Job Results:
Ligand
Structure
Job ID
43a62498dd267475554cbec83e92e777
Job name
NA
Time
2025-02-13 15:08:06
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Histamine H3 receptor (H3R) | 7F61 | 4.03 | |
Target general information Gen name HRH3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histamine receptor 3; HH3R; GPCR97; G-protein coupled receptor 97; G protein-coupled receptor 97 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signals through the inhibition of adenylate cyclase and displays high constitutive activity (spontaneous activity in the absence of agonist). Agonist stimulation of isoform 3 neither modified adenylate cyclase activity nor induced intracellular calcium mobilization. The H3 subclass of histamine receptors could mediate the histamine signals in CNS and peripheral nervous system. Related diseases Immunodeficiency 48 (IMD48) [MIM:269840]: A form of severe immunodeficiency characterized by a selective absence of CD8+ T-cells. {ECO:0000269|PubMed:11123350, ECO:0000269|PubMed:11412303, ECO:0000269|PubMed:18509675, ECO:0000269|PubMed:8124727, ECO:0000269|PubMed:8202713}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Autoimmune disease, multisystem, infantile-onset, 2 (ADMIO2) [MIM:617006]: An autosomal recessive, autoimmune disorder characterized by systemic manifestations including blistering skin disease, uncontrollable bullous pemphigoid, inflammatory colitis, autoimmune hypothyroidism, proteinuria and nephrotic syndrome. {ECO:0000269|PubMed:26783323}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01238; DB06698; DB05381; DB17087; DB05080; DB00768; DB11642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34321.1 Length 301 Aromaticity 0.16 Instability index 32.22 Isoelectric point 9.63 Charge (pH=7) 15.11 2D Binding mode Binding energy (Kcal/mol) -5.49 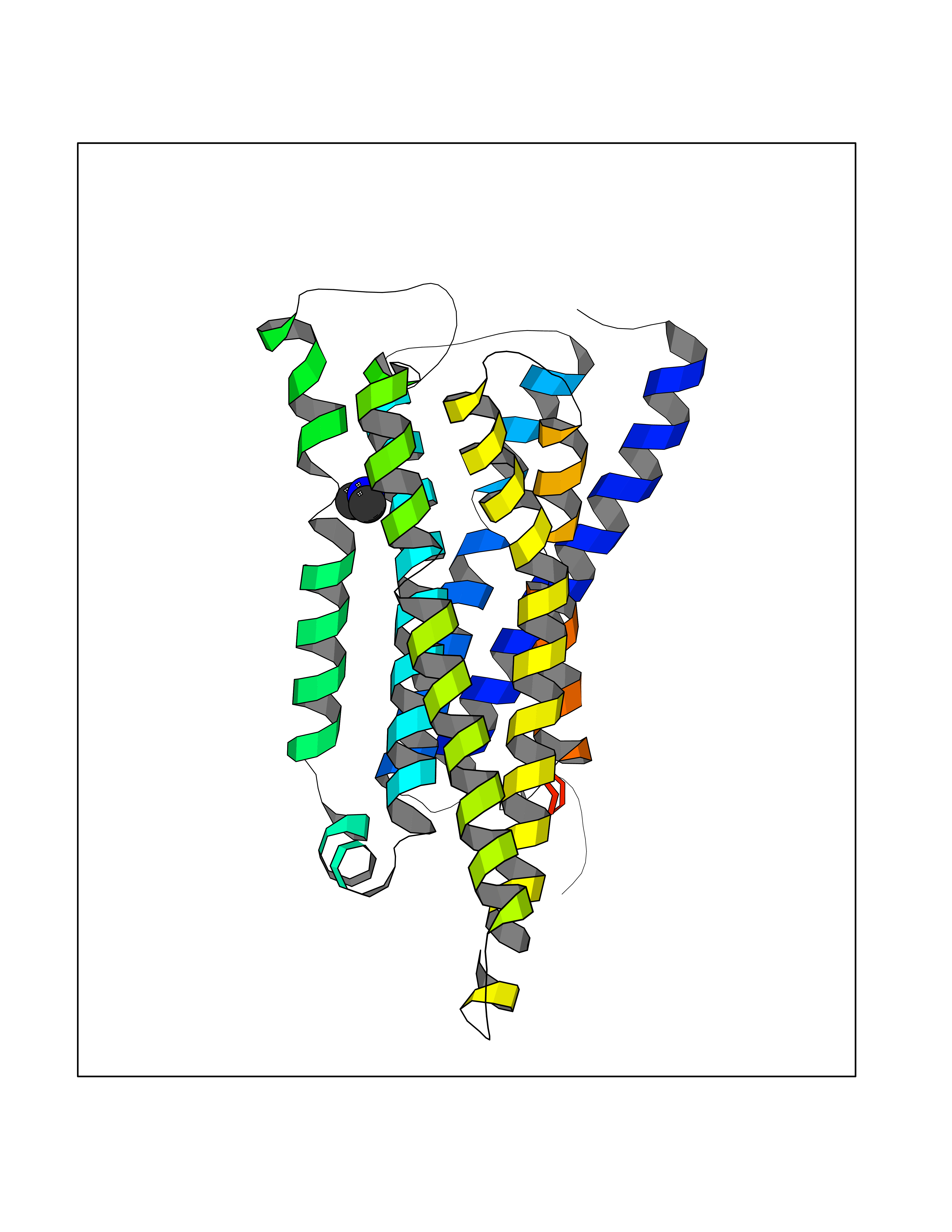 Molscript Map 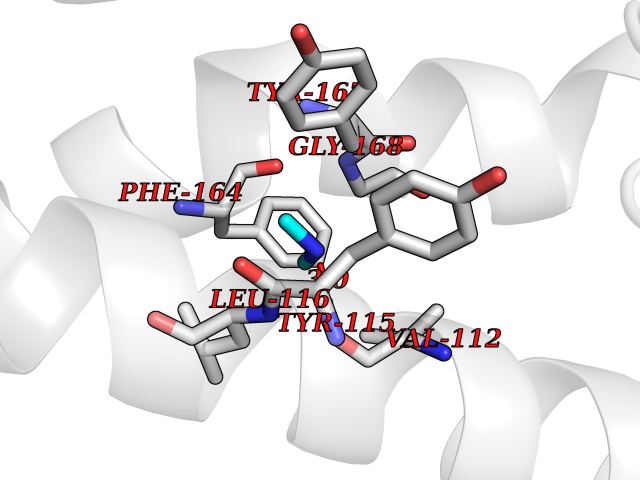 Pymol Map 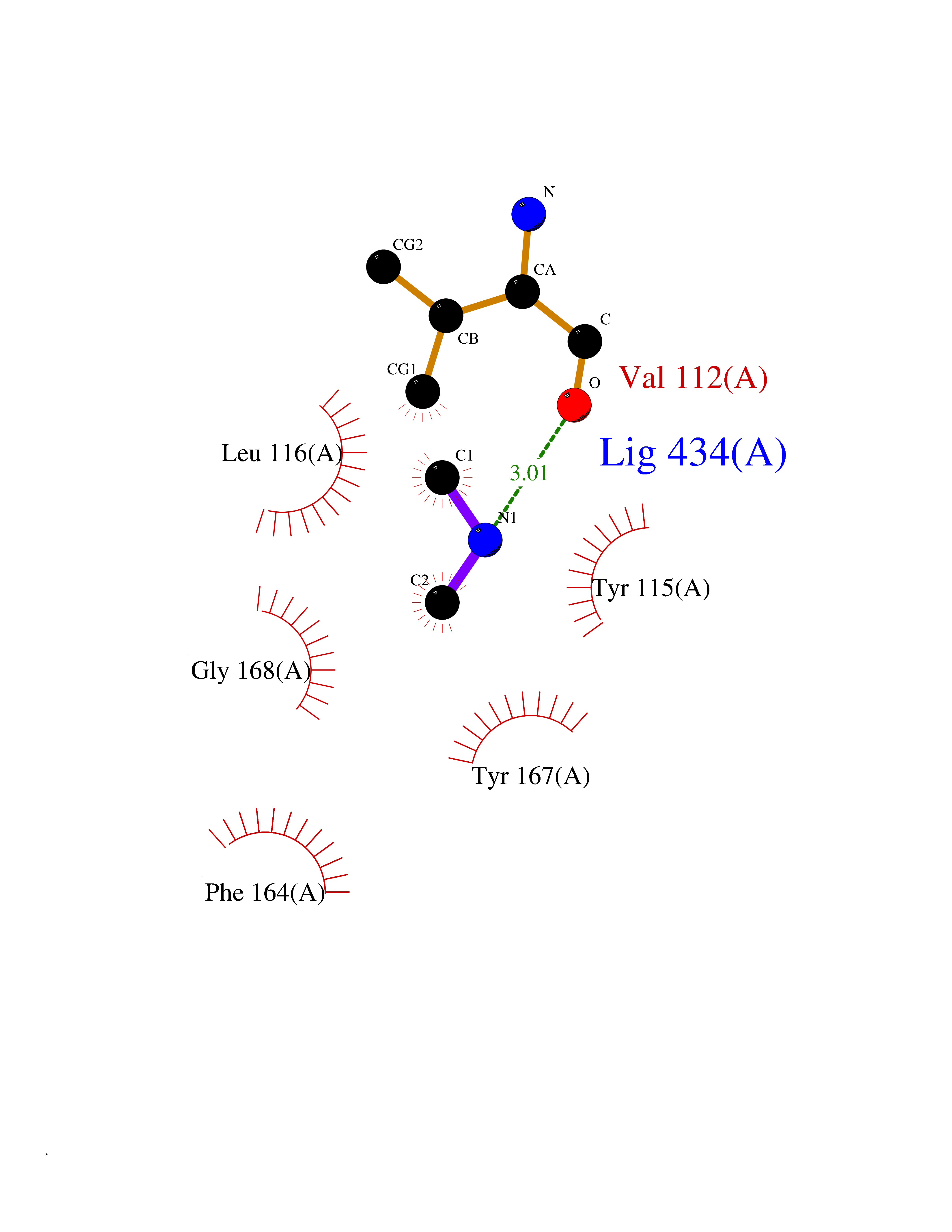 Ligplot Map 3D Binding mode Sequence RGFSAAWTAVLAALMALLIVATVLGNALVMLAFVADSSLRTQNNFFLLNLAISDFLVGAFCIPLYVPYVLTGRWTFGRGLCKLWLVVDYLLCTSKAFNIVLISYDRFLSVTRAVSYRAQQGDTRRAVRKMLLVWVLAFLLYGPAILSWEYLSGGSSIPEGHCYAEFFYNWYFLITASTLEFFTPFLSVTFFNLSIYLNIQRRTRLRLDGAREAAGRFRLSRDRKVAKSLAVIVSIFGLCWAPYTLLMIIRAACHGHCVPDYWYETSFWLLWANSAVNPVLYPLCHHSFRRAFTKLLCPQKL Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Deubiquitinating enzyme 1 (USP1) | 7ZH4 | 4.03 | |
Target general information Gen name USP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hUBP; Ubiquitin-specific-processing protease 1; Ubiquitin thioesterase 1; Ubiquitin carboxyl-terminal hydrolase 1 Protein family Peptidase C19 family Biochemical class Peptidase Function Involved in PCNA-mediated translesion synthesis (TLS) by deubiquitinating monoubiquitinated PCNA. Has almost no deubiquitinating activity by itself and requires the interaction with WDR48 to have a high activity. Negative regulator of DNA damage repair which specifically deubiquitinates monoubiquitinated FANCD2. Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8TAF3; Q8TAF3-1 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Autocatalytic cleavage; DNA damage; DNA repair; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID D Molecular weight (Da) 32426 Length 285 Aromaticity 0.1 Instability index 50.73 Isoelectric point 5.85 Charge (pH=7) -4.67 2D Binding mode Binding energy (Kcal/mol) -5.49 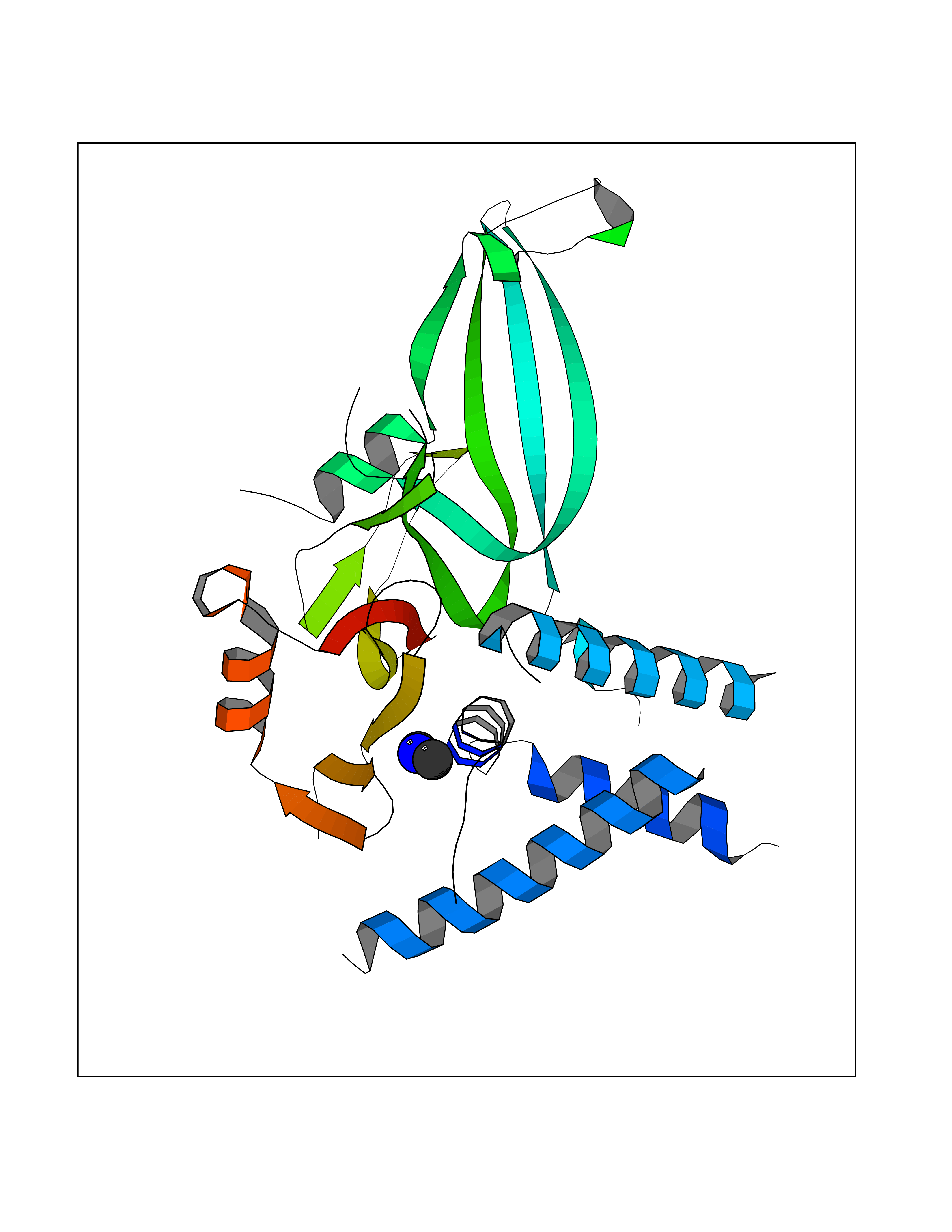 Molscript Map 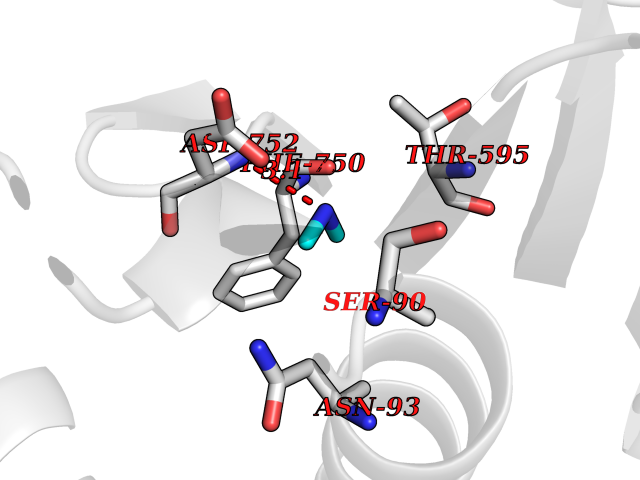 Pymol Map 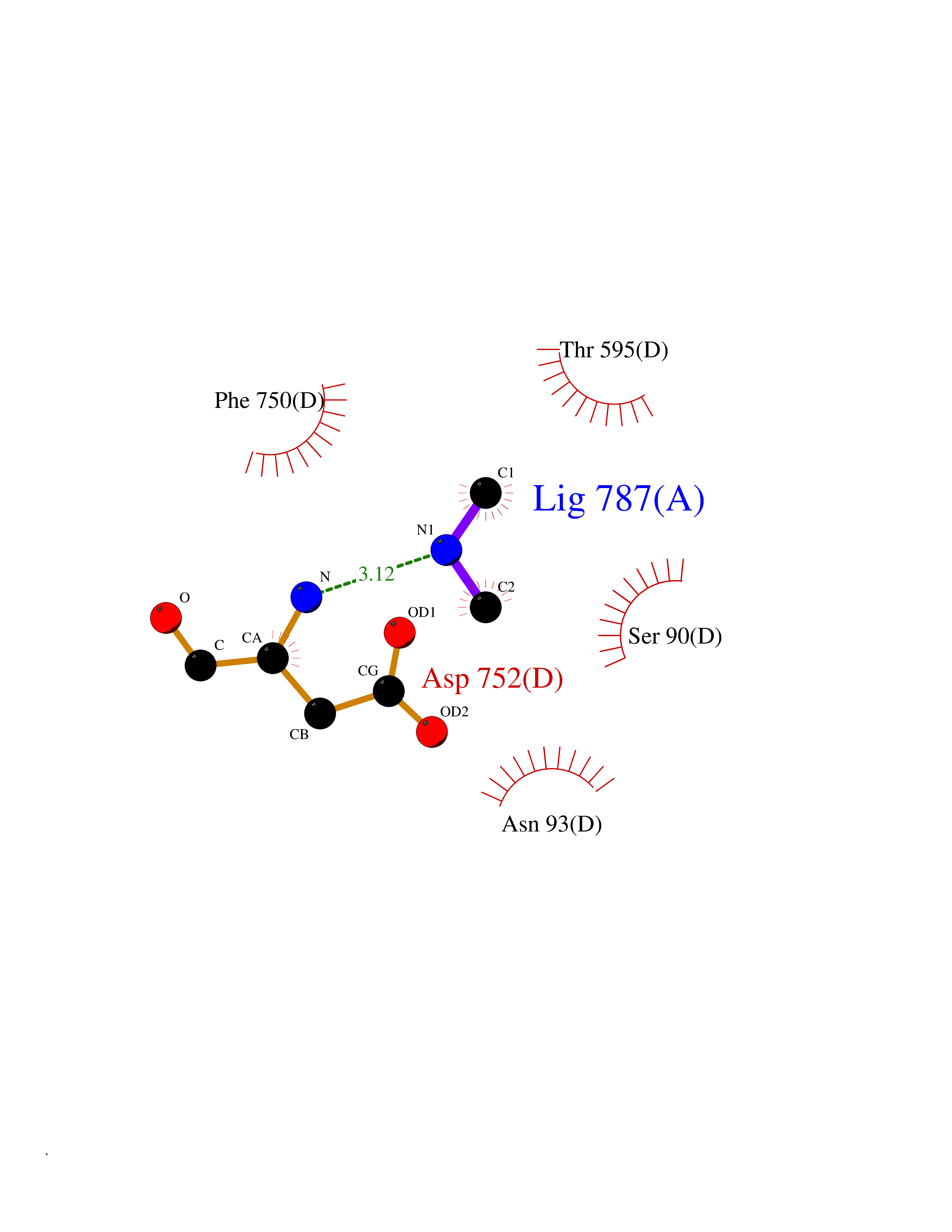 Ligplot Map 3D Binding mode Sequence GLNNLGNTSYLNSILQVLYFCPGFKSGVKHLFNIISRKKYELICSLQSLIISVEQLQASFLLNPLQHDAQEVLQCILGNIQETCQLLKKGFELVEKLFQGQLVLRTRCLECESLTERREDFQDISVPVQEDMKTLRWAISQFASVERIVGEDKYFCENCHHYTEAERSLLFDKMPEVITIHLKCFAASGLSKINTPLLTPLKLSLEEWSTKPTNDSYGLFAVVMHSGITISSGHYTASVKVTYEGKWLLFDDSEVKVTEEKDFLNSLSPSTSPTSTPYLLFYKKL Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Lysine-specific demethylase 4C (KDM4C) | 4XDO | 4.03 | |
Target general information Gen name KDM4C Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms KIAA0780; Jumonji domain-containing protein 2C; JmjC domain-containing histone demethylation protein 3C; JMJD2C; JHDM3C; Gene amplified in squamous cell carcinoma 1 protein; GASC1; GASC-1 protein Protein family JHDM3 histone demethylase family Biochemical class Paired donor oxygen oxidoreductase Function Does not demethylate histone H3 'Lys-4', H3 'Lys-27' nor H4 'Lys-20'. Demethylates trimethylated H3 'Lys-9' and H3 'Lys-36' residue, while it has no activity on mono- and dimethylated residues. Demethylation of Lys residue generates formaldehyde and succinate. Histone demethylase that specifically demethylates 'Lys-9' and 'Lys-36' residues of histone H3, thereby playing a central role in histone code. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Dioxygenase; Iron; Metal-binding; Nucleus; Oxidoreductase; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 39355.6 Length 338 Aromaticity 0.14 Instability index 38.34 Isoelectric point 8.04 Charge (pH=7) 2.41 2D Binding mode Binding energy (Kcal/mol) -5.5 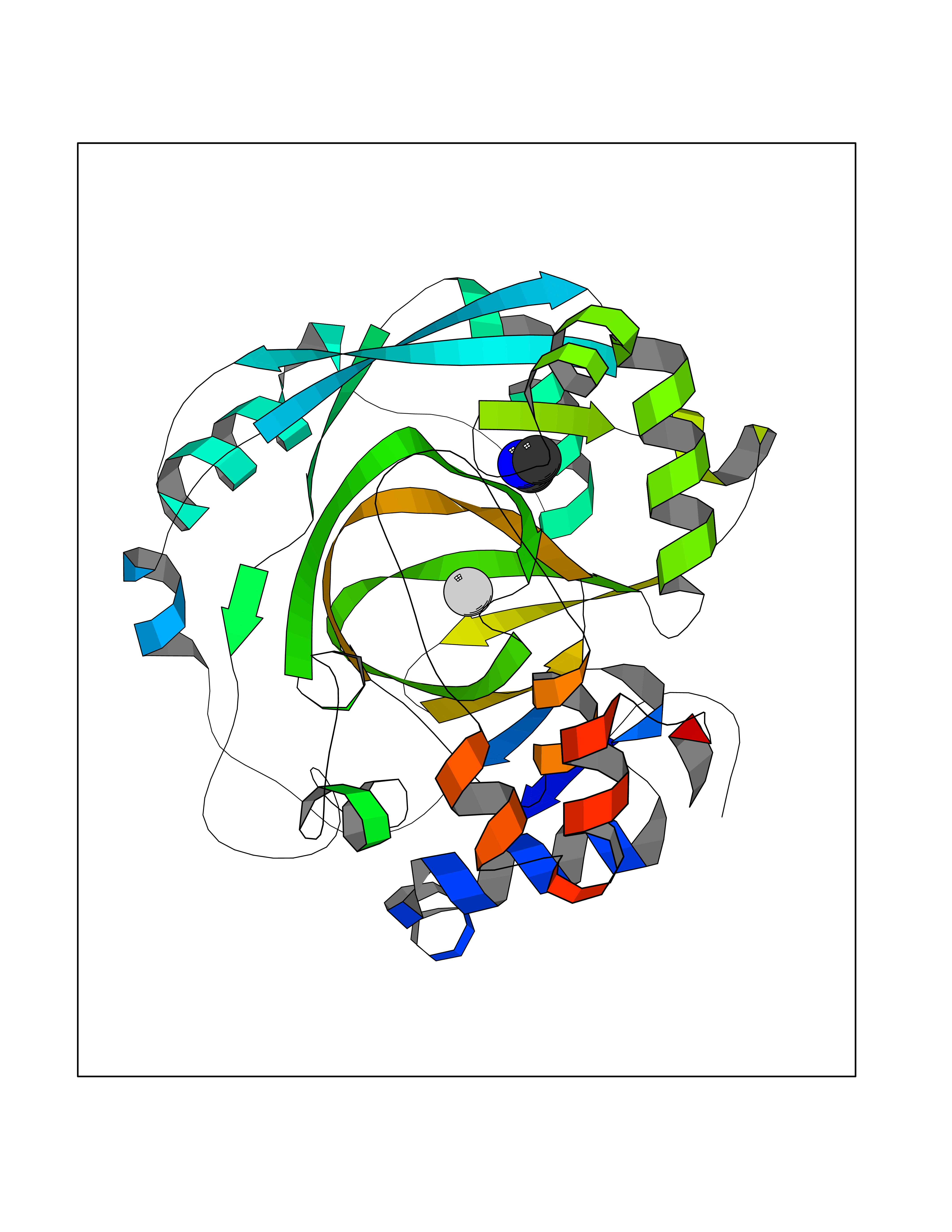 Molscript Map 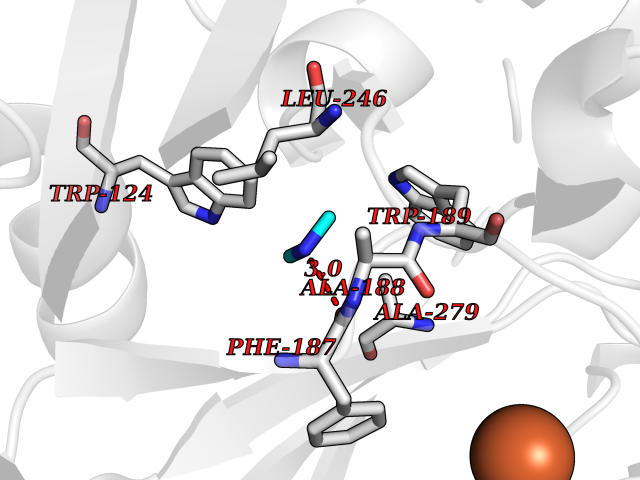 Pymol Map 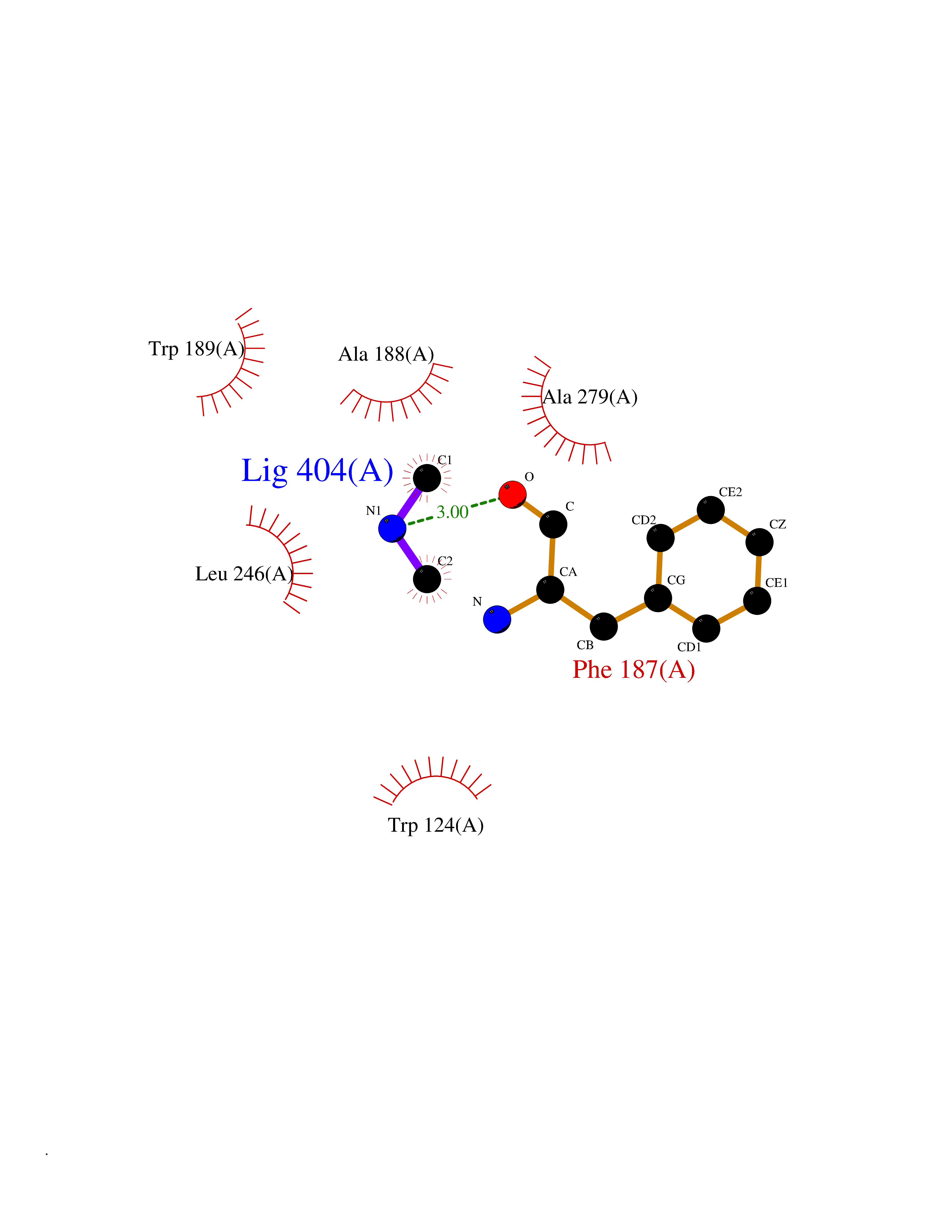 Ligplot Map 3D Binding mode Sequence LNPSCKIMTFRPSMEEFREFNKYLAYMESKGAHRAGLAKVIPPKEWKPRQCYDDIDNLLIPAPIQQMVTGQSGLFTQYNIQKKAMTVKEFRQLANSGKYCTPRYLDYEDLERKYWKNLTFVAPIYGADINGSIYDEGVDEWNIARLNTVLDVVEEECGISIEGVNTPYLYFGMWKTTFAWHTEDMDLYSINYLHFGEPKSWYAIPPEHGKRLERLAQGFFPSSSQGCDAFLRHKMTLISPSVLKKYGIPFDKITQEAGEFMITFPYGYHAGFNHGFNCAESTNFATVRWIDYGKVAKLCTCRKDMVKISMDIFVRKFQPDRYQLWKQGKDIYTIDHTK Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Carbonic anhydrase II (CA-II) | 3K34 | 4.02 | |
Target general information Gen name CA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase C; Carbonic anhydrase 2; Carbonate dehydratase II; CAC Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Can hydrate cyanamide to urea. Involved in the regulation of fluid secretion into the anterior chamber of the eye. Contributes to intracellular pH regulation in the duodenal upper villous epithelium during proton-coupled peptide absorption. Stimulates the chloride-bicarbonate exchange activity of SLC26A6. Essential for bone resorption and osteoclast differentiation. Related diseases Osteopetrosis, autosomal recessive 3 (OPTB3) [MIM:259730]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability. {ECO:0000269|PubMed:15300855, ECO:0000269|PubMed:1542674, ECO:0000269|PubMed:1928091, ECO:0000269|PubMed:8834238, ECO:0000269|PubMed:9143915}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07596; DB03333; DB08418; DB04081; DB08416; DB02479; DB07467; DB03950; DB03594; DB03294; DB04763; DB03270; DB08083; DB06954; DB08659; DB08046; DB02087; DB08156; DB04203; DB04394; DB08782; DB04549; DB02221; DB04180; DB03039; DB02861; DB02429; DB08202; DB04600; DB04601; DB01784; DB03385; DB03697; DB04002; DB07632; DB07050; DB06891; DB08645; DB08765; DB00819; DB03877; DB03262; DB03598; DB04089; DB01964; DB03526; DB04371; DB02220; DB03221; DB02602; DB02535; DB00436; DB00562; DB01194; DB00482; DB00880; DB02679; DB00606; DB02866; DB01144; DB00869; DB08846; DB01031; DB00311; DB08157; DB01942; DB00695; DB00774; DB08165; DB02292; DB03975; DB00703; DB00232; DB02610; DB07742; DB03844; DB02069; DB02986; DB08301; DB07048; DB03596; DB07476; DB01748; DB08155; DB01671; DB07710; DB01325; DB09460; DB09472; DB02894; DB00391; DB08329; DB07363; DB00273; DB01021; DB03904; DB00580; DB14533; DB14548; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Lyase; Membrane; Metal-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29027.4 Length 258 Aromaticity 0.1 Instability index 20.07 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 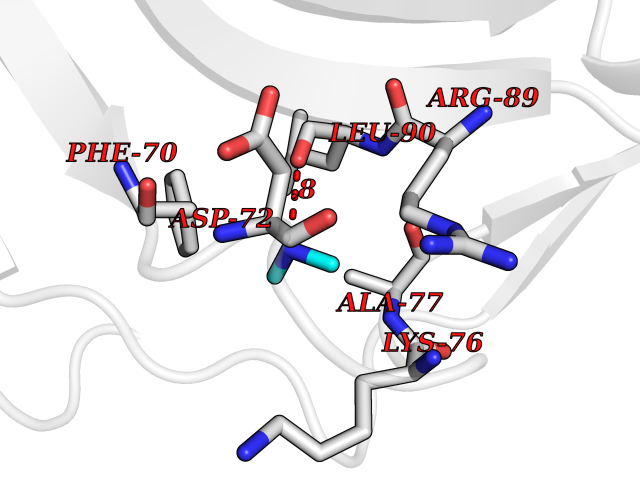 Pymol Map 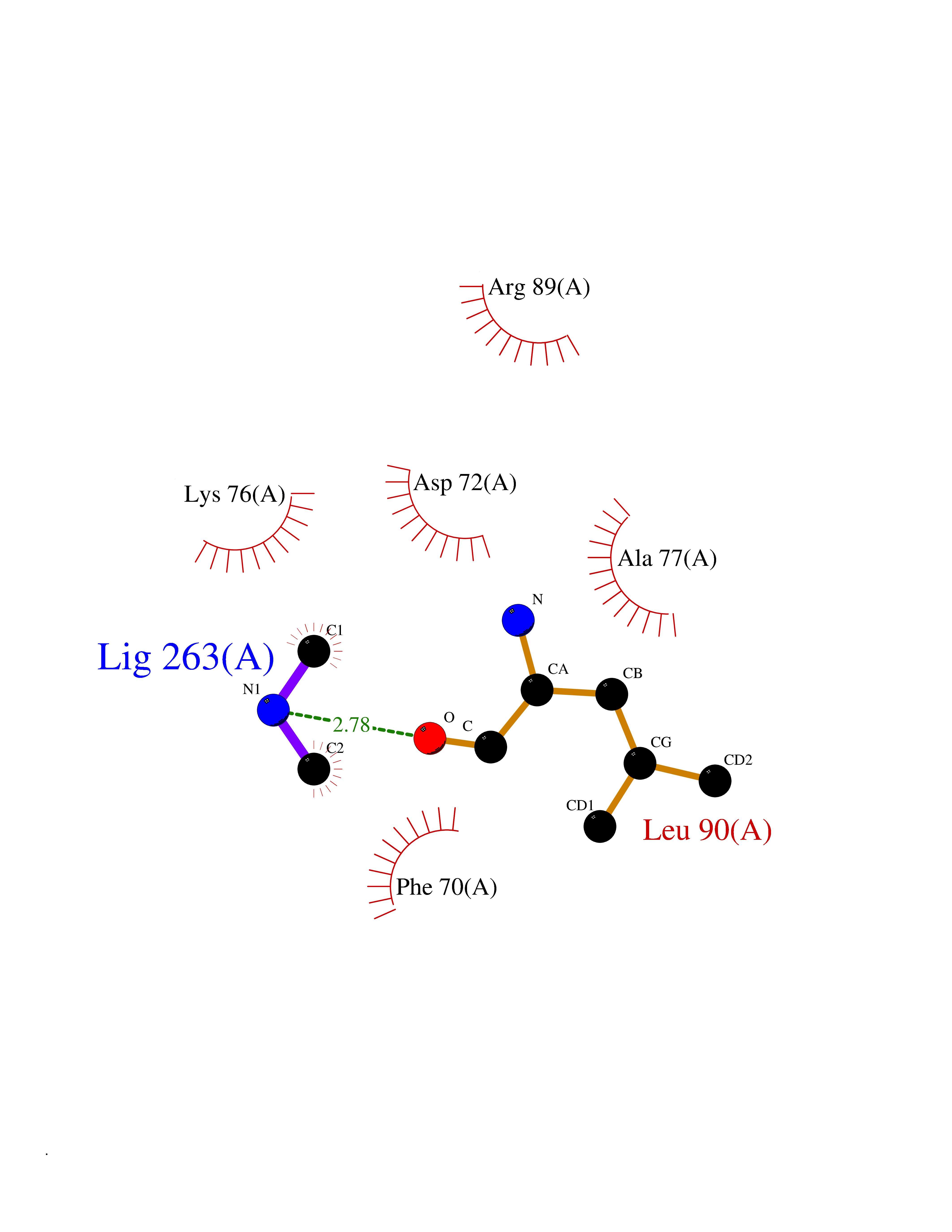 Ligplot Map 3D Binding mode Sequence HHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEPISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Mitochondrial aldehyde dehydrogenase (ALDH2) | 1O04 | 4.02 | |
Target general information Gen name ALDH2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde dehydrogenase, mitochondrial; ALDM; ALDHI; ALDH-E2; ALDH class 2 Protein family Aldehyde dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Second enzyme of the major oxidative pathway of alcohol metabolism. Catalyzes the chemical transformation from acetaldehyde to acetic acid. Additionally, functions as a protector against oxidative stress. Related diseases AMED syndrome, digenic (AMEDS) [MIM:619151]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. AMEDS is an autosomal recessive, digenic form characterized by childhood onset of bone marrow failure resulting in aplastic anemia, in association with global developmental delay, intellectual disability, and poor overall growth with short stature. {ECO:0000269|PubMed:33355142}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. AMEDS patients carry ADH5 biallelic variants and homozygous or heterozygous ALDH2 variant p.Glu504Lys, affecting protein activity. Cellular and animal studies demonstrate that the simultaneous loss of ALDH2 and ADH5 activities leads to an increase of cellular formaldehyde sensitivity and multisystem abnormalities including hematopoietic failure. {ECO:0000269|PubMed:33355142}. Drugs (DrugBank ID) DB01612; DB06770; DB04381; DB02115; DB00822; DB00536; DB00157; DB00435; DB00727; DB09117; DB06154; DB06207 Interacts with NA EC number EC 1.2.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; Dwarfism; Intellectual disability; Mitochondrion; NAD; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 50223.5 Length 462 Aromaticity 0.1 Instability index 33.68 Isoelectric point 5.29 Charge (pH=7) -7.87 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 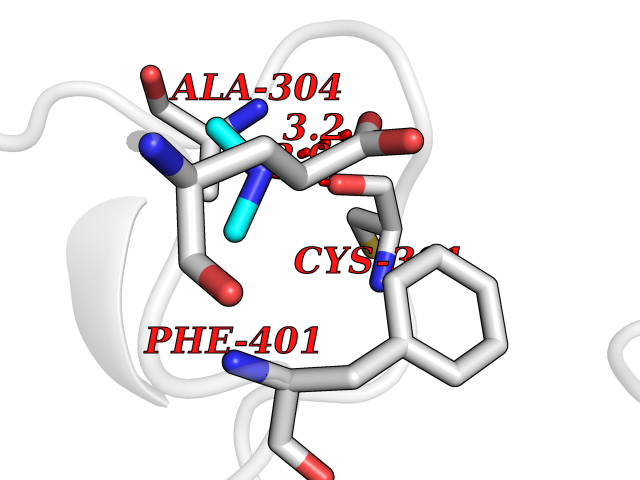 Pymol Map 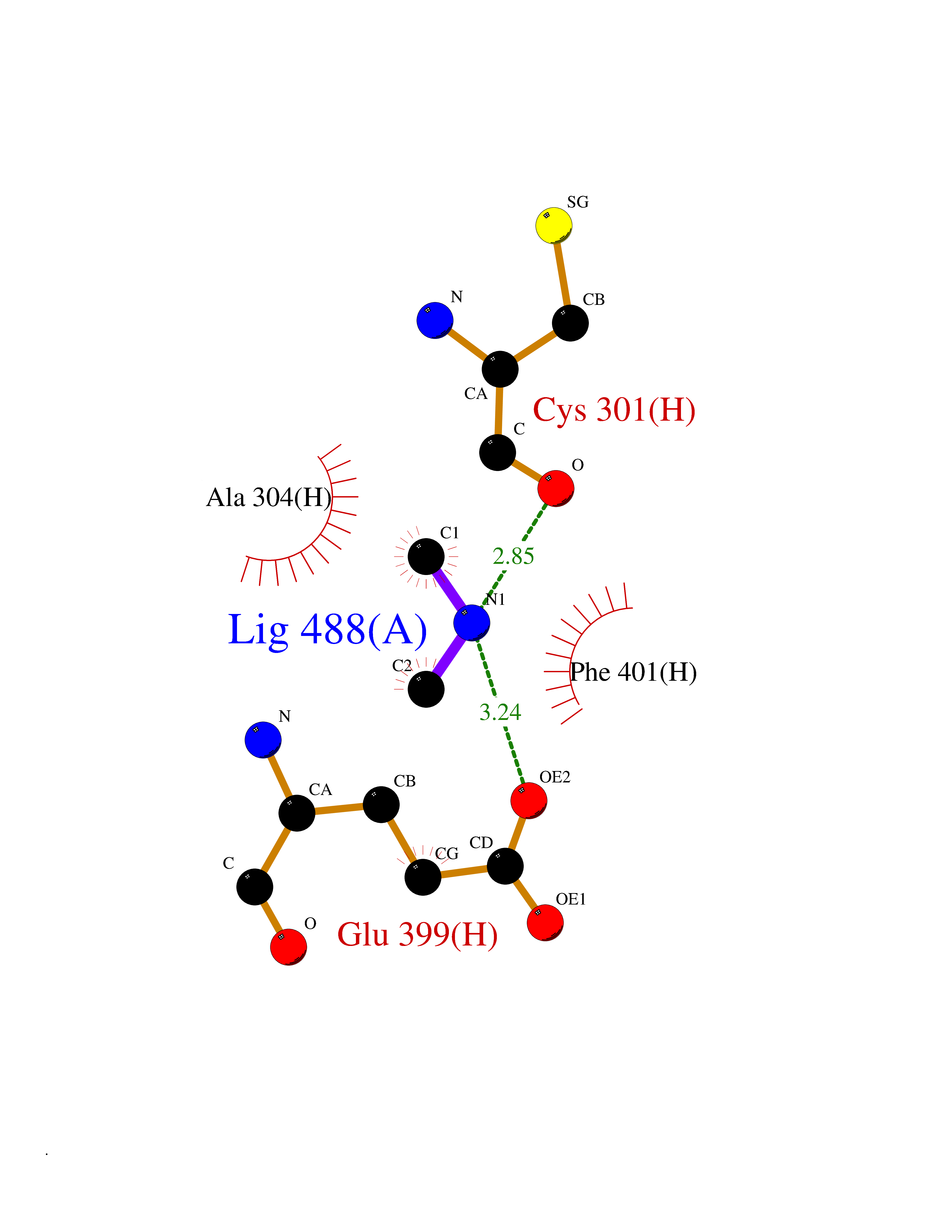 Ligplot Map 3D Binding mode Sequence AVPAPNQQPEVFCNQIFINNEWHDAVSRKTFPTVNPSTGEVICQVAEGDKEDVDKAVKAARAAFQLGSPWRRMDASHRGRLLNRLADLIERDRTYLAALETLDNGKPYVISYLVDLDMVLKCLRYYAGWADKEPVGVCGQIIPWNFPLLMQAWKLGPALATGNVVVMKVAEQTPLTALYVANLIKEAGFPPGVVNIVPGFGPTAGAAIASHEDVDKVAFTGSTEIGRVIQVAAGSSNLKRVTLELGGKSPNIIMSDADMDWAVEQAHFALFFNQGQCSCAGSRTFVQEDIYDEFVERSVARAKSRVVGNPFDSKTEQGPQVDETQFKKILGYINTGKQEGAKLLCGGGIAADRGYFIQPTVFGDVQDGMTIAKEEIFGPVMQILKFKTIEEVVGRANNSTYGLAAAVFTKDLDKANYLSQALQAGTVWVNCYDVFGAQSPFGGYKMSGSGRELGEYGLQAYT Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Bifunctional dihydrofolate reductase-thymidylate synthase | 1J3K | 4.02 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate K1 / Thailand) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydrofolate reductase family; Thymidylate synthase family Biochemical class Oxidoreductase Function Dihydrofolate reductase activity.Thymidylate synthase activity. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01131; DB00205; DB01299 Interacts with NA EC number 1.5.1.3; 2.1.1.45 Uniprot keywords 3D-structure; Methyltransferase; Multifunctional enzyme; NADP; Nucleotide biosynthesis; One-carbon metabolism; Oxidoreductase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61720.6 Length 525 Aromaticity 0.13 Instability index 33.9 Isoelectric point 8.79 Charge (pH=7) 11.61 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map 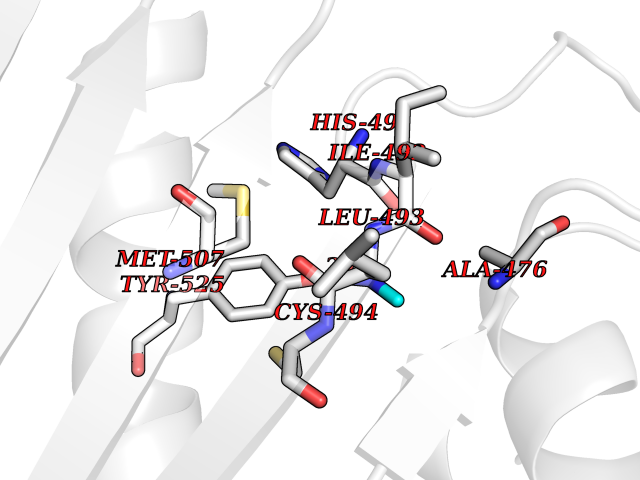 Pymol Map 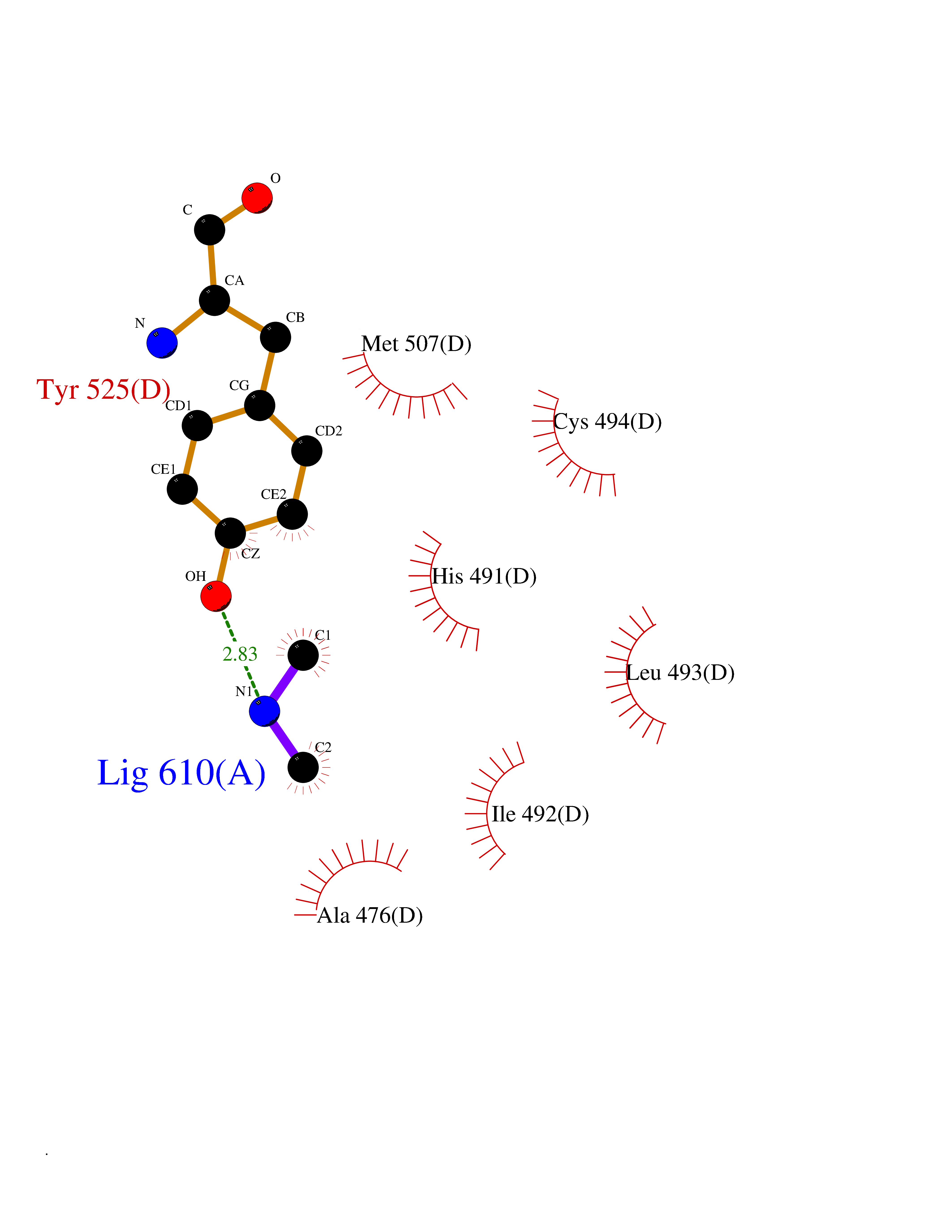 Ligplot Map 3D Binding mode Sequence NSIHPNDFQIYNSLKYKYHPEYQYLNIIYDIMMNGNKQSDRTGVGVLSKFGYIMKFDLSQYFPLLTTKKLFLRGIIEELLWFIRGETNGNTLLNKNVRIWEANGTREFLDNRKLFHREVNDLGPIYGFQWRHFGAEYTNMYDNYENKGVDQLKNIINLIKNDPTSRRILLCAWNVKDLDQMALPPCHILCQFYVFDGKLSCIMYQRSCDLGLGVPFNIASYSIFTHMIAQVCNLQPAQFIHVLGNAHVYNNHIDSLKIQLNRIPYPFPTLKLNPDIKNIEDFTISDFTIQNYVHHEKISMDMAAMMEQVCDVFDIYAICACCKVESKNEGKKNEVFNNYTFRGLGNKGVLPWKCISLDMKYFRAVTTYVNESKYEKLKYKRCKYLPNSKKLQNVVVMGRTNWESIPKKFKPLSNRINVILSRTLKKEDFDEDVYIINKVEDLIVLLGKLNYYKCFILGGSVVYQEFLEKKLIKKIYFTRINSTYECDVFFPEINENEYQIISVSDVYTSNNTTLDFIIYKKTNNK Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Urokinase-type plasminogen activator (PLAU) | 4JNI | 4.02 | |
Target general information Gen name PLAU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UPA; U-plasminogen activator Protein family Peptidase S1 family Biochemical class Peptidase Function Specifically cleaves the zymogen plasminogen to form the active enzyme plasmin. Related diseases Quebec platelet disorder (QPD) [MIM:601709]: An autosomal dominant bleeding disorder due to a gain-of-function defect in fibrinolysis. Although affected individuals do not exhibit systemic fibrinolysis, they show delayed onset bleeding after challenge, such as surgery. The hallmark of the disorder is markedly increased PLAU levels within platelets, which causes intraplatelet plasmin generation and secondary degradation of alpha-granule proteins. {ECO:0000269|PubMed:20007542}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07129; DB07122; DB01905; DB02287; DB03729; DB01725; DB08072; DB07625; DB07626; DB08697; DB03136; DB01977; DB07076; DB03082; DB02705; DB02473; DB02398; DB02551; DB03865; DB06855; DB06856; DB03046; DB04059; DB04172; DB00594; DB03127; DB02526; DB03159; DB05254; DB03782; DB06857; DB16701; DB03876; DB03476 Interacts with Q9UKQ2; P05067; Q03405-1; P05121; P55000 EC number EC 3.4.21.73 Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Direct protein sequencing; Disulfide bond; EGF-like domain; Fibrinolysis; Glycoprotein; Hemostasis; Hydrolase; Kringle; Pharmaceutical; Phosphoprotein; Plasminogen activation; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID U Molecular weight (Da) 25825.3 Length 229 Aromaticity 0.1 Instability index 47.36 Isoelectric point 8.65 Charge (pH=7) 5.38 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 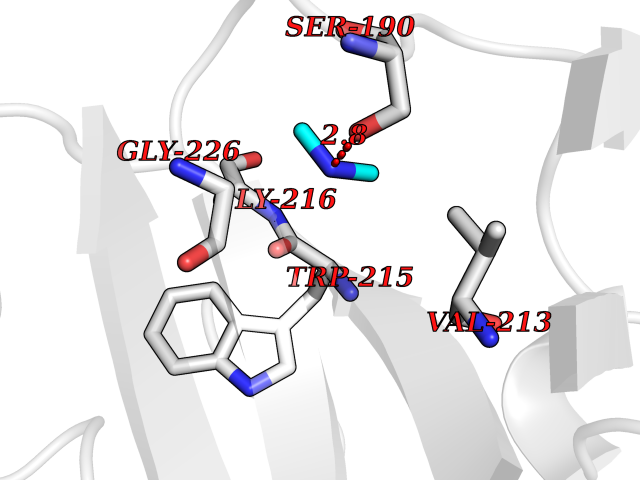 Pymol Map 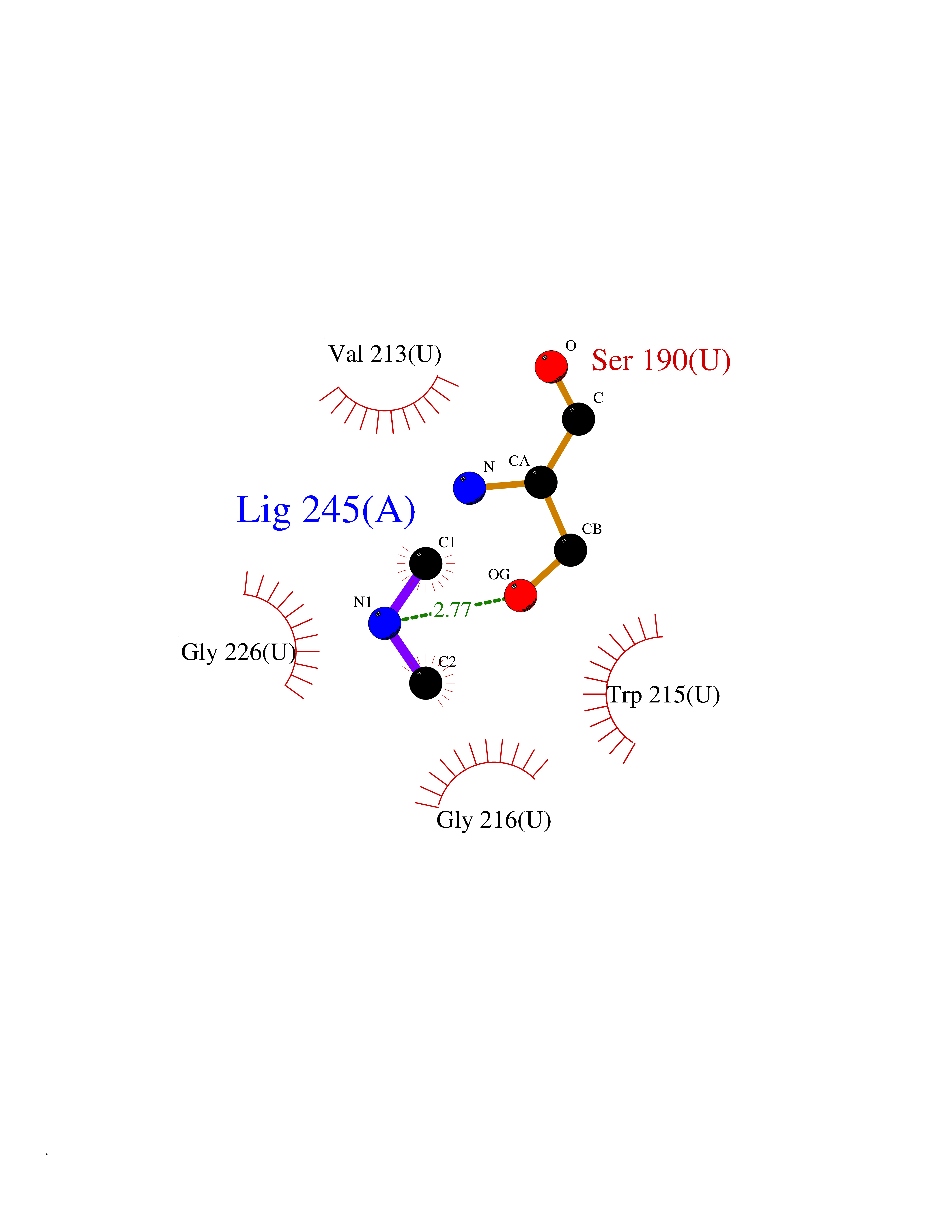 Ligplot Map 3D Binding mode Sequence IIGGEFTTIENQPWFAAIYRRSVTYVCGGSLISPCWVISATHCFPKKEDYIVYLGRSRLNSNTQGEMKFEVENLILHKDYSALAHHNDIALLKIRRCAQPSRTIQTIALPSMYNDPQFGTSCEITGFGKEQSTDYLYPEQLKMTVVKLISHRECQQHYYGSEVTTKMLCAAQWKTDSCQGDSGGPLVCSLQGRMTLTGIVSWGRGCALDKPGVYTRVSHFLPWIRSHTK Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Tartrate-resistant acid phosphatase type 5 | 2BQ8 | 4.02 | |
Target general information Gen name ACP5 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Metallophosphoesterase superfamily, Purple acid phosphatase family Biochemical class Hydrolase Function Acid phosphatase activity.Ferric iron binding.Ferrous iron binding. Related diseases Spondyloenchondrodysplasia with immune dysregulation (SPENCDI) [MIM:607944]: A disease characterized by vertebral and metaphyseal dysplasia, spasticity with cerebral calcifications, and strong predisposition to autoimmune diseases. The skeletal dysplasia is characterized by radiolucent and irregular spondylar and metaphyseal lesions that represent islands of chondroid tissue within bone. {ECO:0000269|PubMed:21217752, ECO:0000269|PubMed:21217755}. The disease is caused by variants affecting the gene represented in this entry. ACP5 inactivating mutations result in a functional excess of phosphorylated osteopontin causing deregulation of osteopontin signaling and consequential autoimmune disease. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.3.2 Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Iron; Lysosome; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID X Molecular weight (Da) 34330.6 Length 304 Aromaticity 0.12 Instability index 42.3 Isoelectric point 9.11 Charge (pH=7) 6.75 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map  Pymol Map 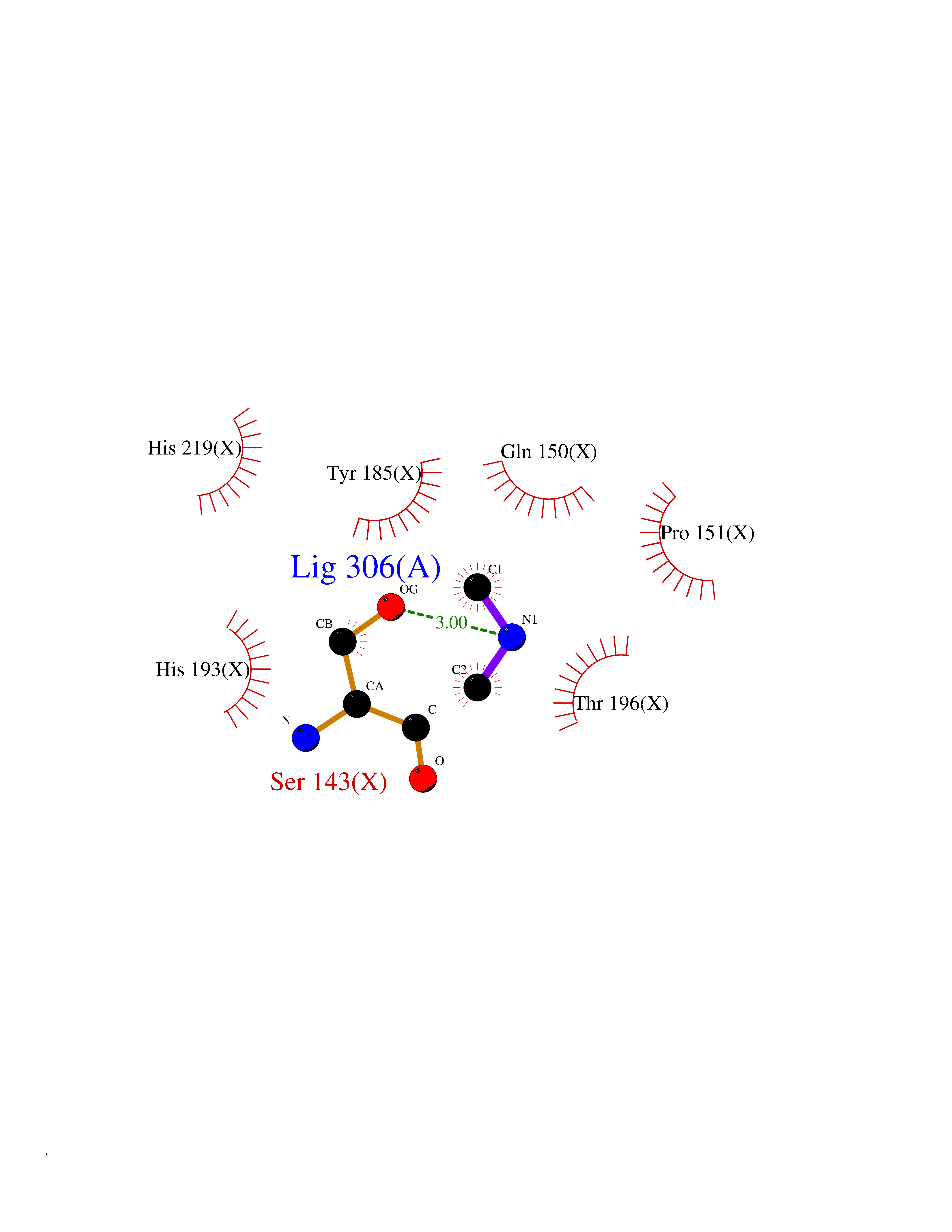 Ligplot Map 3D Binding mode Sequence ATPALRFVAVGDWGGVPNAPFHTAREMANAKEIARTVQILGADFILSLGDNFYFTGVQDINDKRFQETFEDVFSDRSLRKVPWYVLAGNHDHLGNVSAQIAYSKISKRWNFPSPFYRLHFKIPQTNVSVAIFMLDTVTLCGNSDDFLSQQPERPRDVKLARTQLSWLKKQLAAAREDYVLVAGHYPVWSIAEHGPTHCLVKQLRPLLATYGVTAYLCGHDHNLQYLQDENGVGYVLSGAGNFMDPSKRHQRKVPNGYLRFHYGTEDSLGGFAYVEISSKEMTVTYIEASGKSLFKTRLPRRARP Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Alpha-N-acetylglucosaminidase (NAGLU) | 4XWH | 4.02 | |
Target general information Gen name NAGLU Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UFHSD1; NAGLU; NAG; N-acetyl-alpha-glucosaminidase; Alpha-N-acetylglucosaminidase82 kDa form; Alpha-N-acetylglucosaminidase77 kDa form Protein family Glycosyl hydrolase 89 family Biochemical class Glycosylase Function Involved in the degradation of heparan sulfate. Related diseases Mucopolysaccharidosis 3B (MPS3B) [MIM:252920]: A form of mucopolysaccharidosis type 3, an autosomal recessive lysosomal storage disease due to impaired degradation of heparan sulfate. MPS3 is characterized by severe central nervous system degeneration, but only mild somatic disease. Onset of clinical features usually occurs between 2 and 6 years; severe neurologic degeneration occurs in most patients between 6 and 10 years of age, and death occurs typically during the second or third decade of life. {ECO:0000269|PubMed:10094189, ECO:0000269|PubMed:11068184, ECO:0000269|PubMed:11153910, ECO:0000269|PubMed:11286389, ECO:0000269|PubMed:11793481, ECO:0000269|PubMed:11836372, ECO:0000269|PubMed:12202988, ECO:0000269|PubMed:14984474, ECO:0000269|PubMed:15933803, ECO:0000269|PubMed:16151907, ECO:0000269|PubMed:28101780, ECO:0000269|PubMed:9443875, ECO:0000269|PubMed:9443878, ECO:0000269|PubMed:9832037, ECO:0000269|PubMed:9950362}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2V (CMT2V) [MIM:616491]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2V is an autosomal dominant sensory neuropathy with late onset. The main clinical feature is recurrent leg pain that progresses to constant painful paraesthesias in the feet and later the hands. As it evolves, some patients develop a mild sensory ataxia. {ECO:0000269|PubMed:25818867}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06773; DB00141 Interacts with NA EC number EC 3.2.1.50 Uniprot keywords 3D-structure; Charcot-Marie-Tooth disease; Direct protein sequencing; Disease variant; Glycoprotein; Glycosidase; Hydrolase; Lysosome; Mucopolysaccharidosis; Neurodegeneration; Neuropathy; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 80206.3 Length 720 Aromaticity 0.13 Instability index 41.59 Isoelectric point 6.28 Charge (pH=7) -4.09 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map 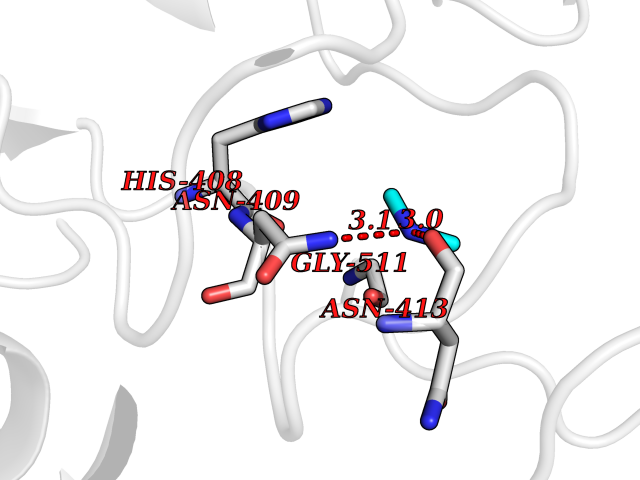 Pymol Map 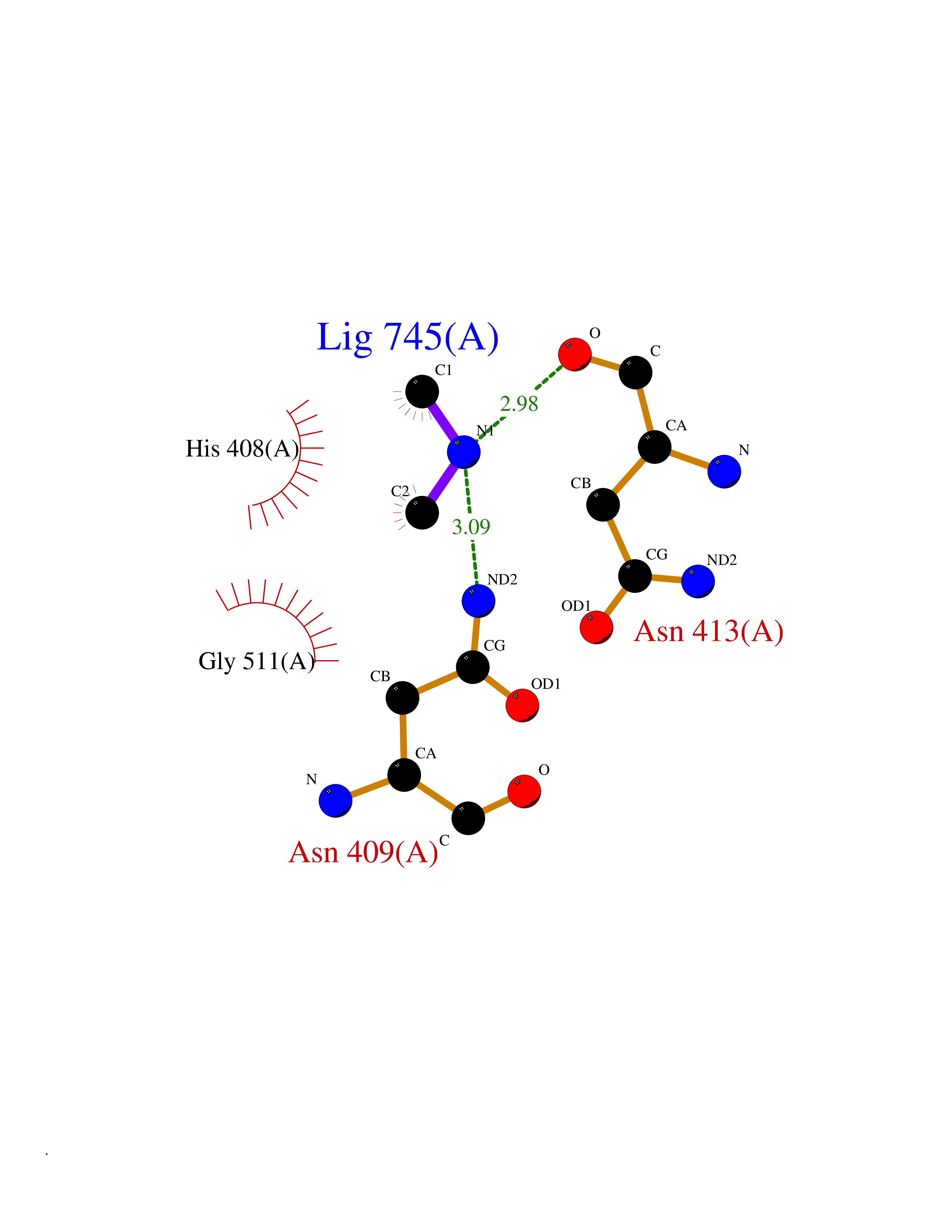 Ligplot Map 3D Binding mode Sequence DEAREAAAVRALVARLLGPGPAADFSVSVERALAAKPGLDTYSLGGGGAARVRVRGSTGVAAAAGLHRYLRDFCGCHVAWSGSQLRLPRPLPAVPGELTEATPNRYRYYQNVCTQSYSFVWWDWARWEREIDWMALNGINLALAWSGQEAIWQRVYLALGLTQAEINEFFTGPAFLAWGRMGNLHTWDGPLPPSWHIKQLYLQHRVLDQMRSFGMTPVLPAFAGHVPEAVTRVFPQVNVTKMGSWGHFNCSYSCSFLLAPEDPIFPIIGSLFLRELIKEFGTDXIYGADTFNEMQPPSSEPSYLAAATTAVYEAMTAVDTEAVWLLQGWLFQHQPQFWGPAQIRAVLGAVPRGRLLVLDLFAESQPVYTRTASFQGQPFIWCMLHNFGGNHGLFGALEAVNGGPEAARLFPNSTMVGTGMAPEGISQNEVVYSLMAELGWRKDPVPDLAAWVTSFAARRYGVSHPDAGAAWRLLLRSVYNCSGEACRGHNRSPLVRRPSLQMNTSIWYNRSDVFEAWRLLLTSAPSLATSPAFRYDLLDLTRQAVQELVSLYYEEARSAYLSKELASLLRAGGVLAYELLPALDEVLASDSRFLLGSWLEQARAAAVSEAEADFYEQNSRYQLTLWGPEGNILDYANKQLAGLVANYYTPRWRLFLEALVDSVAQGIPFQQHQFDKNVFQLEQAFVLSKQRYPSQPRGDTVDLAKKIFLKYYPRWVAGSW Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | S-adenosylmethionine synthase isoform type-1 | 2OBV | 4.02 | |
Target general information Gen name MAT1A Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MATA1;AMS1 Protein family AdoMet synthase family Biochemical class Transferase Function ATP binding.Identical protein binding.Metal ion binding.Methionine adenosyltransferase activity.Selenomethionine adenosyltransferase activity. Related diseases Methionine adenosyltransferase deficiency (MATD) [MIM:250850]: An inborn error of metabolism resulting in isolated hypermethioninemia. Most patients have no clinical abnormalities, although some neurologic symptoms may be present in rare cases with severe loss of methionine adenosyltransferase activity. {ECO:0000269|PubMed:10677294, ECO:0000269|PubMed:7560086, ECO:0000269|PubMed:8770875, ECO:0000269|PubMed:9042912}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03191; DB00118; DB03611; DB00134 Interacts with P05067; P42858; Q00266; P31153 EC number 2.5.1.6 Uniprot keywords 3D-structure; ATP-binding; Disease variant; Disulfide bond; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Potassium; Proteomics identification; Reference proteome; S-nitrosylation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42222.9 Length 381 Aromaticity 0.08 Instability index 41.95 Isoelectric point 6.14 Charge (pH=7) -4.58 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map 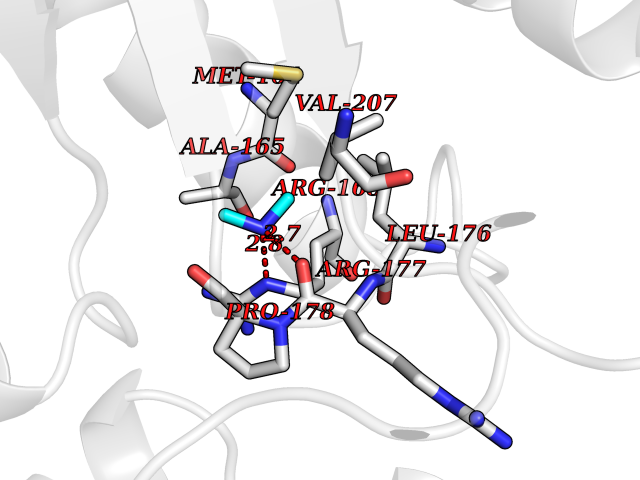 Pymol Map 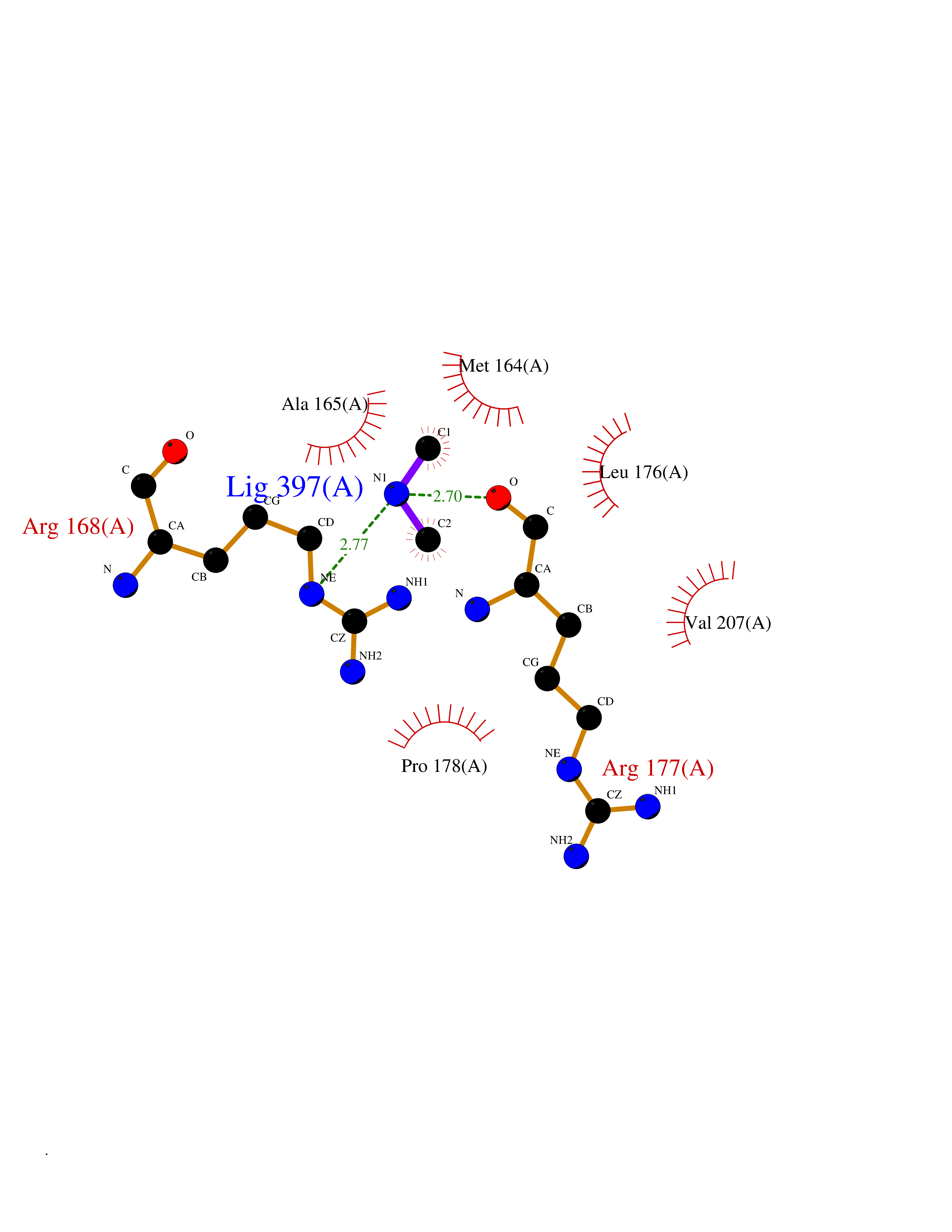 Ligplot Map 3D Binding mode Sequence MGVFMFTSESVGEGHPDKICDQISDAVLDAHLKQDPNAKVACETVCKTGMVLLCGEITSMAMVDYQRVVRDTIKHIGYDDSAKGFDFKTCNVLVALEQQSPDIAQCVHLDRNEEDVGAGDQGLMFGYATDETEECMPLTIILAHKLNARMADLRRSGLLPWLRPDSKTQVTVQYMQDNGAVIPVRIHTIVISVQHNEDITLEEMRRALKEQVIRAVVPAKYLDEDTVYHLQPSGRFVIGGPQGDAGVTGRKIIVDTYGGWGAHGGGAFSGKDYTKVDRSAAYAARWVAKSLVKAGLCRRVLVQVSYAIGVAEPLSISIFTYGTSQKTERELLDVVHKNFDLRPGVIVRDLDLKKPIYQKTACYGHFGRSEFPWEVPRKLVF Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Aminoacylase-1 | 1Q7L | 4.02 | |
Target general information Gen name ACY1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase M20A family Biochemical class Hydrolase Function Aminoacylase activity.Identical protein binding.Metal ion binding.Metallopeptidase activity. Related diseases Aminoacylase-1 deficiency (ACY1D) [MIM:609924]: An enzymatic deficiency resulting in encephalopathy, unspecific psychomotor delay, psychomotor delay with atrophy of the vermis and syringomyelia, marked muscular hypotonia or normal clinical features. Epileptic seizures are a frequent feature. All affected individuals exhibit markedly increased urinary excretion of several N-acetylated amino acids. {ECO:0000269|PubMed:16274666, ECO:0000269|PubMed:16465618, ECO:0000269|PubMed:17562838, ECO:0000269|PubMed:21414403}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06151; DB00128; DB09130 Interacts with Q03154; O75934; Q96HA8; P36639; P36639-2; Q8TCT1; P0CG20; Q96A09; P54274; O43711; Q9UPN9; Q9NZC7-5 EC number 3.5.1.14 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,C Molecular weight (Da) 31172.2 Length 275 Aromaticity 0.11 Instability index 36.46 Isoelectric point 6 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -5.49  Molscript Map 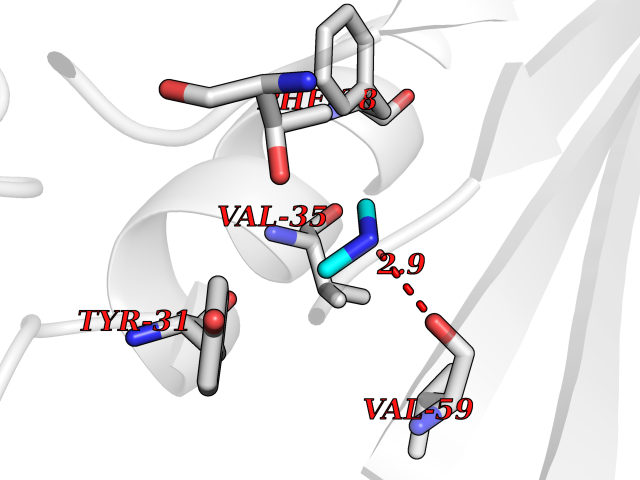 Pymol Map 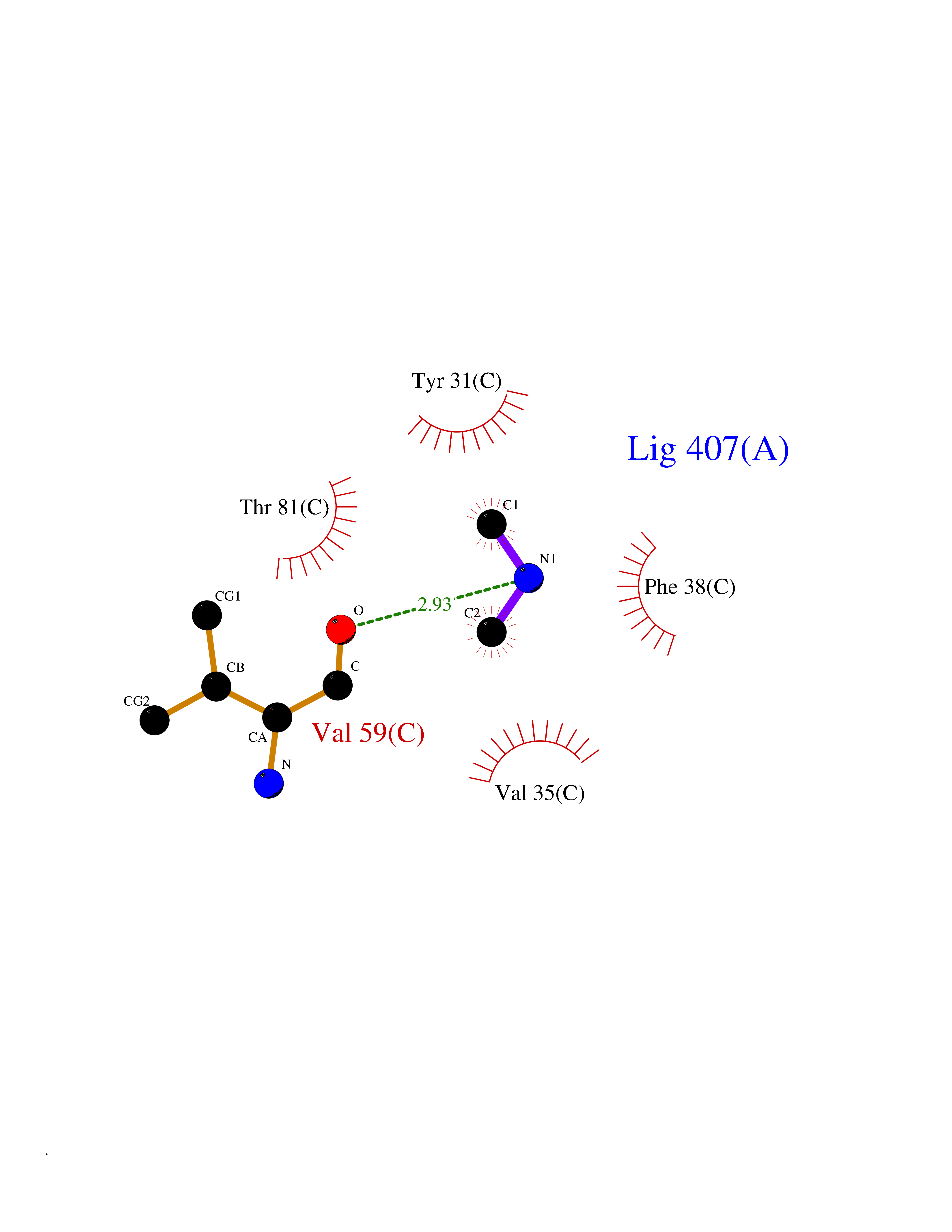 Ligplot Map 3D Binding mode Sequence NPWWAAFSRVCKDMNLTLEPEIMPAAGDNRYIRAVGVPALGFSPMNRTPVLLHDHDERLHEAVFLRGVDIYTRLLPALASVPALPEEHPSVTLFRQYLRIRTVQPKPDYGAAVAFFEETARQLGLGCQKVEVAPGYVVTVLTWPGTNPTLSSILLNSHTDVVPVFKEHWSHDPFEAFKDSEGYIYARGAQDMKCVSIQYLEAVRRLKVEGHRFPRTIHMTFVPDEEVGGHQGMELFVQRPEFHALRAGFALDEGIANPTDAFTVFYSERSPWWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Pectate lyase | 1R76 | 4.02 | |
Target general information Gen name pelA Organism Niveispirillum irakense (Azospirillum irakense) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Lyase Function Lyase activity. Related diseases A chromosomal aberration involving ALK is found in a form of non-Hodgkin lymphoma. Translocation t(2;5)(p23;q35) with NPM1. The resulting chimeric NPM1-ALK protein homodimerize and the kinase becomes constitutively activated. The constitutively active fusion proteins are responsible for 5-10% of non-Hodgkin lymphomas. {ECO:0000269|PubMed:15938644}.; DISEASE: A chromosomal aberration involving ALK is associated with inflammatory myofibroblastic tumors (IMTs). Translocation t(2;11)(p23;p15) with CARS; translocation t(2;4)(p23;q21) with SEC31A. {ECO:0000269|PubMed:12112524, ECO:0000269|PubMed:16161041}.; DISEASE: A chromosomal aberration involving ALK is associated with anaplastic large-cell lymphoma (ALCL). Translocation t(2;17)(p23;q25) with ALO17. {ECO:0000269|PubMed:12112524}.; DISEASE: Neuroblastoma 3 (NBLST3) [MIM:613014]: A common neoplasm of early childhood arising from embryonic cells that form the primitive neural crest and give rise to the adrenal medulla and the sympathetic nervous system. {ECO:0000269|PubMed:18724359, ECO:0000269|PubMed:18923523, ECO:0000269|PubMed:18923525, ECO:0000269|PubMed:21242967, ECO:0000269|PubMed:22932897}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: The ALK signaling pathway plays an important role in glioblastoma, the most common malignant brain tumor of adults and one of the most lethal cancers. It regulates both glioblastoma migration and growth. {ECO:0000269|PubMed:15908427}.; DISEASE: A chromosomal aberration involving ALK is found in one subject with colorectal cancer. Translocation t(2;2)(p23.1;p23.3). A 5 million base pair tandem duplication generates an in-frame WDCP-ALK gene fusion. {ECO:0000269|PubMed:22327622}.; DISEASE: A chromosomal aberration involving ALK has been identified in a subset of patients with non-small-cell lung carcinoma. This aberration leads to the production of a fusion protein between the N-terminus of EML4 et the C-terminus of ALK. It is unclear whether the fusion protein is caused by a simple inversion within 2p (inv(2)(p21p23)) or whether the chromosome translocation involving 2p is more complex. When tested in a heterologous system, the fusion protein EML4-ALK possesses transforming activity that is dependent on ALK catalytic activity, possibly due to spontaneous dimerization mediated by the EML4 moiety, leading to ALK kinase activation. {ECO:0000269|PubMed:17625570}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Lyase; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41907.5 Length 384 Aromaticity 0.08 Instability index 43.72 Isoelectric point 6.11 Charge (pH=7) -3.46 2D Binding mode Binding energy (Kcal/mol) -5.48 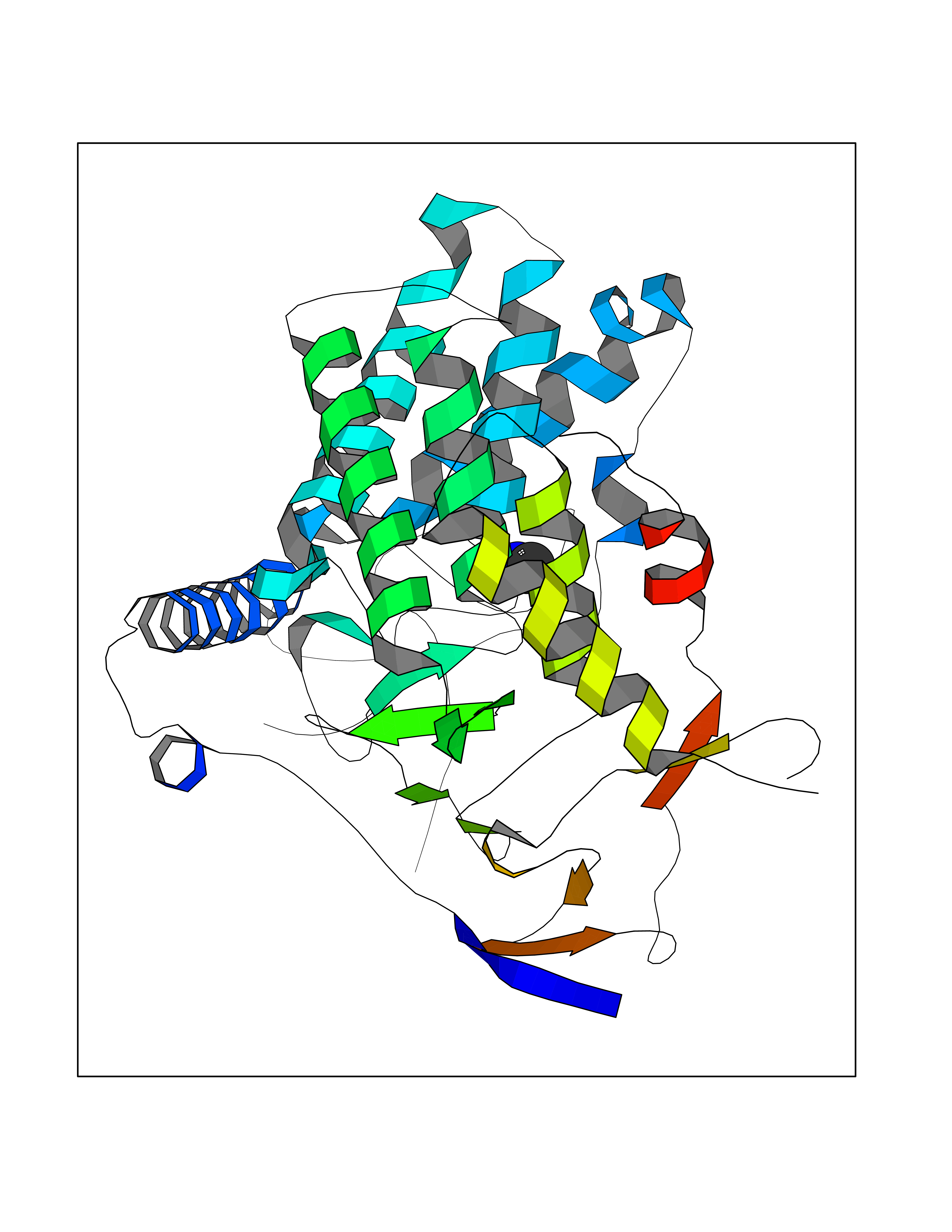 Molscript Map 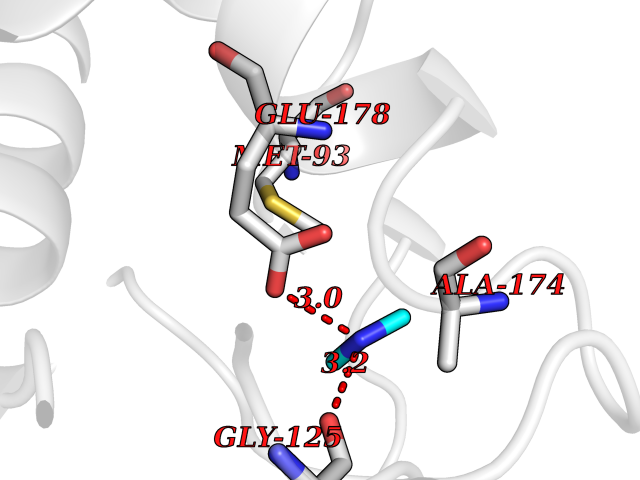 Pymol Map 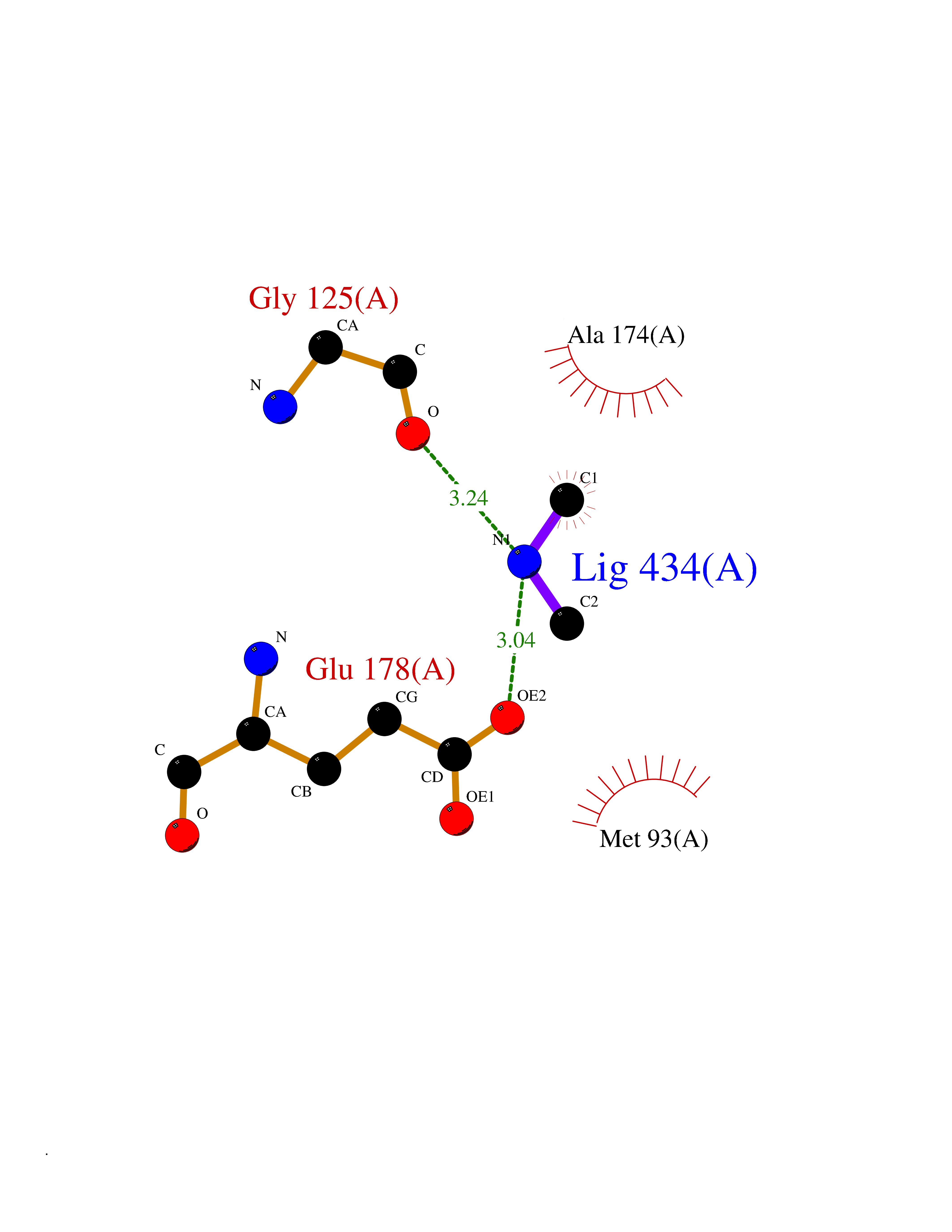 Ligplot Map 3D Binding mode Sequence AVIGMNEAASALTPSRVSSLPDTQRAAWQEYLARSEAQLSRDKASLAAELAPGQPLPPPPAEGKGADTMPLDKPAAWYTSKAARHVADVIVSFQTPAGGWGKNQPRDGALRLPGQHYTGENVAKVKRDRDWHYVGTIDNDATVTEIRFLAQVVSQLAPEEAAPYRDAALKGIEYLLASQFPNGGWPQVWPLEGGYHDAITYNDDALVHVAELLSDIAAGRDGFGFVPPAIRTRALEATNAAIHCIVETQVVQDGKRLGWGQQHDALTLRPTSARNFEPAALSSTESARILLFLMEIEAPSDAVKQAIRGGVAWLNTSVIRDQGAKPLWSRFYSLDGNKPVFGDRDKTIHDDVMGISQERRTGYAWYTTSPQKALSAFTKWEKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Trypanosoma Cruzipain (Trypano CYSP) | 1EWM | 4.02 | |
Target general information Gen name Trypano CYSP Organism Trypanosoma cruzi Uniprot ID TTD ID Synonyms Cruzaine; Major cysteine proteinase Protein family Peptidase C1 family Biochemical class Peptidase Function The cysteine protease may play an important role in the development and differentiation of the parasites at several stages of their life cycle. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02200; DB02051; DB01871; DB01810; DB02128; DB03536; DB04427; DB03691; DB04502; DB03573 Interacts with NA EC number EC 3.4.22.51 Uniprot keywords 3D-structure; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Signal; Thiol protease; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 22703 Length 215 Aromaticity 0.09 Instability index 28.98 Isoelectric point 4.37 Charge (pH=7) -13.9 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 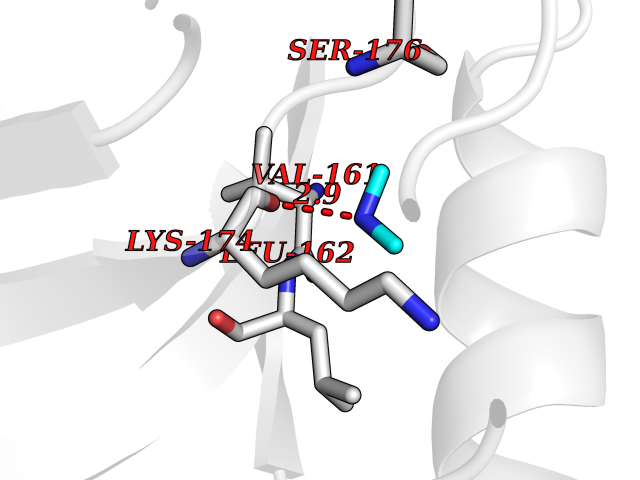 Pymol Map 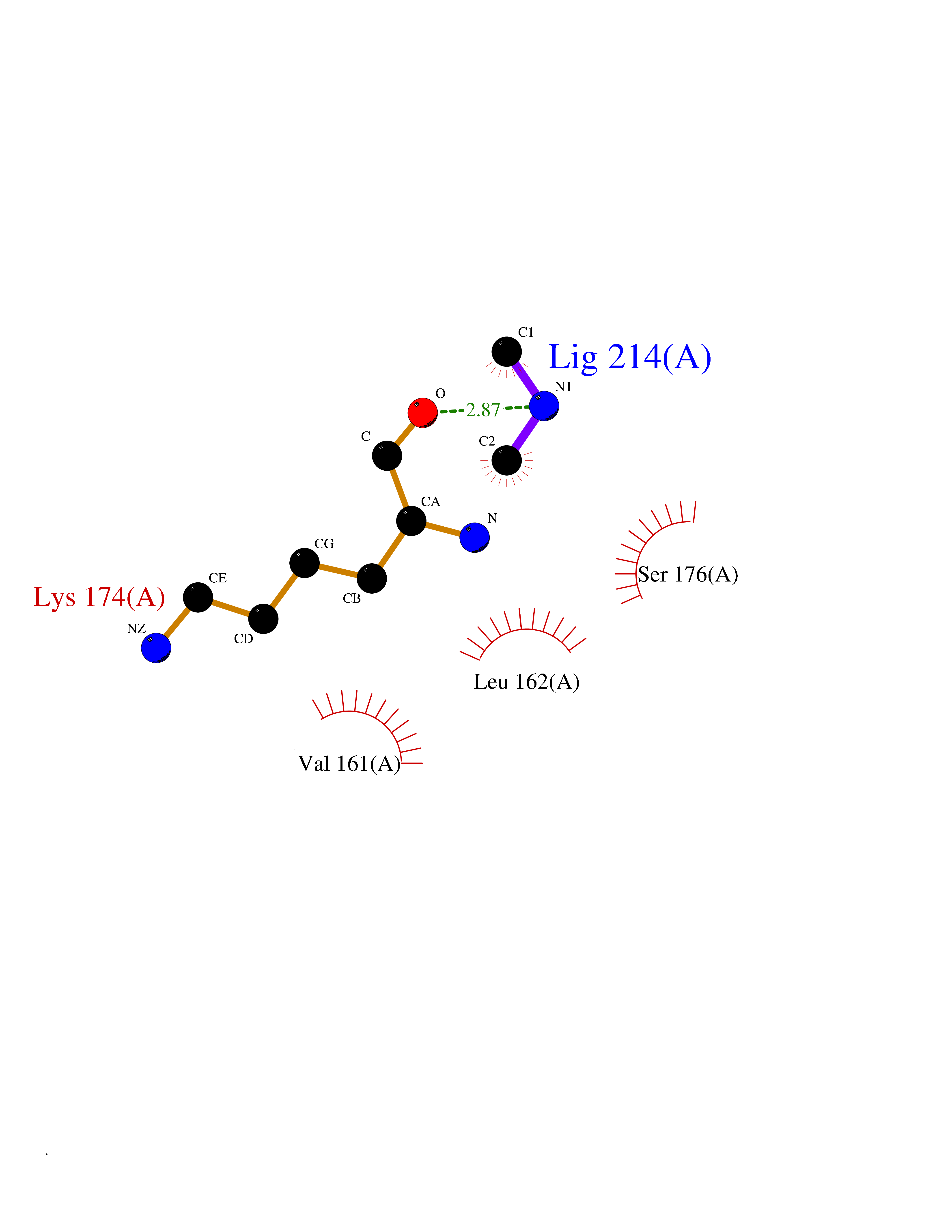 Ligplot Map 3D Binding mode Sequence APAAVDWRARGAVTAVKDQGQCGSCWAFSAIGNVECQWFLAGHPLTNLSEQMLVSCDKTDSGCSGGLMNNAFEWIVQENNGAVYTEDSYPYASGEGISPPCTTSGHTVGATITGHVELPQDEAQIAAWLAVNGPVAVAVDASSWMTYTGGVMTSCVSEQLDHGVLLVGYNDSAAVPYWIIKNSWTTQWGEEGYIRIAKGSNQCLVKEEASSAVVG Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Plasmodium DOXP reductoisomerase (Malaria DXR) | 3AU9 | 4.02 | |
Target general information Gen name Malaria DXR Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID Synonyms IspC; DXR; DXP reductoisomerase; DOXP reductoisomerase; 2-C-Methyl-d-erythritol 4-phosphate synthase; 1-deoxyxylulose-5-phosphate reductoisomerase Protein family DXR family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADP-dependent rearrangement and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol 4-phosphate (MEP). Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.1.267 Uniprot keywords 3D-structure; Apicoplast; Isoprene biosynthesis; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Plastid; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46644.4 Length 410 Aromaticity 0.09 Instability index 36.77 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 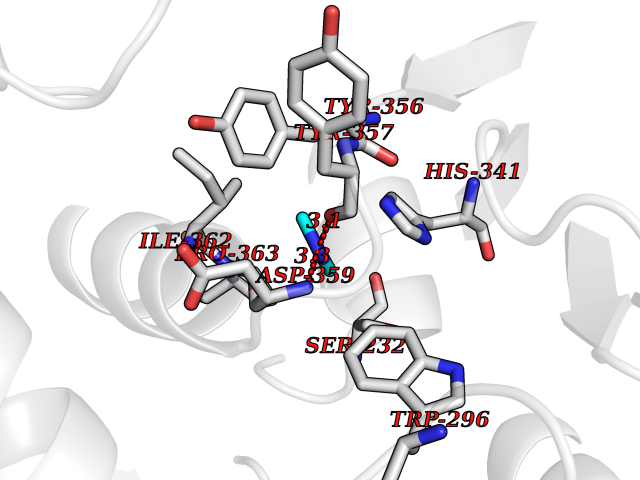 Pymol Map 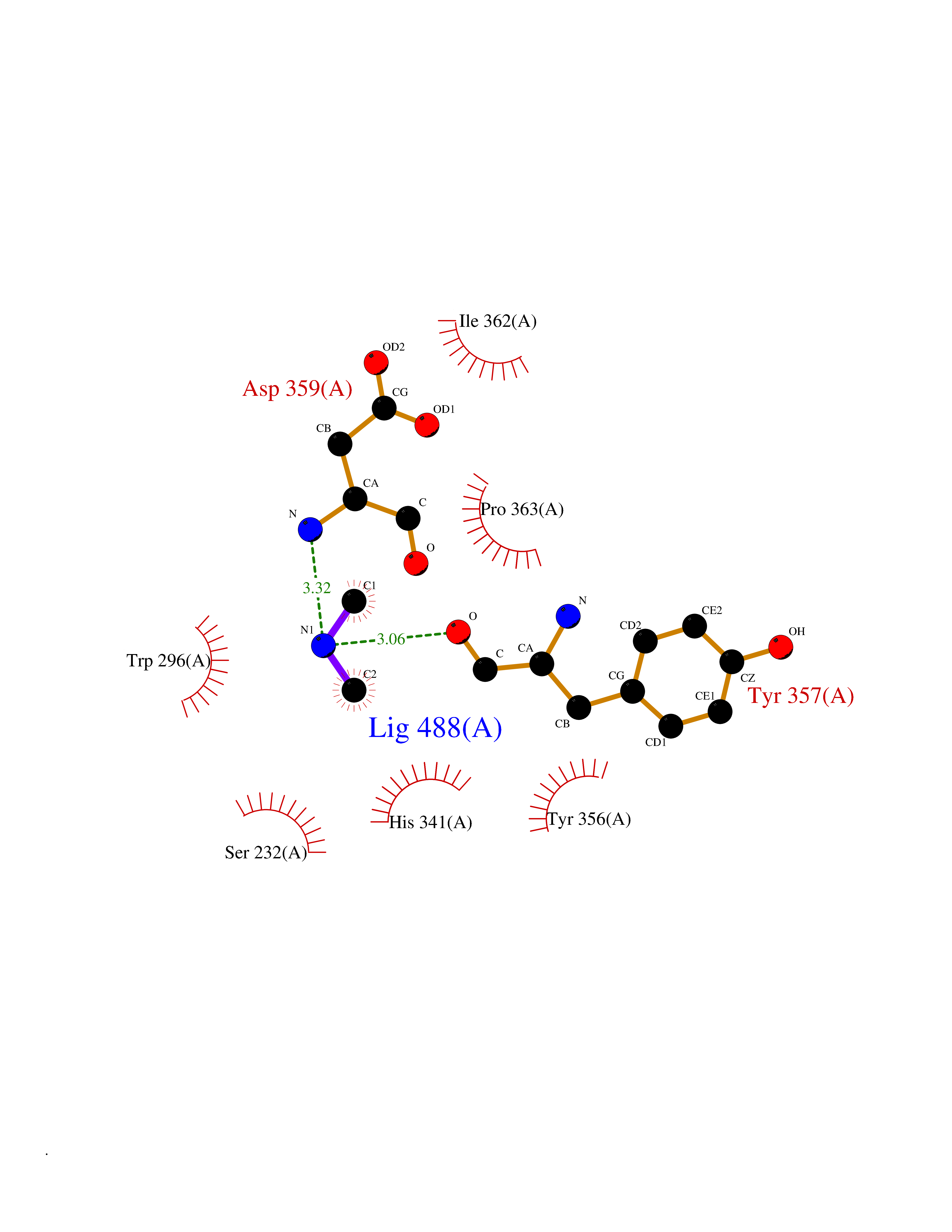 Ligplot Map 3D Binding mode Sequence PINVAIFGSTGSIGTNALNIIRECNKIENVFNVKALYVNKSVNELYEQAREFLPEYLCIHDKSVYEELKELVKNIKDYKPIILCGDEGMKEICSSNSIDKIVIGIDSFQGLYSTMYAIMNNKIVALANKESIVSAGFFLKKLLNIHKNAKIIPVDSEHSAIFQCLDNNKVLKTKCLQDNFSKINNINKIFLCSSGGPFQNLTMDELKNVTSENALKHPKWKMGKKITIDSATMMNKGLEVIETHFLFDVDYNDIEVIVHKECIIHSCVEFIDKSVISQMYYPDMQIPILYSLTWPDRIKTNLKPLDLAQVSTLTFHKPSLEHFPCIKLAYQAGIKGNFYPTVLNASNEIANNLFLNNKIKYFDISSIISQVLESFNSQKVSENSEDLMKQILQIHSWAKDKATDIYNKHN Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Tankyrase-2 (TNKS-2) | 3U9H | 4.02 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -5.48 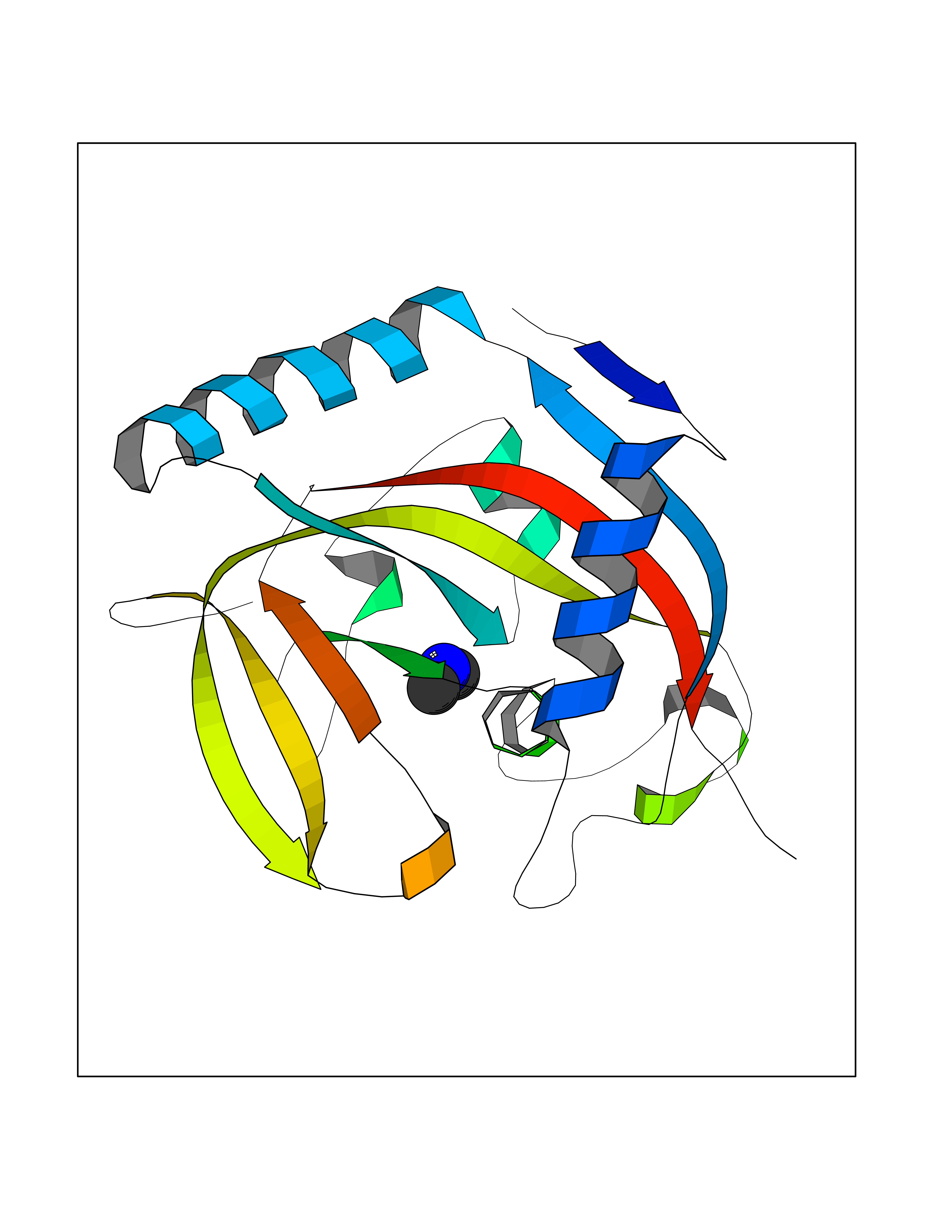 Molscript Map 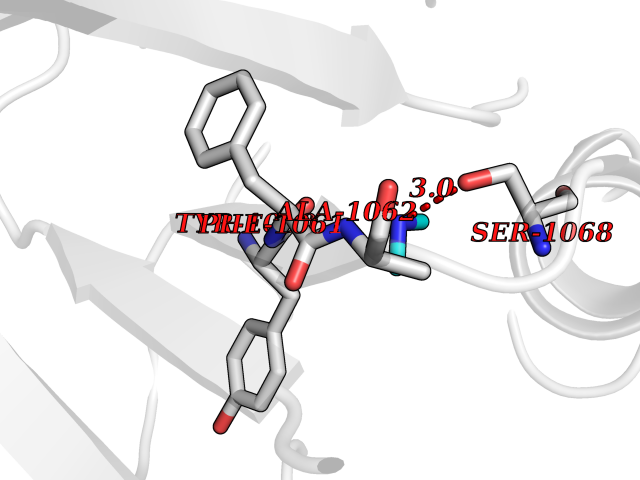 Pymol Map 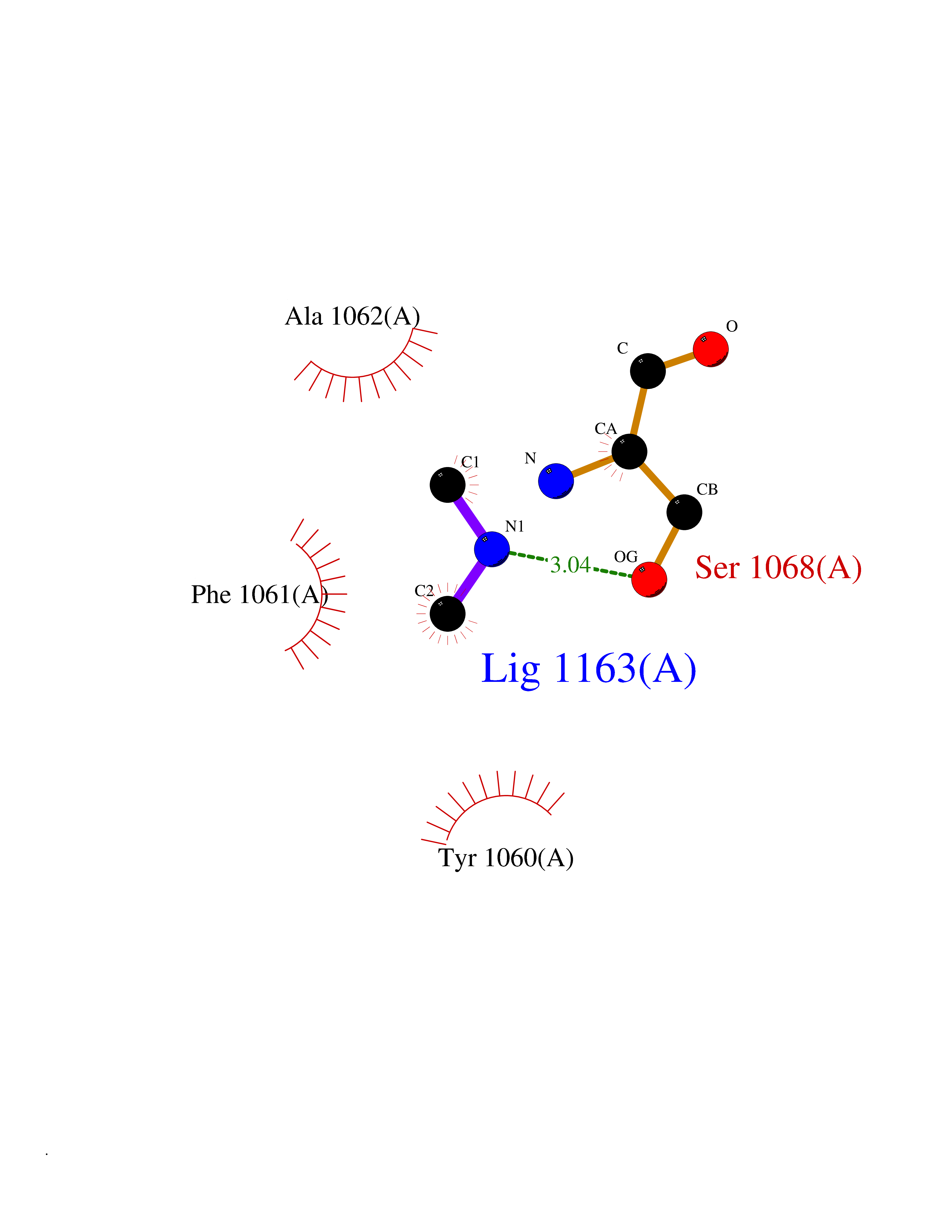 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Lysine-specific demethylase 6B (KDM6B) | 6F6D | 4.02 | |
Target general information Gen name KDM6B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine demethylase 6B; KIAA0346; Jumonji domain-containing protein 3; JmjC domain-containing protein 3; JMJD3 Protein family UTX family Biochemical class NA Function Histone demethylase that specifically demethylates 'Lys-27' of histone H3, thereby playing a central role in histone code. Demethylates trimethylated and dimethylated H3 'Lys-27'. Plays a central role in regulation of posterior development, by regulating HOX gene expression. Involved in inflammatory response by participating in macrophage differentiation in case of inflammation by regulating gene expression and macrophage differentiation. Plays a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression by acting as a link between T-box factors and the SMARCA4-containing SWI/SNF remodeling complex (By similarity). Related diseases Stolerman neurodevelopmental syndrome (NEDSST) [MIM:618505]: An autosomal dominant disorder characterized by global developmental delay, variable intellectual disability, poor language acquisition, and dysmorphic facial features including a prominent nasal bridge and coarse features. Some patients manifest autism spectrum disorder. Musculoskeletal features may be present and include widened and thickened hands and fingers, joint hypermobility, clinodactyly of the fifth fingers, and toe syndactyly. {ECO:0000269|PubMed:31124279}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P03372 EC number EC 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Dioxygenase; Disease variant; Inflammatory response; Intellectual disability; Iron; Isopeptide bond; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 53008.8 Length 467 Aromaticity 0.1 Instability index 44.23 Isoelectric point 8.37 Charge (pH=7) 4.9 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 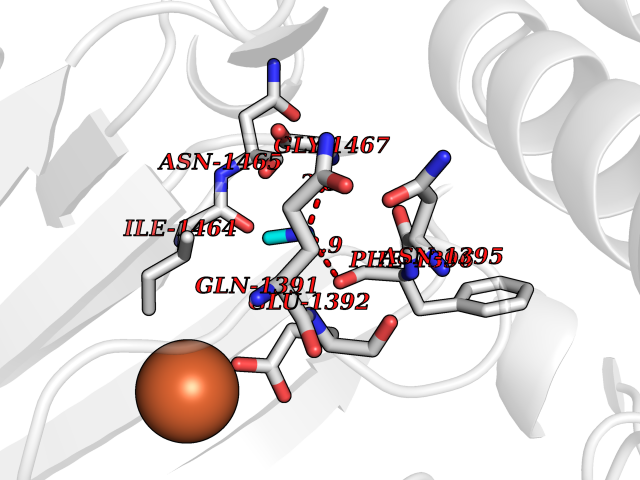 Pymol Map 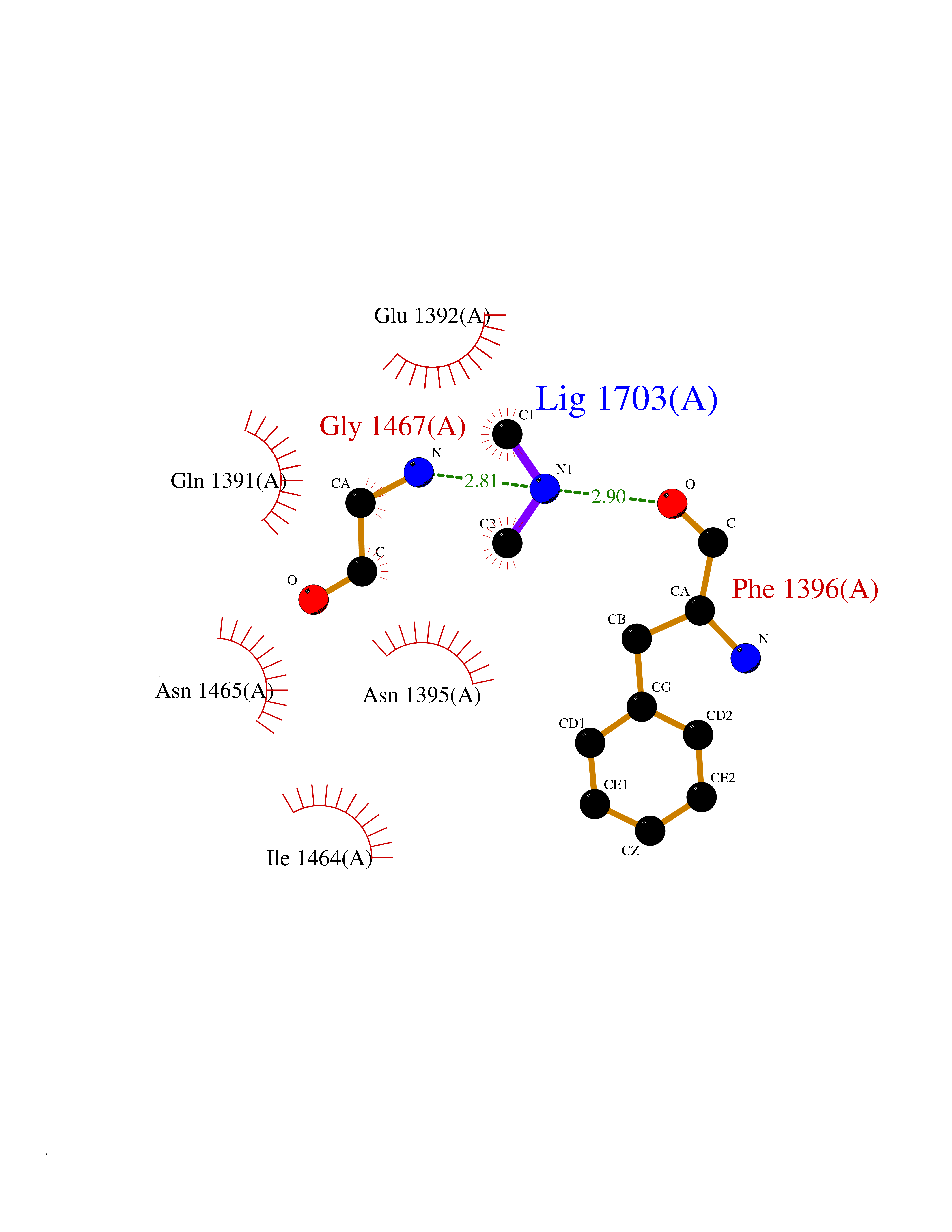 Ligplot Map 3D Binding mode Sequence ESYLSPAQSVKPKINTEEKLPREKLNPPTPSIYLESKRDAFSPVLLQFCTDPRNPITVIRGLAGSLRLNLGLFSTKTLVEASGEHTVEVRTQVQQPSDENWDLTGTRQIWPCESSRSHTTIAKYAQYQASSFQESHIIKFGTNIDLSDAKRWKPQLQELLKLPAFMRVTSTGNMLSHVGHTILGMNTVQLYMKVPGSRTPGHQENNNFCSVNINIGPGDCEWFAVHEHYWETISAFCDRHGVDYLTGSWWPILDDLYASNIPVYRFVQRPGDLVWINAGTVHWVQATGWCNNIAWNVGPLTAYQYQLALERYEWNEVKNVKSIVPMIHVSWNVARTVKISDPDLFKMIKFCLLQSMKHCQVQRESLVRAGKKIAYQGRVKDEPAYYCNECDVEVFNILFVTSENGSRNTYLVHCEGCARRRSAGLQGVVVLEQYRTEELAQAYDAFTLAPRIQLMTKAARKSAPATG Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Alpha-ketoglutarate dehydrogenase (OGDH) | 7WGR | 4.02 | |
Target general information Gen name OGDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OGDC-E1; 2-oxoglutarate dehydrogenase, mitochondrial; 2-oxoglutarate dehydrogenase complex component E1 Protein family Alpha-ketoglutarate dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function The 2-oxoglutarate dehydrogenase complex catalyzes the overall conversion of 2-oxoglutarate to succinyl-CoA and CO(2). The 2-oxoglutarate dehydrogenase complex is mainly active in the mitochondrion. A fraction of the 2-oxoglutarate dehydrogenase complex also localizes in the nucleus and is required for lysine succinylation of histones: associates with KAT2A on chromatin and provides succinyl-CoA to histone succinyltransferase KAT2A. 2-oxoglutarate dehydrogenase (E1) component of the 2-oxoglutarate dehydrogenase complex, which mediates the decarboxylation of alpha-ketoglutarate. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00313; DB09092 Interacts with P54253; P42858 EC number EC 1.2.4.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Calcium; Glycolysis; Isopeptide bond; Magnesium; Metal-binding; Mitochondrion; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Thiamine pyrophosphate; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 192730 Length 1700 Aromaticity 0.09 Instability index 43.47 Isoelectric point 5.97 Charge (pH=7) -29.2 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 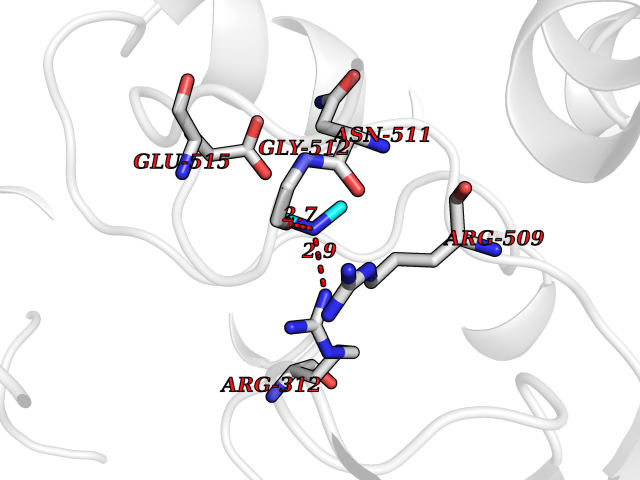 Pymol Map 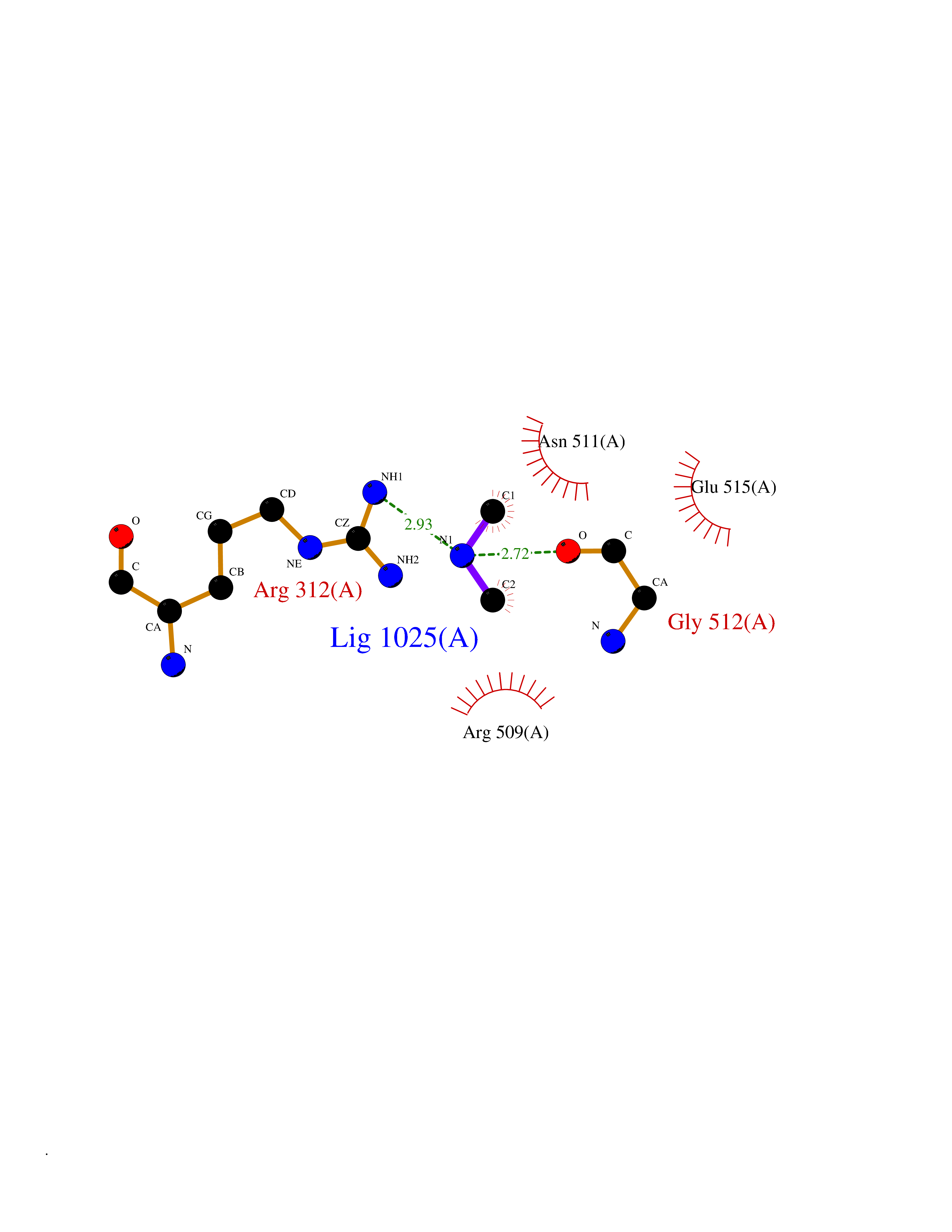 Ligplot Map 3D Binding mode Sequence AVQSLIRAYQIRGHHVAQLDPLGILDADLDSSVPADIISSTDKLGFYGLDESDLDKVFHLPTTTFIGGQESALPLREIIRRLEMAYCQHIGVEFMFINDLEQCQWIRQKFETPGIMQFTNEEKRTLLARLVRSTRFEEFLQRKWSSEKRFGLEGCEVLIPALKTIIDKSSENGVDYVIMGMPHRGRLNVLANVIRKELEQIFCQFDSKLEAADEGSGDNITLSLVANPSHLEAADPVVMGKTKAEQFYCGDTEGKKVMSILLHGDAAFAGQGIVYETFHLSDLPSYTTHGTVHVVVNNQIGFTTDPRMARSSPYPTDVARVVNAPIFHVNSDDPEAVMYVCKVAAEWRSTFHKDVVVDLVCYRRNGHNEMDEPMFTQPLMYKQIRKQKPVLQKYAELLVSQGVVNQPEYEEEISKYDKICEEAFARSKMSCPSTGLTEDILTHIGNVASSVPVENFTIHGGLSRILKTRGEMVKNRTVDWALAEYMAFGSLLKEGIHIRLSGQDVERGTFSHRHHVLHDQNVDKRTCIPMNHLWPNQAPYTVCNSSLSEYGVLGFELGFAMASPNALVLWEAQFGDFHNTAQCIIDQFICPGQAKWVRQNGIVLLLPHGMEGMGPEHSSARPERFLQMCNDDPDVLPDLKEANFDINQLYDCNWVVVNCSTPGNFFHVLRRQILLPFRKPLIIFTPKSLLRHPEARSSFDEMLPGTHFQRVIPEDGPAAQNPENVKRLLFCTGKVYYDLTRERKARDMVGQVAITRIEQLSPFPFDLLLKEVQKYPNAELAWCQEEHKNQGYYDYVKPRLRTTISRAKPVWYAGRDPAAAPATGNKKTHLTELQRLLDTAFDLDVFKNFSAVQSLIRAYQIRGHHVAQLDPLGILDADLDSSVPADIISSTDKLGFYGLDESDLDKVFHLPTTTFIGGQESALPLREIIRRLEMAYCQHIGVEFMFINDLEQCQWIRQKFETPGIMQFTNEEKRTLLARLVRSTRFEEFLQRKWSSEKRFGLEGCEVLIPALKTIIDKSSENGVDYVIMGMPHRGRLNVLANVIRKELEQIFCQFDSKLEAADEGSGDNITLSLVANPSHLEAADPVVMGKTKAEQFYCGDTEGKKVMSILLHGDAAFAGQGIVYETFHLSDLPSYTTHGTVHVVVNNQIGFTTDPRMARSSPYPTDVARVVNAPIFHVNSDDPEAVMYVCKVAAEWRSTFHKDVVVDLVCYRRNGHNEMDEPMFTQPLMYKQIRKQKPVLQKYAELLVSQGVVNQPEYEEEISKYDKICEEAFARSKMSCPSTGLTEDILTHIGNVASSVPVENFTIHGGLSRILKTRGEMVKNRTVDWALAEYMAFGSLLKEGIHIRLSGQDVERGTFSHRHHVLHDQNVDKRTCIPMNHLWPNQAPYTVCNSSLSEYGVLGFELGFAMASPNALVLWEAQFGDFHNTAQCIIDQFICPGQAKWVRQNGIVLLLPHGMEGMGPEHSSARPERFLQMCNDDPDVLPDLKEANFDINQLYDCNWVVVNCSTPGNFFHVLRRQILLPFRKPLIIFTPKSLLRHPEARSSFDEMLPGTHFQRVIPEDGPAAQNPENVKRLLFCTGKVYYDLTRERKARDMVGQVAITRIEQLSPFPFDLLLKEVQKYPNAELAWCQEEHKNQGYYDYVKPRLRTTISRAKPVWYAGRDPAAAPATGNKKTHLTELQRLLDTAFDLDVFKNFS Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Cathepsin D (CTSD) | 4OC6 | 4.02 | |
Target general information Gen name CTSD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CPSD; CD Protein family Peptidase A1 family Biochemical class Peptidase Function Plays a role in APP processing following cleavage and activation by ADAM30 which leads to APP degradation. Involved in the pathogenesis of several diseases such as breast cancer and possibly Alzheimer disease. Acid protease active in intracellular protein breakdown. Related diseases Ceroid lipofuscinosis, neuronal, 10 (CLN10) [MIM:610127]: A form of neuronal ceroid lipofuscinosis with onset at birth or early childhood. Neuronal ceroid lipofuscinoses are progressive neurodegenerative, lysosomal storage diseases characterized by intracellular accumulation of autofluorescent liposomal material, and clinically by seizures, dementia, visual loss, and/or cerebral atrophy. {ECO:0000269|PubMed:16670177, ECO:0000269|PubMed:16685649, ECO:0000269|PubMed:21990111}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03028; DB03096; DB07542; DB08740; DB02216 Interacts with P05067; Q9P1A6-3; I6L9I8; Q9H6S3; Q7Z602; P28799; PRO_0000012695 [P28799]; PRO_0000012696 [P28799]; PRO_0000012697 [P28799]; PRO_0000012698 [P28799]; PRO_0000012699 [P28799]; PRO_0000012700 [P28799]; PRO_0000012701 [P28799]; P68431; Q9Y6F6-3; Q12756; Q5TA79; Q86VF5-3; O15130-2; Q96LB9; P09565; Q9C004; Q8NBJ7; Q9BQG1; P28347-2; P45880; Q15007-2; O00308; Q5W0Z9-4; Q6ZNH5 EC number EC 3.4.23.5 Uniprot keywords 3D-structure; Alzheimer disease; Aspartyl protease; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Neurodegeneration; Neuronal ceroid lipofuscinosis; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37264.2 Length 341 Aromaticity 0.1 Instability index 32.32 Isoelectric point 5.6 Charge (pH=7) -4.86 2D Binding mode Binding energy (Kcal/mol) -5.48  Molscript Map 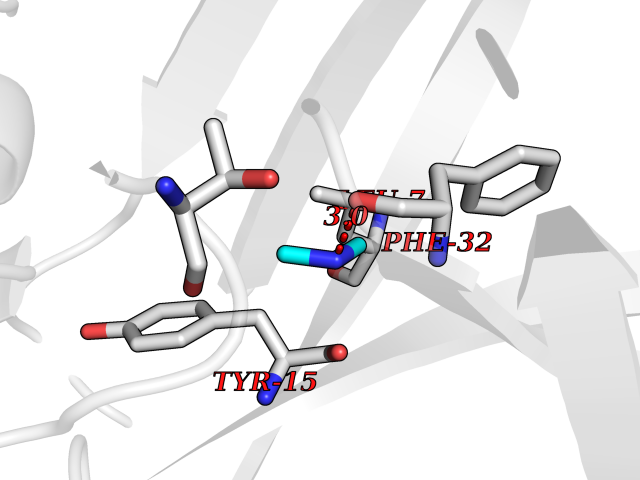 Pymol Map 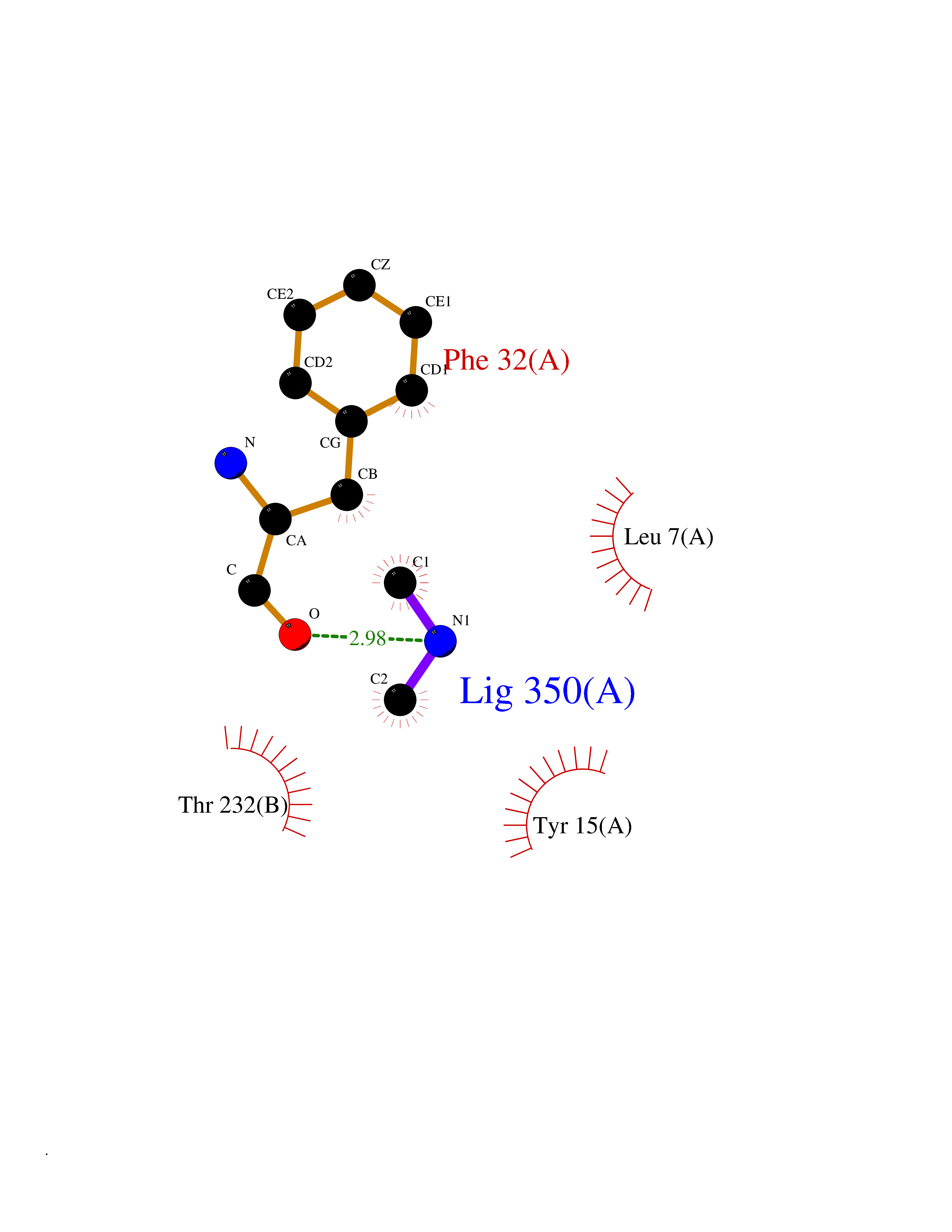 Ligplot Map 3D Binding mode Sequence GPIPEVLKNYMDAQYYGEIGIGTPPQCFTVVFDTGSSNLWVPSIHCKLLDIACWIHHKYNSDKSSTYVKNGTSFDIHYGSGSLSGYLSQDTVSVPCQSGGVKVERQVFGEATKQPGITFIAAKFDGILGMAYPRISVNNVLPVFDNLMQQKLVDQNIFSFYLSRDPDAQPGGELMLGGTDSKYYKGSLSYLNVTRKAYWQVHLDQVEVASGLTLCKEGCEAIVDTGTSLMVGPVDEVRELQKAIGAVPLIQGEYMIPCEKVSTLPAITLKLGGKGYKLSPEDYTLKVSQAGKTLCLSGFMGMDIPPPSGPLWILGDVFIGRYYTVFDRDNNRVGFAEAARL Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Membrane copper amine oxidase (AOC3) | 4BTX | 4.02 | |
Target general information Gen name AOC3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vascular adhesion protein-1; Vascular adhesion protein 1; VAP1; VAP-1; Semicarbazide-sensitive amine oxidase; SSAO; Membrane primary amine oxidase; HPAO; Copper amine oxidase Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Has semicarbazide-sensitive (SSAO) monoamine oxidase activity. May play a role in adipogenesis. Cell adhesion protein that participates in lymphocyte extravasation and recirculation by mediating the binding of lymphocytes to peripheral lymph node vascular endothelial cells in an L-selectin-independent fashion. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB04334; DB01275; DB00780 Interacts with Q3SXY8; O95484; Q7Z7G2; Q96BA8; Q6PI48; Q8TBE3; Q7Z5P4; P42858; O43765; Q16623 EC number EC 1.4.3.21 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell adhesion; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Signal-anchor; TPQ; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 157266 Length 1415 Aromaticity 0.12 Instability index 39.94 Isoelectric point 6 Charge (pH=7) -23.49 2D Binding mode Binding energy (Kcal/mol) -5.48 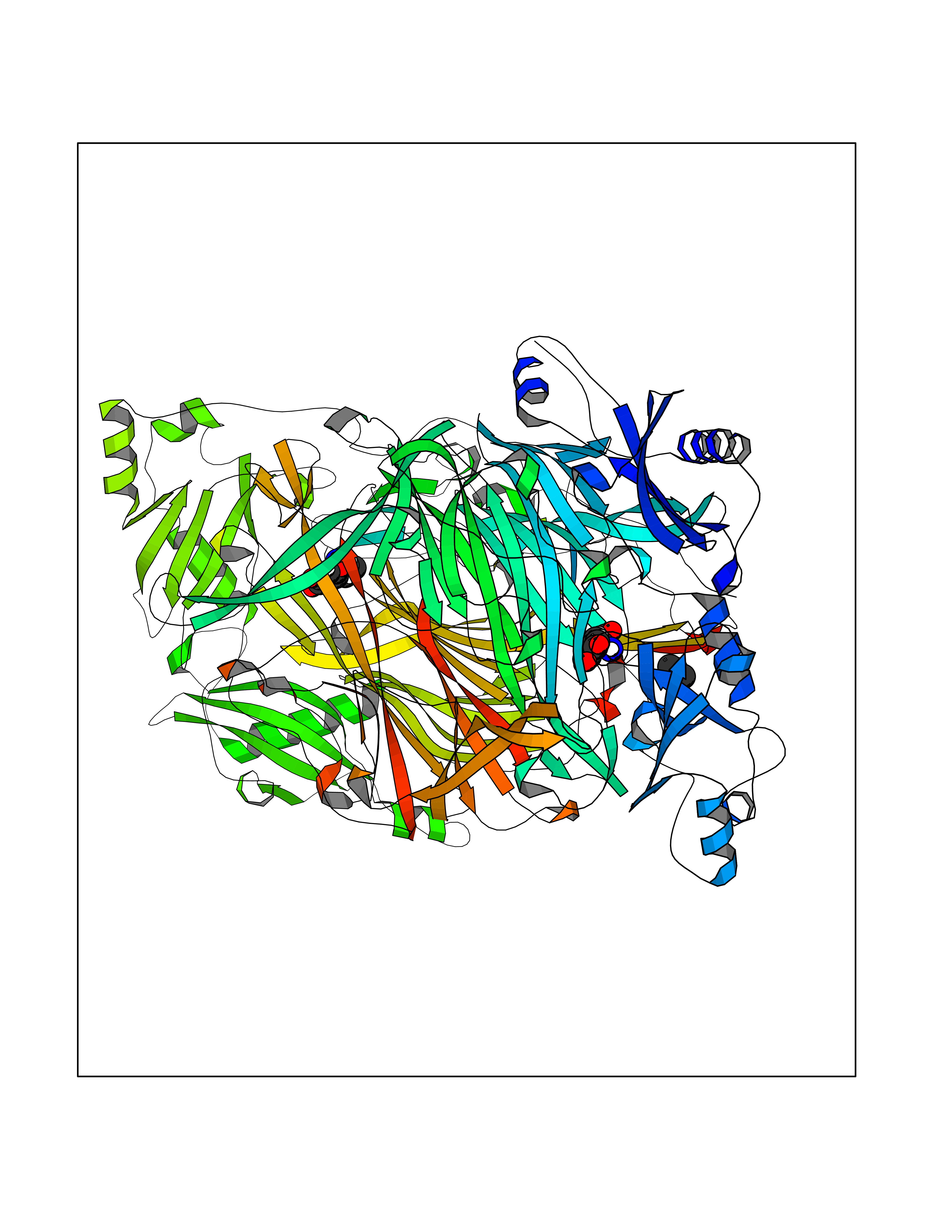 Molscript Map 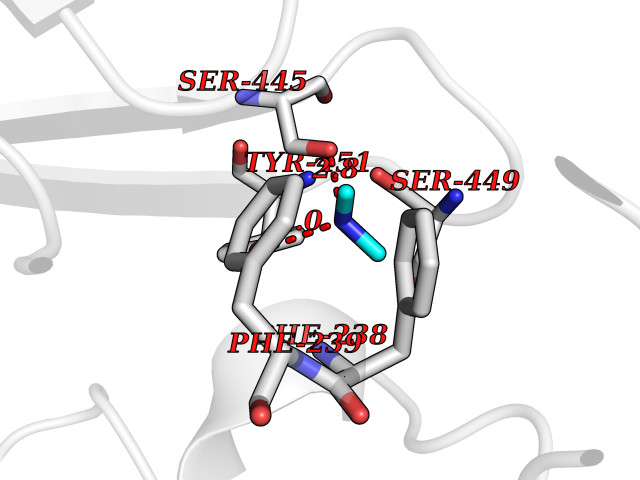 Pymol Map 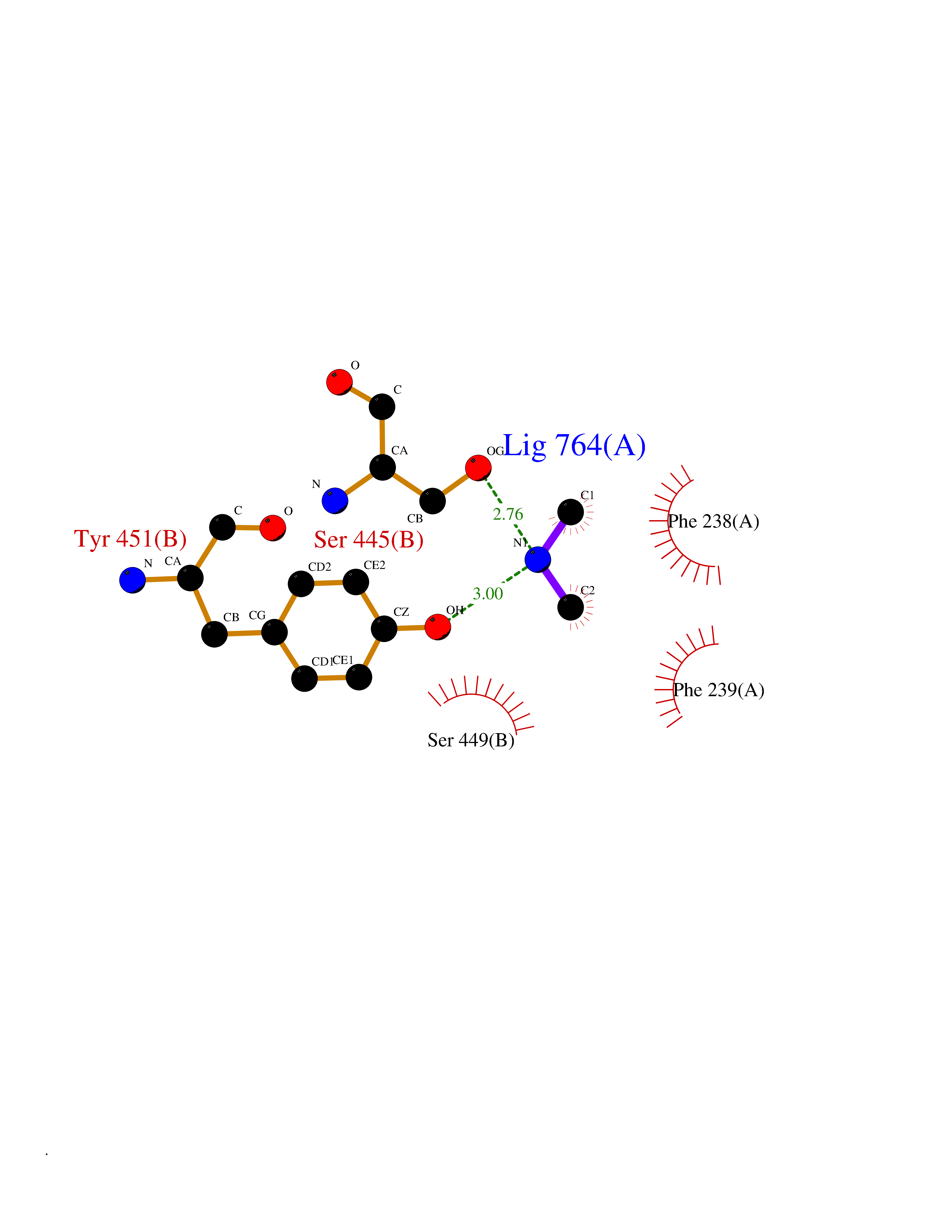 Ligplot Map 3D Binding mode Sequence PGQSQLFADLSREELTAVMRFLTQRLGPGLVDAAQARPSDNCVFSVELQLPPKAAALAHLDRGSPPPAREALAIVFFGRQPQPNVSELVVGPLPHPSYMRDVTVERHGGPLPYHRRPVLFQEYLDIDQMIFNRELPQASGLLHHCCFYKHRGRNLVTMTTAPRGLQSGDRATWFGLYYNISGAGFFLHHVGLELLVNHKALDPARWTIQKVFYQGRYYDSLAQLEAQFEAGLVNVVLIPDNGTGGSWSLKSPVPPGPAPPLQFYPQGPRFSVQGSRVASSLWTFSFGLGAFSGPRIFDVRFQGERLVYEISLQEALAIYGGNSPAAMTTRYVDGGFGMGKYTTPLTRGVDCPYLATYVDWHFLLESQAPKTIRDAFCVFEQNQGLPLRRHHSDLYSHYFGGLAETVLVVRSMSTLLNXDYVWDTVFHPSGAIEIRFYATGYISSAFLFGATGKYGNQVSEHTLGTVHTHSAHFKVDLDVAGLENWVWAEDMVFVPMAVPWSPEHQLQRLQVTRKLLEMEEQAAFLVGSATPRYLYLASNHSNKWGHPRGYRIQMLSFAGEPLPQNSSMARGFSWERYQLAVTQRKEEEPSSSSVFNQNDPWAPTVDFSDFINNETIAGKDLVAWVTAGFLHIPHAEDIPNTVTVGNGVGFFLRPYNFFDEDPSFYSADSIYFRGDQDAGACEVNPLACLPQAAACAPDLPAFSHGGFSHSQLFADLSREELTAVMRFLTQRLGPGLVDAAQARPSDNCVFSVELQLPPKAAALAHLDRGSPPPAREALAIVFFGRQPQPNVSELVVGPLPHPSYMRDVTVERHGGPLPYHRRPVLFQEYLDIDQMIFNRELPQASGLLHHCCFYKHRGRNLVTMTTAPRGLQSGDRATWFGLYYNISGAGFFLHHVGLELLVNHKALDPARWTIQKVFYQGRYYDSLAQLEAQFEAGLVNVVLIPDNGTGGSWSLKSPVPPGPAPPLQFYPQGPRFSVQGSRVASSLWTFSFGLGAFSGPRIFDVRFQGERLVYEISLQEALAIYGGNSPAAMTTRYVDGGFGMGKYTTPLTRGVDCPYLATYVDWHFLLESQAPKTIRDAFCVFEQNQGLPLRRHHSDLYSHYFGGLAETVLVVRSMSTLLNXDYVWDTVFHPSGAIEIRFYATGYISSAFLFGATGKYGNQVSEHTLGTVHTHSAHFKVDLDVAGLENWVWAEDMVFVPMAVPWSPEHQLQRLQVTRKLLEMEEQAAFLVGSATPRYLYLASNHSNKWGHPRGYRIQMLSFAGEPLPQNSSMARGFSWERYQLAVTQRKEEEPSSSSVFNQNDPWAPTVDFSDFINNETIAGKDLVAWVTAGFLHIPHAEDIPNTVTVGNGVGFFLRPYNFFDEDPSFYSADSIYFRGDQDAGACEVNPLACLPQAAACAPDLPAFSHGGFSH Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Complement C1s component (C1S) | 1ELV | 4.02 | |
Target general information Gen name C1S Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complement component 1 subcomponent s; Complement C1s subcomponent; C1-esterase; C1 esterase Protein family Peptidase S1 family Biochemical class Peptidase Function C1r activates C1s so that it can, in turn, activate C2 and C4. C1s B chain is a serine protease that combines with C1q and C1r to form C1, the first component of the classical pathway of the complement system. Related diseases Complement component C1s deficiency (C1SD) [MIM:613783]: A rare defect resulting in C1 deficiency and impaired activation of the complement classical pathway. C1 deficiency generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. {ECO:0000269|PubMed:11390518}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ehlers-Danlos syndrome, periodontal type, 2 (EDSPD2) [MIM:617174]: A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSPD2 is characterized by the association of typical features of Ehlers-Danlos syndrome with gingival recession and severe early-onset periodontal disease, leading to premature loss of permanent teeth. EDSPD2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:27745832}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02371; DB09228; DB09130; DB12831; DB06404; DB14996; DB01593; DB14487; DB14533; DB14548 Interacts with P00736; P09871; P06681; O43889-2; Q9H6H4; P05155 EC number EC 3.4.21.42 Uniprot keywords 3D-structure; Calcium; Complement pathway; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Ehlers-Danlos syndrome; Glycoprotein; Hydrolase; Hydroxylation; Immunity; Innate immunity; Metal-binding; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal; Sushi Protein physicochemical properties Chain ID A Molecular weight (Da) 33278.6 Length 303 Aromaticity 0.1 Instability index 33.69 Isoelectric point 5.16 Charge (pH=7) -7.95 2D Binding mode Binding energy (Kcal/mol) -5.48 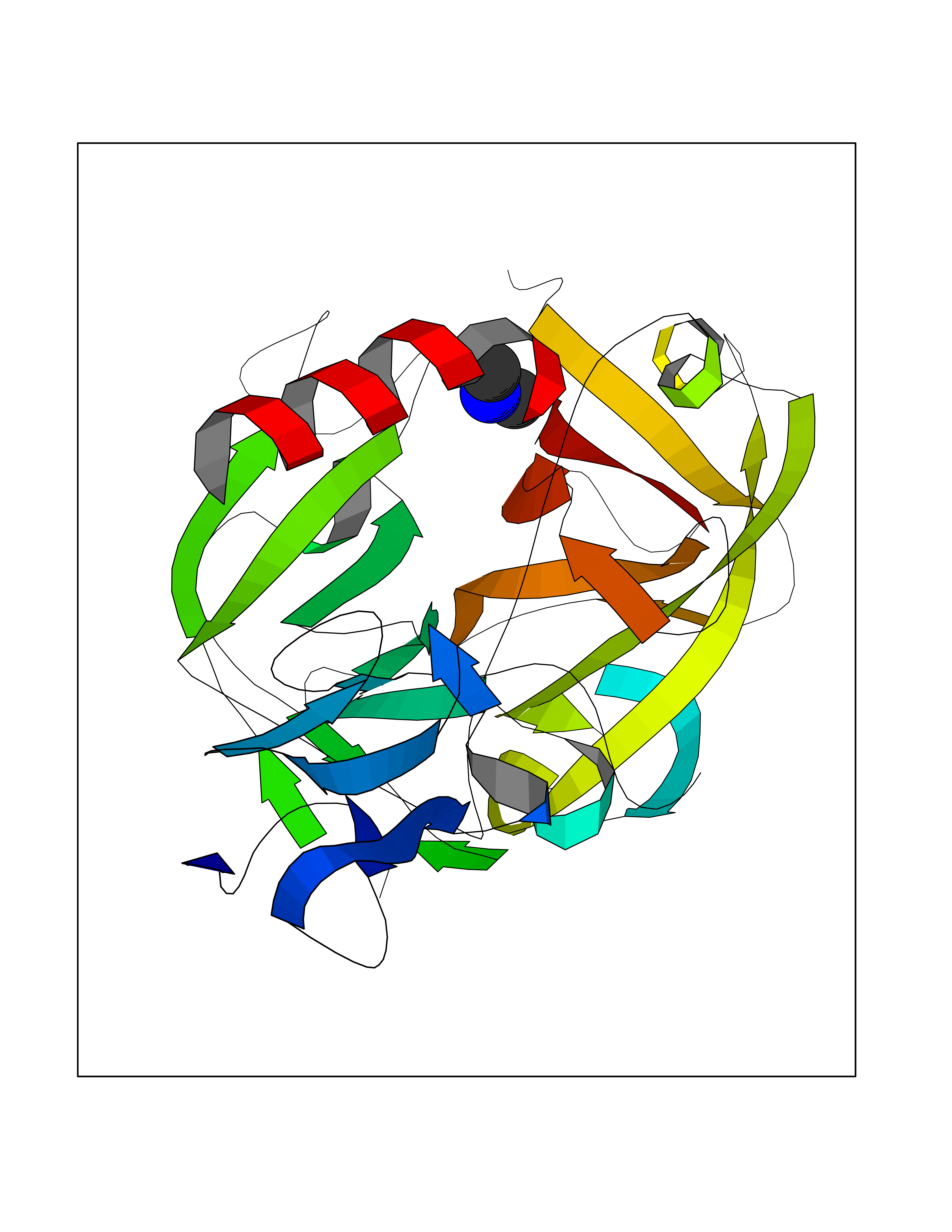 Molscript Map 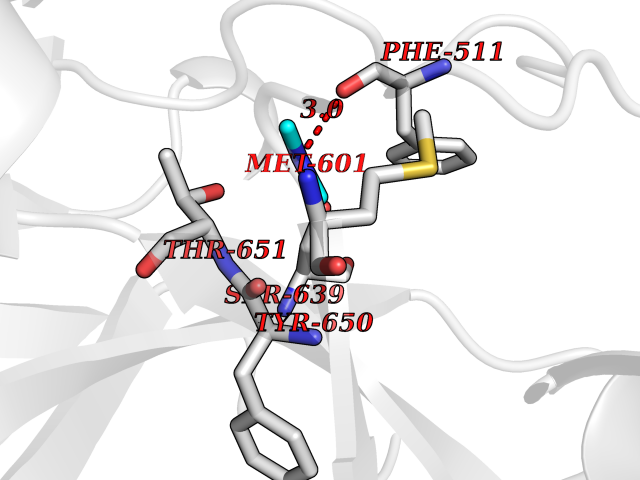 Pymol Map 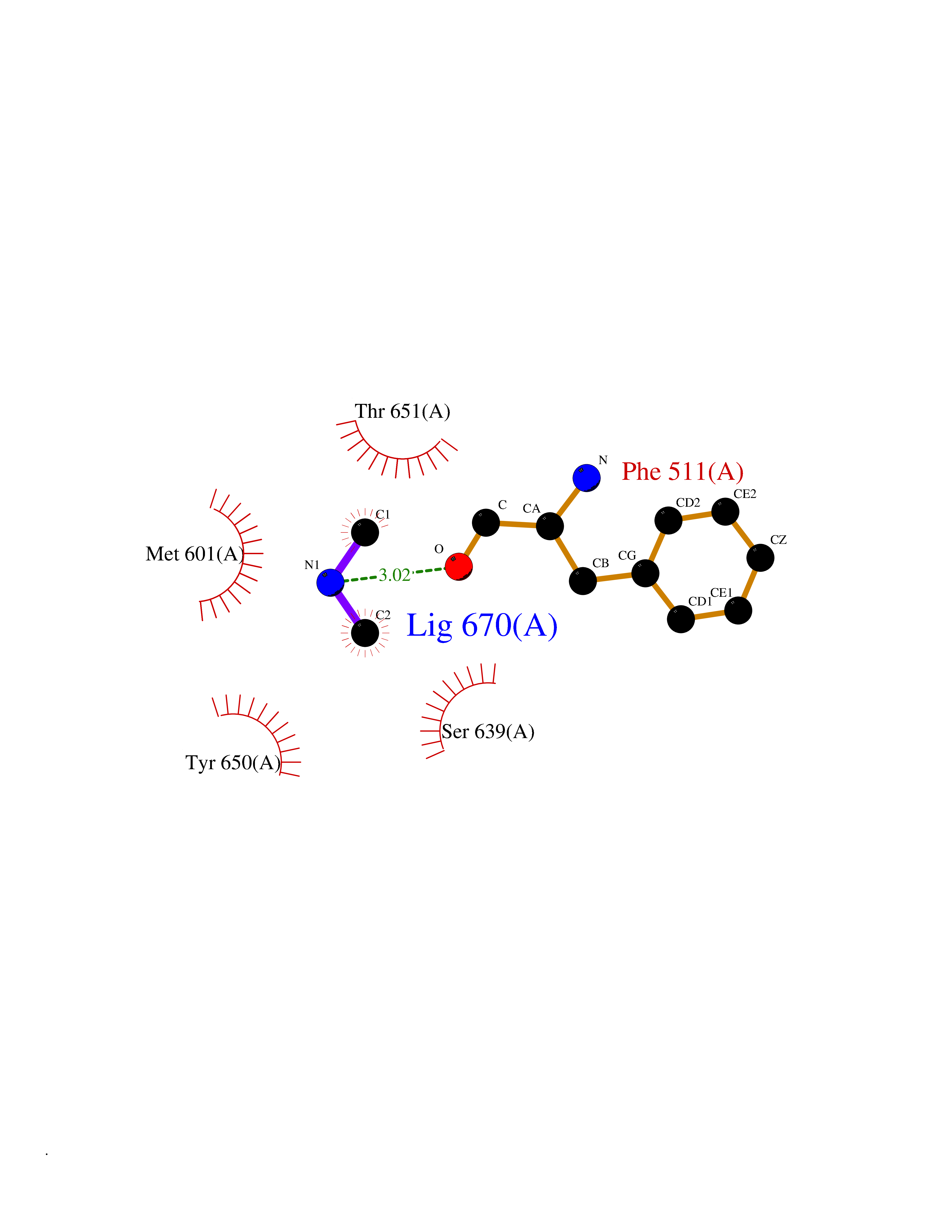 Ligplot Map 3D Binding mode Sequence LDCGIPESIENGKVEDPESTLFGSVIRYTCEEPYYYMEGGGEYHCAGNGSWVNEVLGPELPKCVPVCGVPREPFIIGGSDADIKNFPWQVFFDNPWAGGALINEYWVLTAAHVVEGNREPTMYVGSTSVQKMLTPEHVFIHPGWKLLAVPEGRTNFDNDIALVRLKDPVKMGPTVSPICLPGTSSDYNLMDGDLGLISGWGRTEKRDRAVRLKAARLPVAPLRKCKEVAYVFTPNMICAGGEKGMDSCKGDSGGAFAVQDPNDKTKFYAAGLVSWGPQCGTYGLYTRVKNYVDWIMKTMQENS Hydrogen bonds contact Hydrophobic contact | ||||