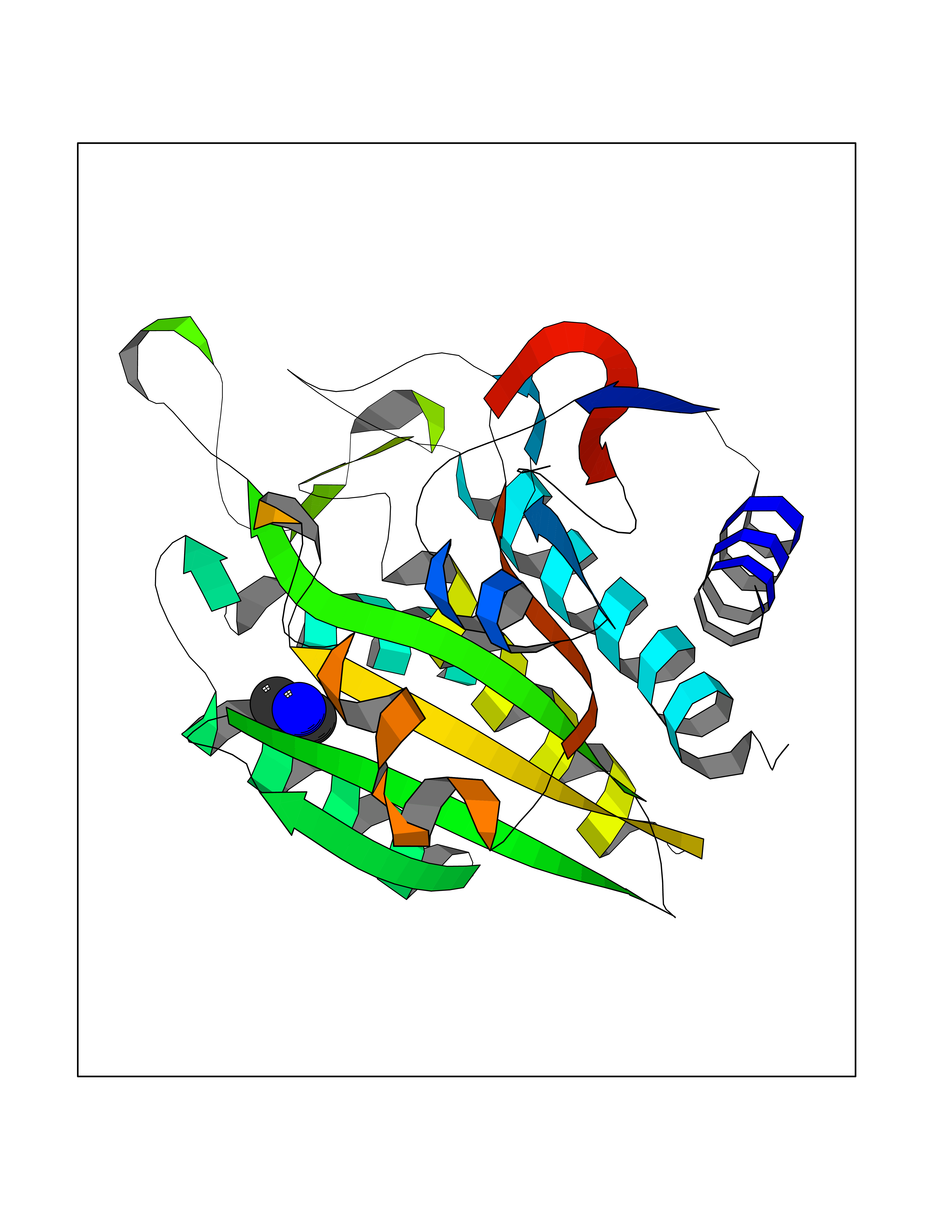 Molscript Map 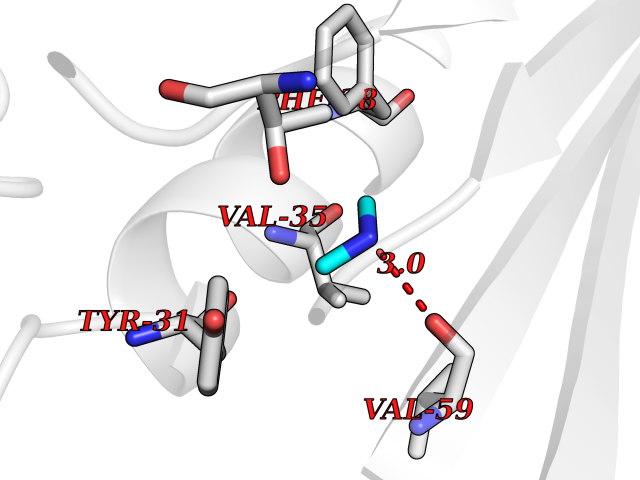 Pymol Map 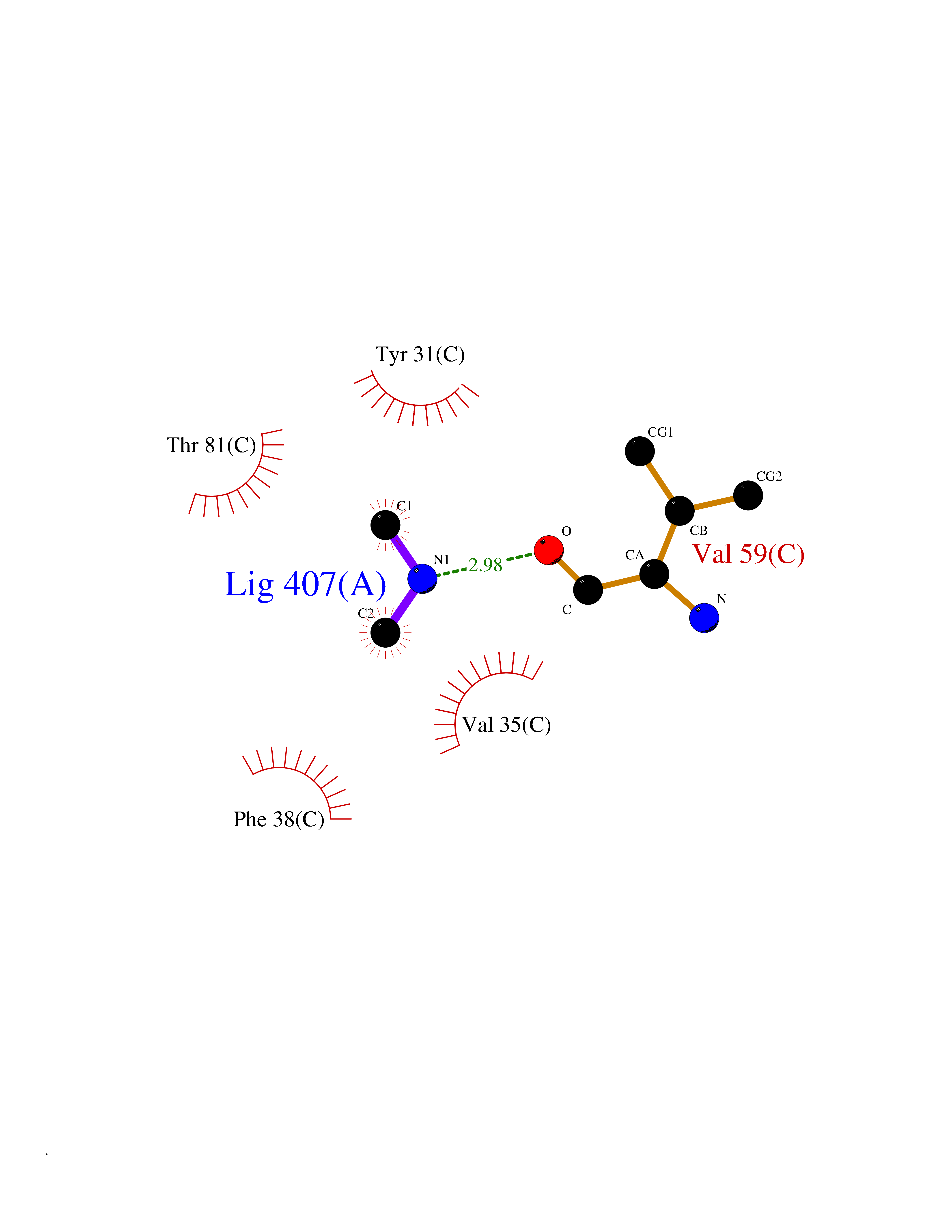 Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Aminoacylase-1 | 1Q7L | 4.02 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -5.49 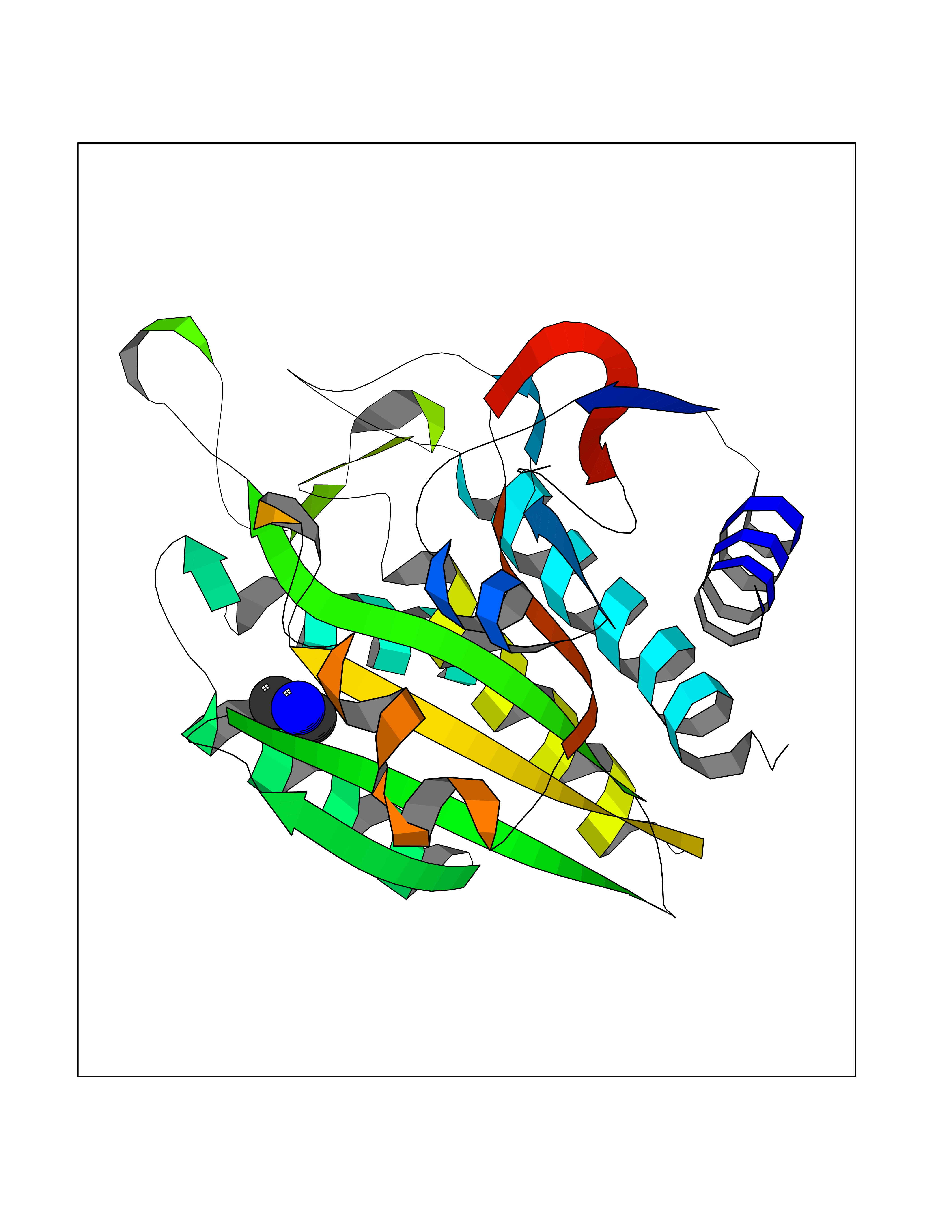 Molscript Map 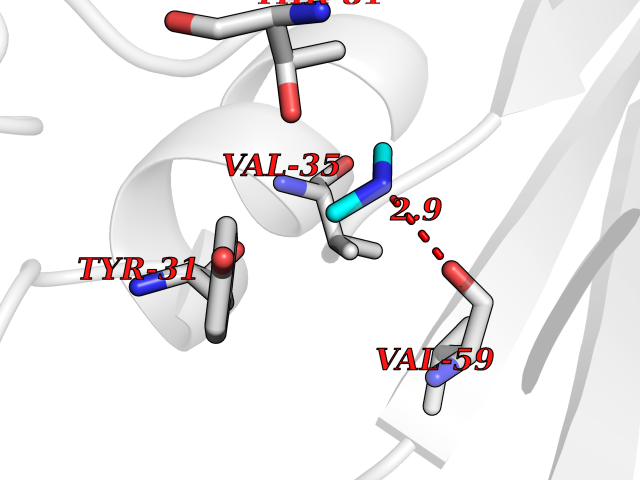 Pymol Map 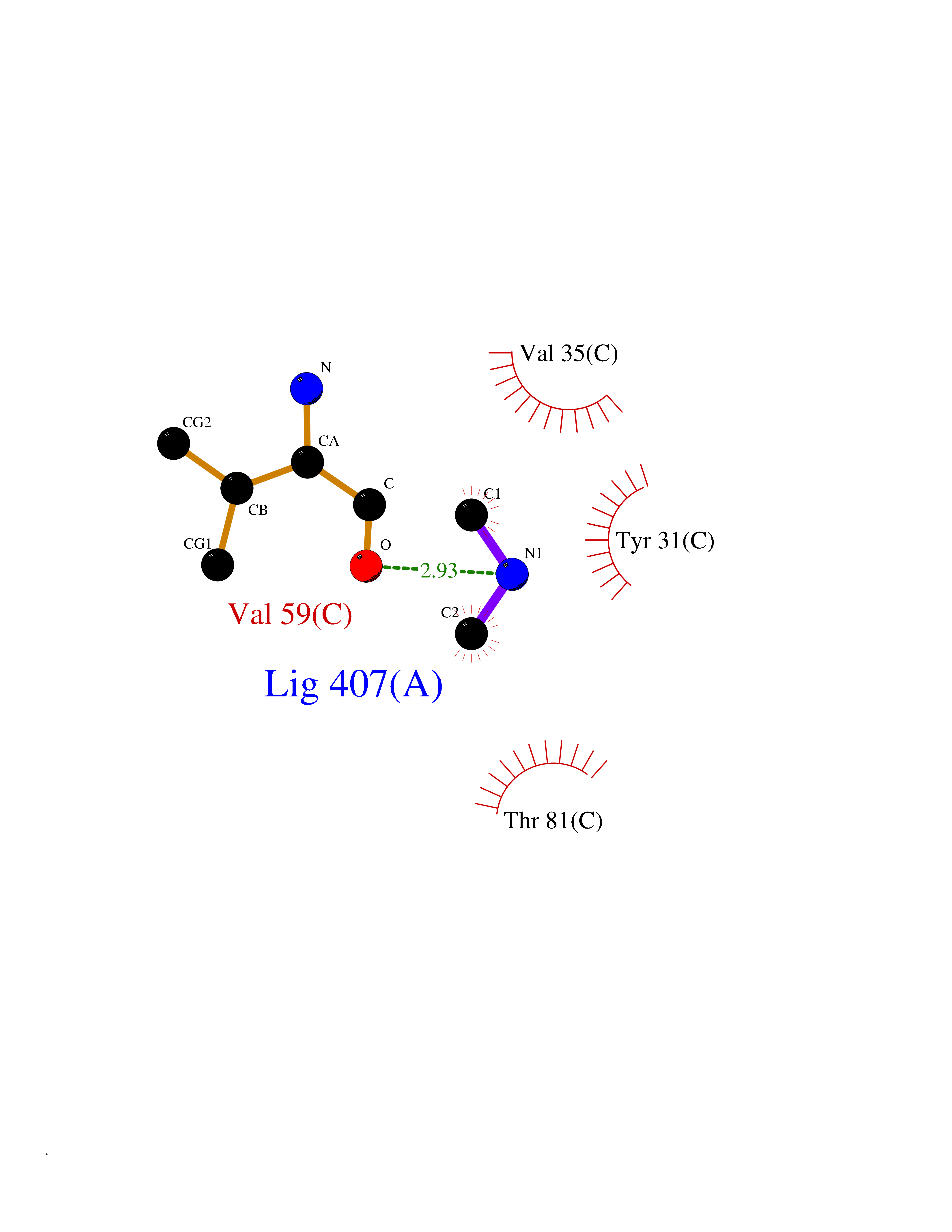 Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Histone deacetylase 2 (HDAC2) | 4LY1 | 4.02 | |
Target general information Gen name HDAC2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HD2 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Forms transcriptional repressor complexes by associating with MAD, SIN3, YY1 and N-COR. Interacts in the late S-phase of DNA-replication with DNMT1 in the other transcriptional repressor complex composed of DNMT1, DMAP1, PCNA, CAF1. Deacetylates TSHZ3 and regulates its transcriptional repressor activity. Component of a RCOR/GFI/KDM1A/HDAC complex that suppresses, via histone deacetylase (HDAC) recruitment, a number of genes implicated in multilineage blood cell development. May be involved in the transcriptional repression of circadian target genes, such as PER1, mediated by CRY1 through histone deacetylation. Involved in MTA1-mediated transcriptional corepression of TFF1 and CDKN1A. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Ventricular tachycardia, catecholaminergic polymorphic, 1, with or without atrial dysfunction and/or dilated cardiomyopathy (CPVT1) [MIM:604772]: An arrhythmogenic disorder characterized by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and cause sudden death. Patients present with recurrent syncope, seizures, or sudden death after physical activity or emotional stress. CPVT1 inheritance is autosomal dominant. {ECO:0000269|PubMed:11157710, ECO:0000269|PubMed:11159936, ECO:0000269|PubMed:11208676, ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:12106942, ECO:0000269|PubMed:14571276, ECO:0000269|PubMed:15046072, ECO:0000269|PubMed:15046073, ECO:0000269|PubMed:15466642, ECO:0000269|PubMed:15544015, ECO:0000269|PubMed:16188589, ECO:0000269|PubMed:24793461, ECO:0000269|PubMed:25372681, ECO:0000269|PubMed:27733687}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ventricular arrhythmias due to cardiac ryanodine receptor calcium release deficiency syndrome (VACRDS) [MIM:115000]: An autosomal dominant arrhythmogenic disorder characterized by syncope, cardiac arrest and/or sudden unexpected death, often in association with physical exertion or acute emotional stress. Patients who survive manifest polymorphic ventricular tachycardia and ventricular fibrillation. Unlike typical catecholaminergic ventricular tachycardia, arrhythmias are not reproducible on exercise stress testing or adrenaline challenge. {ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:17984046, ECO:0000269|PubMed:33536282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB01223; DB01076; DB05015; DB01262; DB11841; DB01095; DB12645; DB00227; DB11830; DB01303; DB06603; DB06819; DB05223; DB00175; DB03766; DB12847; DB06176; DB00641; DB00277; DB09091; DB00313; DB02546 Interacts with Q9C0K0; Q9HCU9; P68400; Q9UER7; P51610; Q13547; Q9UIS9; Q13330; P01106; P06748; P48382; Q96ST3; O95863; Q9HD15; O43463; Q9H3M7; Q92618; Q17R98; Q2HR82; PRO_0000449623 [P0DTD1] EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Cytoplasm; Hydrolase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 42020.5 Length 366 Aromaticity 0.13 Instability index 29.52 Isoelectric point 6.52 Charge (pH=7) -2.16 2D Binding mode Binding energy (Kcal/mol) -5.48 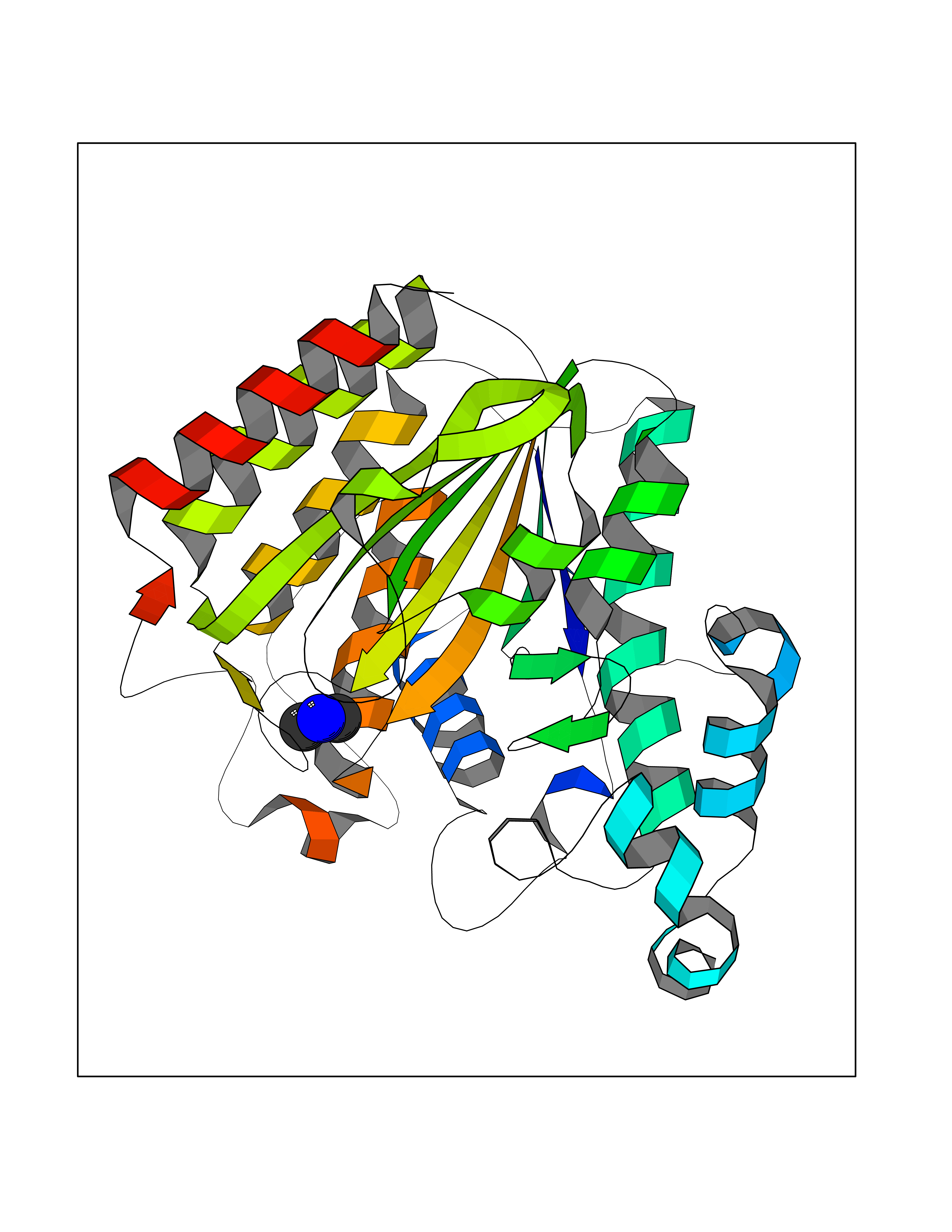 Molscript Map 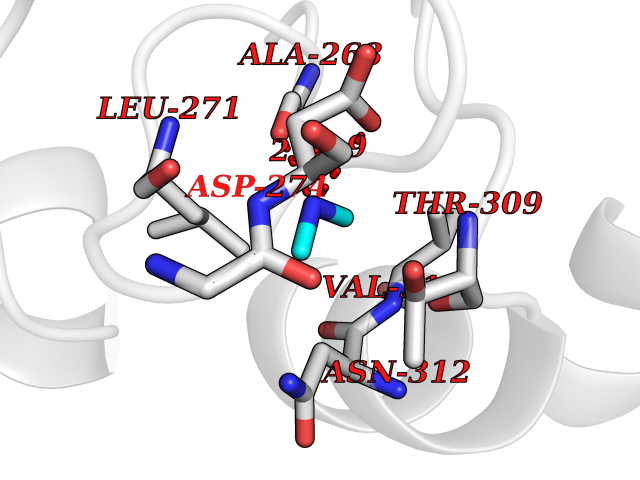 Pymol Map 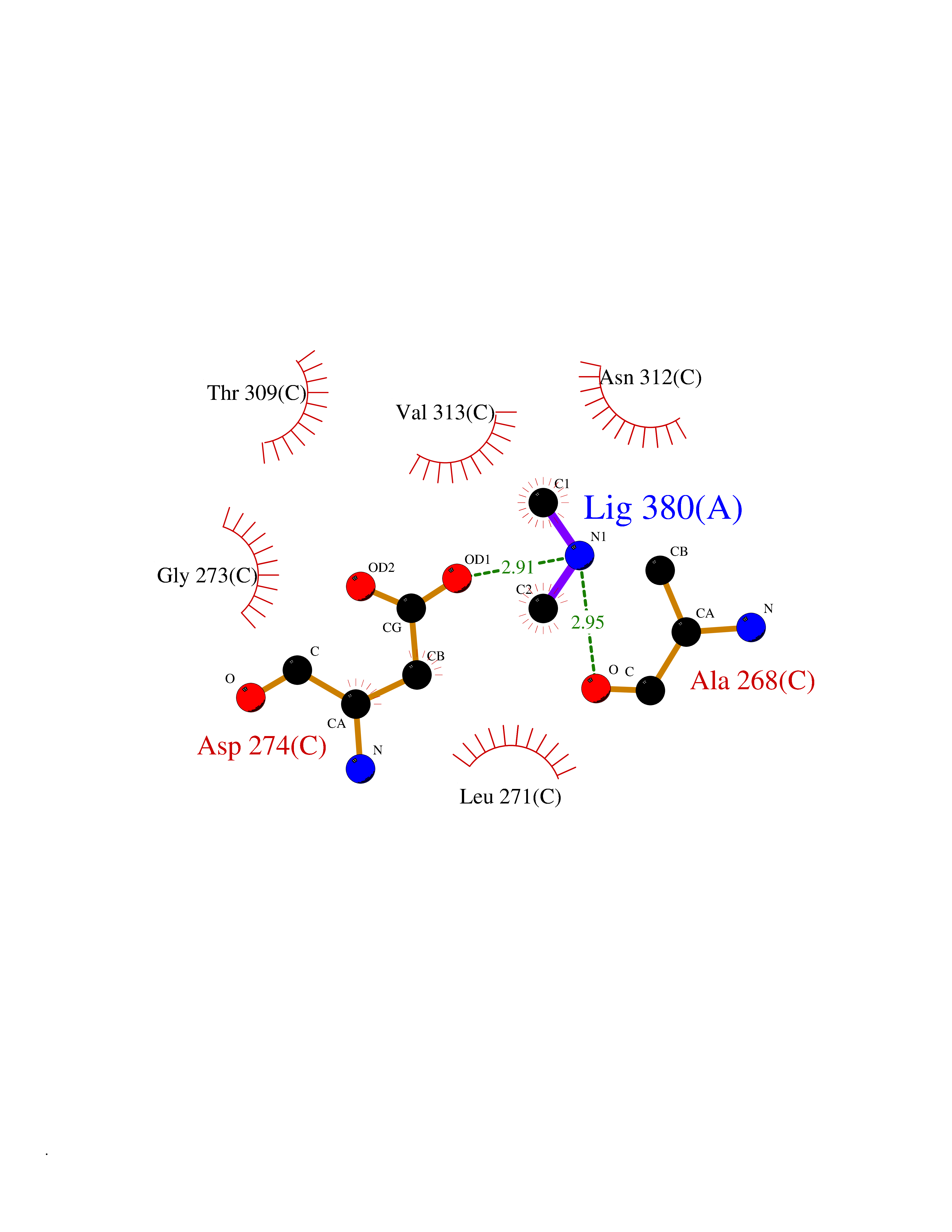 Ligplot Map 3D Binding mode Sequence KKKVCYYYDGDIGNYYYGQGHPMKPHRIRMTHNLLLNYGLYRKMEIYRPHKATAEEMTKYHSDEYIKFLRSIRPDNMSEYSKQMQRFNVGEDCPVFDGLFEFCQLSTGGSVAGAVKLNRQQTDMAVNWAGGLHHAKKSEASGFCYVNDIVLAILELLKYHQRVLYIDIDIHHGDGVEEAFYTTDRVMTVSFHKYGEYFPGTGDLRDIGAGKGKYYAVNFPMRDGIDDESYGQIFKPIISKVMEMYQPSAVVLQCGADSLSGDRLGCFNLTVKGHAKCVEVVKTFNLPLLMLGGGGYTIRNVARCWTYETAVALDCEIPNELPYNDYFEYFGPDFKLHISPSNMTNQNTPEYMEKIKQRLFENLRML Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Pectate lyase | 1R76 | 4.02 | |
Target general information Gen name pelA Organism Niveispirillum irakense (Azospirillum irakense) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Lyase Function Lyase activity. Related diseases A chromosomal aberration involving ALK is found in a form of non-Hodgkin lymphoma. Translocation t(2;5)(p23;q35) with NPM1. The resulting chimeric NPM1-ALK protein homodimerize and the kinase becomes constitutively activated. The constitutively active fusion proteins are responsible for 5-10% of non-Hodgkin lymphomas. {ECO:0000269|PubMed:15938644}.; DISEASE: A chromosomal aberration involving ALK is associated with inflammatory myofibroblastic tumors (IMTs). Translocation t(2;11)(p23;p15) with CARS; translocation t(2;4)(p23;q21) with SEC31A. {ECO:0000269|PubMed:12112524, ECO:0000269|PubMed:16161041}.; DISEASE: A chromosomal aberration involving ALK is associated with anaplastic large-cell lymphoma (ALCL). Translocation t(2;17)(p23;q25) with ALO17. {ECO:0000269|PubMed:12112524}.; DISEASE: Neuroblastoma 3 (NBLST3) [MIM:613014]: A common neoplasm of early childhood arising from embryonic cells that form the primitive neural crest and give rise to the adrenal medulla and the sympathetic nervous system. {ECO:0000269|PubMed:18724359, ECO:0000269|PubMed:18923523, ECO:0000269|PubMed:18923525, ECO:0000269|PubMed:21242967, ECO:0000269|PubMed:22932897}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: The ALK signaling pathway plays an important role in glioblastoma, the most common malignant brain tumor of adults and one of the most lethal cancers. It regulates both glioblastoma migration and growth. {ECO:0000269|PubMed:15908427}.; DISEASE: A chromosomal aberration involving ALK is found in one subject with colorectal cancer. Translocation t(2;2)(p23.1;p23.3). A 5 million base pair tandem duplication generates an in-frame WDCP-ALK gene fusion. {ECO:0000269|PubMed:22327622}.; DISEASE: A chromosomal aberration involving ALK has been identified in a subset of patients with non-small-cell lung carcinoma. This aberration leads to the production of a fusion protein between the N-terminus of EML4 et the C-terminus of ALK. It is unclear whether the fusion protein is caused by a simple inversion within 2p (inv(2)(p21p23)) or whether the chromosome translocation involving 2p is more complex. When tested in a heterologous system, the fusion protein EML4-ALK possesses transforming activity that is dependent on ALK catalytic activity, possibly due to spontaneous dimerization mediated by the EML4 moiety, leading to ALK kinase activation. {ECO:0000269|PubMed:17625570}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Lyase; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41907.5 Length 384 Aromaticity 0.08 Instability index 43.72 Isoelectric point 6.11 Charge (pH=7) -3.46 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 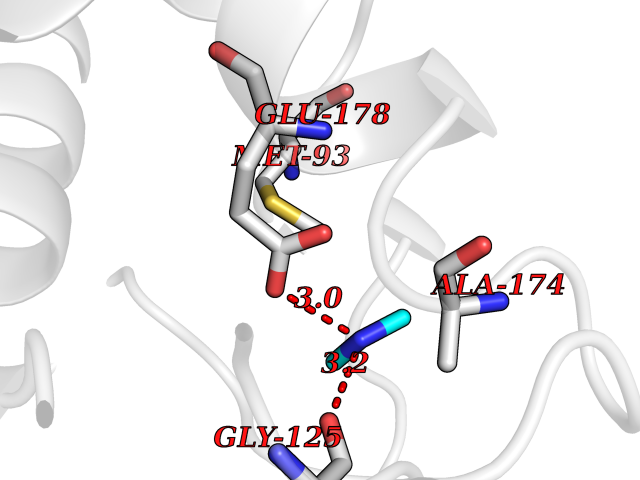 Pymol Map 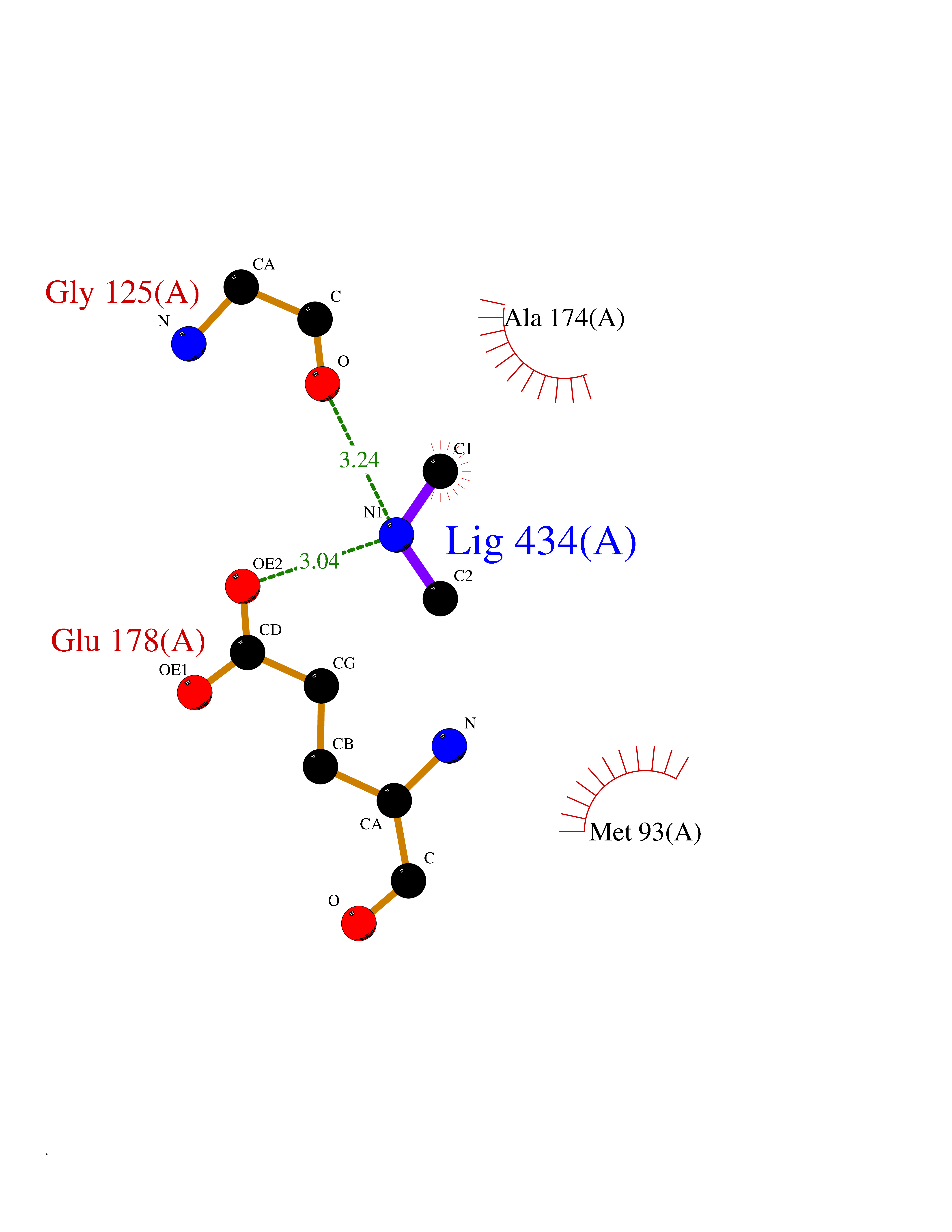 Ligplot Map 3D Binding mode Sequence AVIGMNEAASALTPSRVSSLPDTQRAAWQEYLARSEAQLSRDKASLAAELAPGQPLPPPPAEGKGADTMPLDKPAAWYTSKAARHVADVIVSFQTPAGGWGKNQPRDGALRLPGQHYTGENVAKVKRDRDWHYVGTIDNDATVTEIRFLAQVVSQLAPEEAAPYRDAALKGIEYLLASQFPNGGWPQVWPLEGGYHDAITYNDDALVHVAELLSDIAAGRDGFGFVPPAIRTRALEATNAAIHCIVETQVVQDGKRLGWGQQHDALTLRPTSARNFEPAALSSTESARILLFLMEIEAPSDAVKQAIRGGVAWLNTSVIRDQGAKPLWSRFYSLDGNKPVFGDRDKTIHDDVMGISQERRTGYAWYTTSPQKALSAFTKWEKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Kallikrein-5 (KLK5) | 6QFE | 4.02 | |
Target general information Gen name KLK5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ570/PRO1132; Stratum corneum tryptic enzyme; SCTE; Kallikrein-like protein 2; KLK-L2 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function May be involved in desquamation. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20930; Q9NQG1 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50299.2 Length 454 Aromaticity 0.07 Instability index 40.74 Isoelectric point 9.25 Charge (pH=7) 23.09 2D Binding mode Binding energy (Kcal/mol) -5.49 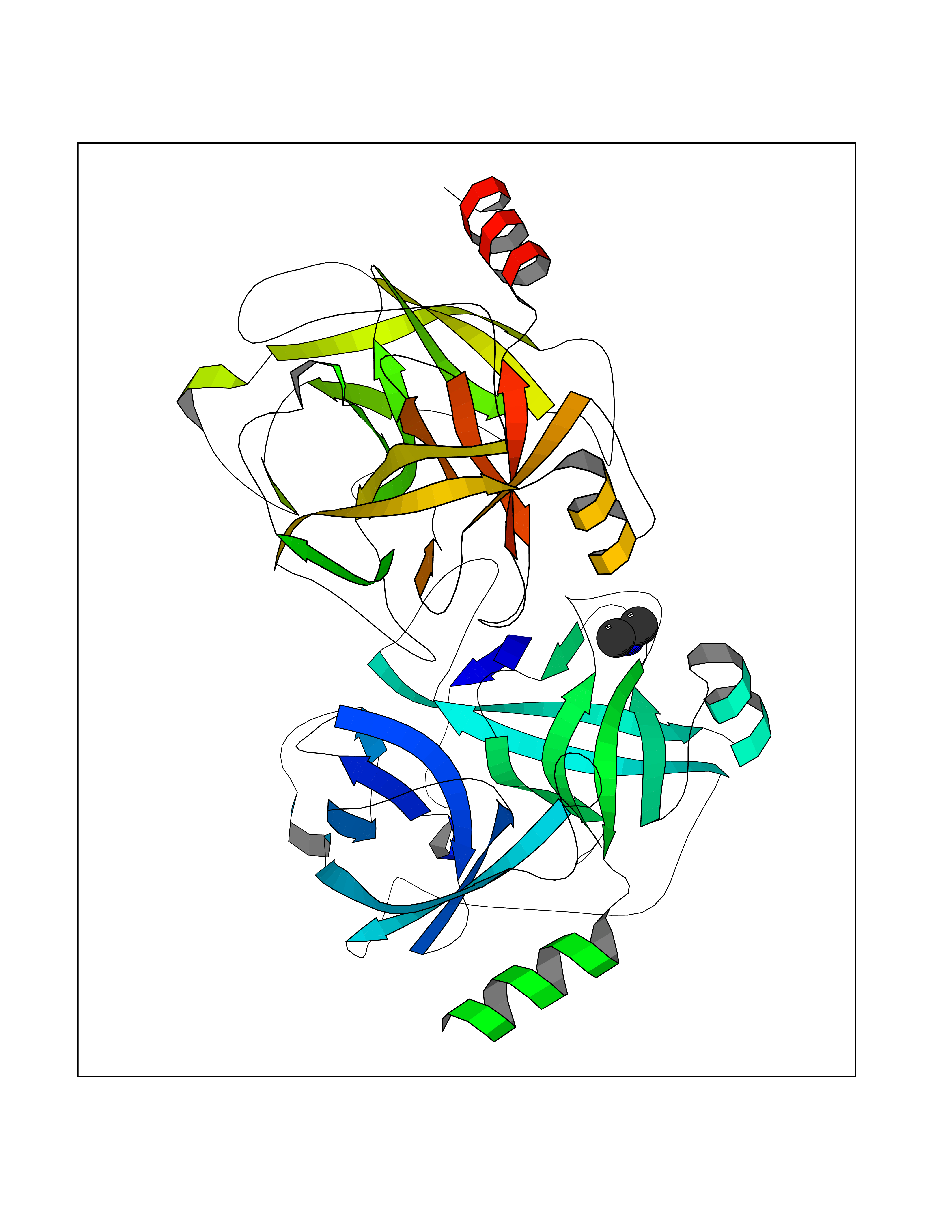 Molscript Map  Pymol Map 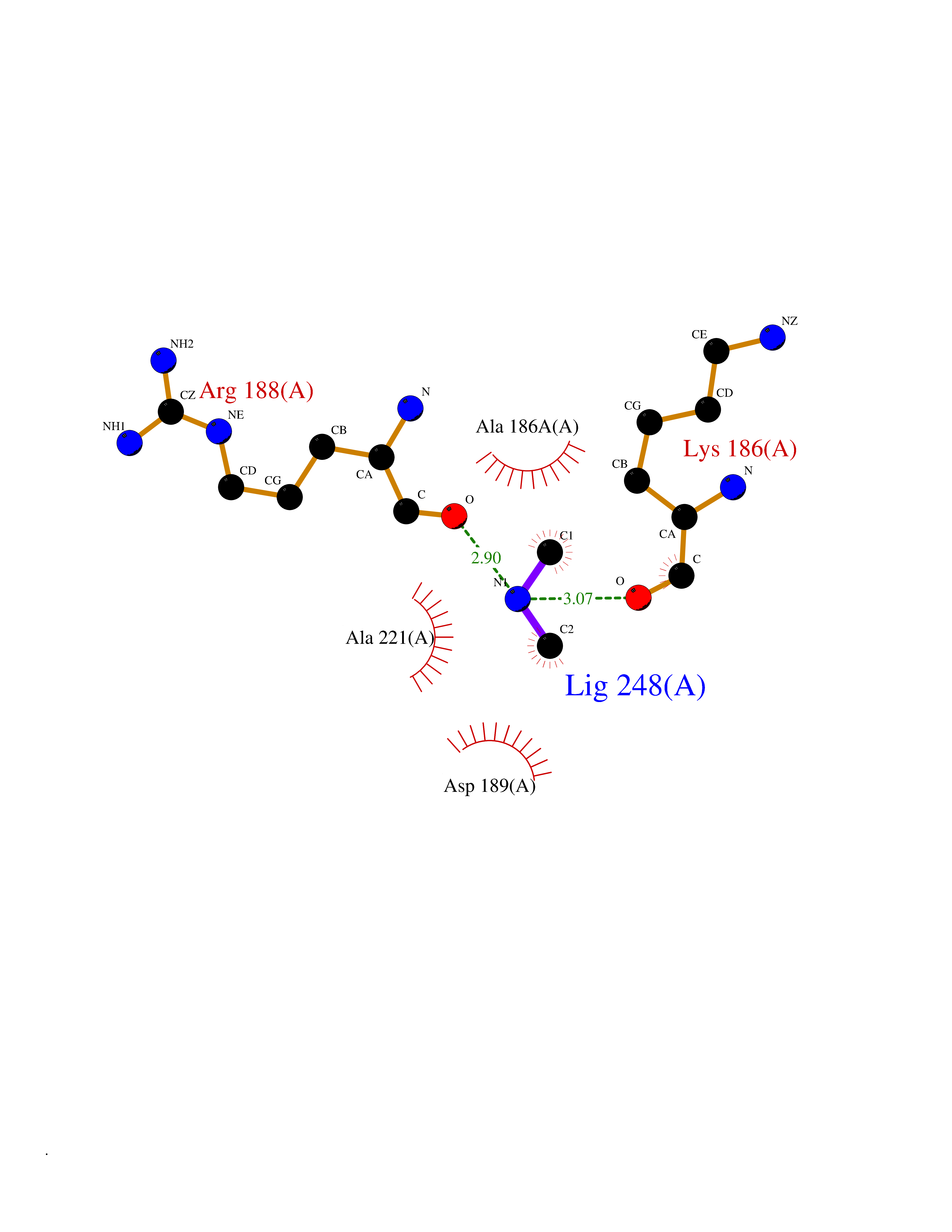 Ligplot Map 3D Binding mode Sequence IINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANSIINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANS Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Tankyrase-2 (TNKS-2) | 3U9H | 4.02 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -5.48 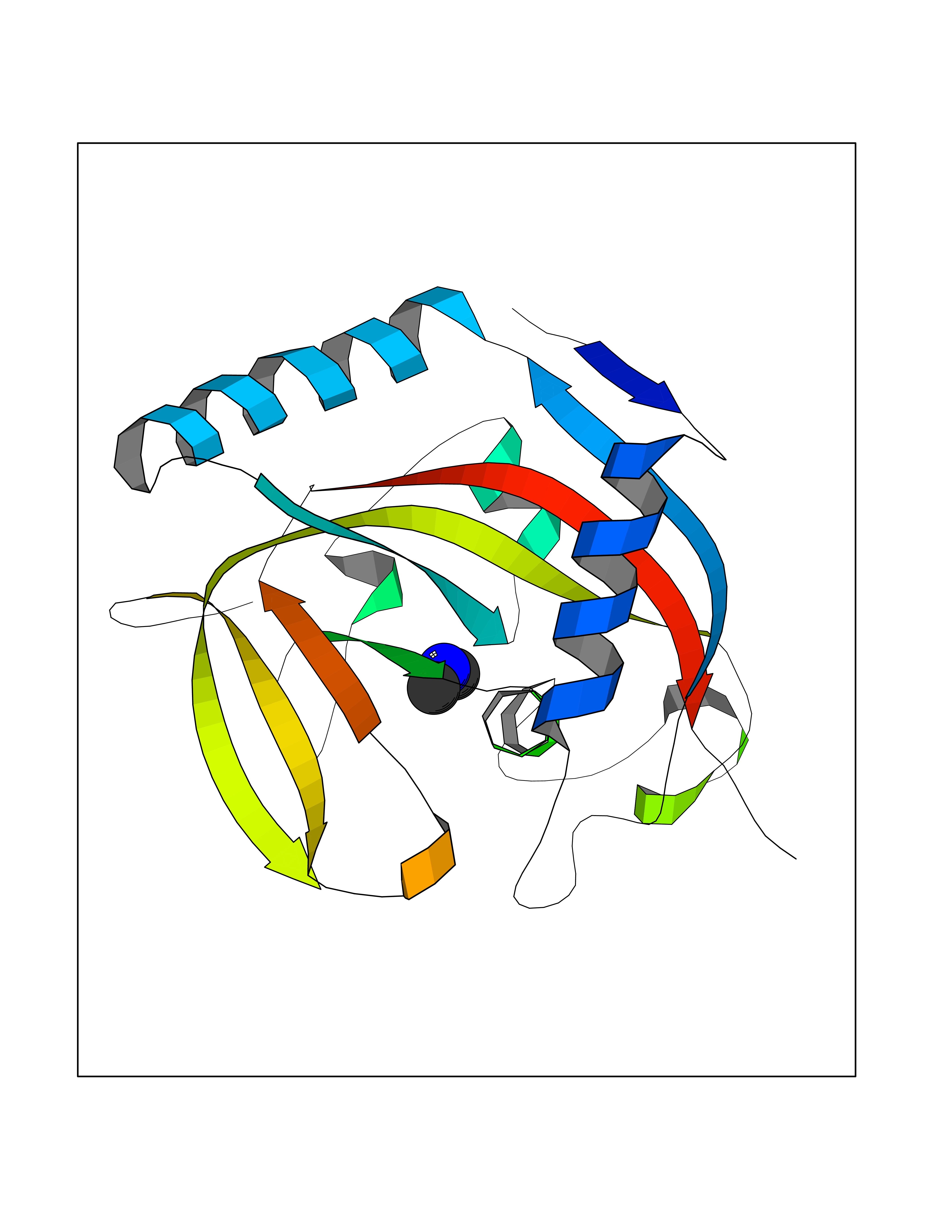 Molscript Map 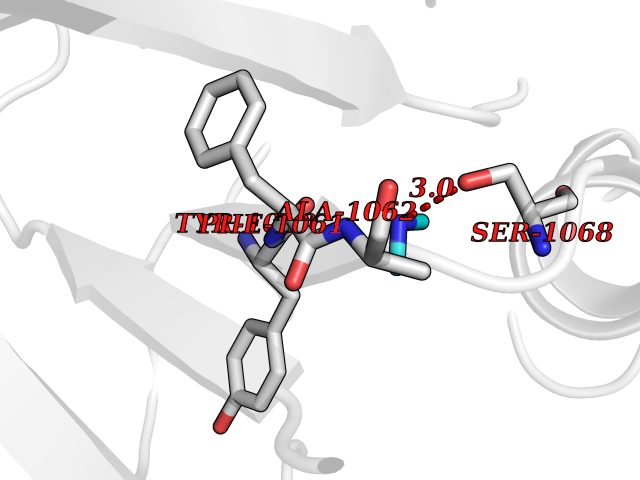 Pymol Map 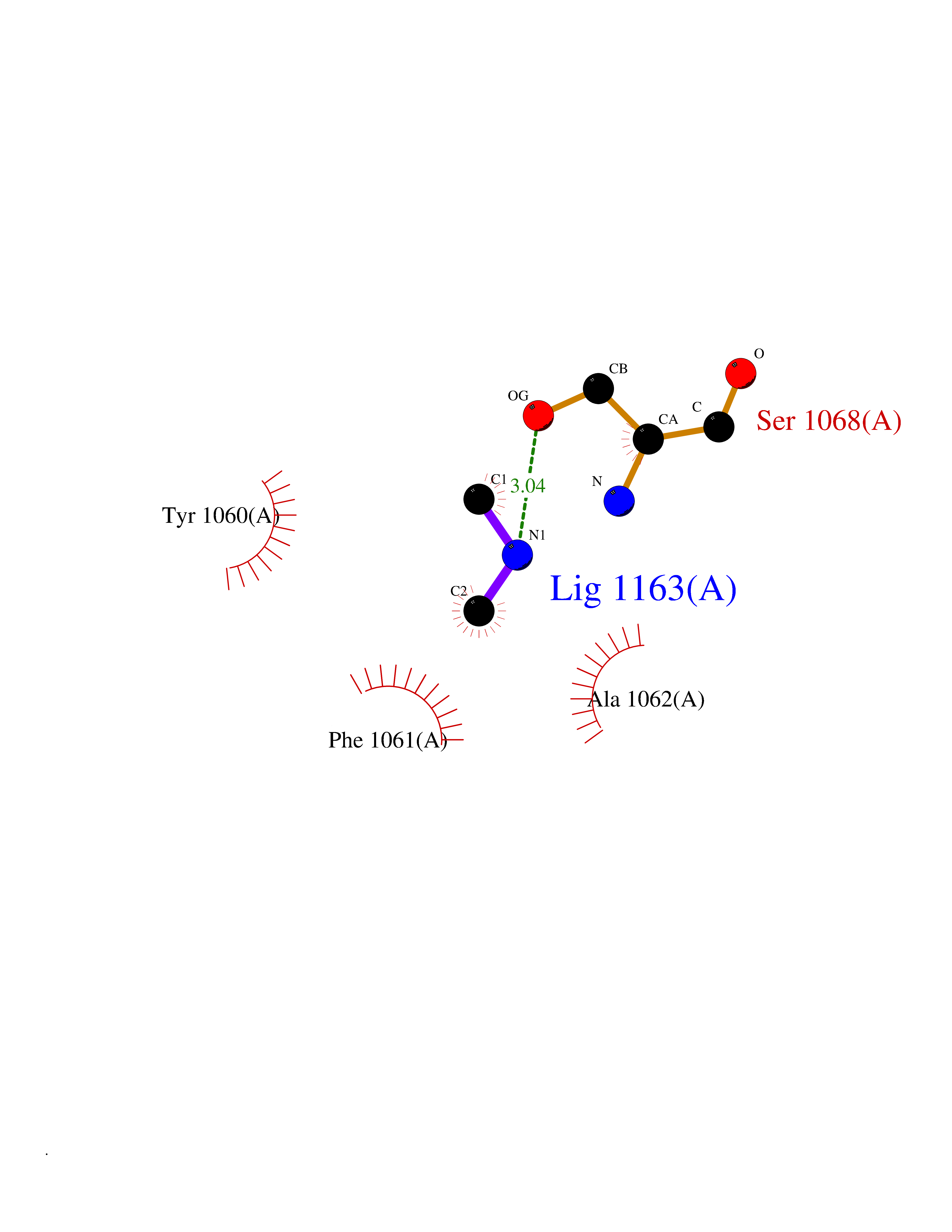 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Complement C1s component (C1S) | 1ELV | 4.02 | |
Target general information Gen name C1S Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complement component 1 subcomponent s; Complement C1s subcomponent; C1-esterase; C1 esterase Protein family Peptidase S1 family Biochemical class Peptidase Function C1r activates C1s so that it can, in turn, activate C2 and C4. C1s B chain is a serine protease that combines with C1q and C1r to form C1, the first component of the classical pathway of the complement system. Related diseases Complement component C1s deficiency (C1SD) [MIM:613783]: A rare defect resulting in C1 deficiency and impaired activation of the complement classical pathway. C1 deficiency generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. {ECO:0000269|PubMed:11390518}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ehlers-Danlos syndrome, periodontal type, 2 (EDSPD2) [MIM:617174]: A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSPD2 is characterized by the association of typical features of Ehlers-Danlos syndrome with gingival recession and severe early-onset periodontal disease, leading to premature loss of permanent teeth. EDSPD2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:27745832}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02371; DB09228; DB09130; DB12831; DB06404; DB14996; DB01593; DB14487; DB14533; DB14548 Interacts with P00736; P09871; P06681; O43889-2; Q9H6H4; P05155 EC number EC 3.4.21.42 Uniprot keywords 3D-structure; Calcium; Complement pathway; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Ehlers-Danlos syndrome; Glycoprotein; Hydrolase; Hydroxylation; Immunity; Innate immunity; Metal-binding; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal; Sushi Protein physicochemical properties Chain ID A Molecular weight (Da) 33278.6 Length 303 Aromaticity 0.1 Instability index 33.69 Isoelectric point 5.16 Charge (pH=7) -7.95 2D Binding mode Binding energy (Kcal/mol) -5.48 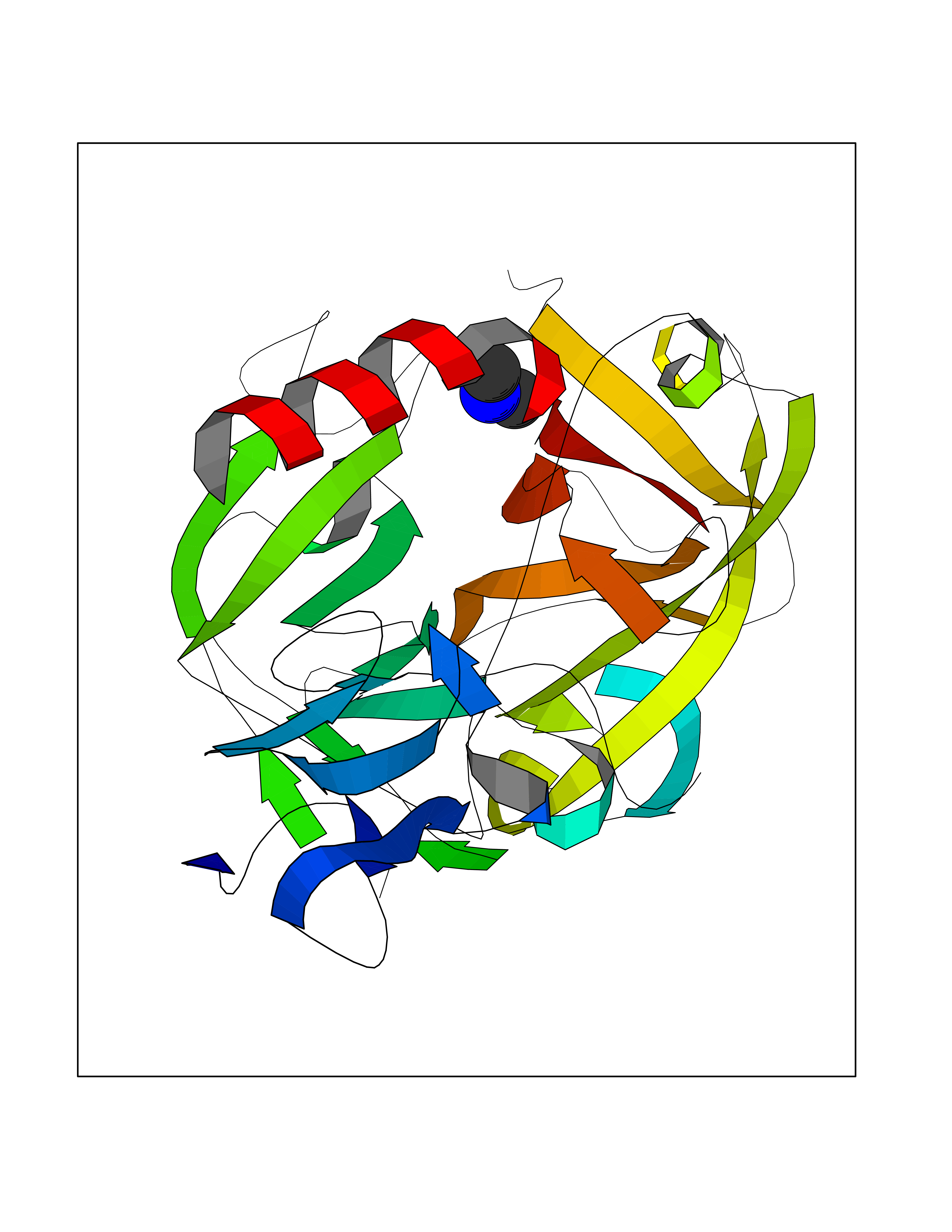 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LDCGIPESIENGKVEDPESTLFGSVIRYTCEEPYYYMEGGGEYHCAGNGSWVNEVLGPELPKCVPVCGVPREPFIIGGSDADIKNFPWQVFFDNPWAGGALINEYWVLTAAHVVEGNREPTMYVGSTSVQKMLTPEHVFIHPGWKLLAVPEGRTNFDNDIALVRLKDPVKMGPTVSPICLPGTSSDYNLMDGDLGLISGWGRTEKRDRAVRLKAARLPVAPLRKCKEVAYVFTPNMICAGGEKGMDSCKGDSGGAFAVQDPNDKTKFYAAGLVSWGPQCGTYGLYTRVKNYVDWIMKTMQENS Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Alcohol dehydrogenase 1B | 1U3U | 4.01 | |
Target general information Gen name ADH1B Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ADH2 Protein family Zinc-containing alcohol dehydrogenase family Biochemical class Oxidoreductase Function Alcohol dehydrogenase activity, zinc-dependent.Ethanol binding.Zinc ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02721; DB03703; DB00898; DB01213; DB09462; DB02481; DB04105; DB00157; DB03461 Interacts with P00326 EC number 1.1.1.105 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Lipid metabolism; Metal-binding; NAD; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39722.9 Length 374 Aromaticity 0.06 Instability index 18.9 Isoelectric point 8.63 Charge (pH=7) 6.96 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 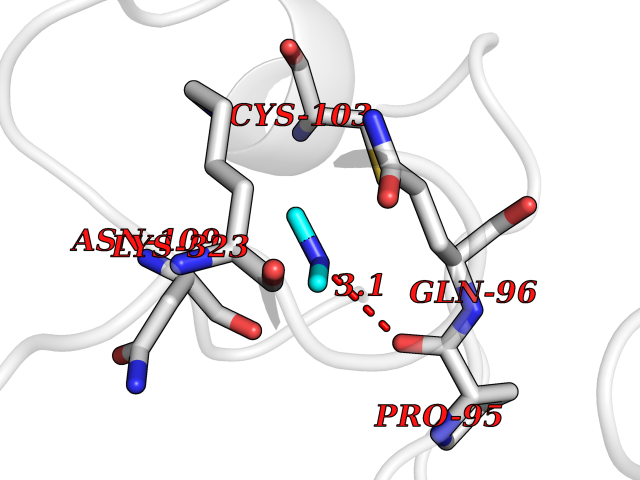 Pymol Map  Ligplot Map 3D Binding mode Sequence STAGKVIKCKAAVLWEVKKPFSIEDVEVAPPKAYEVRIKMVAVGICRTDDHVVSGNLVTPLPVILGHEAAGIVESVGEGVTTVKPGDKVIPLFTPQCGKCRVCKNPESNYCLKNDLGNPRGTLQDGTRRFTCRGKPIHHFLGTSTFSQYTVVDENAVAKIDAASPLEKVCLIGCGFSTGYGSAVNVAKVTPGSTCAVFGLGGVGLSAVMGCKAAGAARIIAVDINKDKFAKAKELGATECINPQDYKKPIQEVLKEMTDGGVDFSFEVIGRLDTMMASLLCCHEACGTSVIVGVPPASQNLSINPMLLLTGRTWKGAVYGGFKSKEGIPKLVADFMAKKFSLDALITHVLPFEKINEGFDLLHSGKSIRTVLTF Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | mRNA-capping enzyme | 2C46 | 4.01 | |
Target general information Gen name RNGTT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CAP1A Protein family Non-receptor class of the protein-tyrosine phosphatase family; Eukaryotic GTase family Biochemical class Transferase Function GTP binding.MRNA guanylyltransferase activity.Polynucleotide 5'-phosphatase activity.Protein tyrosine/serine/threonine phosphatase activity.Protein tyrosine phosphatase activity.RNA guanylyltransferase activity.Triphosphatase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) NA Interacts with Q92624; P16333-1 EC number 2.7.7.50; 3.6.1.74 Uniprot keywords 3D-structure; Alternative splicing; GTP-binding; Host-virus interaction; Hydrolase; mRNA capping; mRNA processing; Multifunctional enzyme; Nucleotide-binding; Nucleotidyltransferase; Nucleus; Protein phosphatase; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 21849.8 Length 189 Aromaticity 0.11 Instability index 53.71 Isoelectric point 5.89 Charge (pH=7) -2.91 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NKIPPRWLNCPRRGQPVAGRFLPLKTMLGPRYDSQVAEENRFHPSMLSNYLKSVKMGLLVDLTNTSRFYDRNDIEKEGIKYIKLQCKGHGECPTTENTETFIRLCERFELIGVHCTHGFNRTGFLICAFLVEKMDWSIEAAVATFAQARPPGIYKGDYLKELFRRYGDIEEAPPPPLLPDWCFEDDEDE Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Quinone-dependent D-lactate dehydrogenase | 1F0X | 4.01 | |
Target general information Gen name dld Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2121;b2133 Protein family Quinone-dependent D-lactate dehydrogenase family Biochemical class Oxidoreductase Function (R)-2-hydroxyglutarate dehydrogenase activity.D-lactate dehydrogenase (cytochrome) activity.Electron carrier activity.FAD binding.Flavin adenine dinucleotide binding.NAD binding.Oxidoreductase activity, acting on the CH-OH group of donors, quinone or similar compound as acceptor.Quinone binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB00756 Interacts with NA EC number 1.1.5.12 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Quinone; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56475.2 Length 502 Aromaticity 0.09 Instability index 32.5 Isoelectric point 5.97 Charge (pH=7) -10.38 2D Binding mode Binding energy (Kcal/mol) -5.47 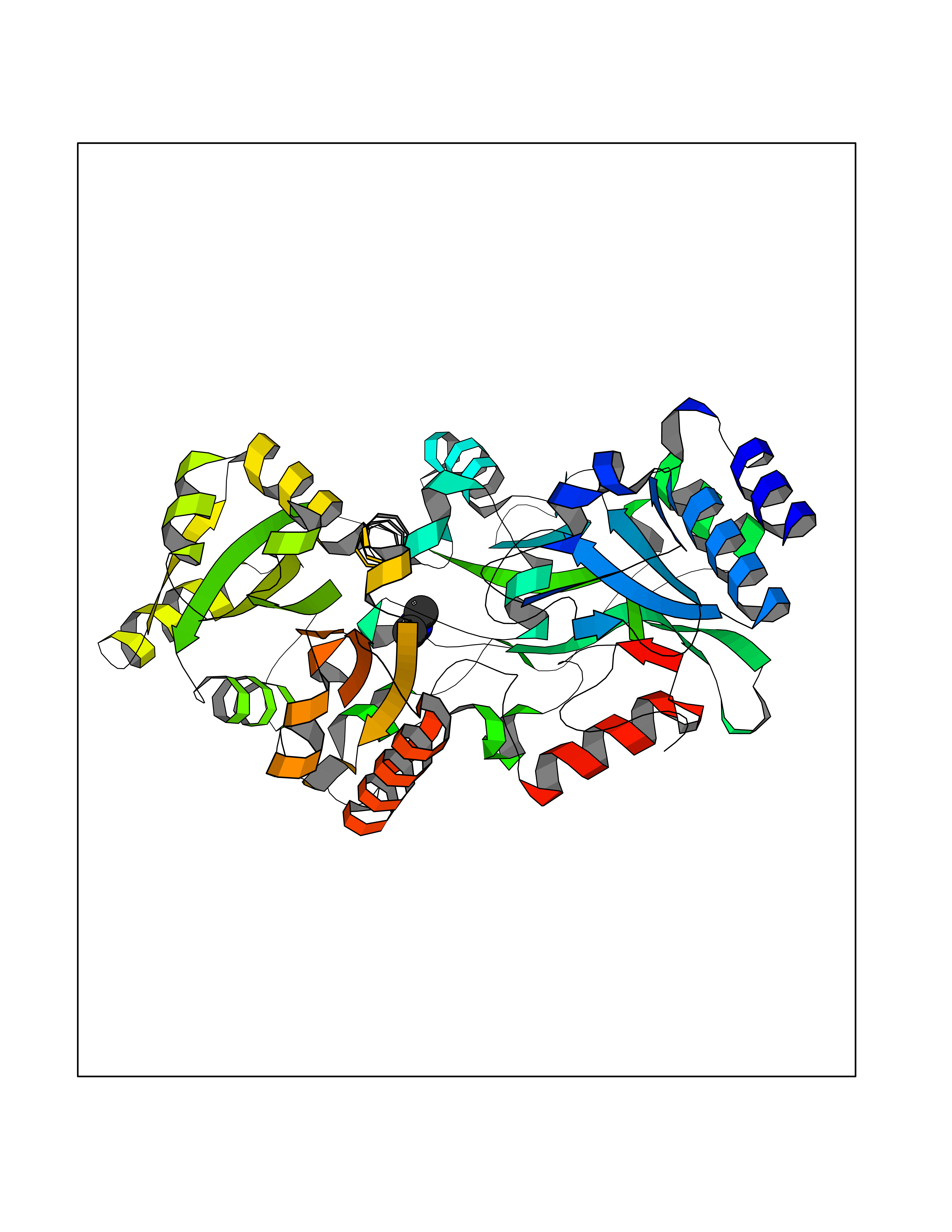 Molscript Map 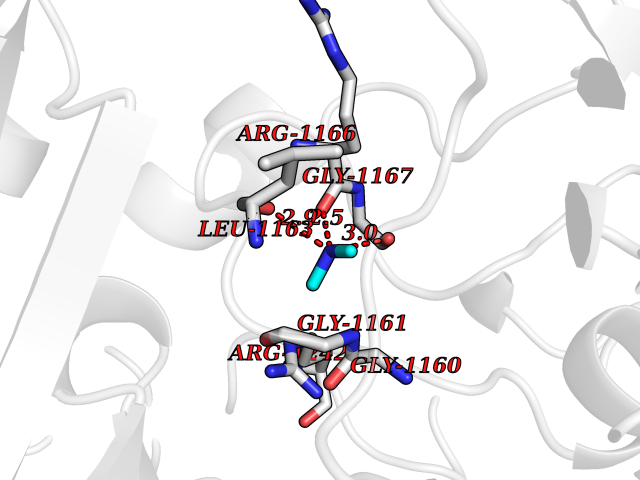 Pymol Map 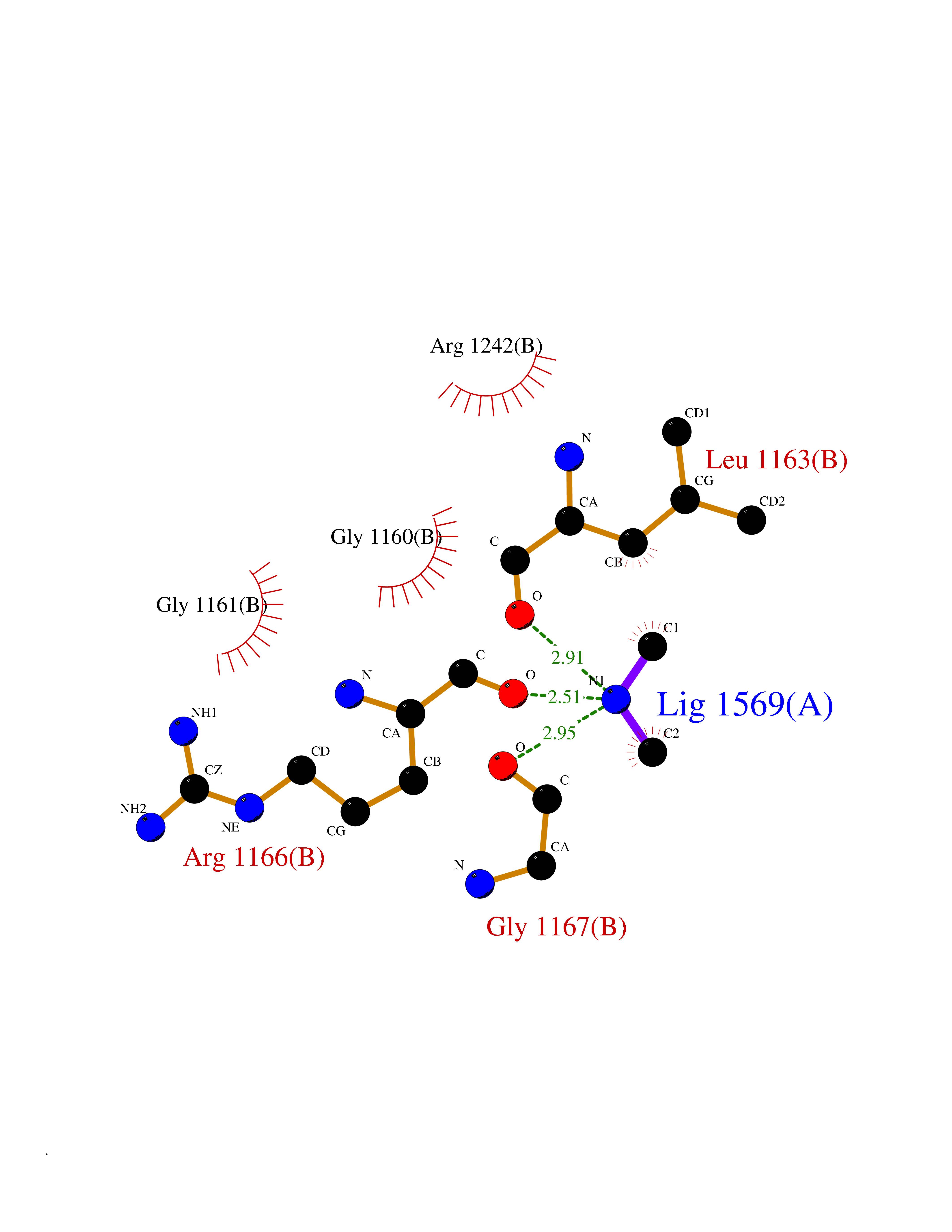 Ligplot Map 3D Binding mode Sequence NKAFLNELARLVGSSHLLTDPAKTARYRKGFRSGQGDALAVVFPGSLLELWRVLKACVTADKIILMQAANTGLTEGSTPNGNDYDRDVVIISTLRLDKLHVLGKGEQVLAYPGTTLYSLEKALKPLGREPHSVIGSSCIGASVIGGICNNSGGSLVQRGPAYTEMSLFARINEDGKLTLVNHLGIDLGETPEQILSKLDDDRIKDDDVRHDGRHAHDYDYVHRVRDIEADTPARYNADPDRLFESSGCAGKLAVFAVRLDTFEAEKNQQVFYIGTNQPEVLTEIRRHILANFENLPVAGEYMHRDIYDIAELPPRMKNWRDKYEHHLLLKMAGDGVGEAKSWLVDYFKQAEGDFFVCTPEEGSKAFLHRFAAAGAAIRYQAVHSDEVEDILALDIALRRNDTEWYEHLPPEIDSQLVHKLYYGHFMCYVFHQDYIVKKGVDVHALKEQMLELLQQRGAQYPAEHNVGHLYKAPETLQKFYRENDPTNSMNPGIGKTSKRKNW Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Dopamine beta-hydroxylase | 4ZEL | 4.01 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -5.47 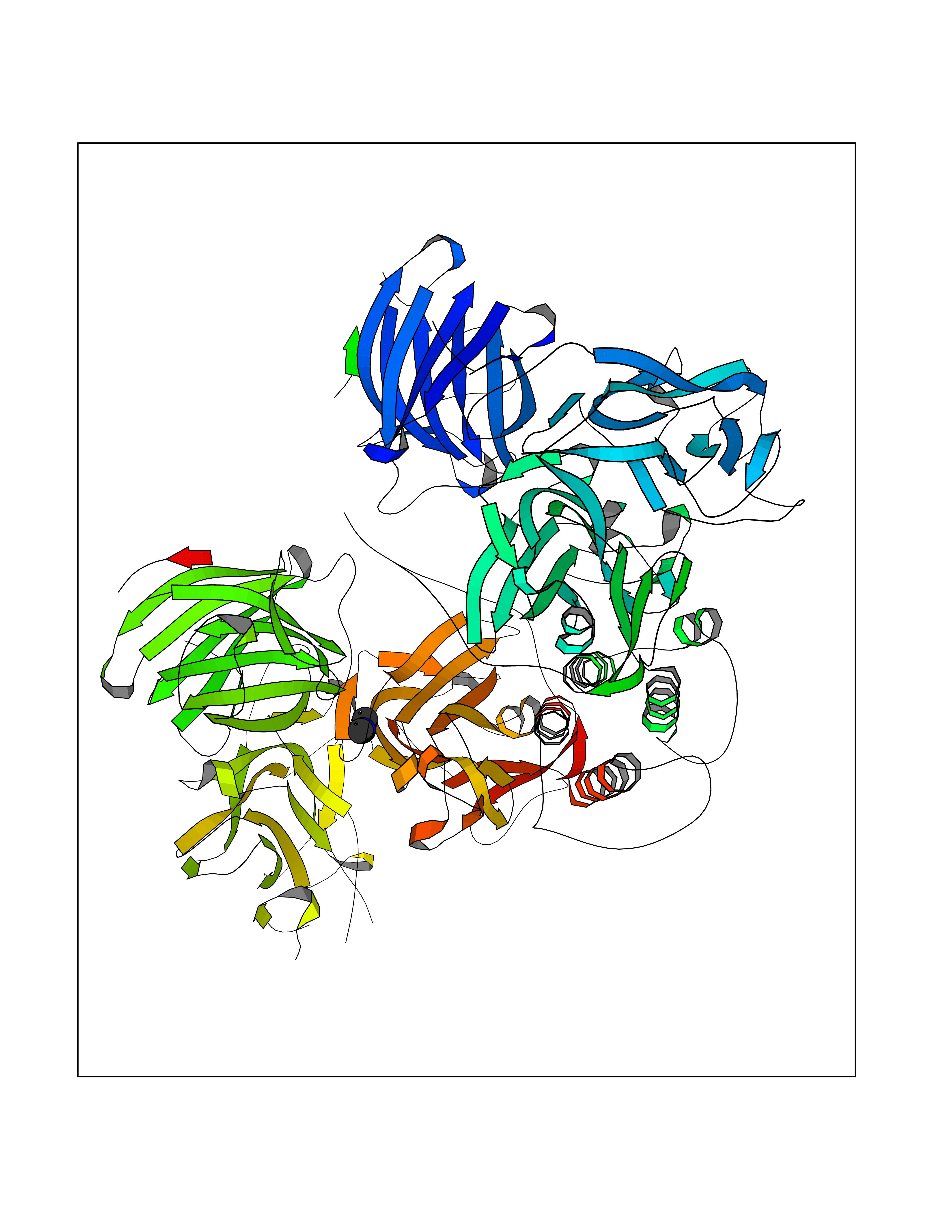 Molscript Map 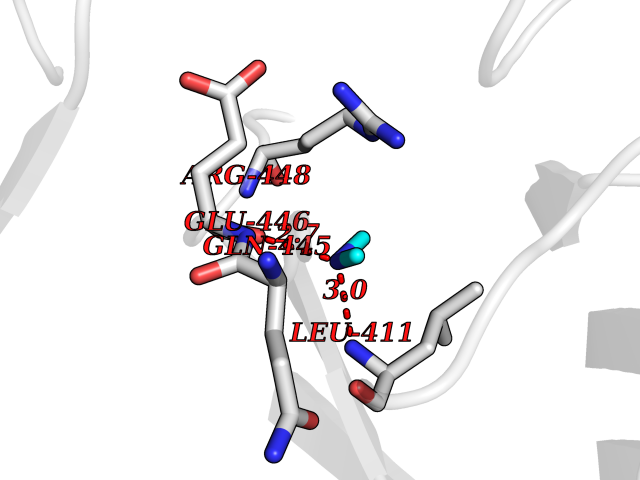 Pymol Map 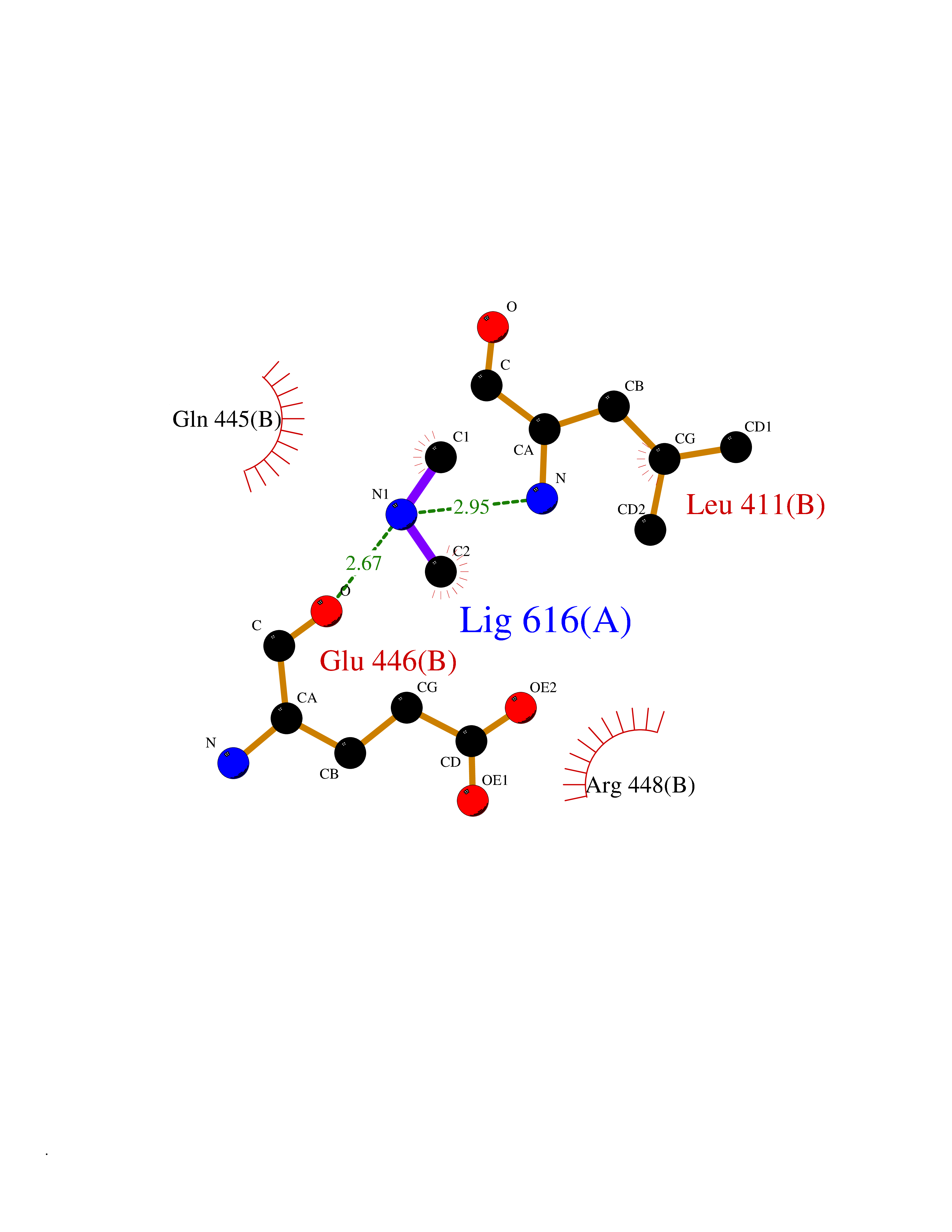 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | DNA topoisomerase II alpha (TOP2A) | 1ZXM | 4.01 | |
Target general information Gen name TOP2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DNA topoisomerase II, alpha isozyme; DNA topoisomerase 2alpha; DNA topoisomerase 2-alpha Protein family Type II topoisomerase family Biochemical class Topoisomerase Function Topoisomerase II makes double-strand breaks. Essential during mitosis and meiosis for proper segregation of daughter chromosomes. May play a role in regulating the period length of ARNTL/BMAL1 transcriptional oscillation. Control of topological states of DNA by transient breakage and subsequent rejoining of DNA strands. Related diseases A chromosomal aberration involving TOP1 is found in a form of therapy-related myelodysplastic syndrome. Translocation t(11;20)(p15;q11) with NUP98. {ECO:0000269|PubMed:10556215}. Drugs (DrugBank ID) DB05706; DB06013; DB05022; DB06263; DB00276; DB06420; DB04975; DB06362; DB00537; DB00970; DB00694; DB06421; DB00380; DB00997; DB05129; DB00467; DB00445; DB00773; DB09047; DB04576; DB01645; DB01177; DB00978; DB04967; DB01204; DB00218; DB01059; DB01165; DB00487; DB01179; DB05920; DB04978; DB01208; DB00444; DB00685; DB00385; DB06042 Interacts with O14497-1; P38398; P35222; Q05655 EC number EC 5.6.2.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Biological rhythms; Cytoplasm; Direct protein sequencing; DNA-binding; Isomerase; Isopeptide bond; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Topoisomerase; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42640.6 Length 373 Aromaticity 0.1 Instability index 33.34 Isoelectric point 8.64 Charge (pH=7) 5.05 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 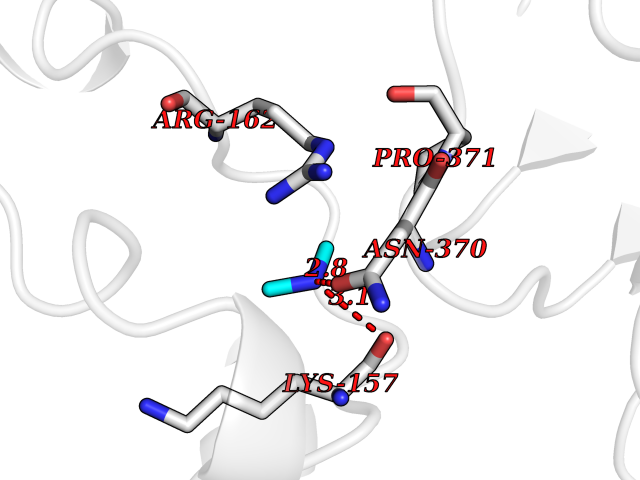 Pymol Map 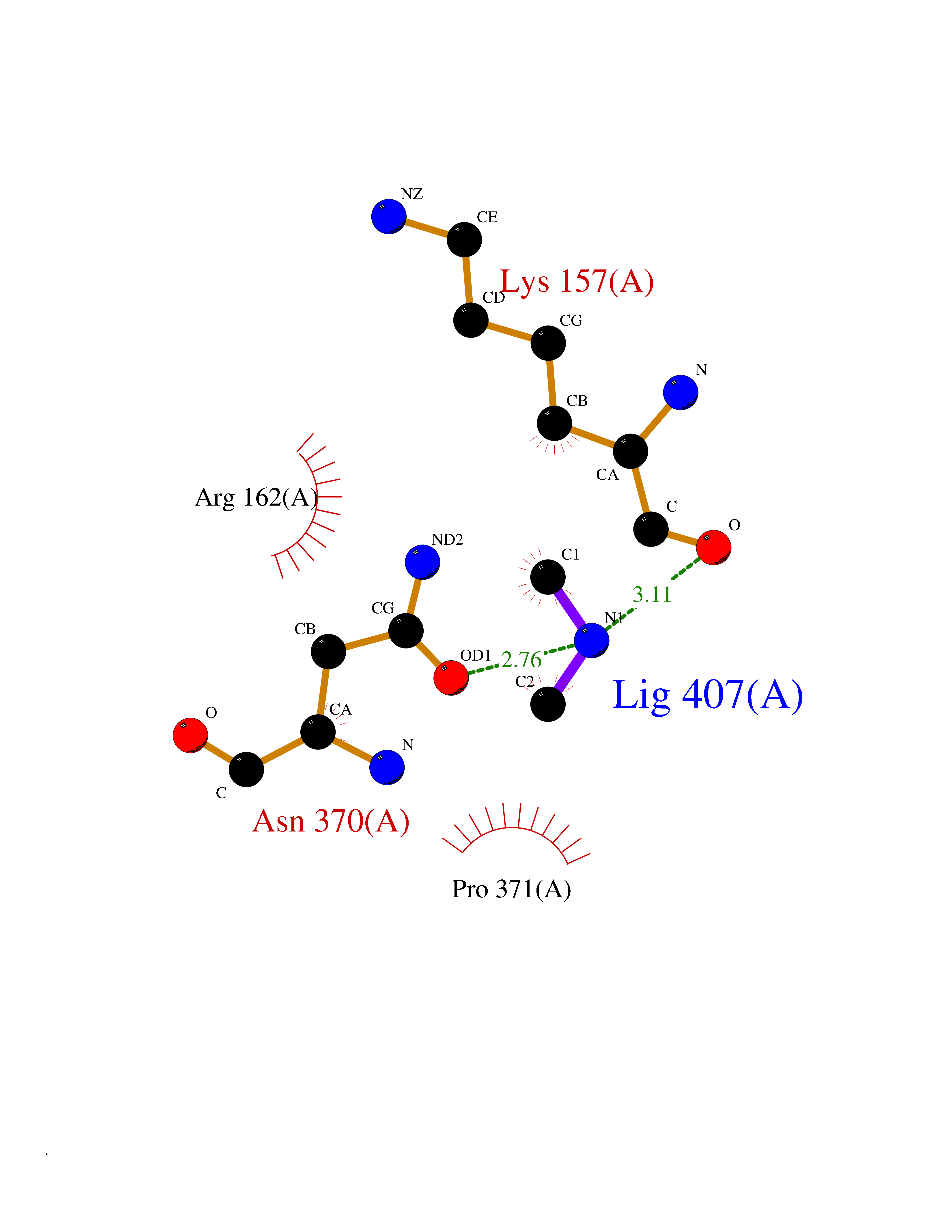 Ligplot Map 3D Binding mode Sequence SVERIYQKKTQLEHILLRPDTYIGSVELVTQQMWVYDEDVGINYREVTFVPGLYKIFDEILVNAADNKQRDPKMSCIRVTIDPENNLISIWNNGKGIPVVEHKVEKMYVPALIFGQLLTSSNYDDDEKKVTGGRNGYGAKLCNIFSTKFTVETASREYKKMFKQTWMDNMGRAGEMELKPFNGEDYTCITFQPDLSKFKMQSLDKDIVALMVRRAYDIAGSTKDVKVFLNGNKLPVKGFRSYVDMYLKDKLDETGNSLKVIHEQVNHRWEVCLTMSEKGFQQISFVNSIATSKGGRHVDYVADQIVTKLVDVVKKKNAVKAHQVKNHMWIFVNALIENPTFDSQTKENMTLQPKSFGSTCQLSEKFIKAAIGC Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Nitric-oxide synthase endothelial (NOS3) | 4D1P | 4.01 | |
Target general information Gen name NOS3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nitric oxide synthase, endothelial; NOSIII; NOS,type III; NOS type III; Endothelial nitric oxide synthase; Endothelial NOS; ENOS; EC-NOS; Constitutive NOS; CNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function NO mediates vascular endothelial growth factor (VEGF)-induced angiogenesis in coronary vessels and promotes blood clotting through the activation of platelets. Produces nitric oxide (NO) which is implicated in vascular smooth muscle relaxation through a cGMP-mediated signal transduction pathway. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB07001; DB02048; DB02911; DB02335; DB01997; DB03332; DB04534; DB07244; DB03100; DB03918; DB02207; DB03065; DB00125; DB02994; DB01833; DB00155; DB00997; DB07388; DB03974; DB02077; DB01821; DB09237; DB01110; DB03144; DB03305; DB01686; DB04559; DB02044; DB08019; DB08018; DB02027; DB02979; DB00435; DB04223; DB06154; DB03910; DB02141; DB03963; DB03707; DB02234; DB04018; DB00360; DB02589 Interacts with P60709; P63010-2; Q8N6T3-3; Q9Y575-3; Q96FT7-4; Q5SZD1; Q16543; Q9UNS2; Q8IUI8; P35222; Q05193; O15287; Q08379; Q71DI3; P69905; P61978; Q12891; Q9UKT9; Q9Y2M5; Q14525; Q6DKI2; P43364-2; Q8N6F8; O94851; A4FUJ8; Q8N594; Q8IVI9; Q6X4W1-6; O15381-5; Q9NV79; Q16549; Q5T2W1; O75925; Q96I34; Q6ZMI0-5; P57052; Q9GZR2; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q7Z699; Q7Z698; P50502; Q9BR01-2; Q9NVV9; Q86WT6-2; Q9H347; P58304; Q9NZC7-5; Q9UNY5; P14079 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cell membrane; Cytoplasm; Cytoskeleton; Direct protein sequencing; FAD; Flavoprotein; FMN; Golgi apparatus; Heme; Iron; Lipoprotein; Membrane; Metal-binding; Myristate; NADP; Oxidoreductase; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 90790.1 Length 803 Aromaticity 0.11 Instability index 50.67 Isoelectric point 6.03 Charge (pH=7) -9.56 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 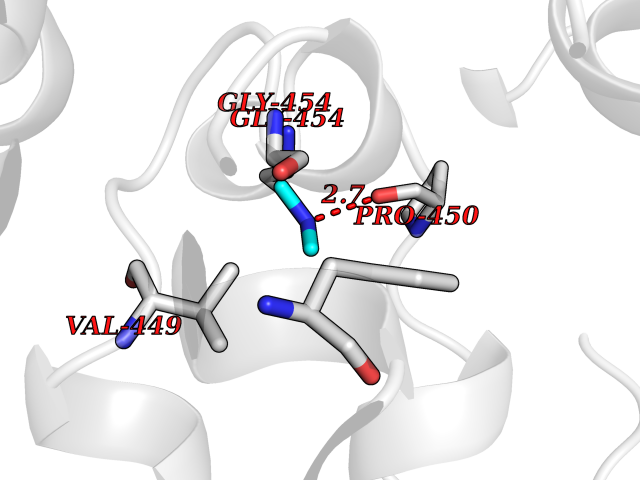 Pymol Map 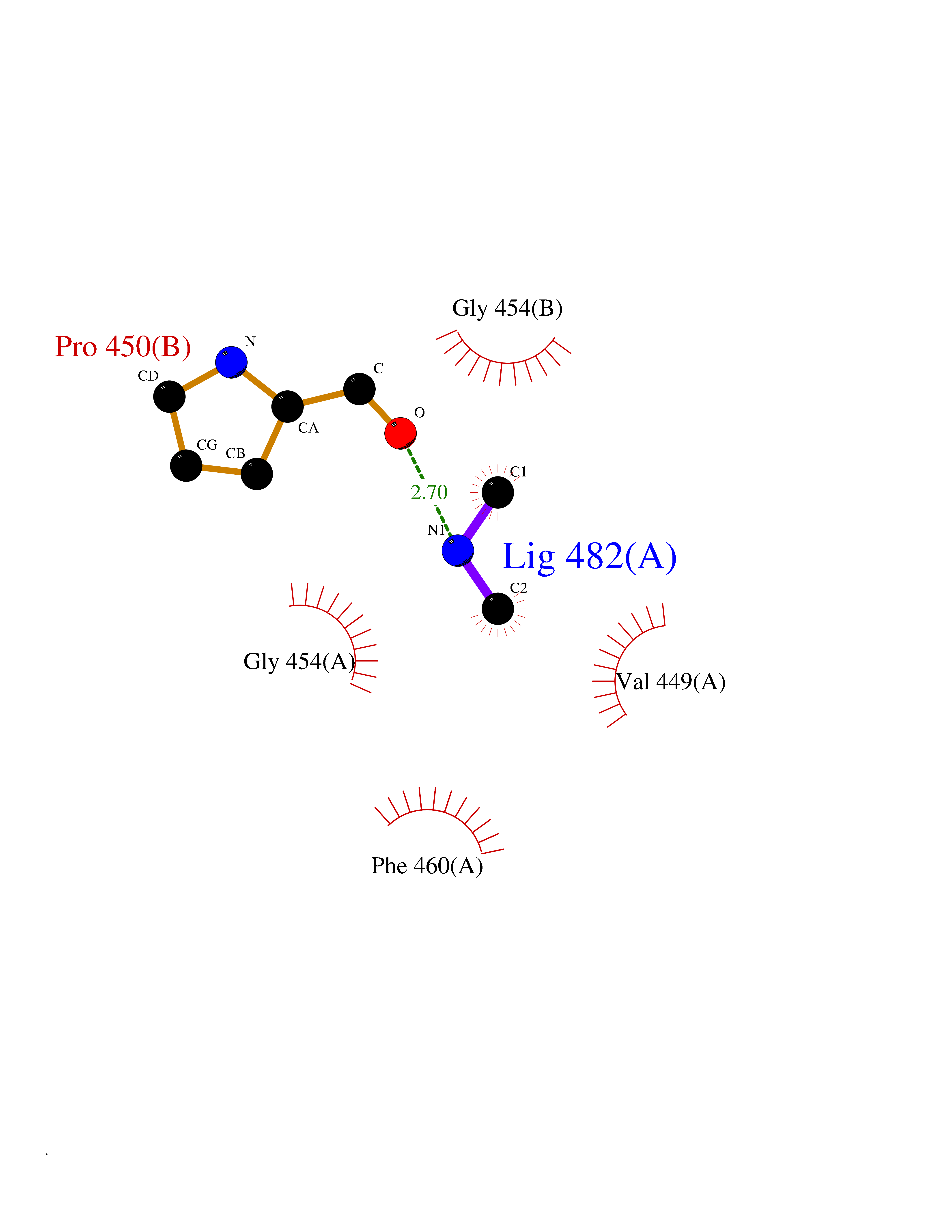 Ligplot Map 3D Binding mode Sequence FPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPWKFPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPW Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Isovaleryl-CoA dehydrogenase, mitochondrial | 1IVH | 4.01 | |
Target general information Gen name IVD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Isovaleryl-CoA dehydrogenase activity. Related diseases Isovaleric acidemia (IVA) [MIM:243500]: A metabolic disorder characterized by retarded psychomotor development, a peculiar odor resembling sweaty feet, an aversion to dietary protein, and pernicious vomiting, leading to acidosis and coma. The acute neonatal form leads to massive metabolic acidosis from the first days of life and rapid death. {ECO:0000269|PubMed:2063866, ECO:0000269|PubMed:22004070, ECO:0000269|PubMed:22350545, ECO:0000269|PubMed:23587913, ECO:0000269|PubMed:28535199, ECO:0000269|PubMed:9665741}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04036; DB03147 Interacts with Q08043; Q9Y4H4 EC number 1.3.8.1; 1.3.8.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84454.2 Length 774 Aromaticity 0.08 Instability index 30.01 Isoelectric point 6.85 Charge (pH=7) -0.77 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 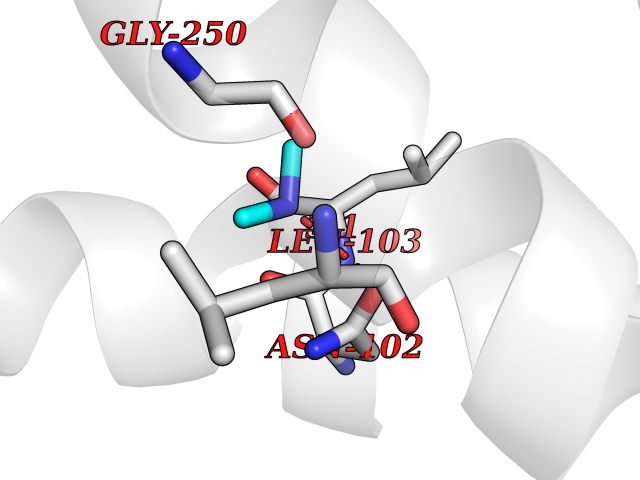 Pymol Map 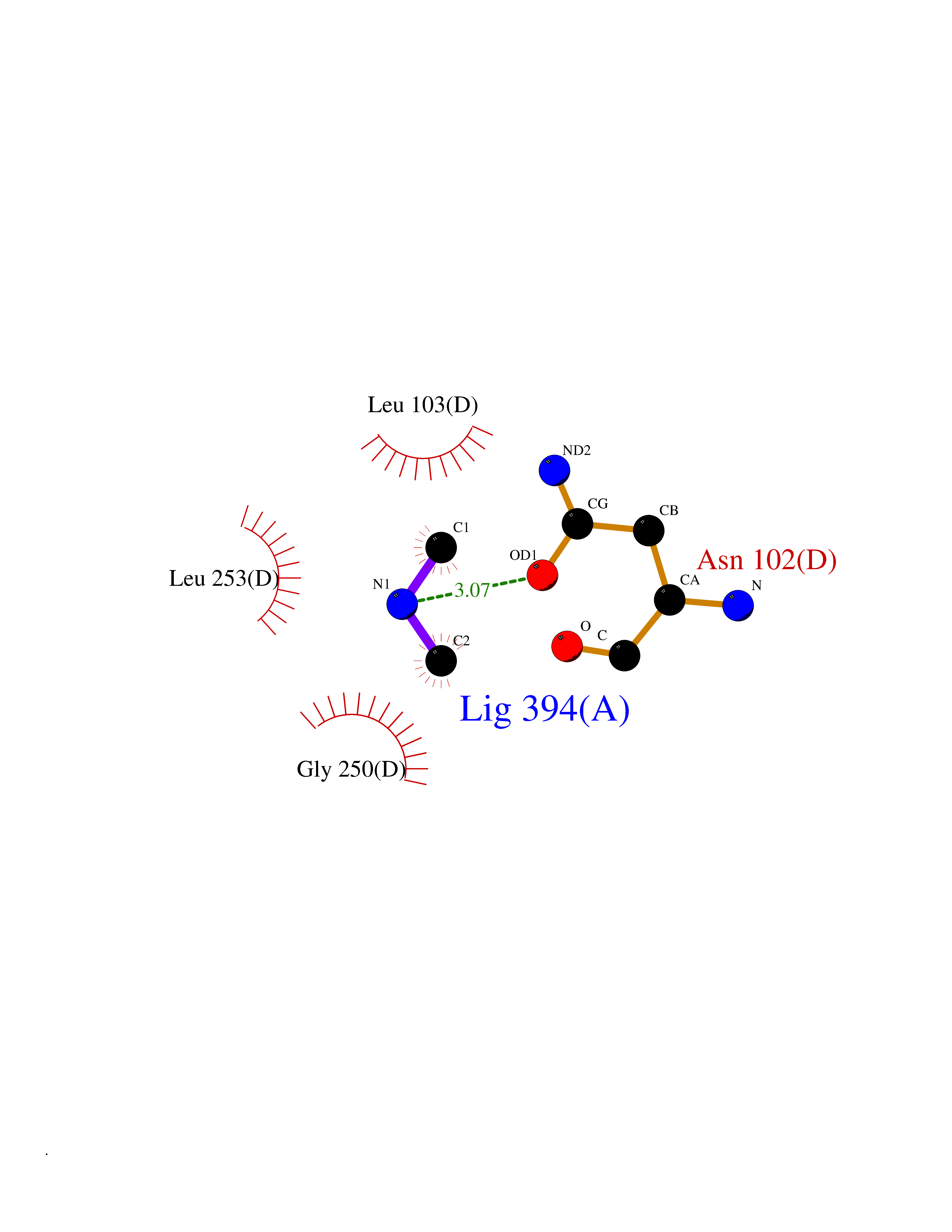 Ligplot Map 3D Binding mode Sequence VDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNADVDDAINGLSEEQRQLRQTMAKFLQEHLAPKAQEIDRSNEFKNLREFWKQLGNLGVLGITAPVQYGGSGLGYLEHVLVMEEISRASGAVGLSYGAHSNLCINQLVRNGNEAQKEKYLPKLISGEYIGALAMSEPNAGSDVVSMKLKAEKKGNHYILNGNKFWITNGPDADVLIVYAKTDLAAVPASRGITAFIVEKGMPGFSTSKKLDKLGMRGSNTCELIFEDCKIPAANILGHENKGVYVLMSGLDLERLVLAGGPLGLMQAVLDHTIPYLHVREAFGQKIGHFQLMQGKMADMYTRLMACRQYVYNVAKACDEGHCTAKDCAGVILYSAECATQVALDGIQCFGGNGYINDFPMGRFLRDAKLYEIGAGTSEVRRLVIGRAFNAD Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.01 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -5.47  Molscript Map 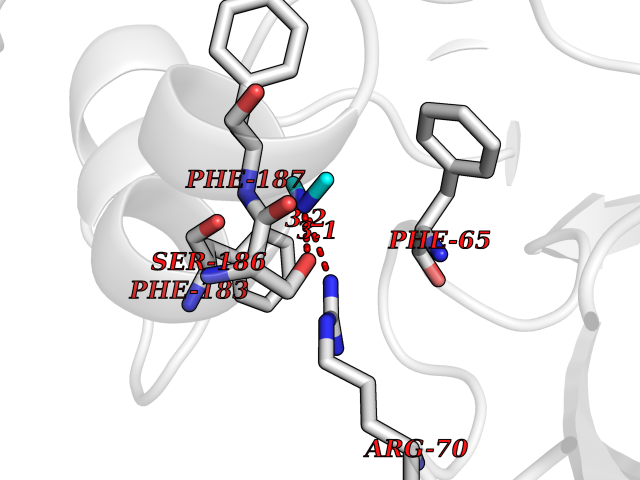 Pymol Map 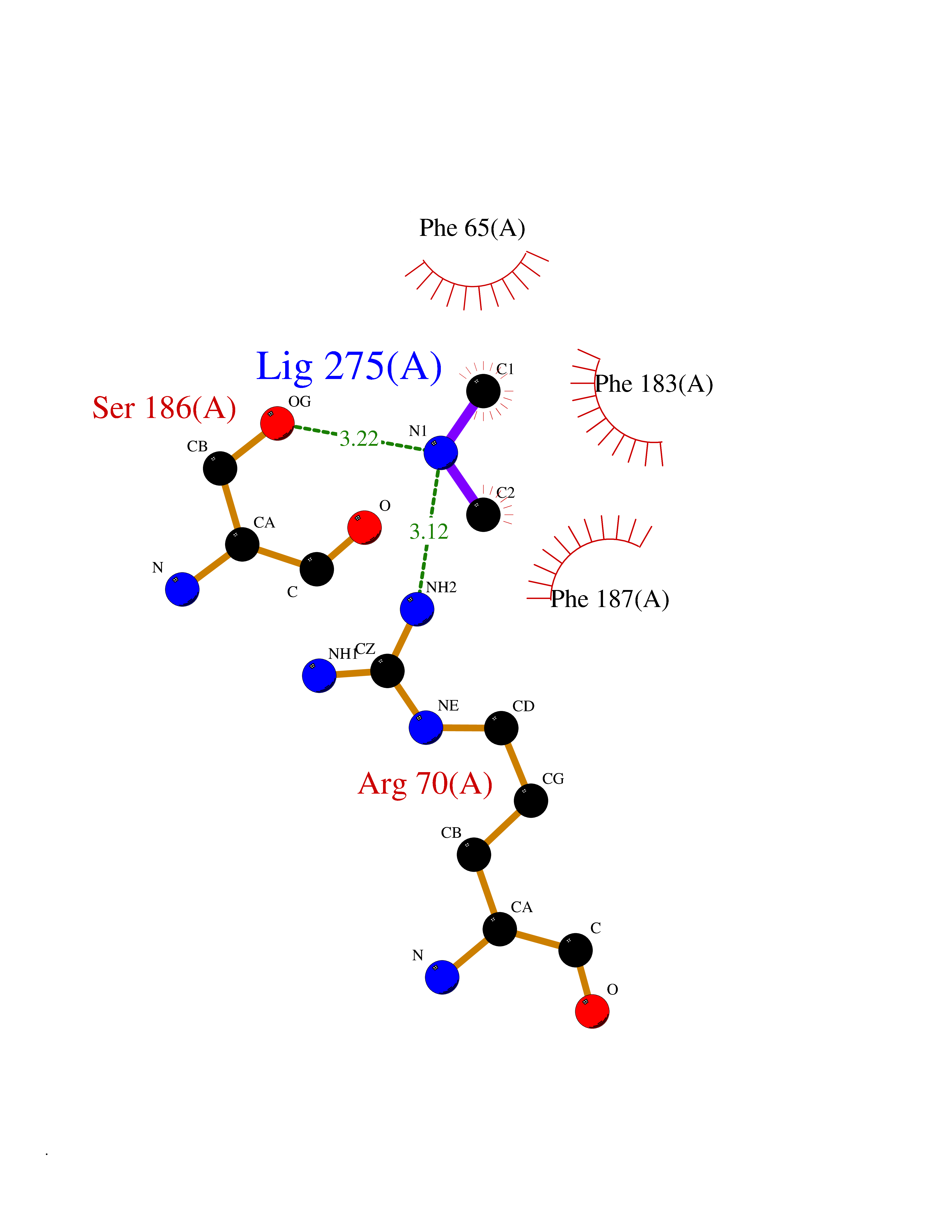 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Kynureninase (KYNU) | 3E9K | 4.01 | |
Target general information Gen name KYNU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms L-kynurenine hydrolase; KYNU Protein family Kynureninase family Biochemical class Carbon-carbon bonds hydrolase Function Catalyzes the cleavage of L-kynurenine (L-Kyn) and L-3- hydroxykynurenine (L-3OHKyn) into anthranilic acid (AA) and 3- hydroxyanthranilic acid (3-OHAA), respectively. Has a preference for the L-3-hydroxy form. Also has cysteine-conjugate-beta-lyase activity. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160; DB07069; DB00114 Interacts with Q8WUE5; P56545-3; Q9NVL1-2; P61968; P59942; Q8TDC0; P78356-2; Q86WH2 EC number EC 3.7.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Proteomics identification; Pyridine nucleotide biosynthesis; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 50204.5 Length 446 Aromaticity 0.09 Instability index 39.91 Isoelectric point 6.36 Charge (pH=7) -4.59 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 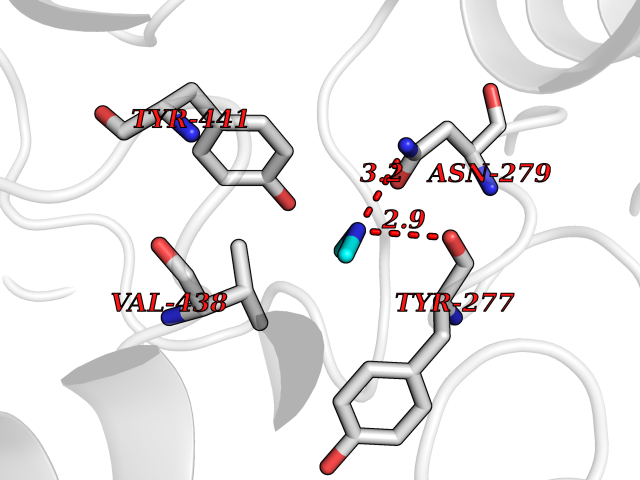 Pymol Map 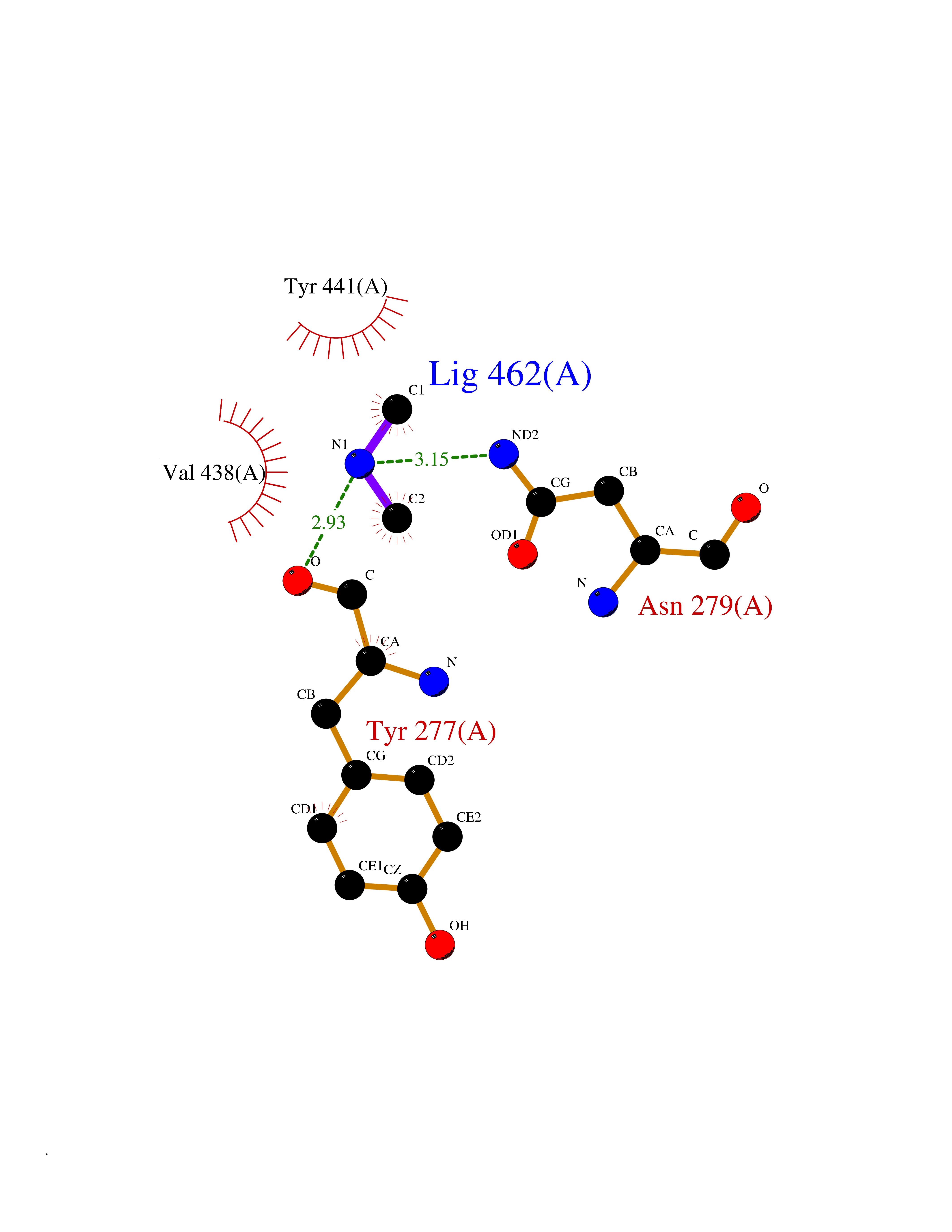 Ligplot Map 3D Binding mode Sequence LELPADTVQRIAAELKCHPTDERVALHLDEEDKLRHFREXFYIPKIQDLPPVDLSLVNKDENAIYFLGNSLGLQPKMVKTYLEEELDKWAKIAAYGHEVGKRPWITGDESIVGLMKDIVGANEKEIALMNALTVNLHLLMLSFFKPTPKRYKILLEAKAFPSDHYAIESQLQLHGLNIEESMRMIKPREGEETLRIEDILEVIEKEGDSIAVILFSGVHFYTGQHFNIPAITKAGQAKGCYVGFDLAHAVGNVELYLHDWGVDFACWCSYKYLNAGAGGIAGAFIHEKHAHTIKPALVGWFGHELSTRFKMDNKLQLIPGVCGFRISNPPILLVCSLHASLEIFKQATMKALRKKSVLLTGYLEYLIKHNYGVVNIITPSHVEERGCQLTITFSVPNKDVFQELEKRGVVCDKRNPNGIRVAPVPLYNSFHDVYKFTNLLTSILDS Hydrogen bonds contact Hydrophobic contact | ||||