Job Results:
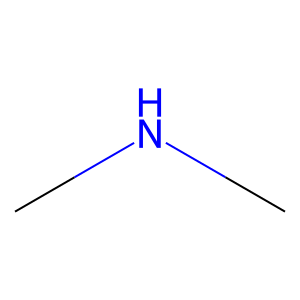
Ligand
Structure
Job ID
8da1e79bf3c5af0a1c99e2e2a8101bb5
Job name
NA
Time
2025-02-13 15:08:06
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Cerebroside-sulfatase (ARSA) | 1E2S | 4.03 | |
Target general information Gen name ARSA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cerebrosidesulfatase; Arylsulfatase A component C; ASA; ARSA Protein family Sulfatase family Biochemical class Sulfuric ester hydrolase Function Hydrolyzes cerebroside sulfate. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB03821; DB01800; DB01141; DB04786 Interacts with P50995; Q6P5X5; Q13554-3; O60826; Q96D98; Q9H0I2; Q12951-2; Q16512; P28069; O75360; Q9BQY4; Q15645; O95231 EC number EC 3.1.6.8 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Ichthyosis; Leukodystrophy; Lipid metabolism; Lysosome; Metachromatic leukodystrophy; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID P Molecular weight (Da) 51173.7 Length 481 Aromaticity 0.09 Instability index 47.42 Isoelectric point 5.59 Charge (pH=7) -12.84 2D Binding mode Binding energy (Kcal/mol) -5.5 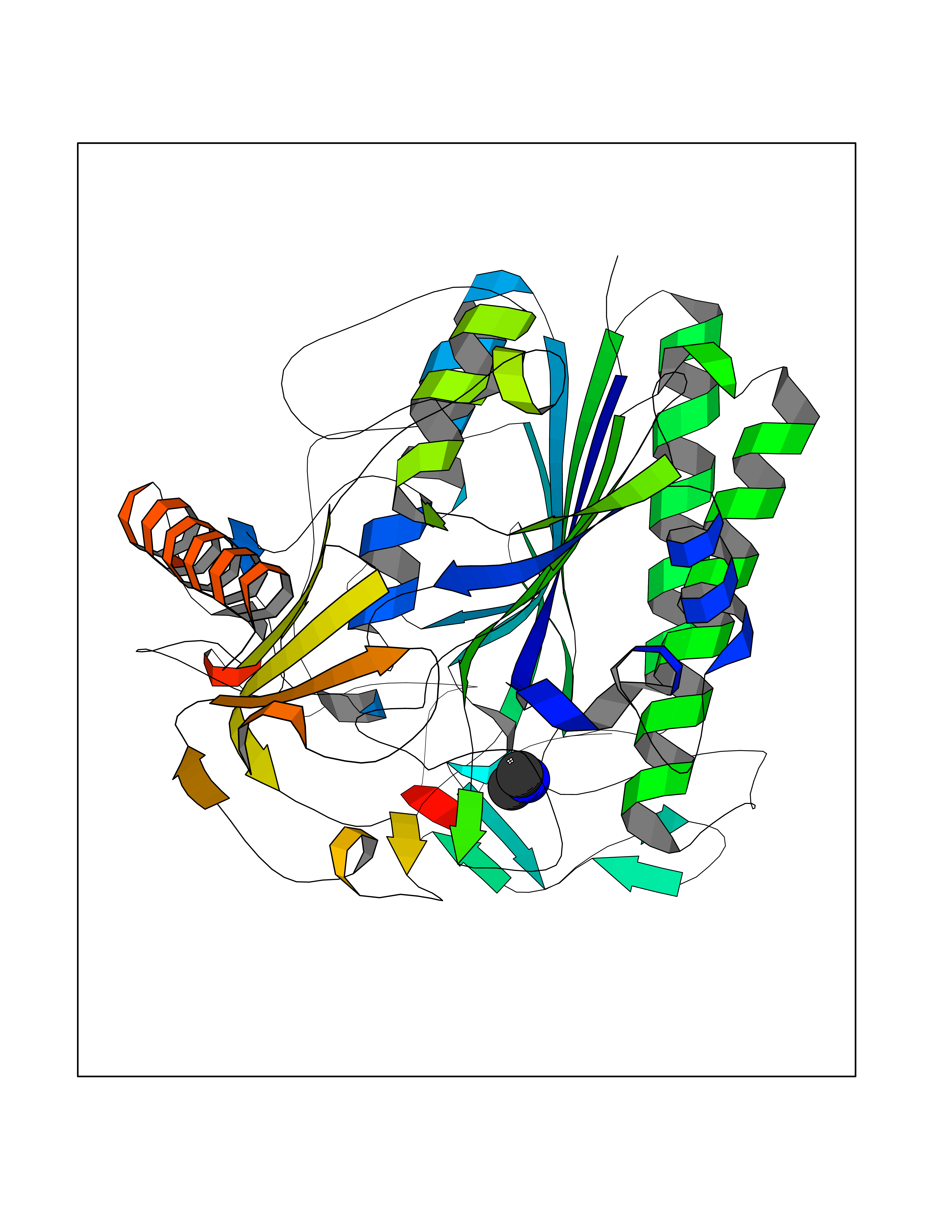 Molscript Map 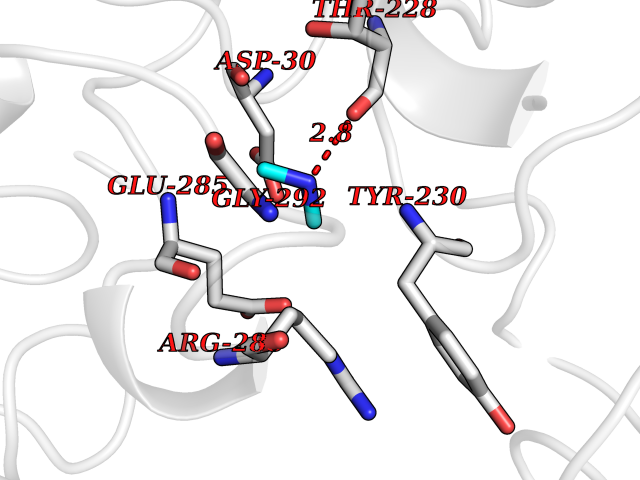 Pymol Map 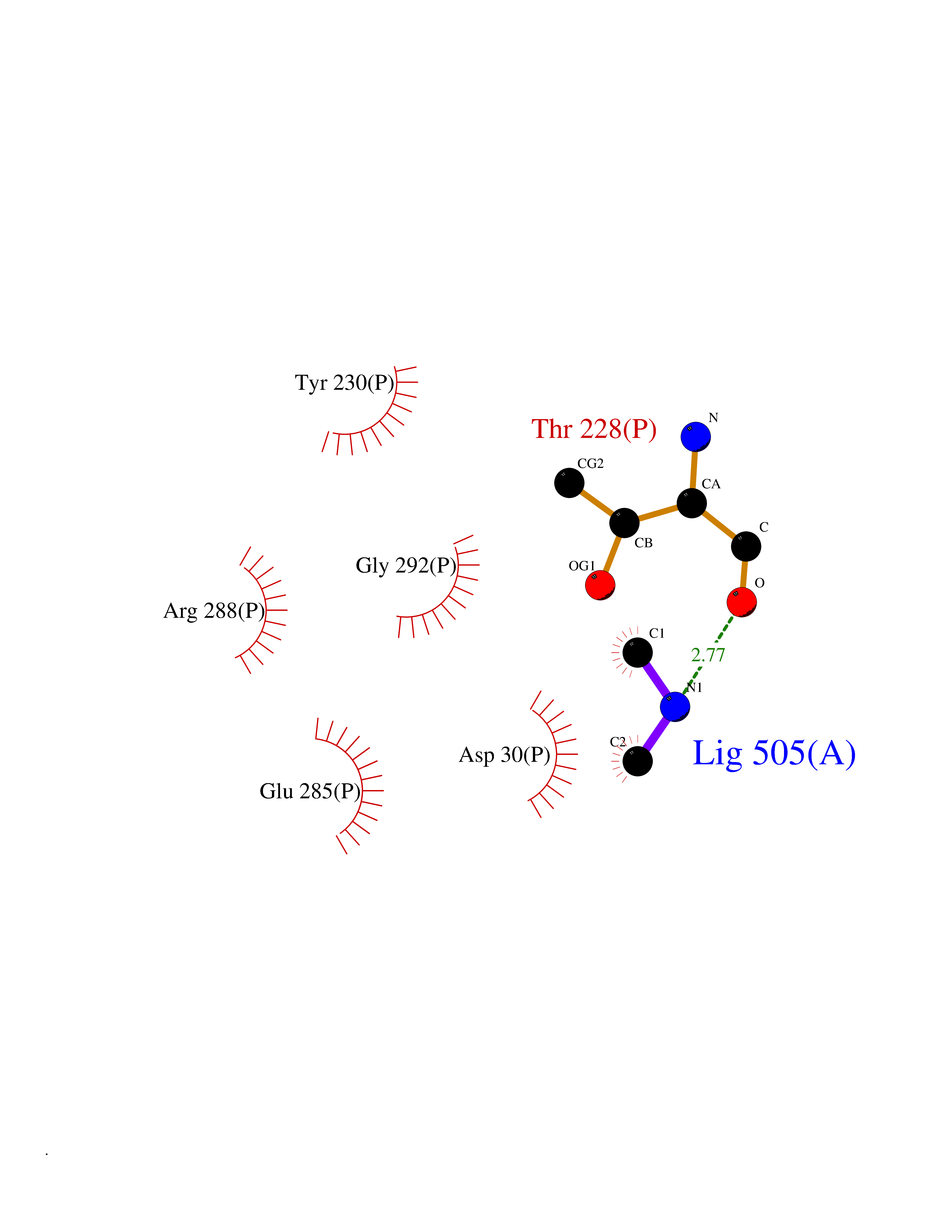 Ligplot Map 3D Binding mode Sequence RPPNIVLIFADDLGYGDLGCYGHPSSTTPNLDQLAAGGLRFTDFYVPVSLATPSRAALLTGRLPVRMGMYPGVLVPSSRGGLPLEEVTVAEVLAARGYLTGMAGKWHLGVGPEGAFLPPHQGFHRFLGIPYSHDQGPCQNLTCFPPATPCDGGCDQGLVPIPLLANLSVEAQPPWLPGLEARYMAFAHDLMADAQRQDRPFFLYYASHHTHYPQFSGQSFAERSGRGPFGDSLMELDAAVGTLMTAIGDLGLLEETLVIFTADNGPETMRMSRGGCSGLLRCGKGTTYEGGVREPALAFWPGHIAPGVTHELASSLDLLPTLAALAGAPLPNVTLDGFDLSPLLLGTGKSPRQSLFFYPSYPDEVRGVFAVRTGKYKAHFFTQGSAHSDTTADPACHASSSLTAHEPPLLYDLSKDPGENYNLLGATPEVLQALKQLQLLKAQLDAAVTFGPSQVARGEDPALQICCHPGCTPRPACCHCP Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Enoyl-[acyl-carrier-protein] reductase [NADH] FabI | 2PD4 | 4.02 | |
Target general information Gen name fabI Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID NA Synonyms HP_0195 Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class Oxidoreductase Function Enoyl-[acyl-carrier-protein] reductase (NADH) activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB04393; DB08604 Interacts with NA EC number 1.3.1.9 Uniprot keywords 3D-structure; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27671.5 Length 256 Aromaticity 0.09 Instability index 24 Isoelectric point 8.49 Charge (pH=7) 2.38 2D Binding mode Binding energy (Kcal/mol) -5.48 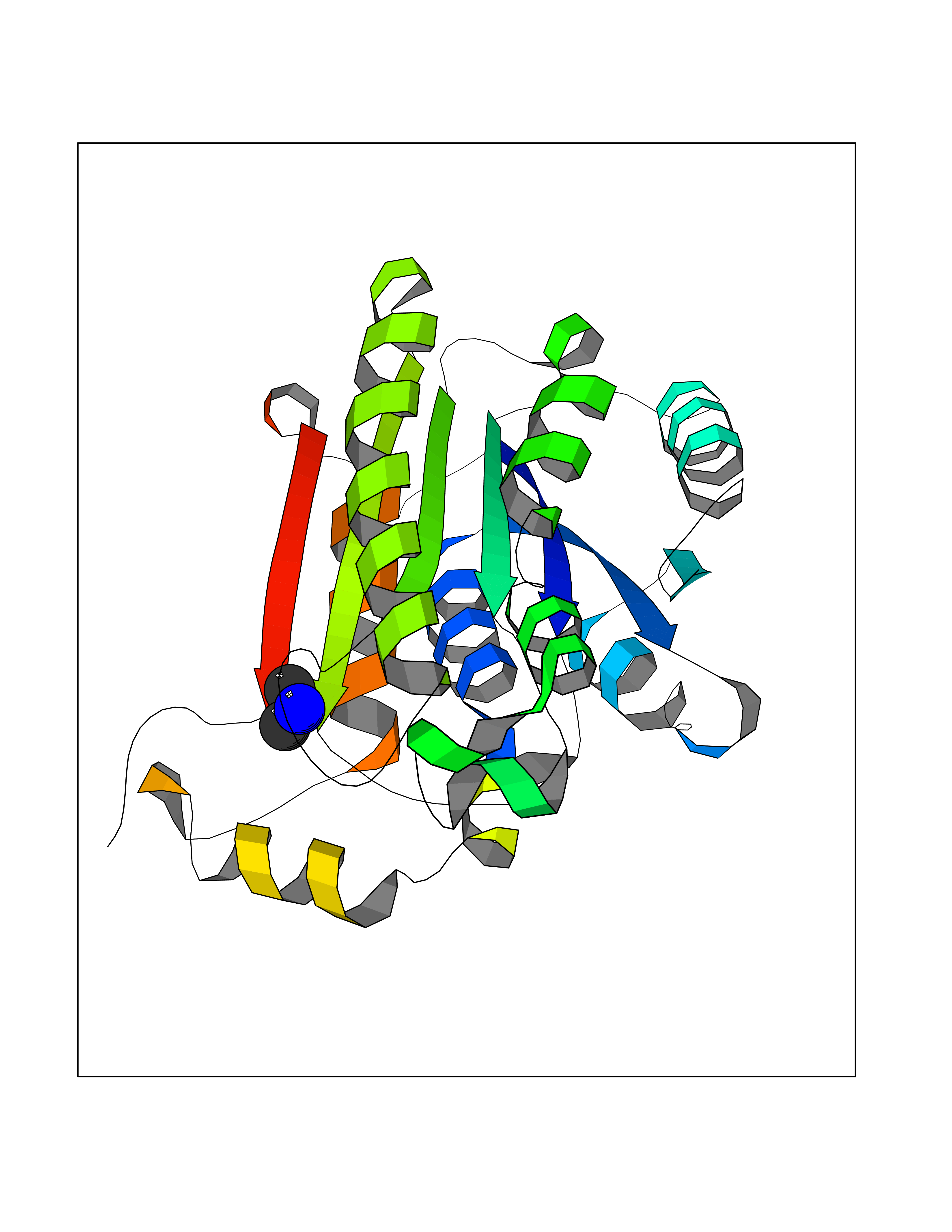 Molscript Map 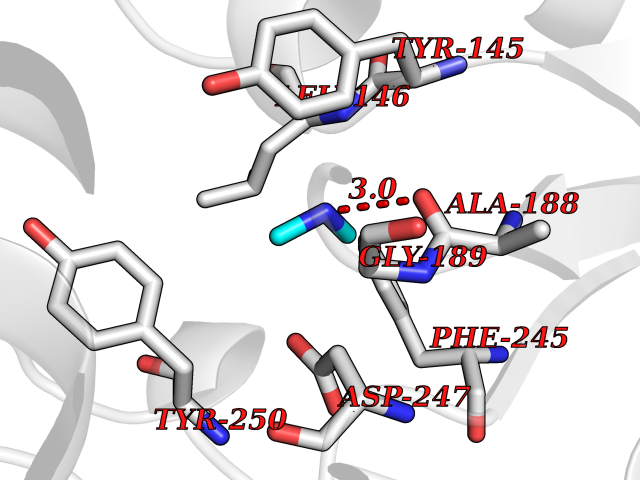 Pymol Map 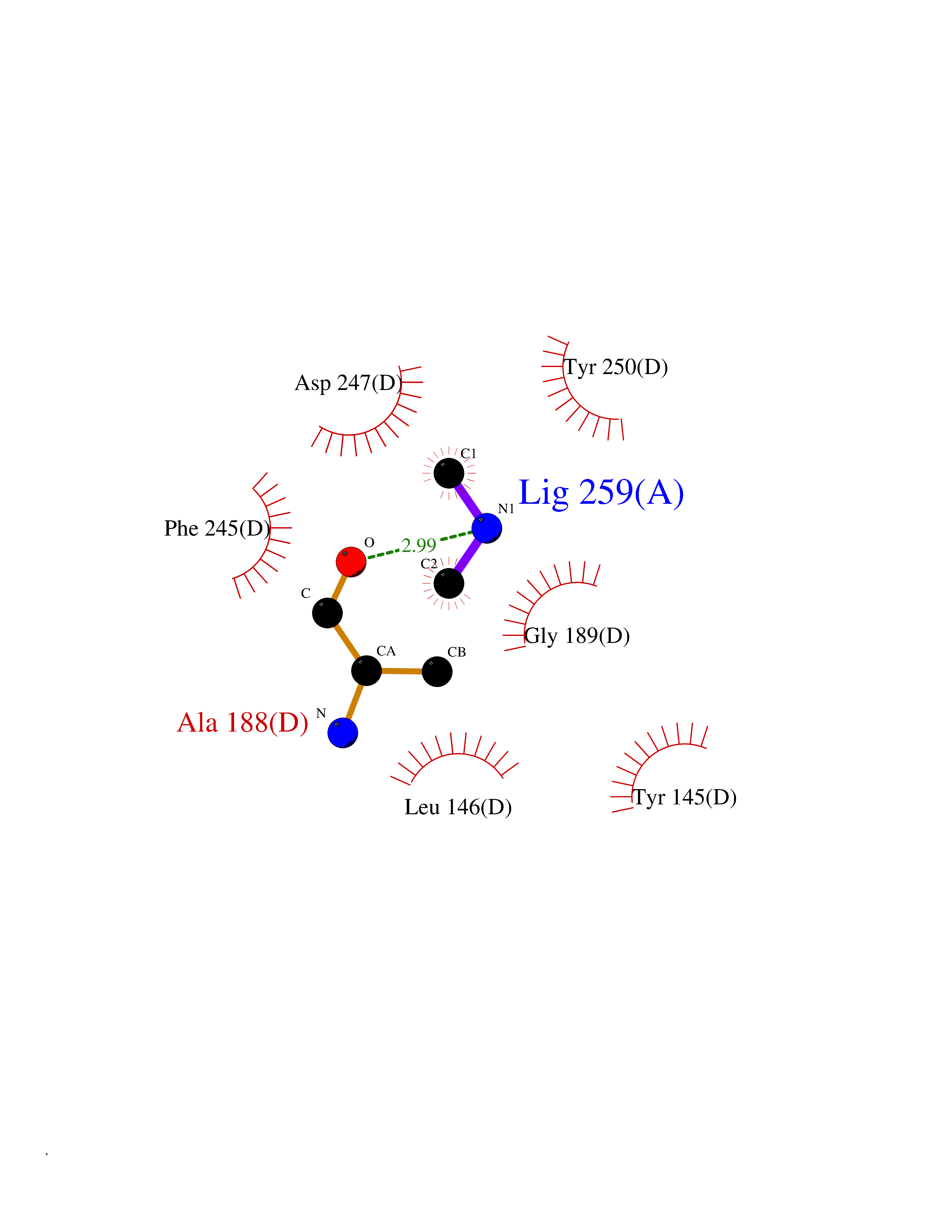 Ligplot Map 3D Binding mode Sequence GFLKGKKGLIVGVANNKSIAYGIAQSCFNQGATLAFTYLNESLEKRVRPIAQELNSPYVYELDVSKEEHFKSLYNSVKKDLGSLDFIVHSVAFAPKEALEGSLLETSKSAFNTAMEISVYSLIELTNTLKPLLNNGASVLTLSYLGSTKYMAHYNVMGLAKAALESAVRYLAVDLGKHHIRVNALSAGPIRTLASSGIADFRMILKWNEINAPLRKNVSLEEVGNAGMYLLSSLSSGVSGEVHFVDAGYHVMGMGA Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Urokinase-type plasminogen activator (PLAU) | 4JNI | 4.02 | |
Target general information Gen name PLAU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UPA; U-plasminogen activator Protein family Peptidase S1 family Biochemical class Peptidase Function Specifically cleaves the zymogen plasminogen to form the active enzyme plasmin. Related diseases Quebec platelet disorder (QPD) [MIM:601709]: An autosomal dominant bleeding disorder due to a gain-of-function defect in fibrinolysis. Although affected individuals do not exhibit systemic fibrinolysis, they show delayed onset bleeding after challenge, such as surgery. The hallmark of the disorder is markedly increased PLAU levels within platelets, which causes intraplatelet plasmin generation and secondary degradation of alpha-granule proteins. {ECO:0000269|PubMed:20007542}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07129; DB07122; DB01905; DB02287; DB03729; DB01725; DB08072; DB07625; DB07626; DB08697; DB03136; DB01977; DB07076; DB03082; DB02705; DB02473; DB02398; DB02551; DB03865; DB06855; DB06856; DB03046; DB04059; DB04172; DB00594; DB03127; DB02526; DB03159; DB05254; DB03782; DB06857; DB16701; DB03876; DB03476 Interacts with Q9UKQ2; P05067; Q03405-1; P05121; P55000 EC number EC 3.4.21.73 Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Direct protein sequencing; Disulfide bond; EGF-like domain; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Phosphoprotein; Plasminogen activation; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID U Molecular weight (Da) 25825.3 Length 229 Aromaticity 0.1 Instability index 47.36 Isoelectric point 8.65 Charge (pH=7) 5.38 2D Binding mode Binding energy (Kcal/mol) -5.48 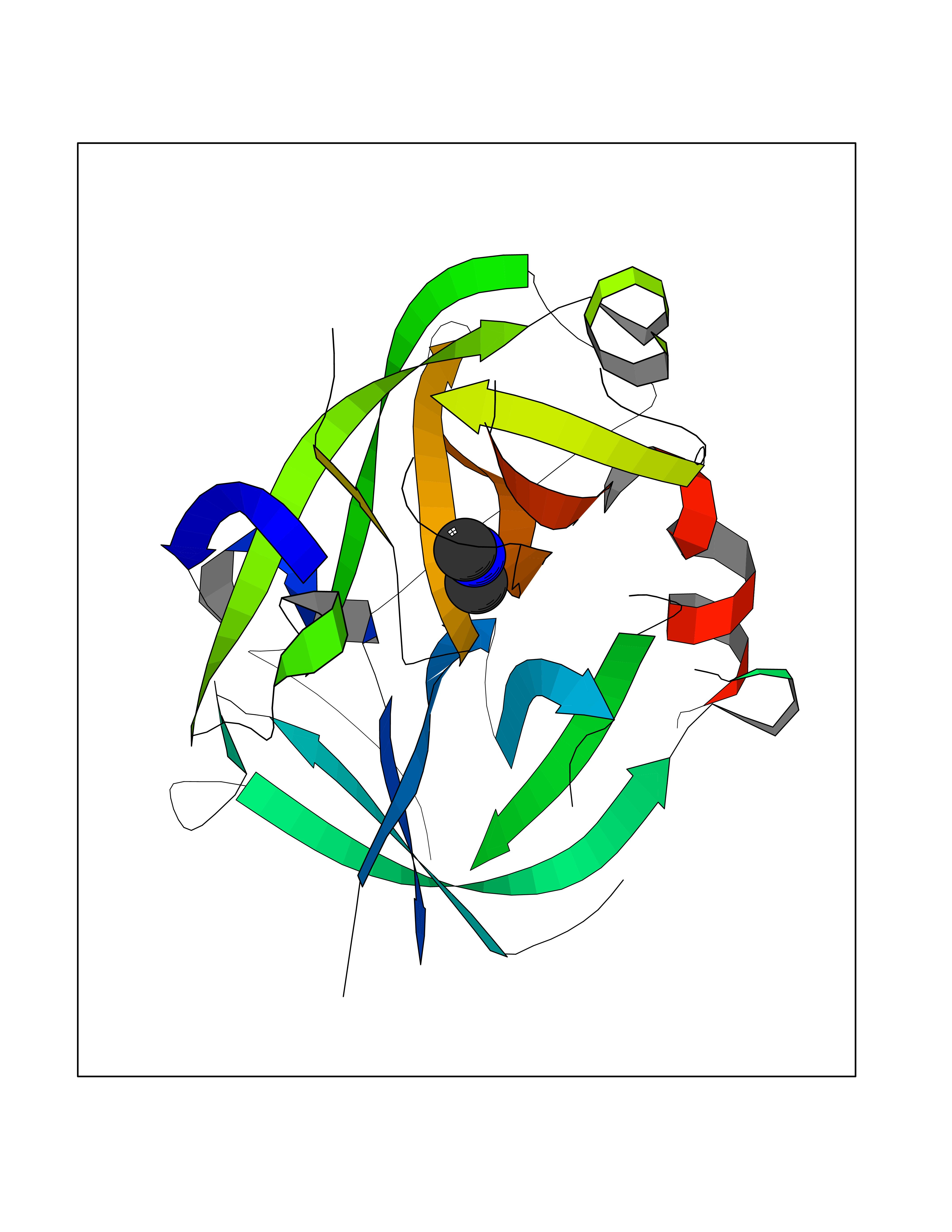 Molscript Map 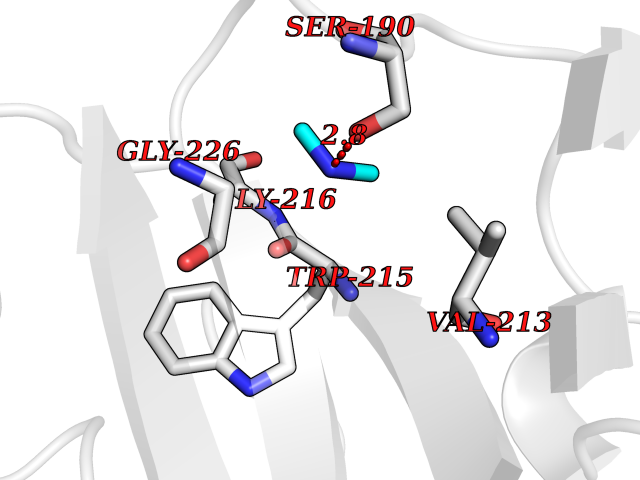 Pymol Map 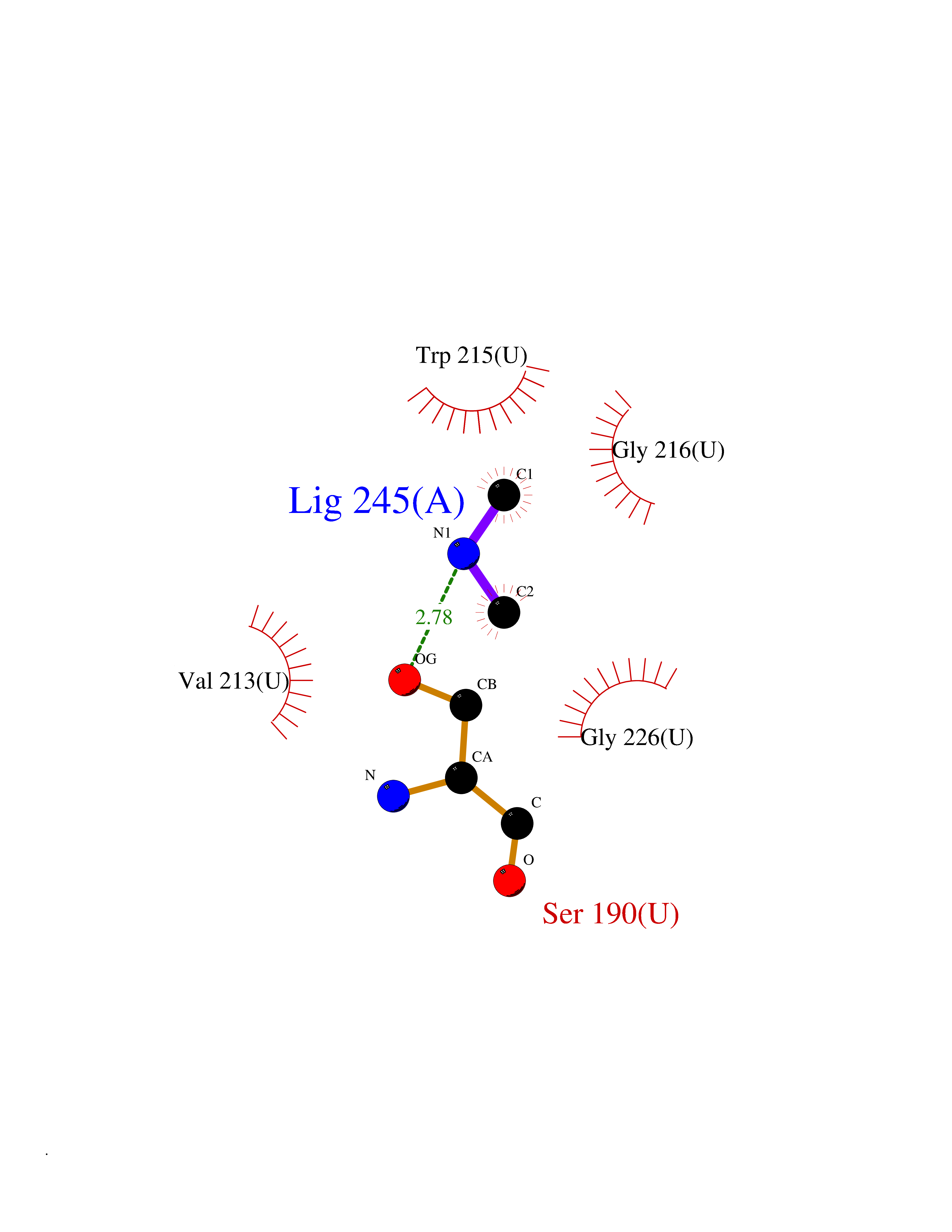 Ligplot Map 3D Binding mode Sequence IIGGEFTTIENQPWFAAIYRRSVTYVCGGSLISPCWVISATHCFPKKEDYIVYLGRSRLNSNTQGEMKFEVENLILHKDYSALAHHNDIALLKIRRCAQPSRTIQTIALPSMYNDPQFGTSCEITGFGKEQSTDYLYPEQLKMTVVKLISHRECQQHYYGSEVTTKMLCAAQWKTDSCQGDSGGPLVCSLQGRMTLTGIVSWGRGCALDKPGVYTRVSHFLPWIRSHTK Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Mitochondrial aldehyde dehydrogenase (ALDH2) | 1O04 | 4.02 | |
Target general information Gen name ALDH2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde dehydrogenase, mitochondrial; ALDM; ALDHI; ALDH-E2; ALDH class 2 Protein family Aldehyde dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Second enzyme of the major oxidative pathway of alcohol metabolism. Catalyzes the chemical transformation from acetaldehyde to acetic acid. Additionally, functions as a protector against oxidative stress. Related diseases AMED syndrome, digenic (AMEDS) [MIM:619151]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. AMEDS is an autosomal recessive, digenic form characterized by childhood onset of bone marrow failure resulting in aplastic anemia, in association with global developmental delay, intellectual disability, and poor overall growth with short stature. {ECO:0000269|PubMed:33355142}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. AMEDS patients carry ADH5 biallelic variants and homozygous or heterozygous ALDH2 variant p.Glu504Lys, affecting protein activity. Cellular and animal studies demonstrate that the simultaneous loss of ALDH2 and ADH5 activities leads to an increase of cellular formaldehyde sensitivity and multisystem abnormalities including hematopoietic failure. {ECO:0000269|PubMed:33355142}. Drugs (DrugBank ID) DB01612; DB06770; DB04381; DB02115; DB00822; DB00536; DB00157; DB00435; DB00727; DB09117; DB06154; DB06207 Interacts with NA EC number EC 1.2.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; Dwarfism; Intellectual disability; Mitochondrion; NAD; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 50223.5 Length 462 Aromaticity 0.1 Instability index 33.68 Isoelectric point 5.29 Charge (pH=7) -7.87 2D Binding mode Binding energy (Kcal/mol) -5.48 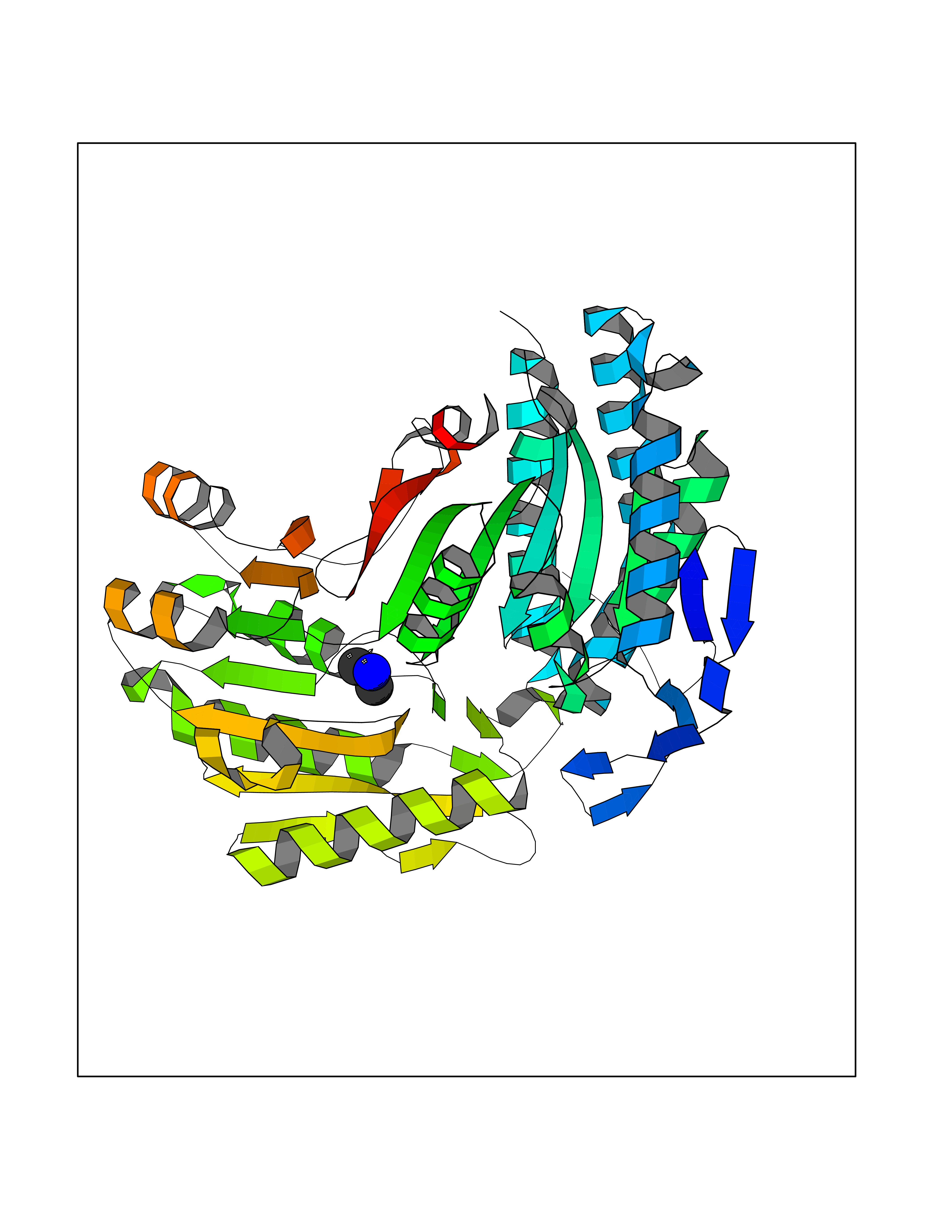 Molscript Map 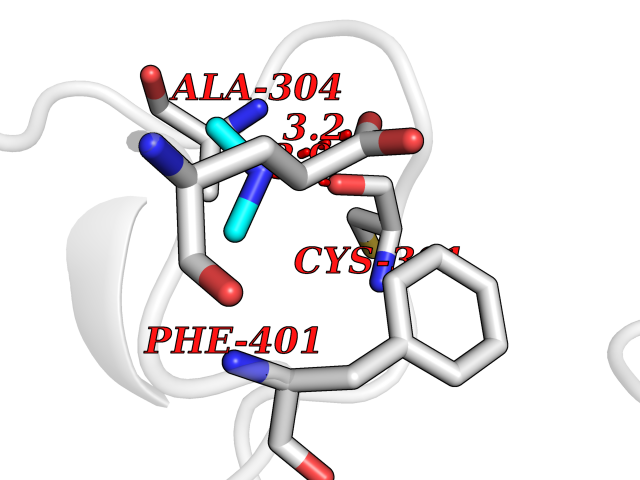 Pymol Map 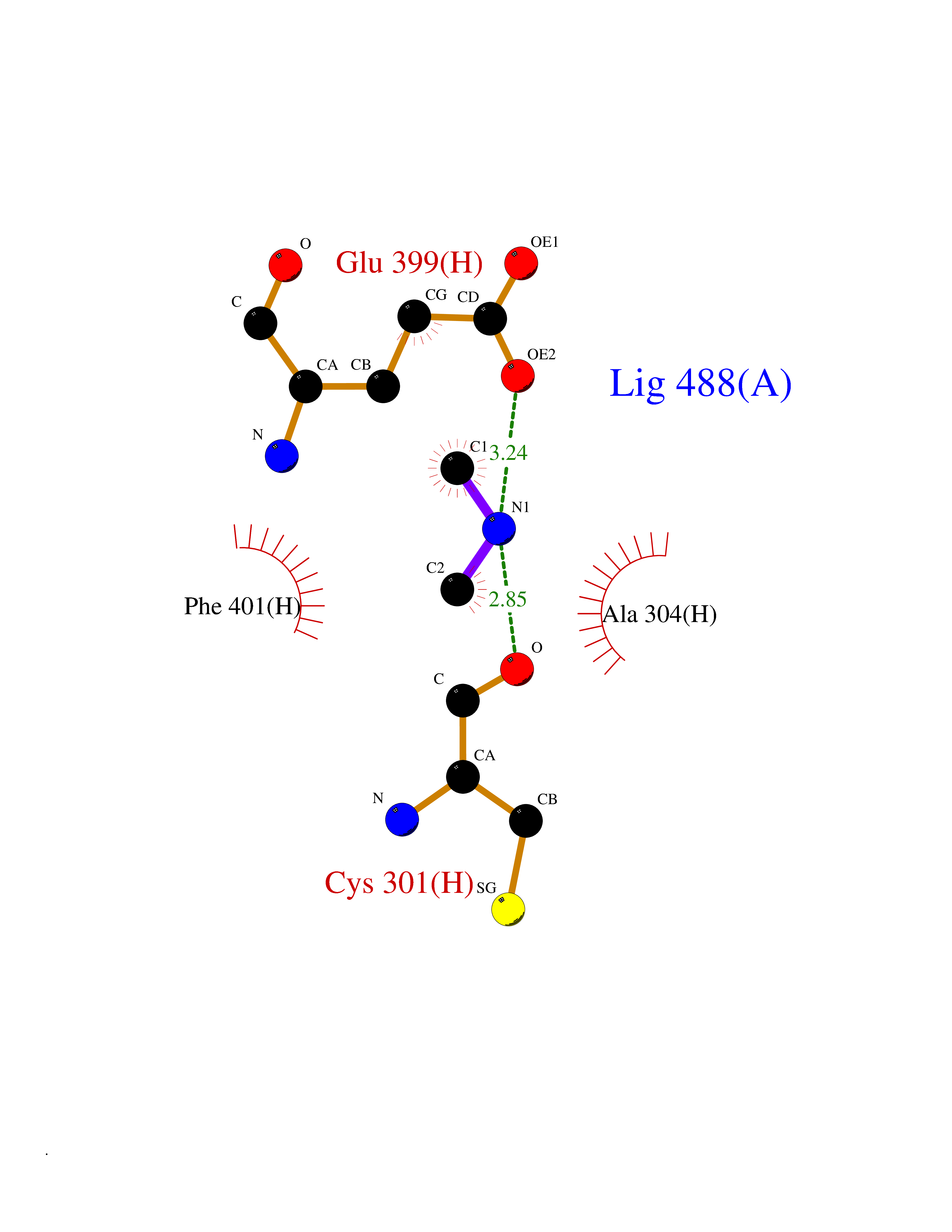 Ligplot Map 3D Binding mode Sequence AVPAPNQQPEVFCNQIFINNEWHDAVSRKTFPTVNPSTGEVICQVAEGDKEDVDKAVKAARAAFQLGSPWRRMDASHRGRLLNRLADLIERDRTYLAALETLDNGKPYVISYLVDLDMVLKCLRYYAGWADKEPVGVCGQIIPWNFPLLMQAWKLGPALATGNVVVMKVAEQTPLTALYVANLIKEAGFPPGVVNIVPGFGPTAGAAIASHEDVDKVAFTGSTEIGRVIQVAAGSSNLKRVTLELGGKSPNIIMSDADMDWAVEQAHFALFFNQGQCSCAGSRTFVQEDIYDEFVERSVARAKSRVVGNPFDSKTEQGPQVDETQFKKILGYINTGKQEGAKLLCGGGIAADRGYFIQPTVFGDVQDGMTIAKEEIFGPVMQILKFKTIEEVVGRANNSTYGLAAAVFTKDLDKANYLSQALQAGTVWVNCYDVFGAQSPFGGYKMSGSGRELGEYGLQAYT Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Tartrate-resistant acid phosphatase type 5 | 2BQ8 | 4.02 | |
Target general information Gen name ACP5 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Metallophosphoesterase superfamily, Purple acid phosphatase family Biochemical class Hydrolase Function Acid phosphatase activity.Ferric iron binding.Ferrous iron binding. Related diseases Spondyloenchondrodysplasia with immune dysregulation (SPENCDI) [MIM:607944]: A disease characterized by vertebral and metaphyseal dysplasia, spasticity with cerebral calcifications, and strong predisposition to autoimmune diseases. The skeletal dysplasia is characterized by radiolucent and irregular spondylar and metaphyseal lesions that represent islands of chondroid tissue within bone. {ECO:0000269|PubMed:21217752, ECO:0000269|PubMed:21217755}. The disease is caused by variants affecting the gene represented in this entry. ACP5 inactivating mutations result in a functional excess of phosphorylated osteopontin causing deregulation of osteopontin signaling and consequential autoimmune disease. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.3.2 Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Iron; Lysosome; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID X Molecular weight (Da) 34330.6 Length 304 Aromaticity 0.12 Instability index 42.3 Isoelectric point 9.11 Charge (pH=7) 6.75 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 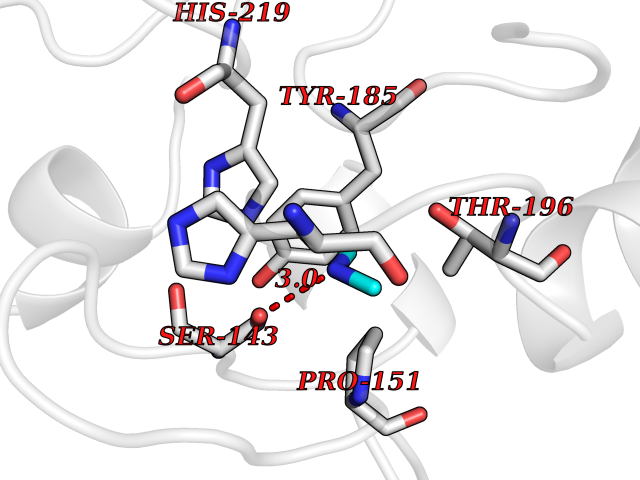 Pymol Map 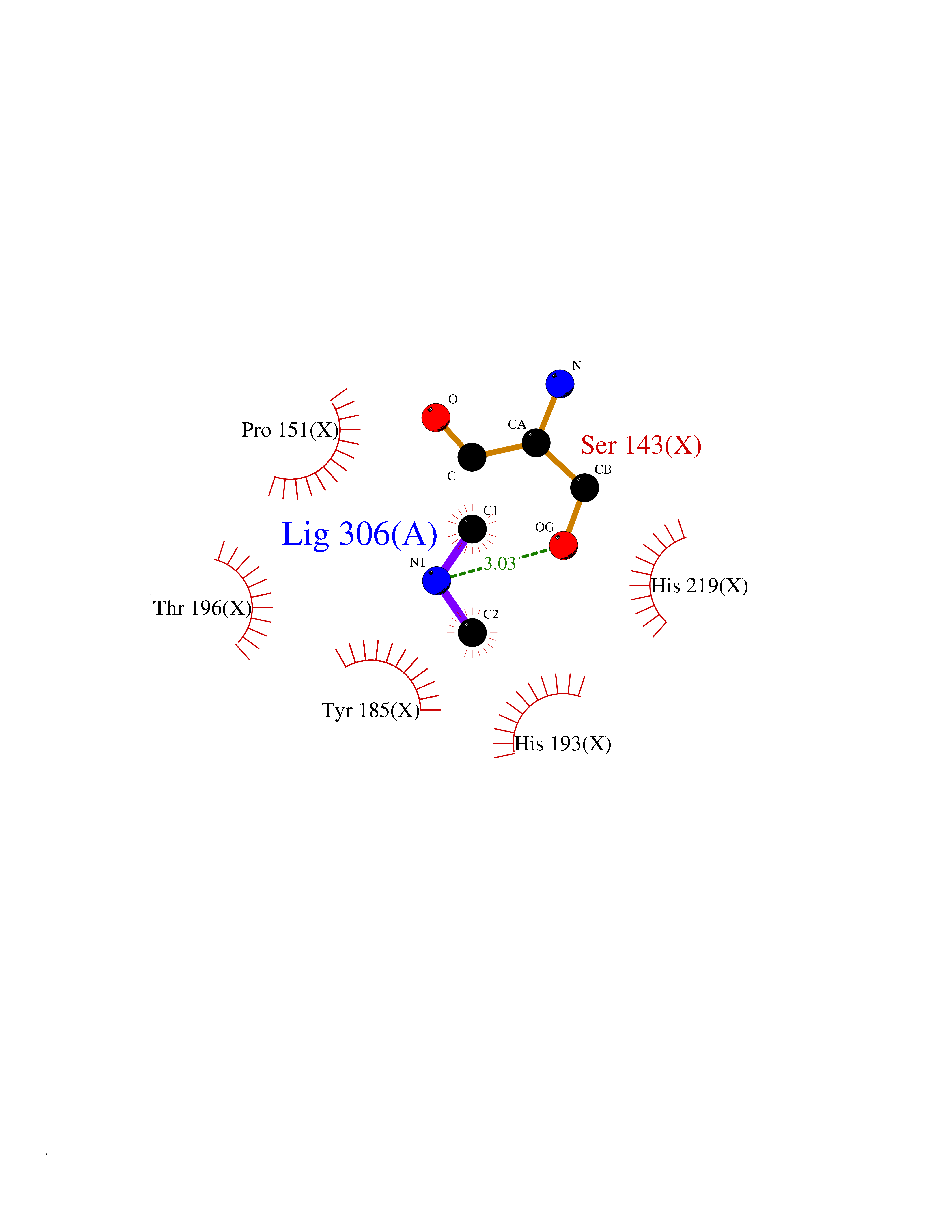 Ligplot Map 3D Binding mode Sequence ATPALRFVAVGDWGGVPNAPFHTAREMANAKEIARTVQILGADFILSLGDNFYFTGVQDINDKRFQETFEDVFSDRSLRKVPWYVLAGNHDHLGNVSAQIAYSKISKRWNFPSPFYRLHFKIPQTNVSVAIFMLDTVTLCGNSDDFLSQQPERPRDVKLARTQLSWLKKQLAAAREDYVLVAGHYPVWSIAEHGPTHCLVKQLRPLLATYGVTAYLCGHDHNLQYLQDENGVGYVLSGAGNFMDPSKRHQRKVPNGYLRFHYGTEDSLGGFAYVEISSKEMTVTYIEASGKSLFKTRLPRRARP Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Flavodoxin/ferredoxin--NADP reductase | 1FDR | 4.02 | |
Target general information Gen name fpr Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms mvrA;b3924;JW3895 Protein family Ferredoxin--NADP reductase type 1 family Biochemical class Flavoprotein Function FAD binding.Ferredoxin-NADP+ reductase activity.Oxidoreductase activity. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.18.1.2; 1.19.1.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; NADP; Nucleotide-binding; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 27346.2 Length 244 Aromaticity 0.08 Instability index 30.68 Isoelectric point 7.25 Charge (pH=7) 0.42 2D Binding mode Binding energy (Kcal/mol) -5.48 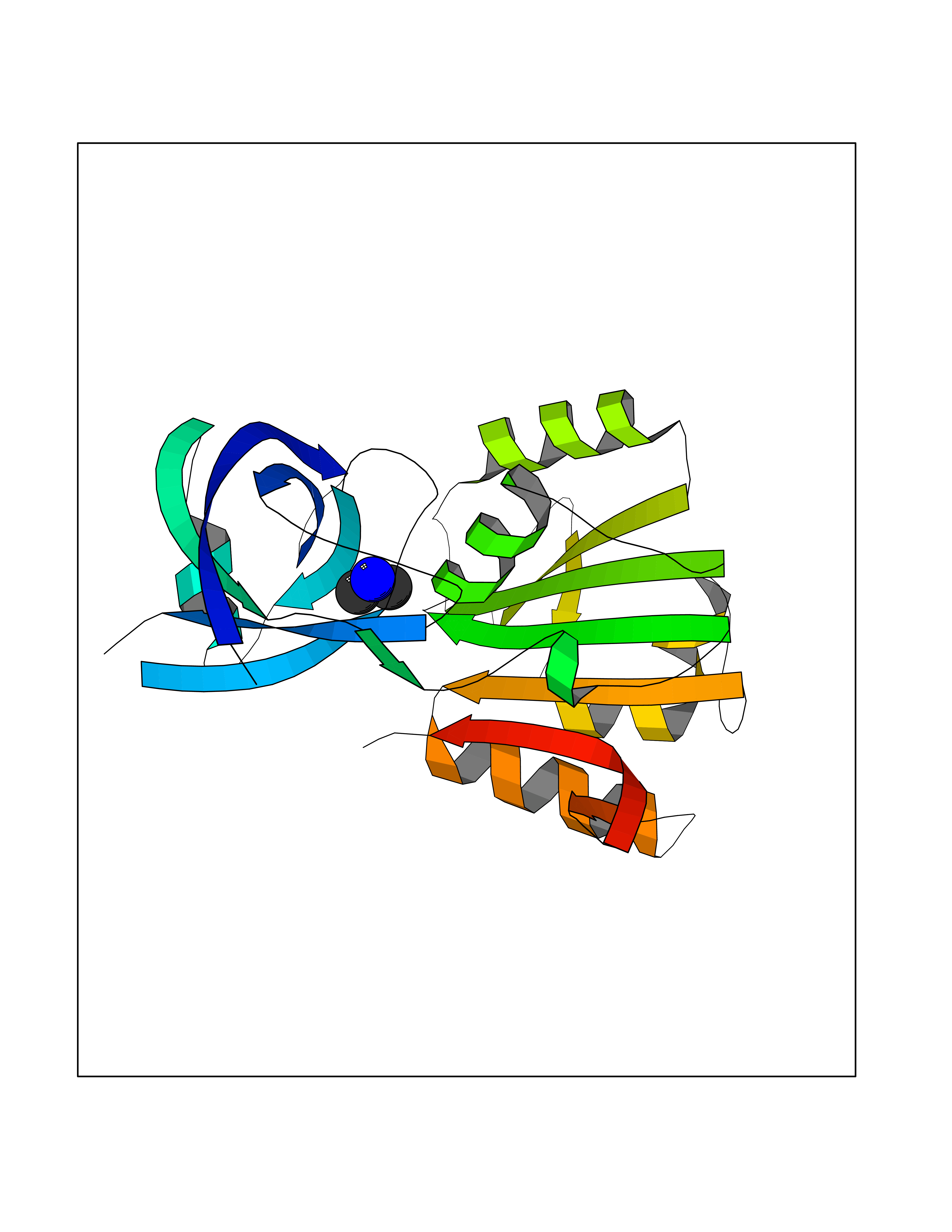 Molscript Map 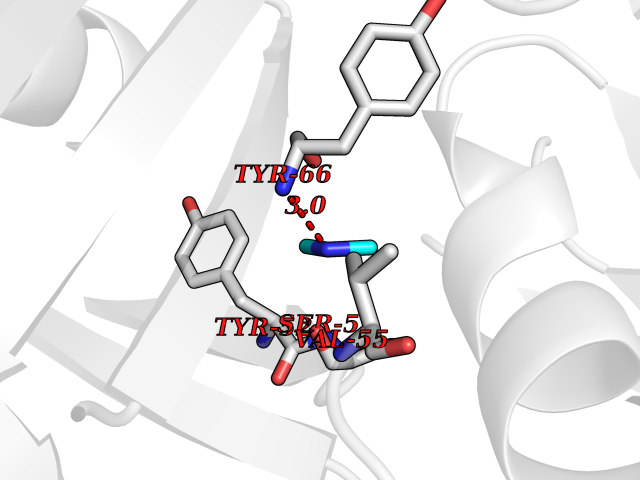 Pymol Map 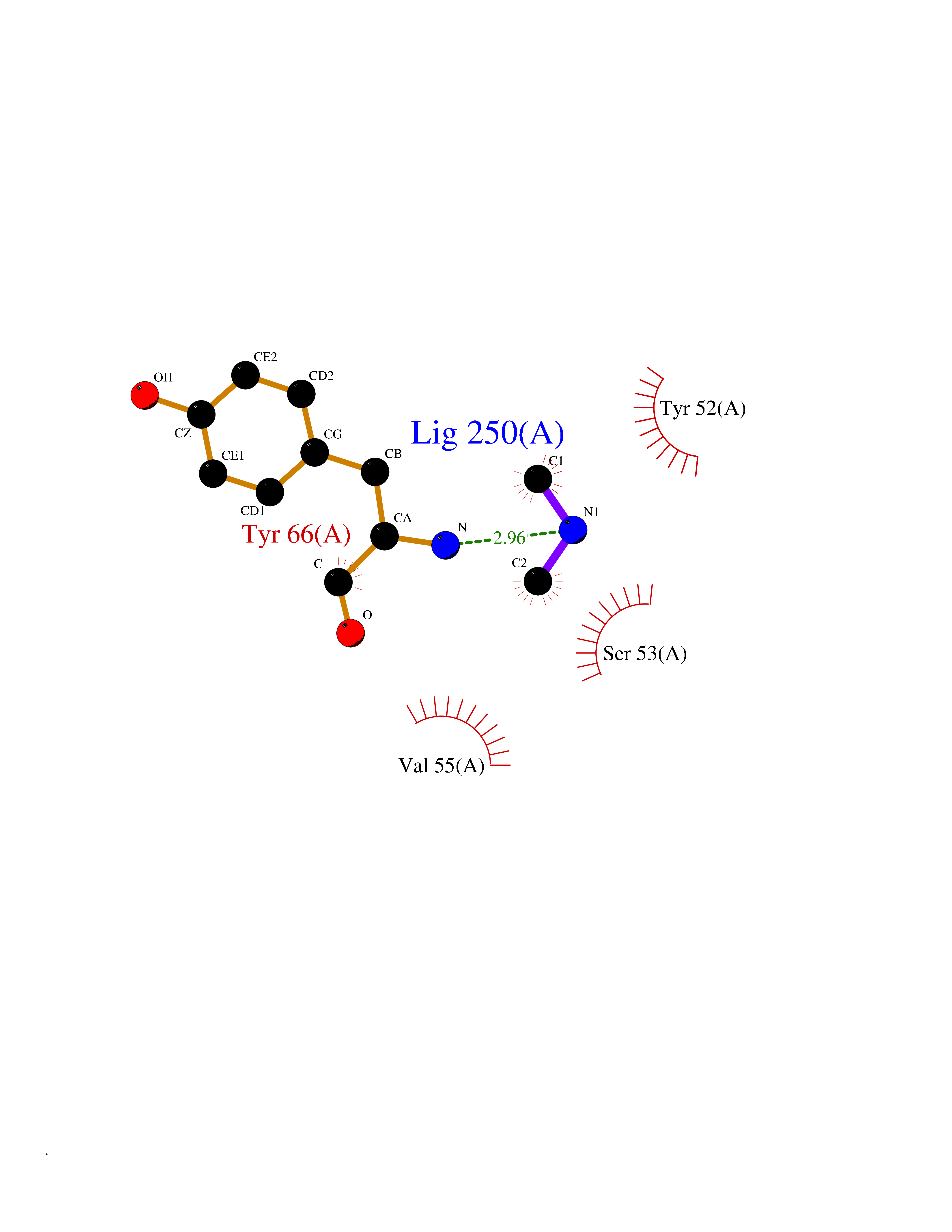 Ligplot Map 3D Binding mode Sequence ADWVTGKVTKVQNWTDALFSLTVHAPVLPFTAGQFTKLGLEIRVQRAYSYVNSPDNPDLEFYLVTVPDGKLSPRLAALKPGDEVQVVSEAAGFFVLDEVPHCETLWMLATGTAIGPYLSILRLGKDLDRFKNLVLVHAARYAADLSYLPLMQELEKRYEGKLRIQTVVSRETAAGSLTGRIPALIESGELESTIGLPMNKETSHVMLCGNPQMVRDTQQLLKETRQMTKHLRRRPGHMTAEHYW Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 4.02 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -5.48 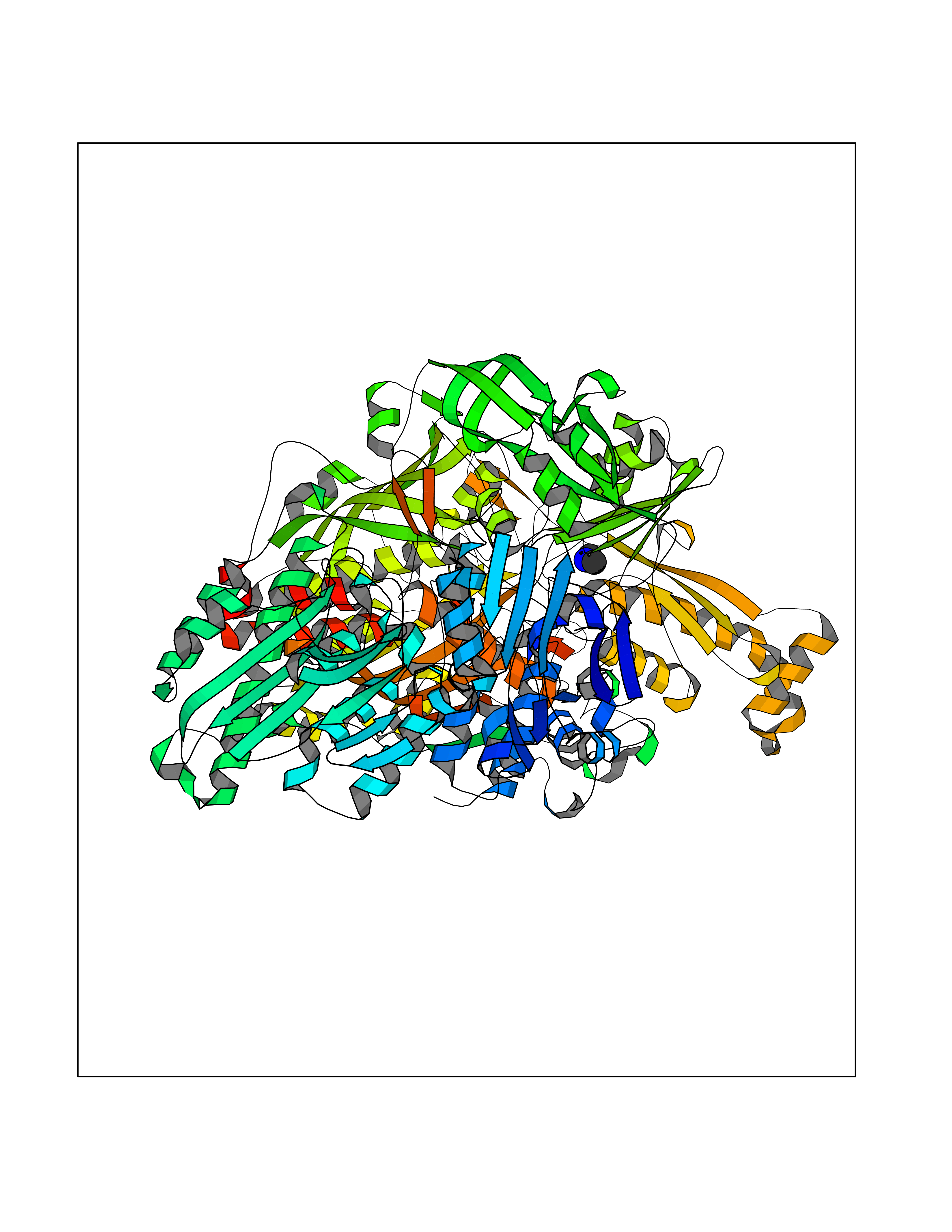 Molscript Map 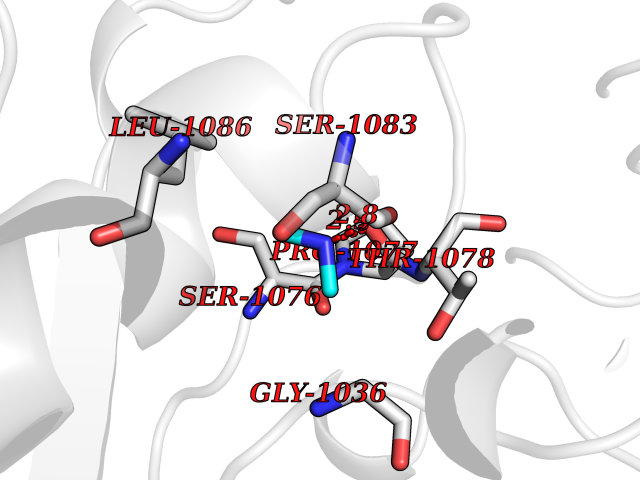 Pymol Map 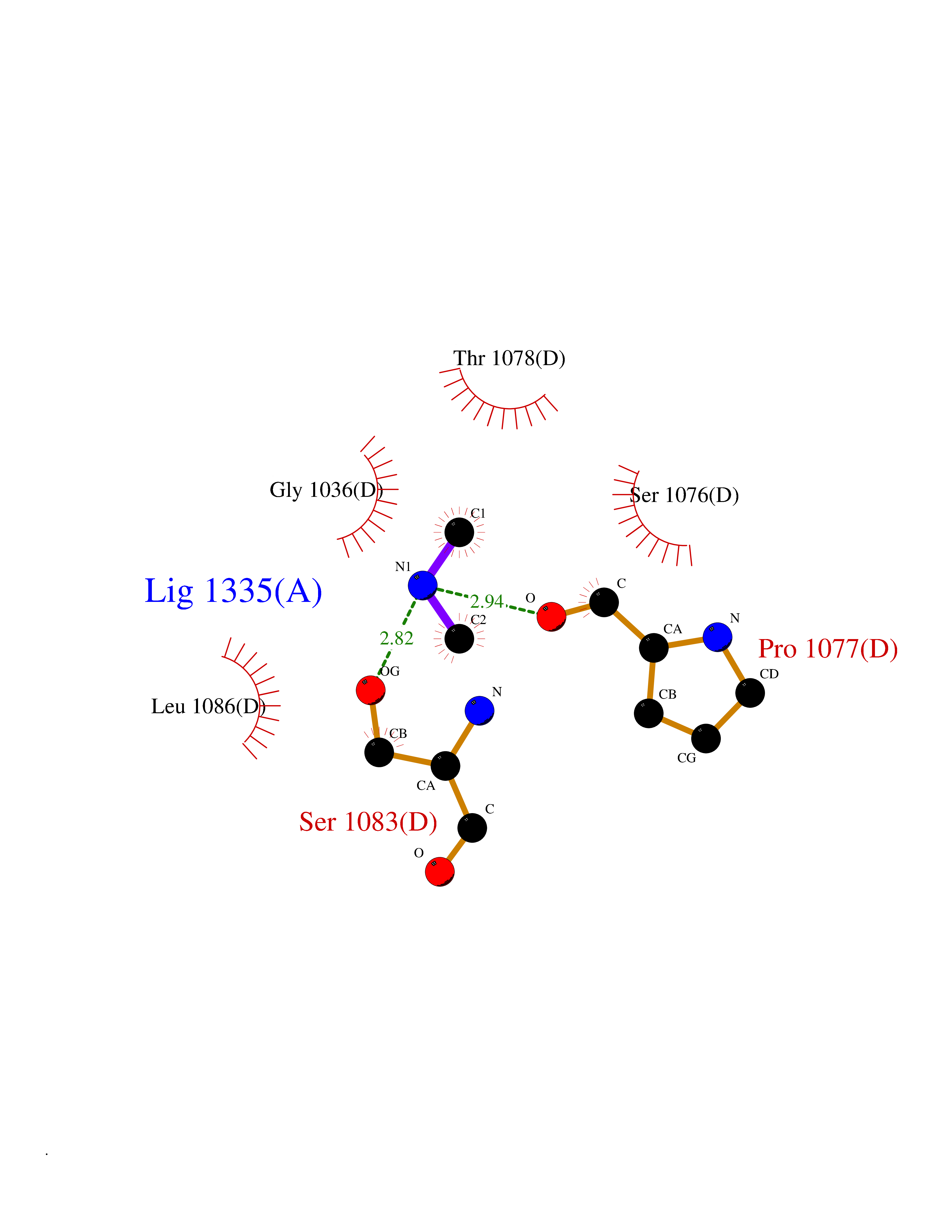 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | S-adenosylmethionine synthase isoform type-1 | 2OBV | 4.02 | |
Target general information Gen name MAT1A Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MATA1;AMS1 Protein family AdoMet synthase family Biochemical class Transferase Function ATP binding.Identical protein binding.Metal ion binding.Methionine adenosyltransferase activity.Selenomethionine adenosyltransferase activity. Related diseases Methionine adenosyltransferase deficiency (MATD) [MIM:250850]: An inborn error of metabolism resulting in isolated hypermethioninemia. Most patients have no clinical abnormalities, although some neurologic symptoms may be present in rare cases with severe loss of methionine adenosyltransferase activity. {ECO:0000269|PubMed:10677294, ECO:0000269|PubMed:7560086, ECO:0000269|PubMed:8770875, ECO:0000269|PubMed:9042912}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03191; DB00118; DB03611; DB00134 Interacts with P05067; P42858; Q00266; P31153 EC number 2.5.1.6 Uniprot keywords 3D-structure; ATP-binding; Disease variant; Disulfide bond; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Potassium; Proteomics identification; Reference proteome; S-nitrosylation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42222.9 Length 381 Aromaticity 0.08 Instability index 41.95 Isoelectric point 6.14 Charge (pH=7) -4.58 2D Binding mode Binding energy (Kcal/mol) -5.48 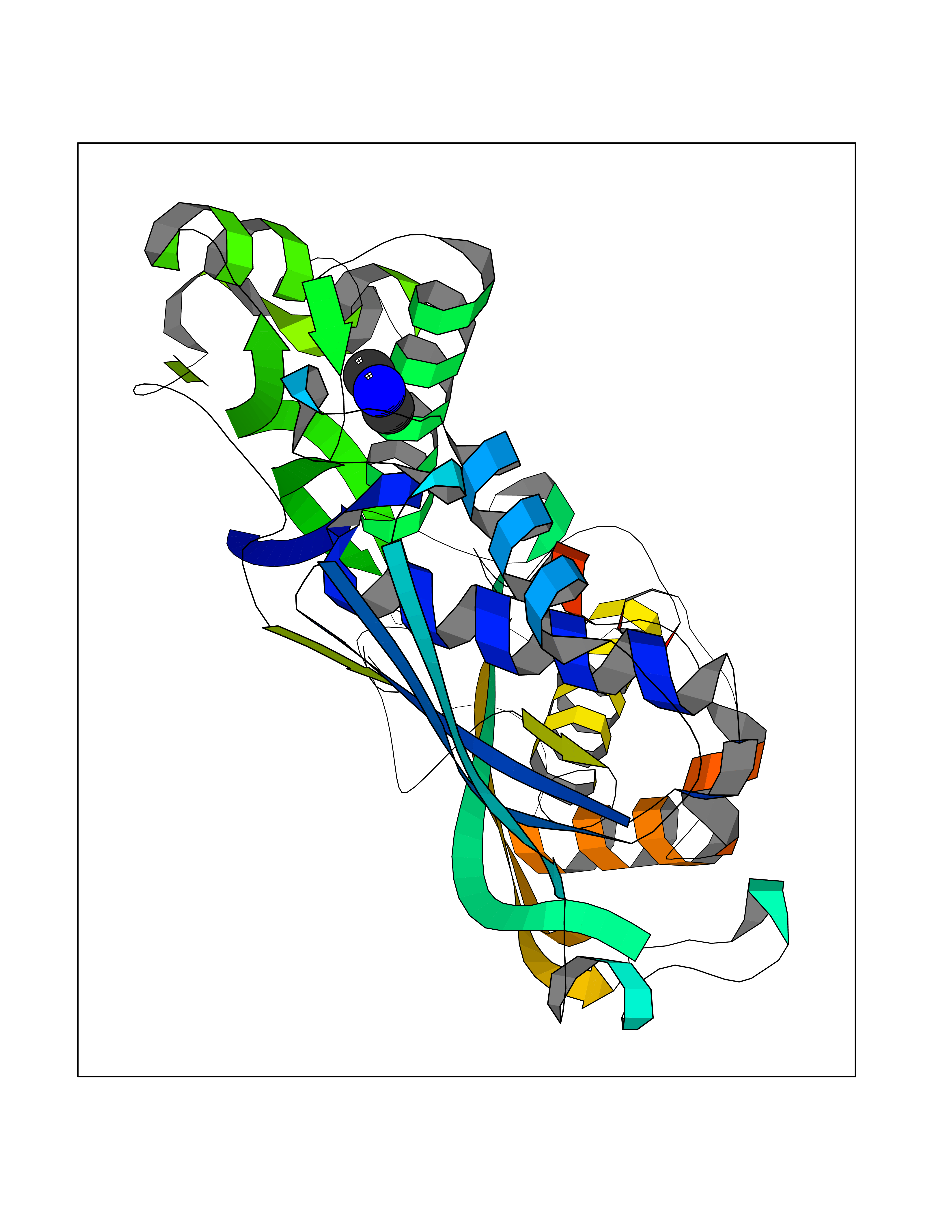 Molscript Map 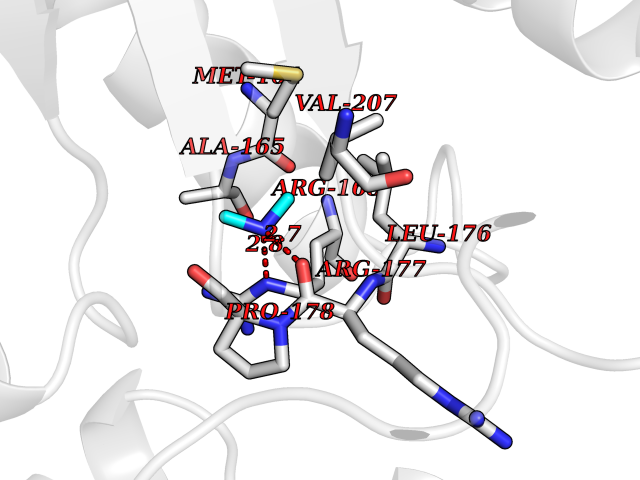 Pymol Map 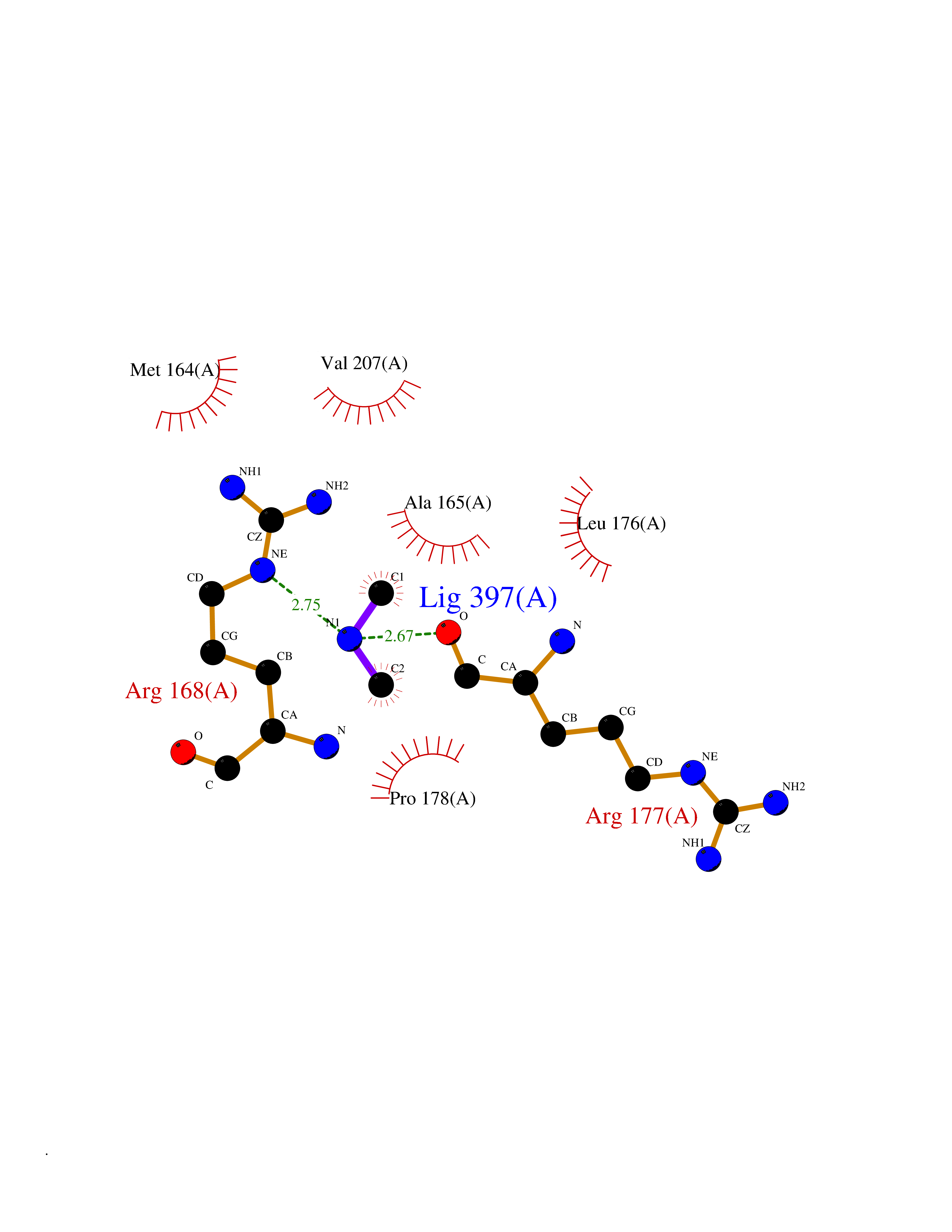 Ligplot Map 3D Binding mode Sequence MGVFMFTSESVGEGHPDKICDQISDAVLDAHLKQDPNAKVACETVCKTGMVLLCGEITSMAMVDYQRVVRDTIKHIGYDDSAKGFDFKTCNVLVALEQQSPDIAQCVHLDRNEEDVGAGDQGLMFGYATDETEECMPLTIILAHKLNARMADLRRSGLLPWLRPDSKTQVTVQYMQDNGAVIPVRIHTIVISVQHNEDITLEEMRRALKEQVIRAVVPAKYLDEDTVYHLQPSGRFVIGGPQGDAGVTGRKIIVDTYGGWGAHGGGAFSGKDYTKVDRSAAYAARWVAKSLVKAGLCRRVLVQVSYAIGVAEPLSISIFTYGTSQKTERELLDVVHKNFDLRPGVIVRDLDLKKPIYQKTACYGHFGRSEFPWEVPRKLVF Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Kynureninase (KYNU) | 3E9K | 4.02 | |
Target general information Gen name KYNU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms L-kynurenine hydrolase; KYNU Protein family Kynureninase family Biochemical class Carbon-carbon bonds hydrolase Function Catalyzes the cleavage of L-kynurenine (L-Kyn) and L-3- hydroxykynurenine (L-3OHKyn) into anthranilic acid (AA) and 3- hydroxyanthranilic acid (3-OHAA), respectively. Has a preference for the L-3-hydroxy form. Also has cysteine-conjugate-beta-lyase activity. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160; DB07069; DB00114 Interacts with Q8WUE5; P56545-3; Q9NVL1-2; P61968; P59942; Q8TDC0; P78356-2; Q86WH2 EC number EC 3.7.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Proteomics identification; Pyridine nucleotide biosynthesis; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 50204.5 Length 446 Aromaticity 0.09 Instability index 39.91 Isoelectric point 6.36 Charge (pH=7) -4.59 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map 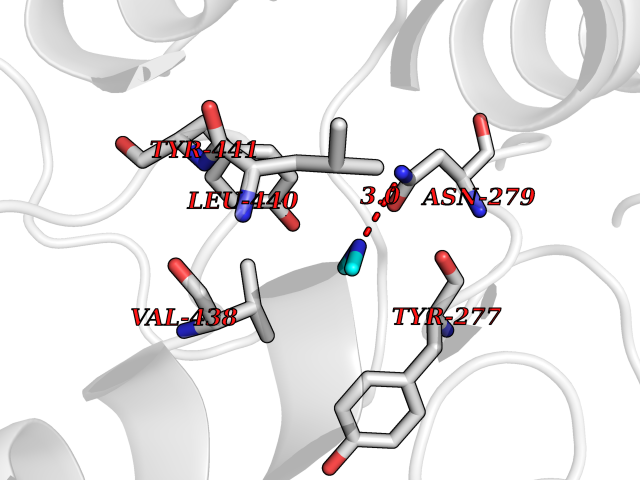 Pymol Map 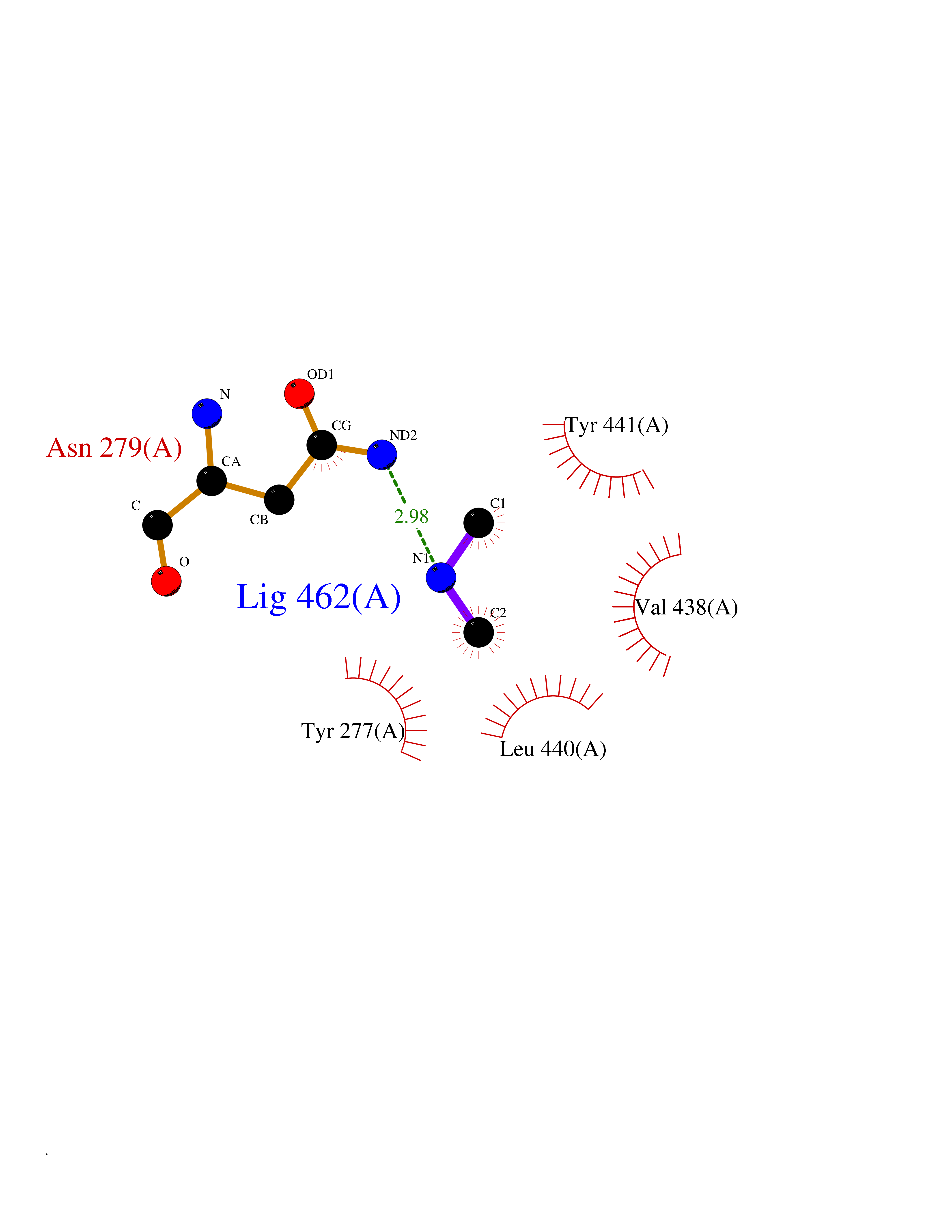 Ligplot Map 3D Binding mode Sequence LELPADTVQRIAAELKCHPTDERVALHLDEEDKLRHFREXFYIPKIQDLPPVDLSLVNKDENAIYFLGNSLGLQPKMVKTYLEEELDKWAKIAAYGHEVGKRPWITGDESIVGLMKDIVGANEKEIALMNALTVNLHLLMLSFFKPTPKRYKILLEAKAFPSDHYAIESQLQLHGLNIEESMRMIKPREGEETLRIEDILEVIEKEGDSIAVILFSGVHFYTGQHFNIPAITKAGQAKGCYVGFDLAHAVGNVELYLHDWGVDFACWCSYKYLNAGAGGIAGAFIHEKHAHTIKPALVGWFGHELSTRFKMDNKLQLIPGVCGFRISNPPILLVCSLHASLEIFKQATMKALRKKSVLLTGYLEYLIKHNYGVVNIITPSHVEERGCQLTITFSVPNKDVFQELEKRGVVCDKRNPNGIRVAPVPLYNSFHDVYKFTNLLTSILDS Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Lanosterol 14-alpha-demethylase (EC 1.14.13.70) | 4G3J | 4.02 | |
Target general information Gen name Tb11.02.4080 Organism Trypanosoma brucei brucei (strain 927/4 GUTat10.1) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class NA Function NA Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with NA EC number 1.14.13.70 Uniprot keywords 3D-structure; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID D Molecular weight (Da) 50691.4 Length 448 Aromaticity 0.08 Instability index 54.09 Isoelectric point 6.99 Charge (pH=7) -0.03 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 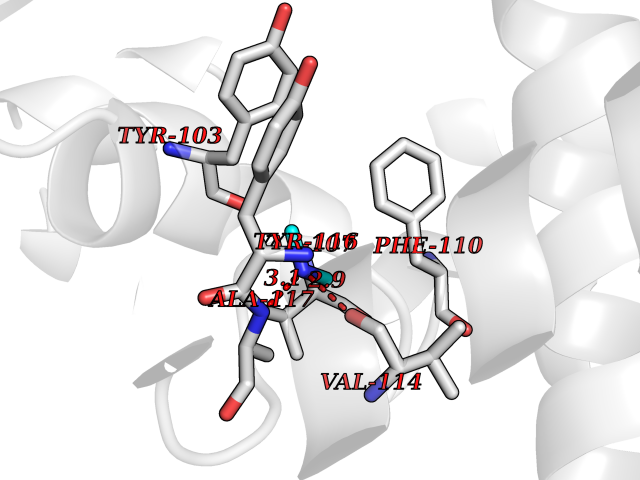 Pymol Map 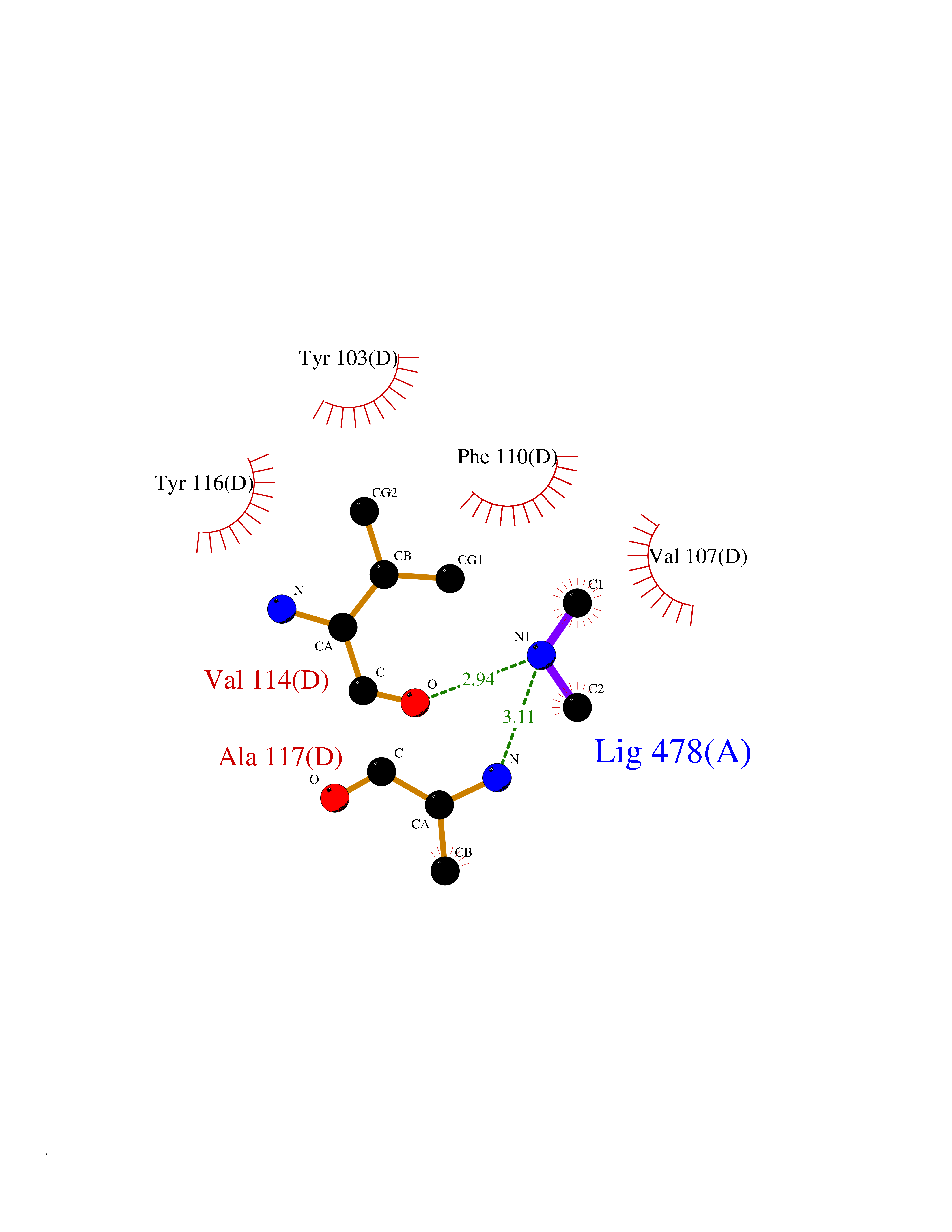 Ligplot Map 3D Binding mode Sequence GKLPPVYPVTVPILGHIIQFGKSPLGFMQECKRQLKSGIFTINIVGKRVTIVGDPHEHSRFFLPRNEVLSPREVYSFMVPVFGEGVAYAAPYPRMREQLNFLAEELTIAKFQNFVPAIQHEVRKFMAANWDKDEGEINLLEDCSTMIINTACQCLFGEDLRKRLDARRFAQLLAKMESSLIPAAVFLPILLKLPLPQSARCHEARTELQKILSEIIIARKEEEVNKDSSTSDLLSGLLSAVYRDGTPMSLHEVCGMIVAAMFAGQHTSSITTTWSMLHLMHPANVKHLEALRKEIEEFPAQLNYNNVMDEMPFAERCARESIRRDPPLLMLMRKVMADVKVGSYVVPKGDIIACSPLLSHHDEEAFPEPRRWDPERDEKVEGAFIGFGAGVHKCIGQKFGLLQVKTILATAFRSYDFQLLRDEVPDPDYHTMVVGPTASQCRVKYIRR Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Histone deacetylase 2 (HDAC2) | 4LY1 | 4.02 | |
Target general information Gen name HDAC2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HD2 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Forms transcriptional repressor complexes by associating with MAD, SIN3, YY1 and N-COR. Interacts in the late S-phase of DNA-replication with DNMT1 in the other transcriptional repressor complex composed of DNMT1, DMAP1, PCNA, CAF1. Deacetylates TSHZ3 and regulates its transcriptional repressor activity. Component of a RCOR/GFI/KDM1A/HDAC complex that suppresses, via histone deacetylase (HDAC) recruitment, a number of genes implicated in multilineage blood cell development. May be involved in the transcriptional repression of circadian target genes, such as PER1, mediated by CRY1 through histone deacetylation. Involved in MTA1-mediated transcriptional corepression of TFF1 and CDKN1A. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Ventricular tachycardia, catecholaminergic polymorphic, 1, with or without atrial dysfunction and/or dilated cardiomyopathy (CPVT1) [MIM:604772]: An arrhythmogenic disorder characterized by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and cause sudden death. Patients present with recurrent syncope, seizures, or sudden death after physical activity or emotional stress. CPVT1 inheritance is autosomal dominant. {ECO:0000269|PubMed:11157710, ECO:0000269|PubMed:11159936, ECO:0000269|PubMed:11208676, ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:12106942, ECO:0000269|PubMed:14571276, ECO:0000269|PubMed:15046072, ECO:0000269|PubMed:15046073, ECO:0000269|PubMed:15466642, ECO:0000269|PubMed:15544015, ECO:0000269|PubMed:16188589, ECO:0000269|PubMed:24793461, ECO:0000269|PubMed:25372681, ECO:0000269|PubMed:27733687}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ventricular arrhythmias due to cardiac ryanodine receptor calcium release deficiency syndrome (VACRDS) [MIM:115000]: An autosomal dominant arrhythmogenic disorder characterized by syncope, cardiac arrest and/or sudden unexpected death, often in association with physical exertion or acute emotional stress. Patients who survive manifest polymorphic ventricular tachycardia and ventricular fibrillation. Unlike typical catecholaminergic ventricular tachycardia, arrhythmias are not reproducible on exercise stress testing or adrenaline challenge. {ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:17984046, ECO:0000269|PubMed:33536282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB01223; DB01076; DB05015; DB01262; DB11841; DB01095; DB12645; DB00227; DB11830; DB01303; DB06603; DB06819; DB05223; DB00175; DB03766; DB12847; DB06176; DB00641; DB00277; DB09091; DB00313; DB02546 Interacts with Q9C0K0; Q9HCU9; P68400; Q9UER7; P51610; Q13547; Q9UIS9; Q13330; P01106; P06748; P48382; Q96ST3; O95863; Q9HD15; O43463; Q9H3M7; Q92618; Q17R98; Q2HR82; PRO_0000449623 [P0DTD1] EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Cytoplasm; Hydrolase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 42020.5 Length 366 Aromaticity 0.13 Instability index 29.52 Isoelectric point 6.52 Charge (pH=7) -2.16 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 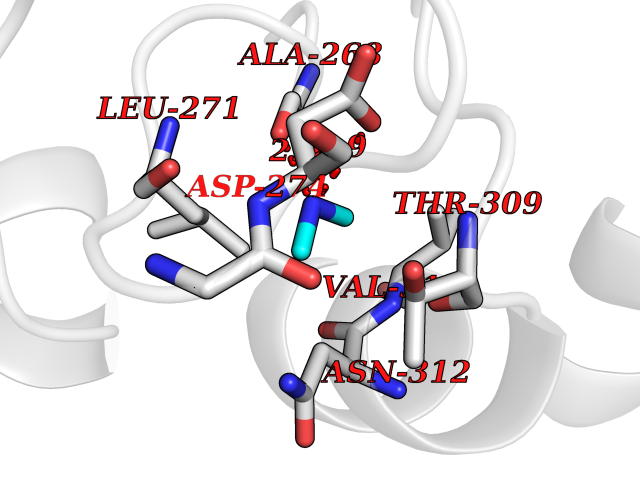 Pymol Map 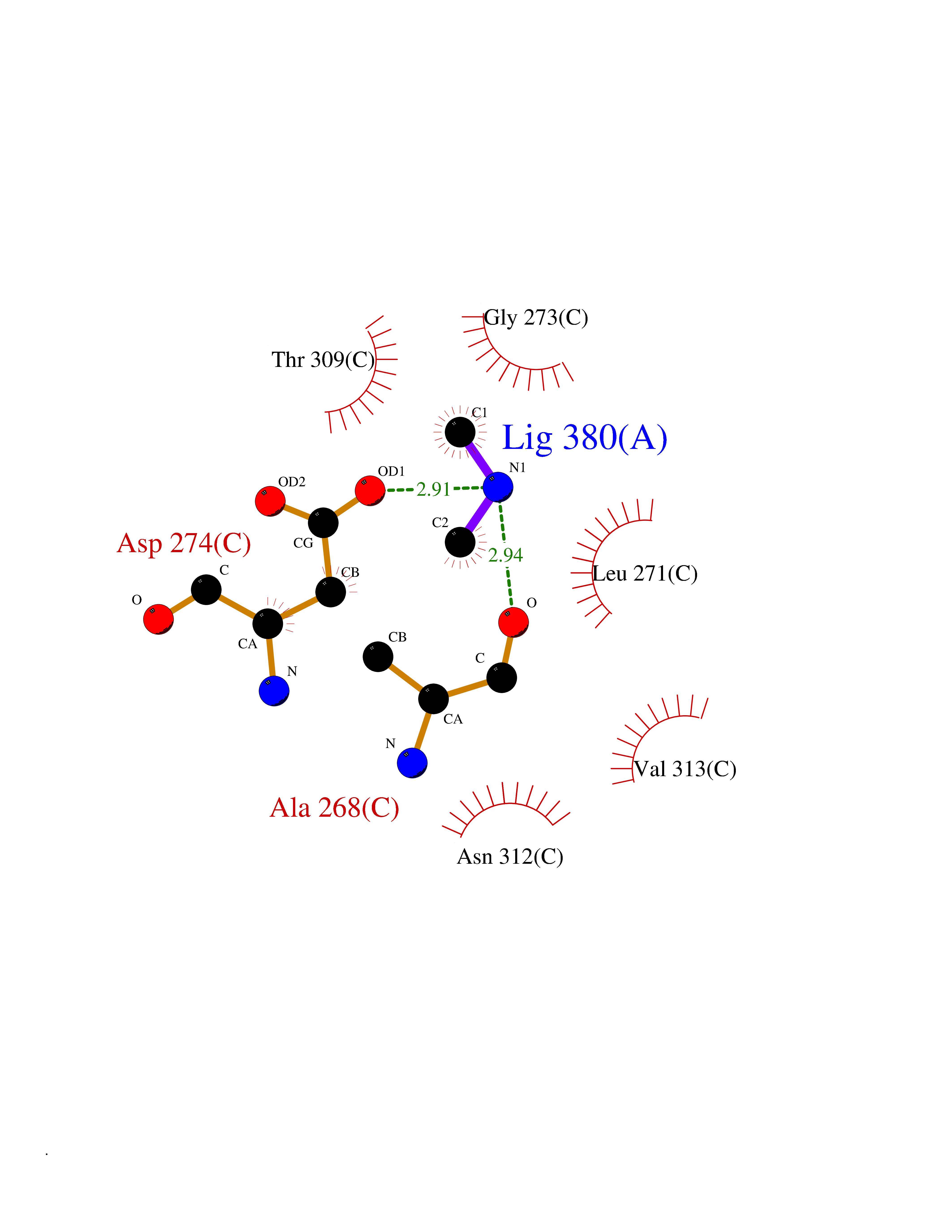 Ligplot Map 3D Binding mode Sequence KKKVCYYYDGDIGNYYYGQGHPMKPHRIRMTHNLLLNYGLYRKMEIYRPHKATAEEMTKYHSDEYIKFLRSIRPDNMSEYSKQMQRFNVGEDCPVFDGLFEFCQLSTGGSVAGAVKLNRQQTDMAVNWAGGLHHAKKSEASGFCYVNDIVLAILELLKYHQRVLYIDIDIHHGDGVEEAFYTTDRVMTVSFHKYGEYFPGTGDLRDIGAGKGKYYAVNFPMRDGIDDESYGQIFKPIISKVMEMYQPSAVVLQCGADSLSGDRLGCFNLTVKGHAKCVEVVKTFNLPLLMLGGGGYTIRNVARCWTYETAVALDCEIPNELPYNDYFEYFGPDFKLHISPSNMTNQNTPEYMEKIKQRLFENLRML Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Pectate lyase | 1R76 | 4.02 | |
Target general information Gen name pelA Organism Niveispirillum irakense (Azospirillum irakense) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Lyase Function Lyase activity. Related diseases A chromosomal aberration involving ALK is found in a form of non-Hodgkin lymphoma. Translocation t(2;5)(p23;q35) with NPM1. The resulting chimeric NPM1-ALK protein homodimerize and the kinase becomes constitutively activated. The constitutively active fusion proteins are responsible for 5-10% of non-Hodgkin lymphomas. {ECO:0000269|PubMed:15938644}.; DISEASE: A chromosomal aberration involving ALK is associated with inflammatory myofibroblastic tumors (IMTs). Translocation t(2;11)(p23;p15) with CARS; translocation t(2;4)(p23;q21) with SEC31A. {ECO:0000269|PubMed:12112524, ECO:0000269|PubMed:16161041}.; DISEASE: A chromosomal aberration involving ALK is associated with anaplastic large-cell lymphoma (ALCL). Translocation t(2;17)(p23;q25) with ALO17. {ECO:0000269|PubMed:12112524}.; DISEASE: Neuroblastoma 3 (NBLST3) [MIM:613014]: A common neoplasm of early childhood arising from embryonic cells that form the primitive neural crest and give rise to the adrenal medulla and the sympathetic nervous system. {ECO:0000269|PubMed:18724359, ECO:0000269|PubMed:18923523, ECO:0000269|PubMed:18923525, ECO:0000269|PubMed:21242967, ECO:0000269|PubMed:22932897}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: The ALK signaling pathway plays an important role in glioblastoma, the most common malignant brain tumor of adults and one of the most lethal cancers. It regulates both glioblastoma migration and growth. {ECO:0000269|PubMed:15908427}.; DISEASE: A chromosomal aberration involving ALK is found in one subject with colorectal cancer. Translocation t(2;2)(p23.1;p23.3). A 5 million base pair tandem duplication generates an in-frame WDCP-ALK gene fusion. {ECO:0000269|PubMed:22327622}.; DISEASE: A chromosomal aberration involving ALK has been identified in a subset of patients with non-small-cell lung carcinoma. This aberration leads to the production of a fusion protein between the N-terminus of EML4 et the C-terminus of ALK. It is unclear whether the fusion protein is caused by a simple inversion within 2p (inv(2)(p21p23)) or whether the chromosome translocation involving 2p is more complex. When tested in a heterologous system, the fusion protein EML4-ALK possesses transforming activity that is dependent on ALK catalytic activity, possibly due to spontaneous dimerization mediated by the EML4 moiety, leading to ALK kinase activation. {ECO:0000269|PubMed:17625570}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Lyase; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41907.5 Length 384 Aromaticity 0.08 Instability index 43.72 Isoelectric point 6.11 Charge (pH=7) -3.46 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 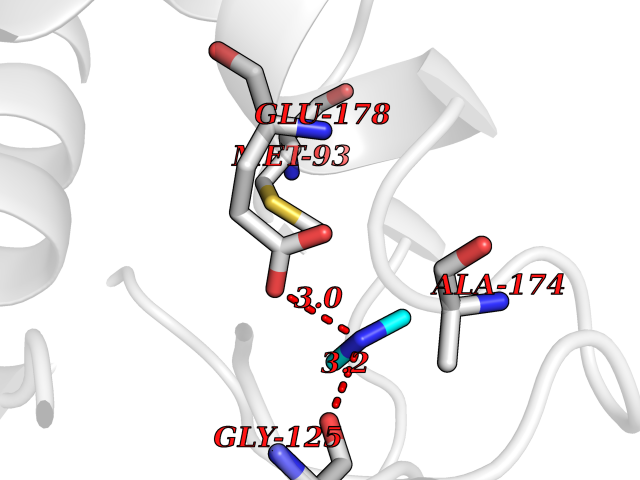 Pymol Map 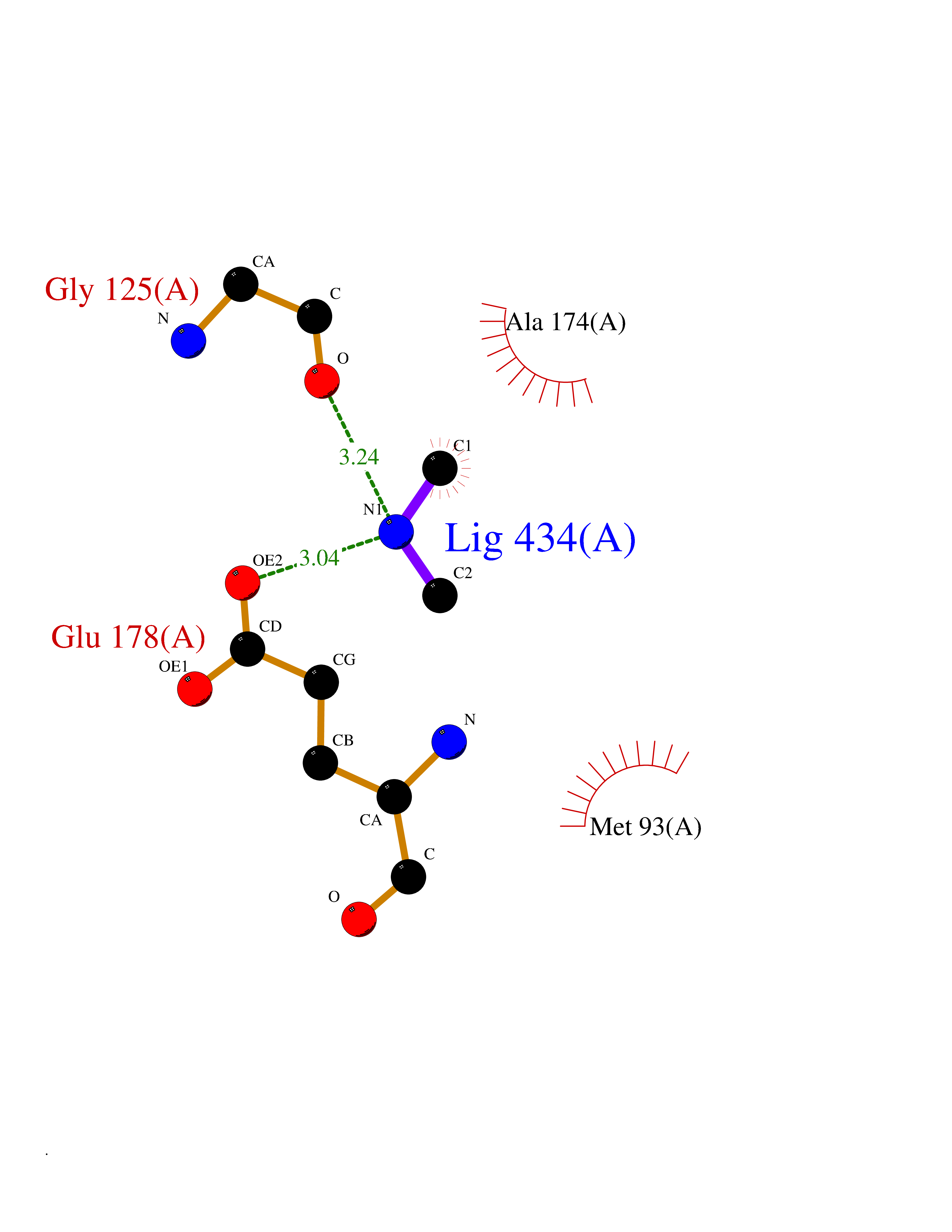 Ligplot Map 3D Binding mode Sequence AVIGMNEAASALTPSRVSSLPDTQRAAWQEYLARSEAQLSRDKASLAAELAPGQPLPPPPAEGKGADTMPLDKPAAWYTSKAARHVADVIVSFQTPAGGWGKNQPRDGALRLPGQHYTGENVAKVKRDRDWHYVGTIDNDATVTEIRFLAQVVSQLAPEEAAPYRDAALKGIEYLLASQFPNGGWPQVWPLEGGYHDAITYNDDALVHVAELLSDIAAGRDGFGFVPPAIRTRALEATNAAIHCIVETQVVQDGKRLGWGQQHDALTLRPTSARNFEPAALSSTESARILLFLMEIEAPSDAVKQAIRGGVAWLNTSVIRDQGAKPLWSRFYSLDGNKPVFGDRDKTIHDDVMGISQERRTGYAWYTTSPQKALSAFTKWEKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Tankyrase-2 (TNKS-2) | 3U9H | 4.02 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -5.48 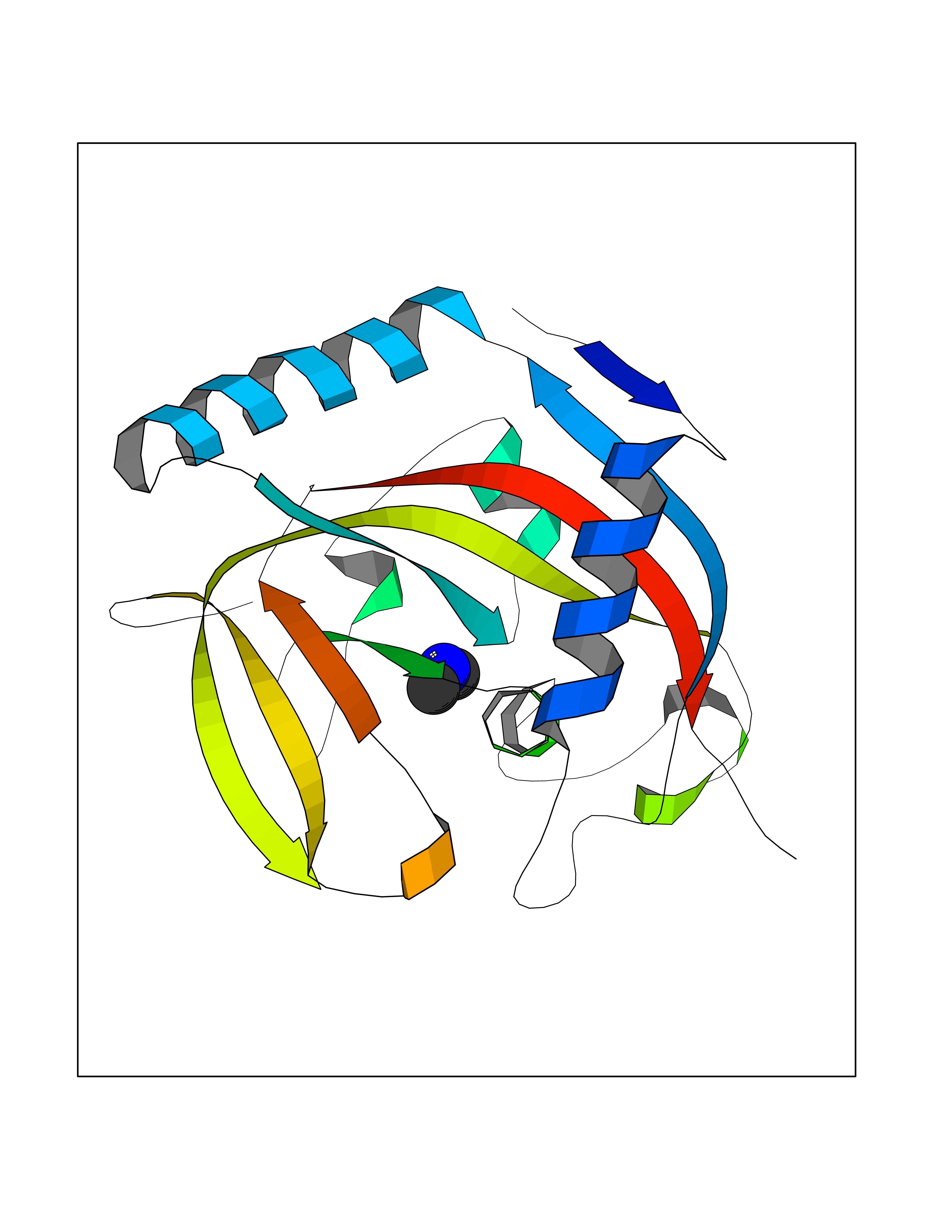 Molscript Map 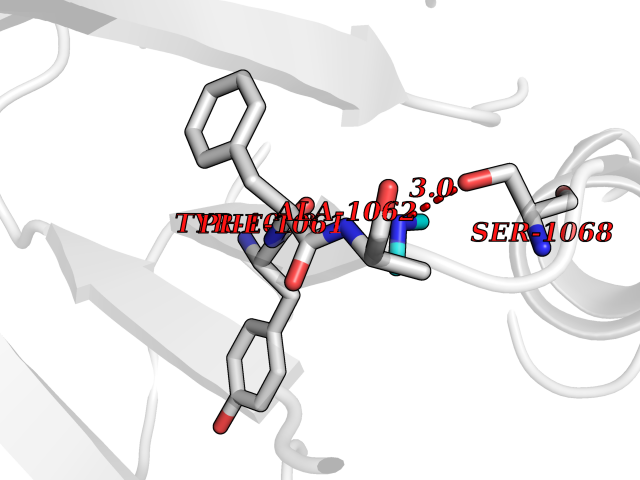 Pymol Map 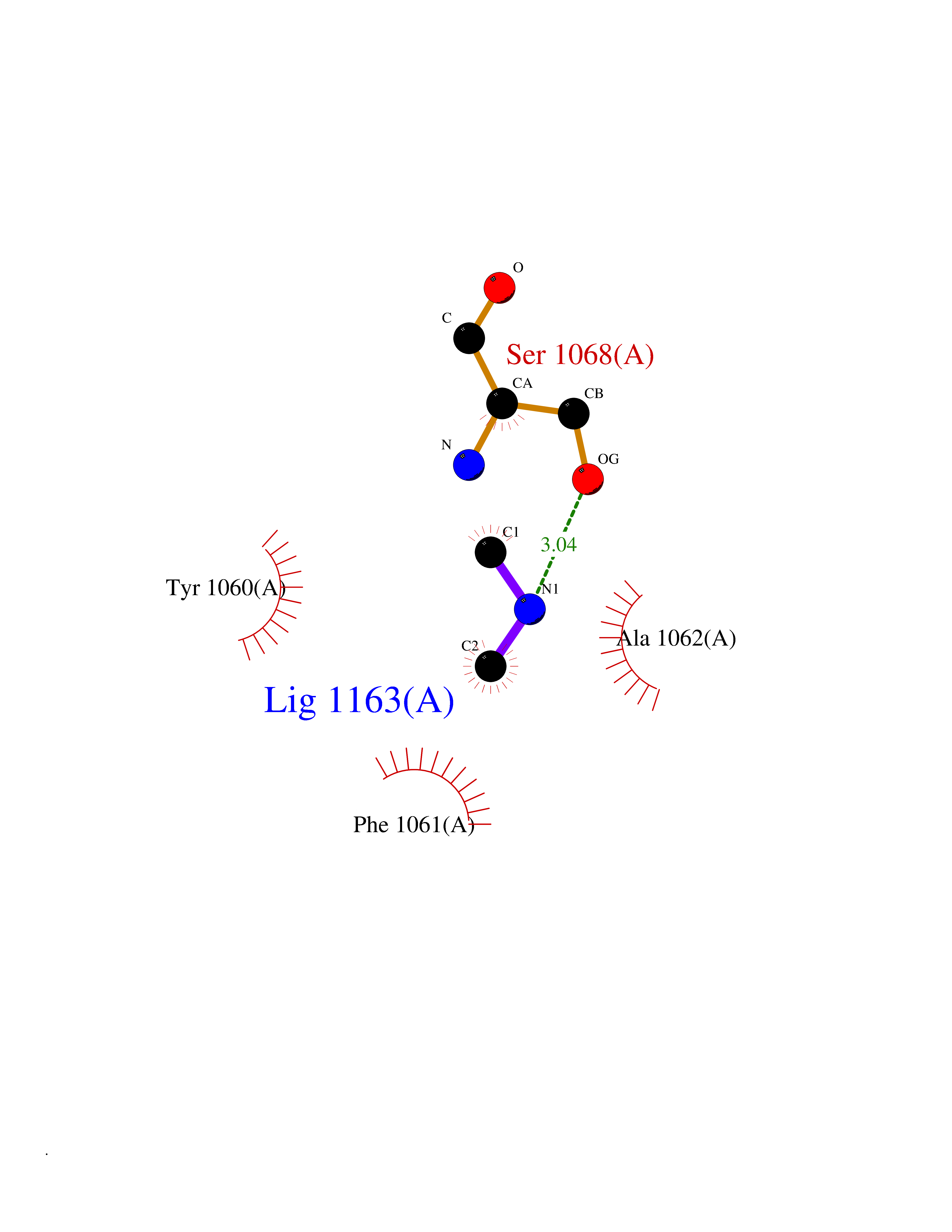 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Alpha-ketoglutarate dehydrogenase (OGDH) | 7WGR | 4.02 | |
Target general information Gen name OGDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OGDC-E1; 2-oxoglutarate dehydrogenase, mitochondrial; 2-oxoglutarate dehydrogenase complex component E1 Protein family Alpha-ketoglutarate dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function The 2-oxoglutarate dehydrogenase complex catalyzes the overall conversion of 2-oxoglutarate to succinyl-CoA and CO(2). The 2-oxoglutarate dehydrogenase complex is mainly active in the mitochondrion. A fraction of the 2-oxoglutarate dehydrogenase complex also localizes in the nucleus and is required for lysine succinylation of histones: associates with KAT2A on chromatin and provides succinyl-CoA to histone succinyltransferase KAT2A. 2-oxoglutarate dehydrogenase (E1) component of the 2-oxoglutarate dehydrogenase complex, which mediates the decarboxylation of alpha-ketoglutarate. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00313; DB09092 Interacts with P54253; P42858 EC number EC 1.2.4.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Calcium; Glycolysis; Isopeptide bond; Magnesium; Metal-binding; Mitochondrion; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Thiamine pyrophosphate; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 192730 Length 1700 Aromaticity 0.09 Instability index 43.47 Isoelectric point 5.97 Charge (pH=7) -29.2 2D Binding mode Binding energy (Kcal/mol) -5.48 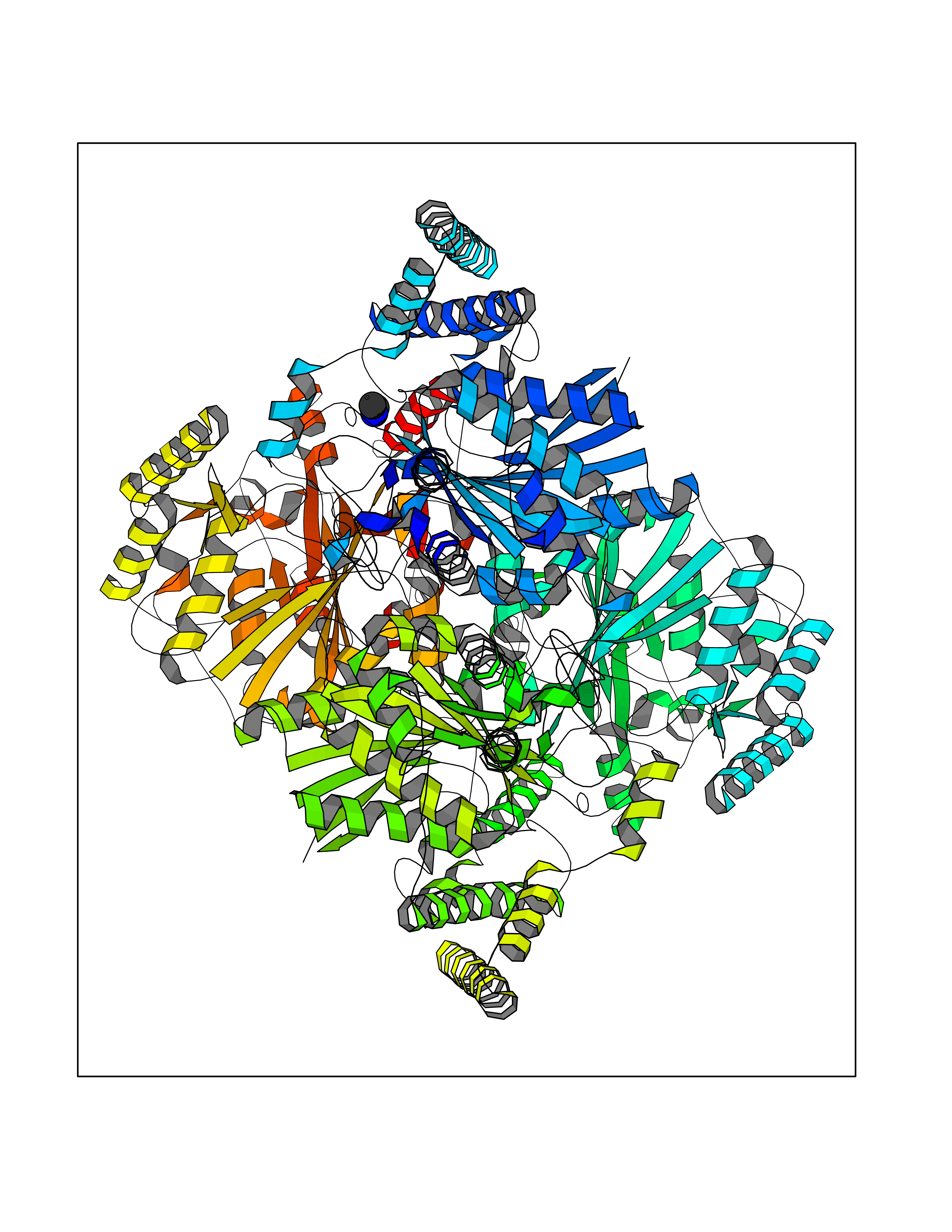 Molscript Map 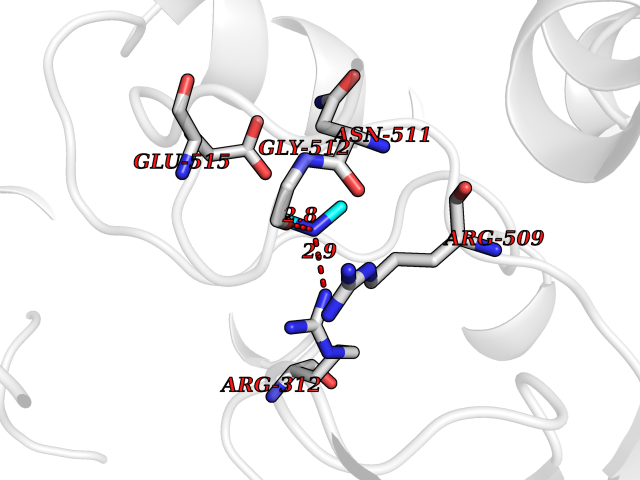 Pymol Map 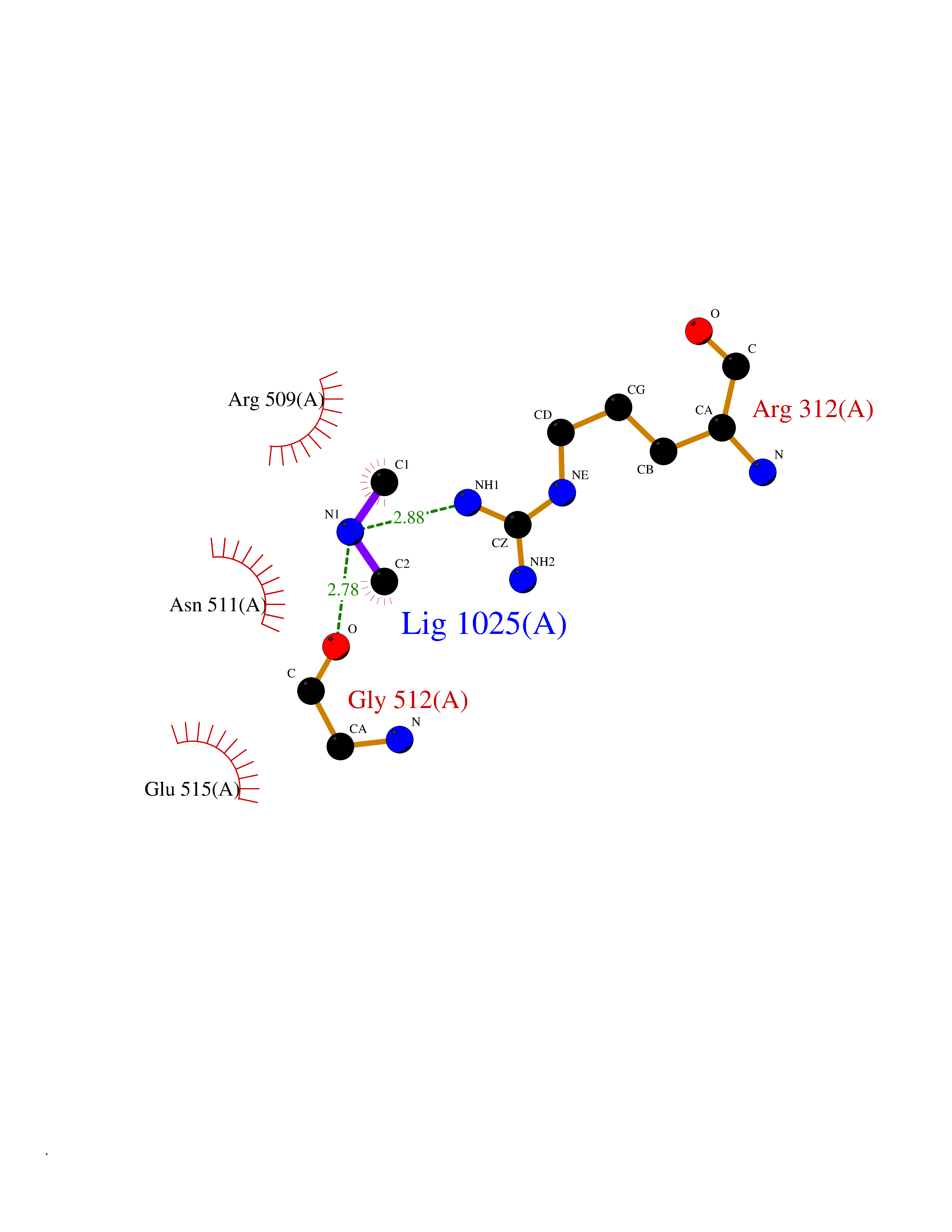 Ligplot Map 3D Binding mode Sequence AVQSLIRAYQIRGHHVAQLDPLGILDADLDSSVPADIISSTDKLGFYGLDESDLDKVFHLPTTTFIGGQESALPLREIIRRLEMAYCQHIGVEFMFINDLEQCQWIRQKFETPGIMQFTNEEKRTLLARLVRSTRFEEFLQRKWSSEKRFGLEGCEVLIPALKTIIDKSSENGVDYVIMGMPHRGRLNVLANVIRKELEQIFCQFDSKLEAADEGSGDNITLSLVANPSHLEAADPVVMGKTKAEQFYCGDTEGKKVMSILLHGDAAFAGQGIVYETFHLSDLPSYTTHGTVHVVVNNQIGFTTDPRMARSSPYPTDVARVVNAPIFHVNSDDPEAVMYVCKVAAEWRSTFHKDVVVDLVCYRRNGHNEMDEPMFTQPLMYKQIRKQKPVLQKYAELLVSQGVVNQPEYEEEISKYDKICEEAFARSKMSCPSTGLTEDILTHIGNVASSVPVENFTIHGGLSRILKTRGEMVKNRTVDWALAEYMAFGSLLKEGIHIRLSGQDVERGTFSHRHHVLHDQNVDKRTCIPMNHLWPNQAPYTVCNSSLSEYGVLGFELGFAMASPNALVLWEAQFGDFHNTAQCIIDQFICPGQAKWVRQNGIVLLLPHGMEGMGPEHSSARPERFLQMCNDDPDVLPDLKEANFDINQLYDCNWVVVNCSTPGNFFHVLRRQILLPFRKPLIIFTPKSLLRHPEARSSFDEMLPGTHFQRVIPEDGPAAQNPENVKRLLFCTGKVYYDLTRERKARDMVGQVAITRIEQLSPFPFDLLLKEVQKYPNAELAWCQEEHKNQGYYDYVKPRLRTTISRAKPVWYAGRDPAAAPATGNKKTHLTELQRLLDTAFDLDVFKNFSAVQSLIRAYQIRGHHVAQLDPLGILDADLDSSVPADIISSTDKLGFYGLDESDLDKVFHLPTTTFIGGQESALPLREIIRRLEMAYCQHIGVEFMFINDLEQCQWIRQKFETPGIMQFTNEEKRTLLARLVRSTRFEEFLQRKWSSEKRFGLEGCEVLIPALKTIIDKSSENGVDYVIMGMPHRGRLNVLANVIRKELEQIFCQFDSKLEAADEGSGDNITLSLVANPSHLEAADPVVMGKTKAEQFYCGDTEGKKVMSILLHGDAAFAGQGIVYETFHLSDLPSYTTHGTVHVVVNNQIGFTTDPRMARSSPYPTDVARVVNAPIFHVNSDDPEAVMYVCKVAAEWRSTFHKDVVVDLVCYRRNGHNEMDEPMFTQPLMYKQIRKQKPVLQKYAELLVSQGVVNQPEYEEEISKYDKICEEAFARSKMSCPSTGLTEDILTHIGNVASSVPVENFTIHGGLSRILKTRGEMVKNRTVDWALAEYMAFGSLLKEGIHIRLSGQDVERGTFSHRHHVLHDQNVDKRTCIPMNHLWPNQAPYTVCNSSLSEYGVLGFELGFAMASPNALVLWEAQFGDFHNTAQCIIDQFICPGQAKWVRQNGIVLLLPHGMEGMGPEHSSARPERFLQMCNDDPDVLPDLKEANFDINQLYDCNWVVVNCSTPGNFFHVLRRQILLPFRKPLIIFTPKSLLRHPEARSSFDEMLPGTHFQRVIPEDGPAAQNPENVKRLLFCTGKVYYDLTRERKARDMVGQVAITRIEQLSPFPFDLLLKEVQKYPNAELAWCQEEHKNQGYYDYVKPRLRTTISRAKPVWYAGRDPAAAPATGNKKTHLTELQRLLDTAFDLDVFKNFS Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Cathepsin D (CTSD) | 4OC6 | 4.02 | |
Target general information Gen name CTSD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CPSD; CD Protein family Peptidase A1 family Biochemical class Peptidase Function Plays a role in APP processing following cleavage and activation by ADAM30 which leads to APP degradation. Involved in the pathogenesis of several diseases such as breast cancer and possibly Alzheimer disease. Acid protease active in intracellular protein breakdown. Related diseases Ceroid lipofuscinosis, neuronal, 10 (CLN10) [MIM:610127]: A form of neuronal ceroid lipofuscinosis with onset at birth or early childhood. Neuronal ceroid lipofuscinoses are progressive neurodegenerative, lysosomal storage diseases characterized by intracellular accumulation of autofluorescent liposomal material, and clinically by seizures, dementia, visual loss, and/or cerebral atrophy. {ECO:0000269|PubMed:16670177, ECO:0000269|PubMed:16685649, ECO:0000269|PubMed:21990111}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03028; DB03096; DB07542; DB08740; DB02216 Interacts with P05067; Q9P1A6-3; I6L9I8; Q9H6S3; Q7Z602; P28799; PRO_0000012695 [P28799]; PRO_0000012696 [P28799]; PRO_0000012697 [P28799]; PRO_0000012698 [P28799]; PRO_0000012699 [P28799]; PRO_0000012700 [P28799]; PRO_0000012701 [P28799]; P68431; Q9Y6F6-3; Q12756; Q5TA79; Q86VF5-3; O15130-2; Q96LB9; P09565; Q9C004; Q8NBJ7; Q9BQG1; P28347-2; P45880; Q15007-2; O00308; Q5W0Z9-4; Q6ZNH5 EC number EC 3.4.23.5 Uniprot keywords 3D-structure; Alzheimer disease; Aspartyl protease; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Neurodegeneration; Neuronal ceroid lipofuscinosis; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37264.2 Length 341 Aromaticity 0.1 Instability index 32.32 Isoelectric point 5.6 Charge (pH=7) -4.86 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 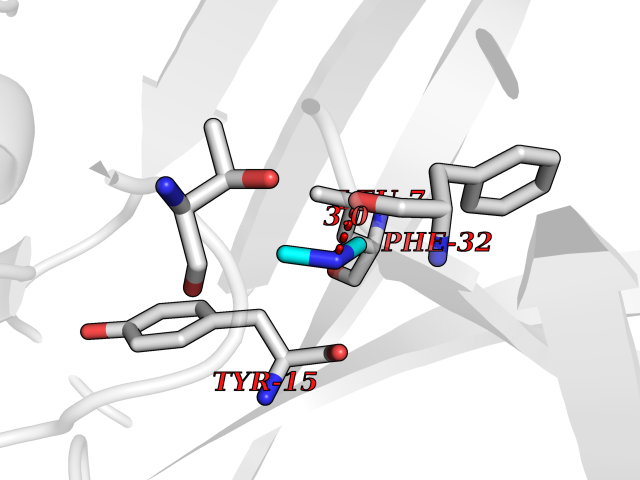 Pymol Map 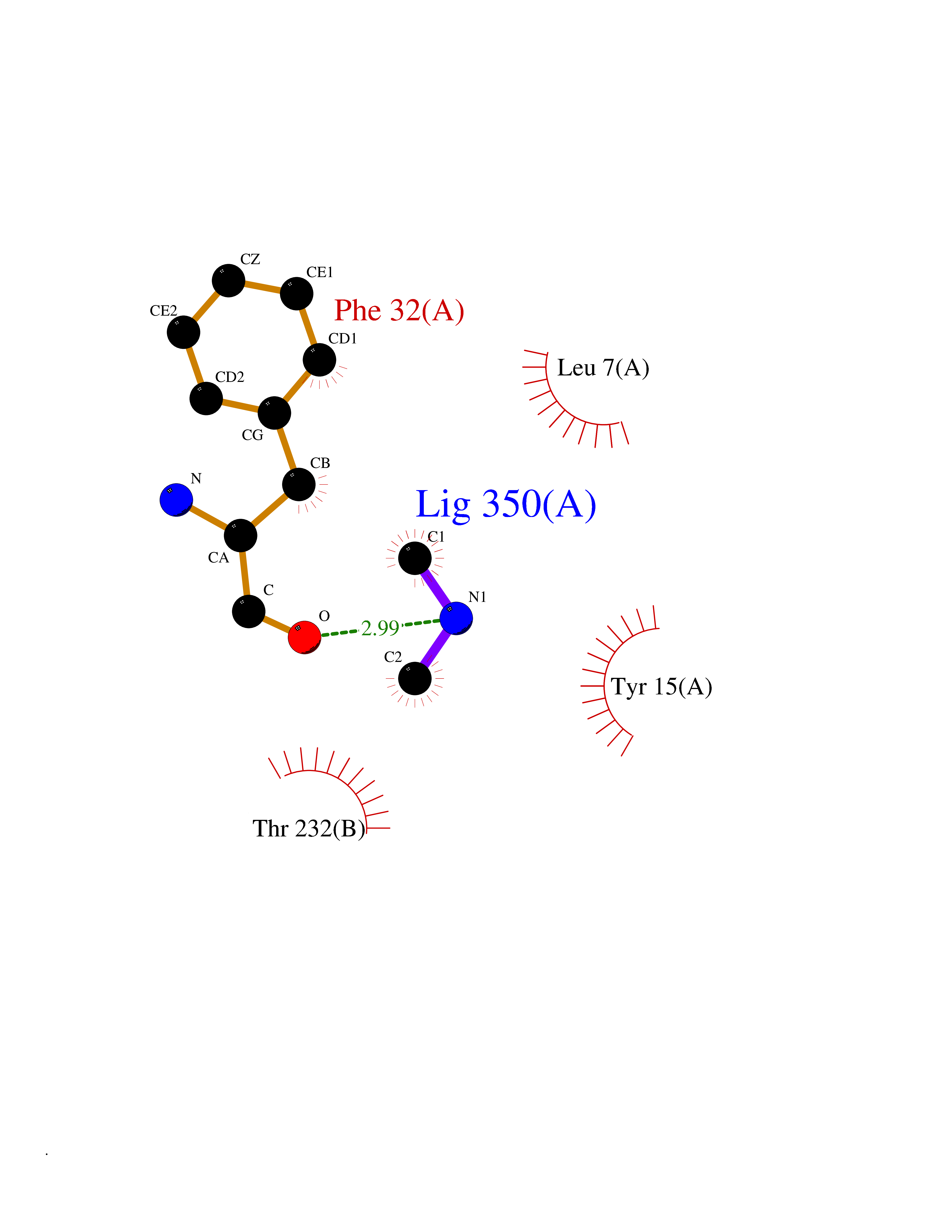 Ligplot Map 3D Binding mode Sequence GPIPEVLKNYMDAQYYGEIGIGTPPQCFTVVFDTGSSNLWVPSIHCKLLDIACWIHHKYNSDKSSTYVKNGTSFDIHYGSGSLSGYLSQDTVSVPCQSGGVKVERQVFGEATKQPGITFIAAKFDGILGMAYPRISVNNVLPVFDNLMQQKLVDQNIFSFYLSRDPDAQPGGELMLGGTDSKYYKGSLSYLNVTRKAYWQVHLDQVEVASGLTLCKEGCEAIVDTGTSLMVGPVDEVRELQKAIGAVPLIQGEYMIPCEKVSTLPAITLKLGGKGYKLSPEDYTLKVSQAGKTLCLSGFMGMDIPPPSGPLWILGDVFIGRYYTVFDRDNNRVGFAEAARL Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Histamine H3 receptor (H3R) | 7F61 | 4.02 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -5.49 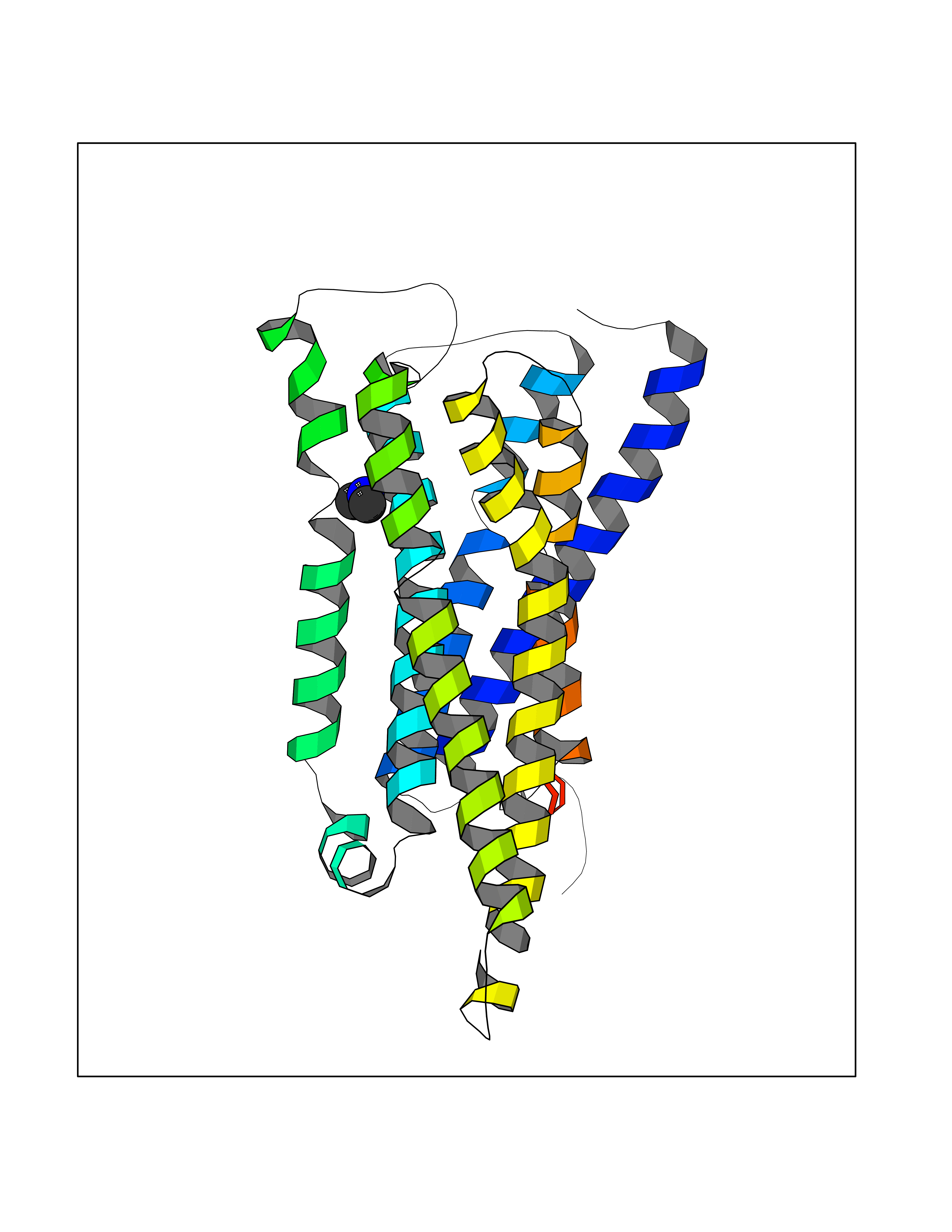 Molscript Map  Pymol Map 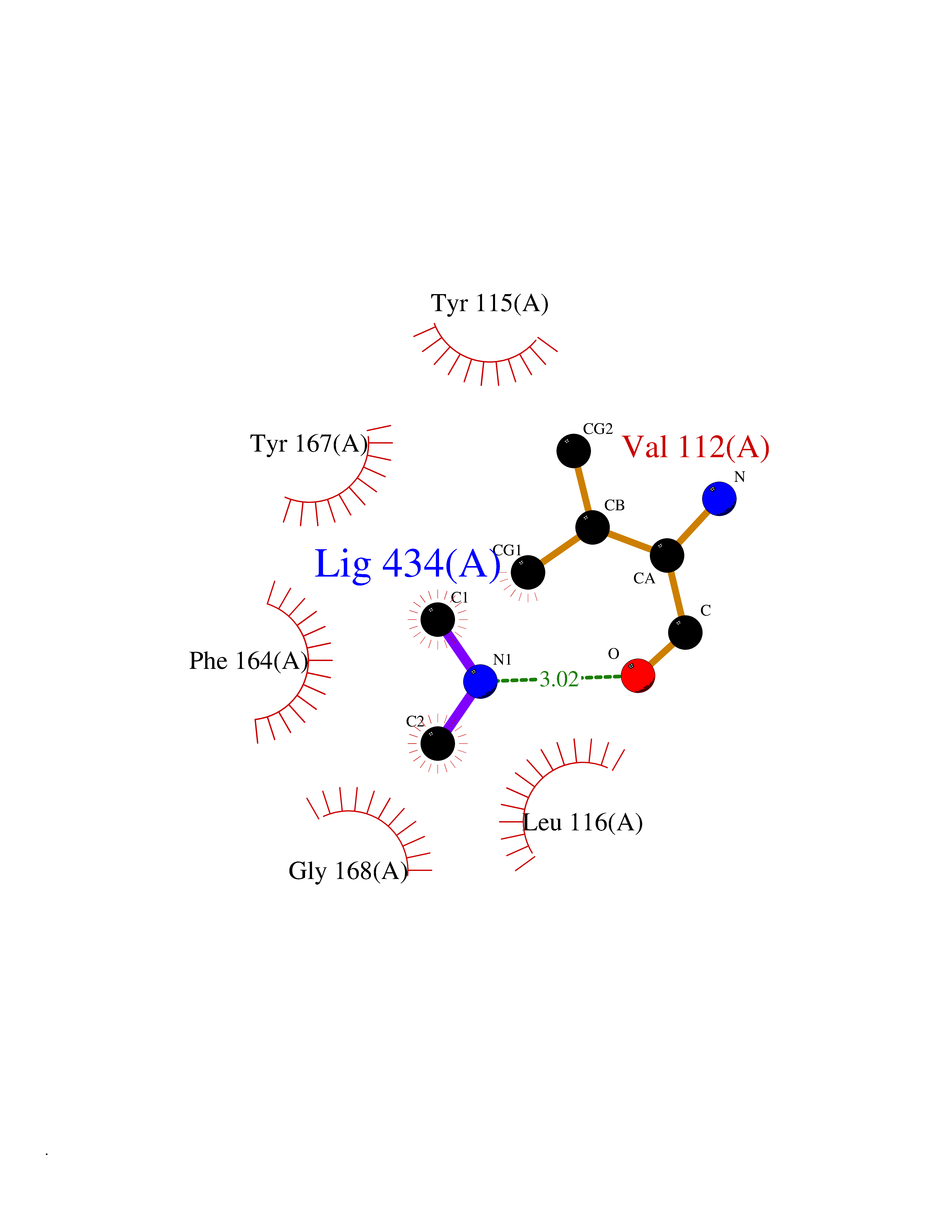 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Membrane copper amine oxidase (AOC3) | 4BTX | 4.02 | |
Target general information Gen name AOC3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vascular adhesion protein-1; Vascular adhesion protein 1; VAP1; VAP-1; Semicarbazide-sensitive amine oxidase; SSAO; Membrane primary amine oxidase; HPAO; Copper amine oxidase Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Has semicarbazide-sensitive (SSAO) monoamine oxidase activity. May play a role in adipogenesis. Cell adhesion protein that participates in lymphocyte extravasation and recirculation by mediating the binding of lymphocytes to peripheral lymph node vascular endothelial cells in an L-selectin-independent fashion. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB04334; DB01275; DB00780 Interacts with Q3SXY8; O95484; Q7Z7G2; Q96BA8; Q6PI48; Q8TBE3; Q7Z5P4; P42858; O43765; Q16623 EC number EC 1.4.3.21 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell adhesion; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Signal-anchor; TPQ; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 157266 Length 1415 Aromaticity 0.12 Instability index 39.94 Isoelectric point 6 Charge (pH=7) -23.49 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 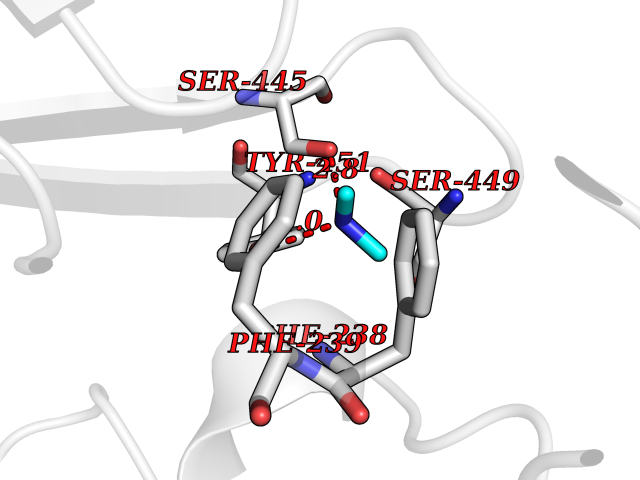 Pymol Map 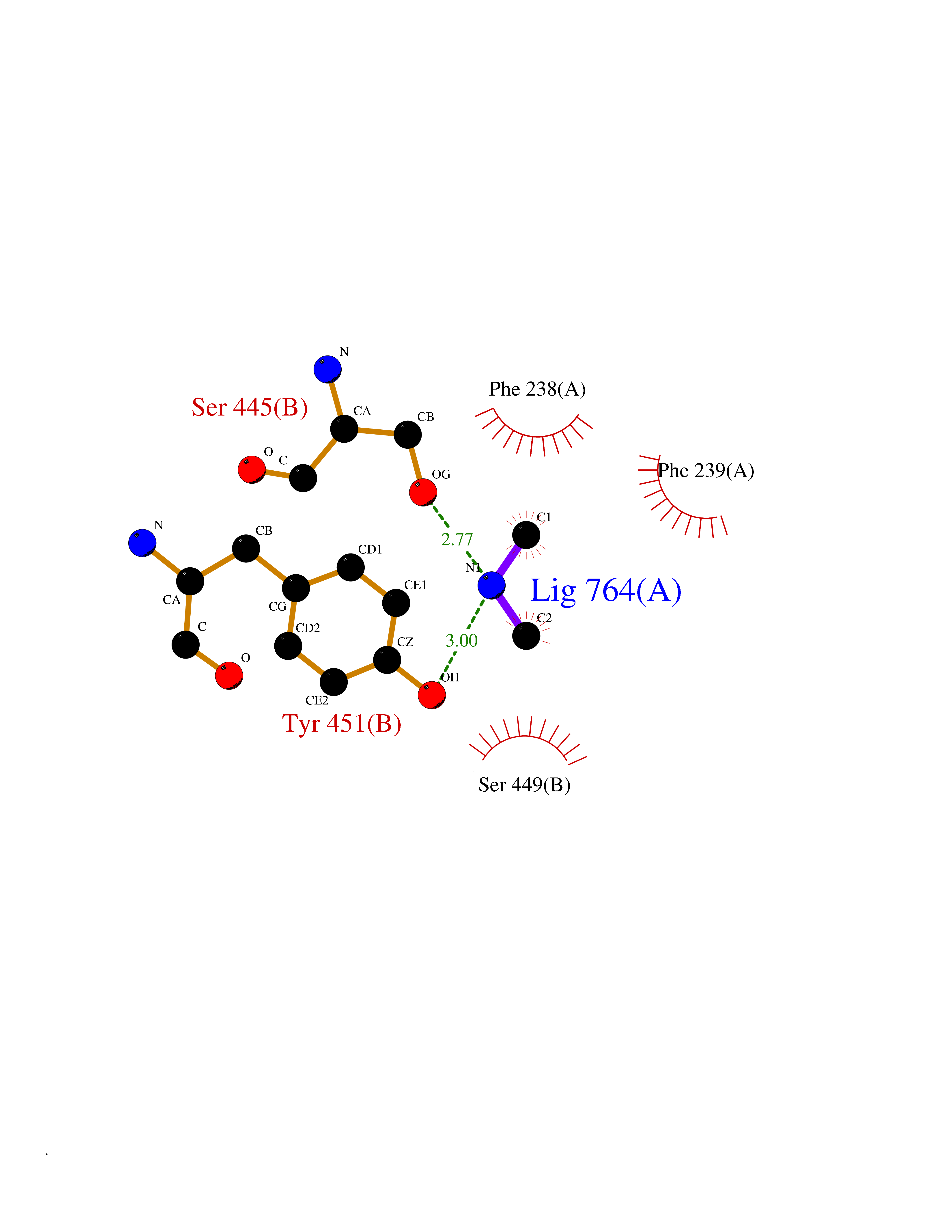 Ligplot Map 3D Binding mode Sequence PGQSQLFADLSREELTAVMRFLTQRLGPGLVDAAQARPSDNCVFSVELQLPPKAAALAHLDRGSPPPAREALAIVFFGRQPQPNVSELVVGPLPHPSYMRDVTVERHGGPLPYHRRPVLFQEYLDIDQMIFNRELPQASGLLHHCCFYKHRGRNLVTMTTAPRGLQSGDRATWFGLYYNISGAGFFLHHVGLELLVNHKALDPARWTIQKVFYQGRYYDSLAQLEAQFEAGLVNVVLIPDNGTGGSWSLKSPVPPGPAPPLQFYPQGPRFSVQGSRVASSLWTFSFGLGAFSGPRIFDVRFQGERLVYEISLQEALAIYGGNSPAAMTTRYVDGGFGMGKYTTPLTRGVDCPYLATYVDWHFLLESQAPKTIRDAFCVFEQNQGLPLRRHHSDLYSHYFGGLAETVLVVRSMSTLLNXDYVWDTVFHPSGAIEIRFYATGYISSAFLFGATGKYGNQVSEHTLGTVHTHSAHFKVDLDVAGLENWVWAEDMVFVPMAVPWSPEHQLQRLQVTRKLLEMEEQAAFLVGSATPRYLYLASNHSNKWGHPRGYRIQMLSFAGEPLPQNSSMARGFSWERYQLAVTQRKEEEPSSSSVFNQNDPWAPTVDFSDFINNETIAGKDLVAWVTAGFLHIPHAEDIPNTVTVGNGVGFFLRPYNFFDEDPSFYSADSIYFRGDQDAGACEVNPLACLPQAAACAPDLPAFSHGGFSHSQLFADLSREELTAVMRFLTQRLGPGLVDAAQARPSDNCVFSVELQLPPKAAALAHLDRGSPPPAREALAIVFFGRQPQPNVSELVVGPLPHPSYMRDVTVERHGGPLPYHRRPVLFQEYLDIDQMIFNRELPQASGLLHHCCFYKHRGRNLVTMTTAPRGLQSGDRATWFGLYYNISGAGFFLHHVGLELLVNHKALDPARWTIQKVFYQGRYYDSLAQLEAQFEAGLVNVVLIPDNGTGGSWSLKSPVPPGPAPPLQFYPQGPRFSVQGSRVASSLWTFSFGLGAFSGPRIFDVRFQGERLVYEISLQEALAIYGGNSPAAMTTRYVDGGFGMGKYTTPLTRGVDCPYLATYVDWHFLLESQAPKTIRDAFCVFEQNQGLPLRRHHSDLYSHYFGGLAETVLVVRSMSTLLNXDYVWDTVFHPSGAIEIRFYATGYISSAFLFGATGKYGNQVSEHTLGTVHTHSAHFKVDLDVAGLENWVWAEDMVFVPMAVPWSPEHQLQRLQVTRKLLEMEEQAAFLVGSATPRYLYLASNHSNKWGHPRGYRIQMLSFAGEPLPQNSSMARGFSWERYQLAVTQRKEEEPSSSSVFNQNDPWAPTVDFSDFINNETIAGKDLVAWVTAGFLHIPHAEDIPNTVTVGNGVGFFLRPYNFFDEDPSFYSADSIYFRGDQDAGACEVNPLACLPQAAACAPDLPAFSHGGFSH Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Beta-galactosidase (GLB1) | 3THD | 4.02 | |
Target general information Gen name GLB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lactase; Elastin receptor 1; ELNR1; Acid beta-galactosidase Protein family Glycosyl hydrolase 35 family Biochemical class NA Function Isoform 1: Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans. Related diseases GM1-gangliosidosis 1 (GM1G1) [MIM:230500]: An autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1-gangliosidosis type 1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life. {ECO:0000269|PubMed:10338095, ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:10839995, ECO:0000269|PubMed:1487238, ECO:0000269|PubMed:15365997, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15791924, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816, ECO:0000269|Ref.28, ECO:0000269|Ref.31}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 2 (GM1G2) [MIM:230600]: A gangliosidosis characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:12644936, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 3 (GM1G3) [MIM:230650]: A gangliosidosis with a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15986423, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8198123, ECO:0000269|Ref.28, ECO:0000269|Ref.30}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 4B (MPS4B) [MIM:253010]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:12393180, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:7586649}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04465 Interacts with Q8NBJ4; Q3KNW5; Q9BRI3; P30825 EC number EC 3.2.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Gangliosidosis; Glycoprotein; Glycosidase; Hydrolase; Lysosome; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 67980.6 Length 605 Aromaticity 0.13 Instability index 40.91 Isoelectric point 5.81 Charge (pH=7) -9.05 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map 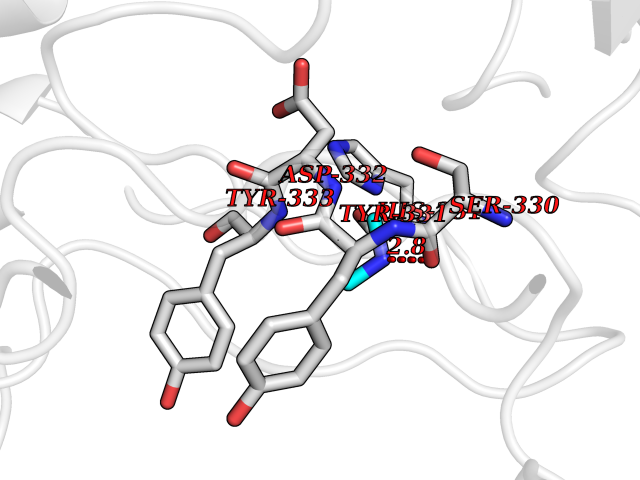 Pymol Map 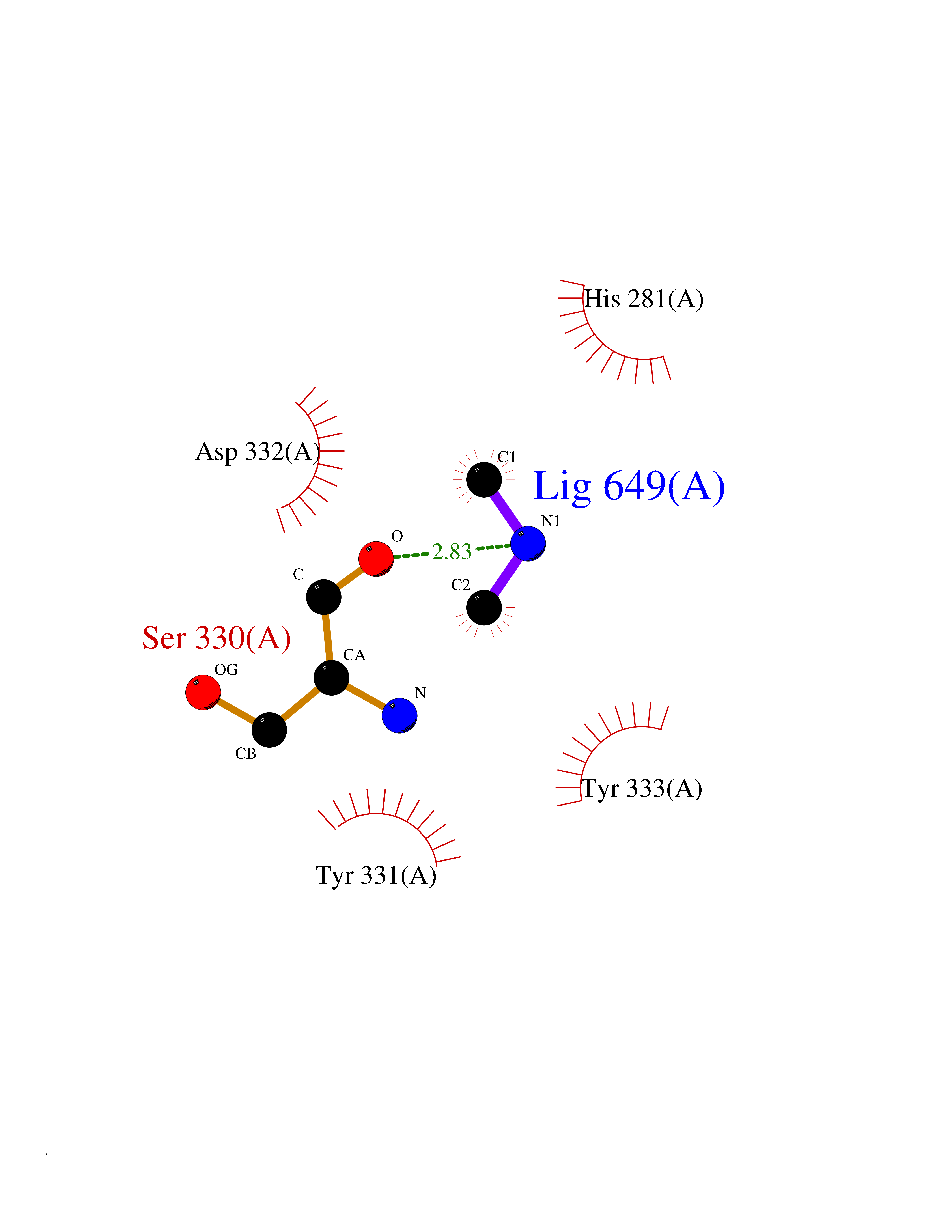 Ligplot Map 3D Binding mode Sequence QRMFEIDYSRDSFLKDGQPFRYISGSIHYSRVPRFYWKDRLLKMKMAGLNAIQTYVPWNFHEPWPGQYQFSEDHDVEYFLRLAHELGLLVILRPGPYICAEWEMGGLPAWLLEKESILLRSSDPDYLAAVDKWLGVLLPKMKPLLYQNGGPVITVQVENEYGSYFACDFDYLRFLQKRFRHHLGDDVVLFTTDGAHKTFLKCGALQGLYTTVDFGTGSNITDAFLSQRKCEPKGPLINSEFYTGWLDHWGQPHSTIKTEAVASSLYDILARGASVNLYMFIGGTNFAYWNGANSPYAAQPTSYDYDAPLSEAGDLTEKYFALRNIIQKFEKVPEGPIPPSTPKFAYGKVTLEKLKTVGAALDILCPSGPIKSLYPLTFIQVKQHYGFVLYRTTLPQDCSNPAPLSSPLNGVHDRAYVAVDGIPQGVLERNNVITLNITGKAGATLDLLVENMGRVNYGAYINDFKGLVSNLTLSSNILTDWTIFPLDTEDAVRSHLGGWGHRNYTLPAFYMGNFSIPSGIPDLPQDTFIQFPGWTKGQVWINGFNLGRYWPARGPQLTLFVPQHILMTSAPNTITVLELEWAPCSSDDPELCAVTFVDRPVIGSS Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Complement C1s component (C1S) | 1ELV | 4.02 | |
Target general information Gen name C1S Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complement component 1 subcomponent s; Complement C1s subcomponent; C1-esterase; C1 esterase Protein family Peptidase S1 family Biochemical class Peptidase Function C1r activates C1s so that it can, in turn, activate C2 and C4. C1s B chain is a serine protease that combines with C1q and C1r to form C1, the first component of the classical pathway of the complement system. Related diseases Complement component C1s deficiency (C1SD) [MIM:613783]: A rare defect resulting in C1 deficiency and impaired activation of the complement classical pathway. C1 deficiency generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. {ECO:0000269|PubMed:11390518}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ehlers-Danlos syndrome, periodontal type, 2 (EDSPD2) [MIM:617174]: A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSPD2 is characterized by the association of typical features of Ehlers-Danlos syndrome with gingival recession and severe early-onset periodontal disease, leading to premature loss of permanent teeth. EDSPD2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:27745832}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02371; DB09228; DB09130; DB12831; DB06404; DB14996; DB01593; DB14487; DB14533; DB14548 Interacts with P00736; P09871; P06681; O43889-2; Q9H6H4; P05155 EC number EC 3.4.21.42 Uniprot keywords 3D-structure; Calcium; Complement pathway; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Ehlers-Danlos syndrome; Glycoprotein; Hydrolase; Hydroxylation; Immunity; Innate immunity; Metal-binding; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal; Sushi Protein physicochemical properties Chain ID A Molecular weight (Da) 33278.6 Length 303 Aromaticity 0.1 Instability index 33.69 Isoelectric point 5.16 Charge (pH=7) -7.95 2D Binding mode Binding energy (Kcal/mol) -5.48 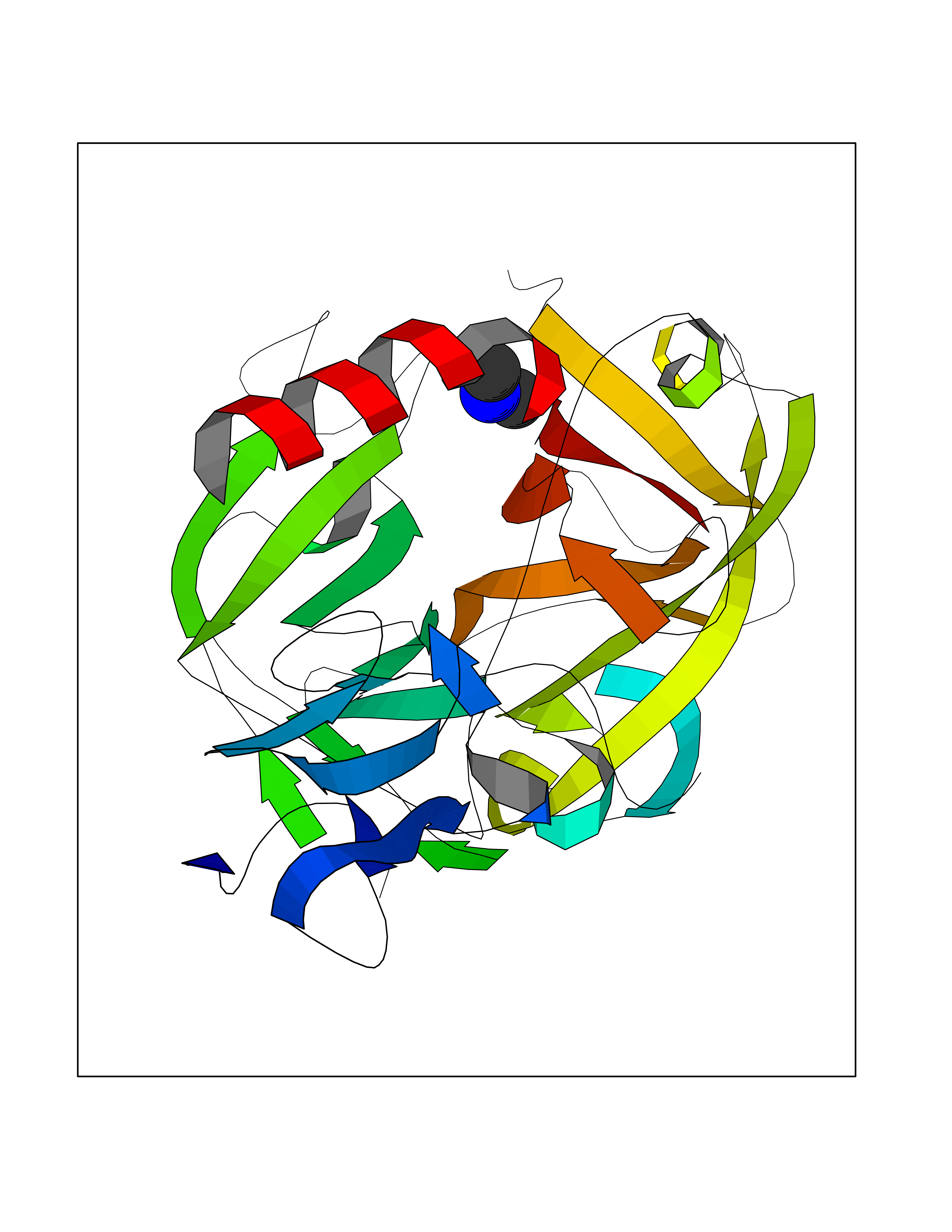 Molscript Map 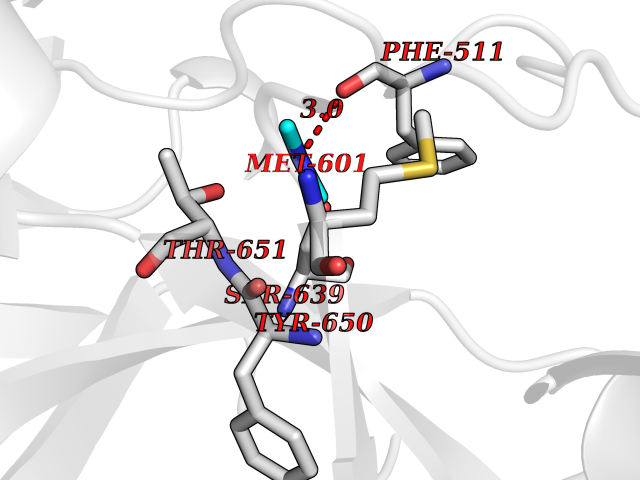 Pymol Map 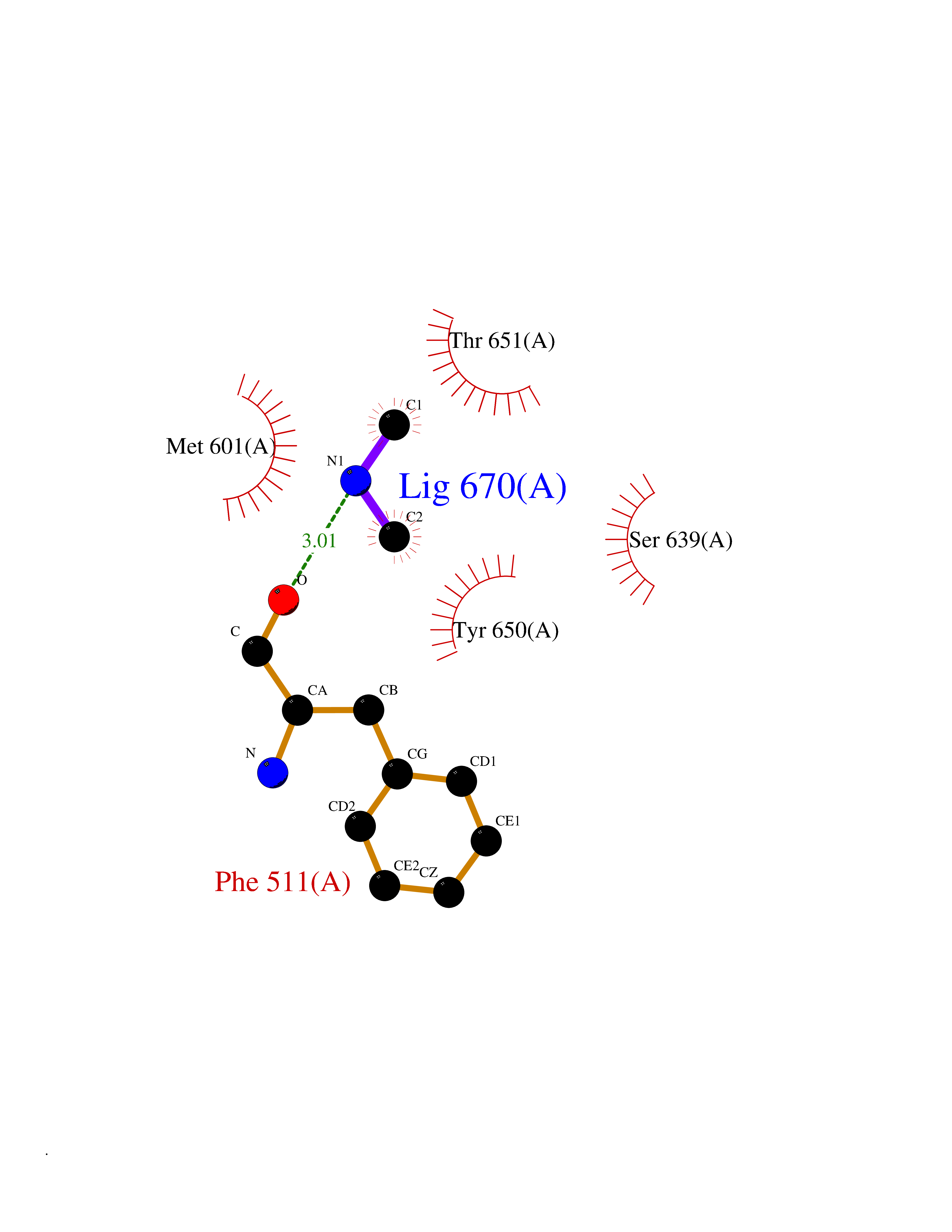 Ligplot Map 3D Binding mode Sequence LDCGIPESIENGKVEDPESTLFGSVIRYTCEEPYYYMEGGGEYHCAGNGSWVNEVLGPELPKCVPVCGVPREPFIIGGSDADIKNFPWQVFFDNPWAGGALINEYWVLTAAHVVEGNREPTMYVGSTSVQKMLTPEHVFIHPGWKLLAVPEGRTNFDNDIALVRLKDPVKMGPTVSPICLPGTSSDYNLMDGDLGLISGWGRTEKRDRAVRLKAARLPVAPLRKCKEVAYVFTPNMICAGGEKGMDSCKGDSGGAFAVQDPNDKTKFYAAGLVSWGPQCGTYGLYTRVKNYVDWIMKTMQENS Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Cathepsin G (CTSG) | 1KYN | 4.02 | |
Target general information Gen name CTSG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CG Protein family Peptidase S1 family Biochemical class Peptidase Function Cleaves complement C3. Has antibacterial activity against the Gram-negative bacterium P. aeruginosa, antibacterial activity is inhibited by LPS from P. aeruginosa, Z-Gly-Leu-Phe-CH2Cl and phenylmethylsulfonyl fluoride. Serine protease with trypsin- and chymotrypsin-like specificity. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04016; DB02360 Interacts with A8MQ03; Q15323; Q7Z3S9; Q9NRD5 EC number EC 3.4.21.20 Uniprot keywords 3D-structure; Antibiotic; Antimicrobial; Cell membrane; Chemotaxis; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Membrane; Nucleus; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 25353.8 Length 223 Aromaticity 0.06 Instability index 66.1 Isoelectric point 11.51 Charge (pH=7) 23.24 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 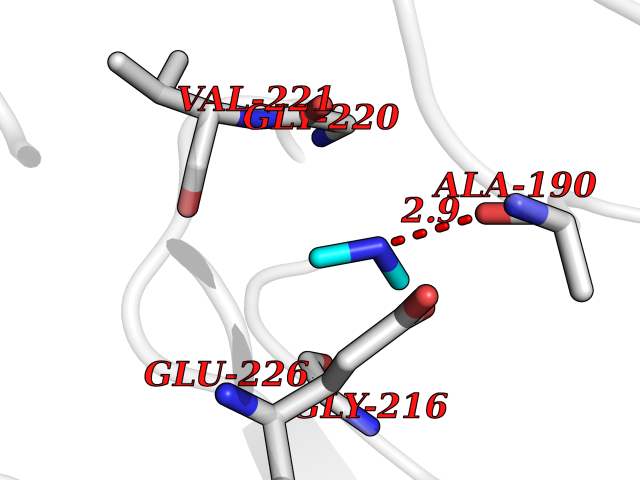 Pymol Map 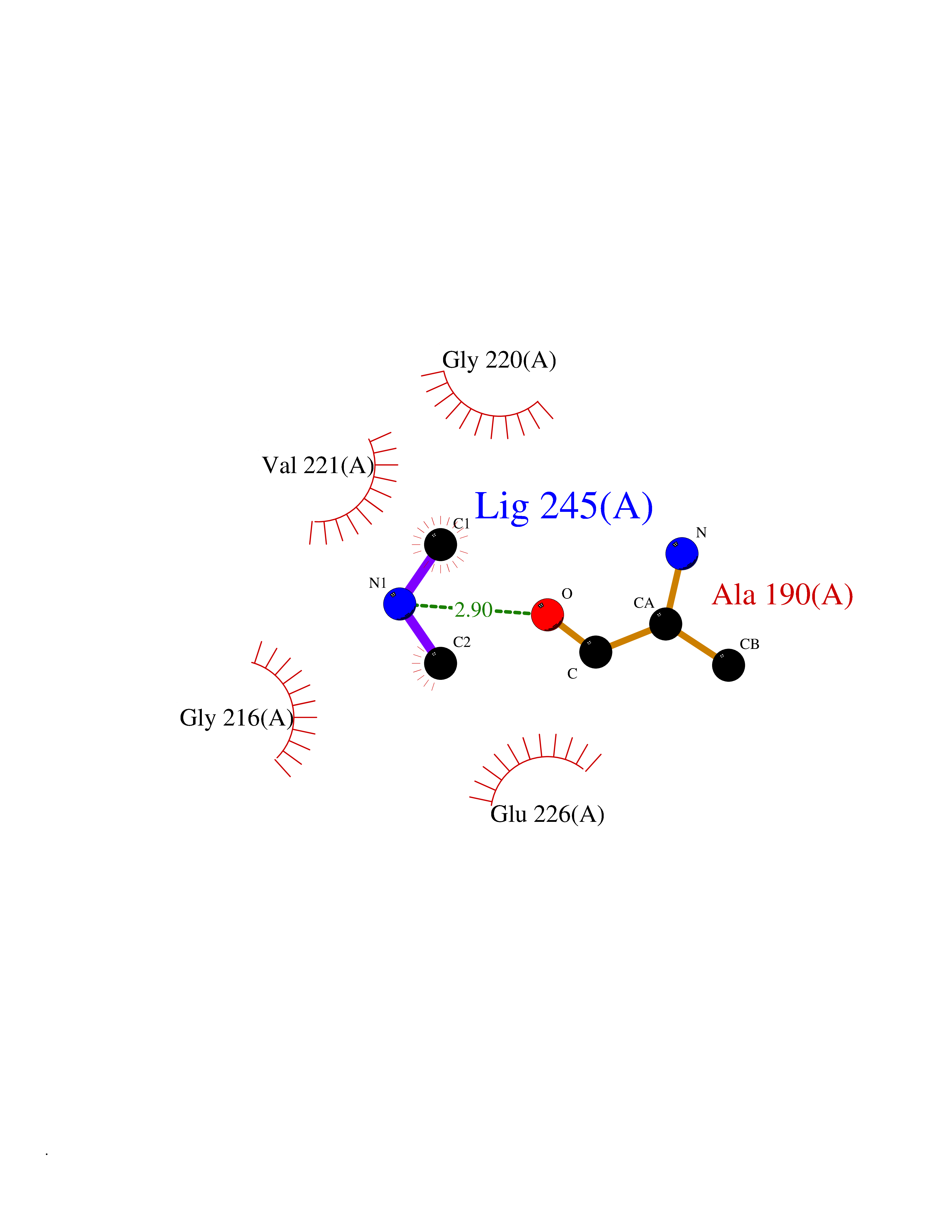 Ligplot Map 3D Binding mode Sequence IIGGRESRPHSRPYMAYLQIQSPAGQSRCGGFLVREDFVLTAAHCWGSNINVTLGAHNIQRRENTQQHITARRAIRHPQYNQRTIQNDIMLLQLSRRVRRNRNVNPVALPRAQEGLRPGTLCTVAGWGRVSMRRGTDTLREVQLRVQRDRQCLRIFGSYDPRRQICVGDRRERKAAFKGDSGGPLLCNNVAHGIVSYGKSSGVPPEVFTRVSSFLPWIRTTMR Hydrogen bonds contact Hydrophobic contact | ||||