Job Results:
Ligand
Structure
Job ID
215921f481371e928c7811c49ba9b3ae
Job name
NA
Time
2025-01-23 16:39:47
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 7.22 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -9.84  Molscript Map 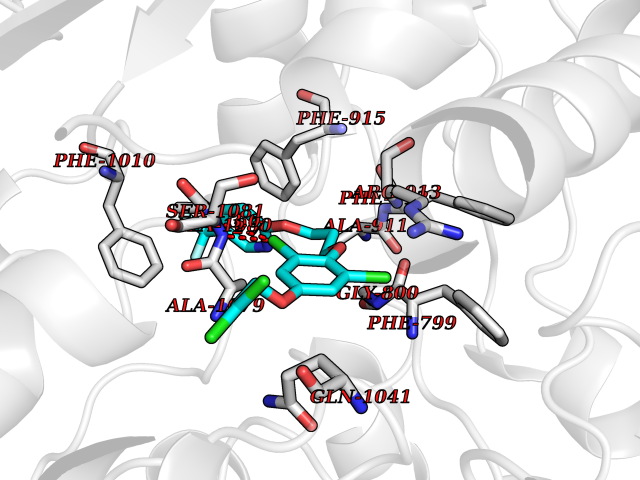 Pymol Map 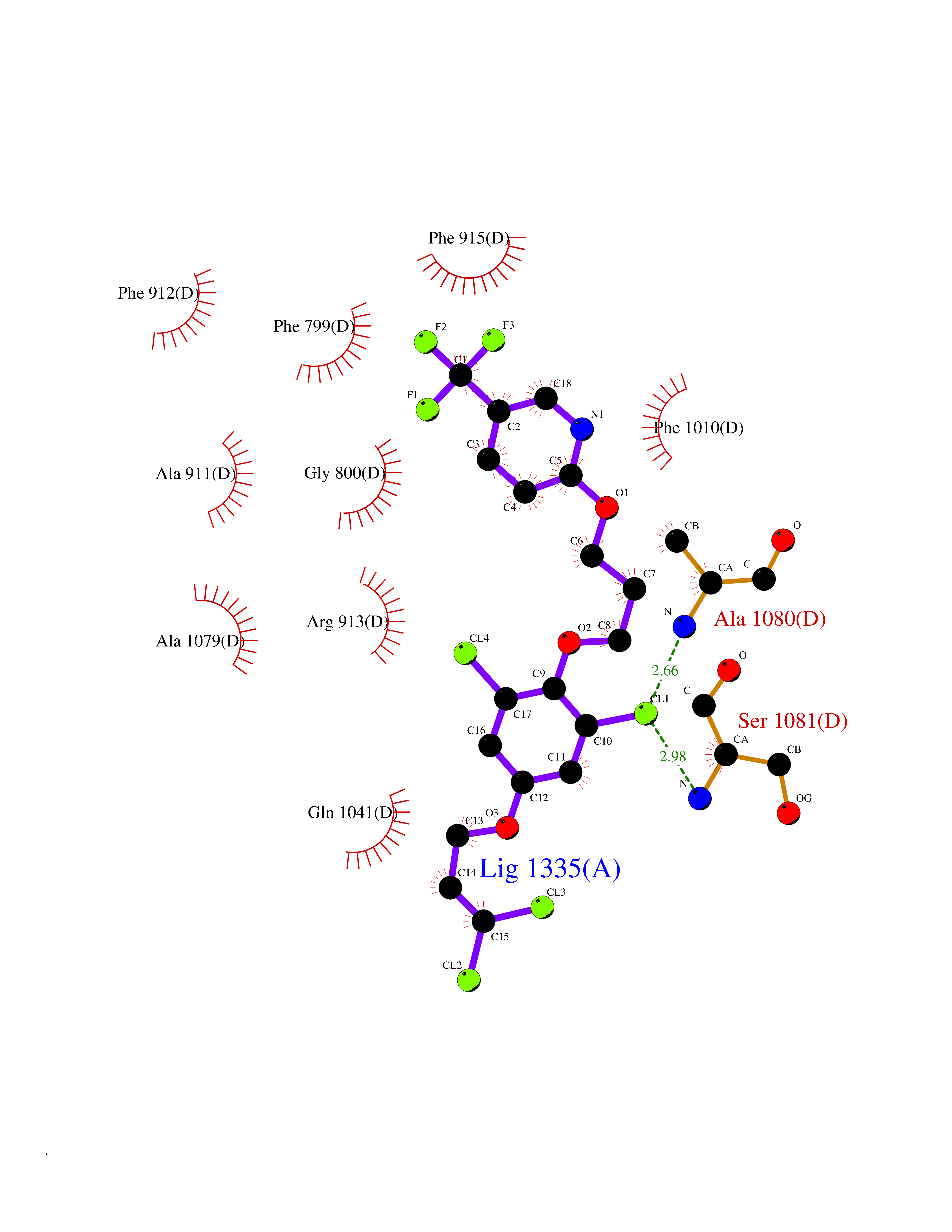 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 7.22 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -9.85 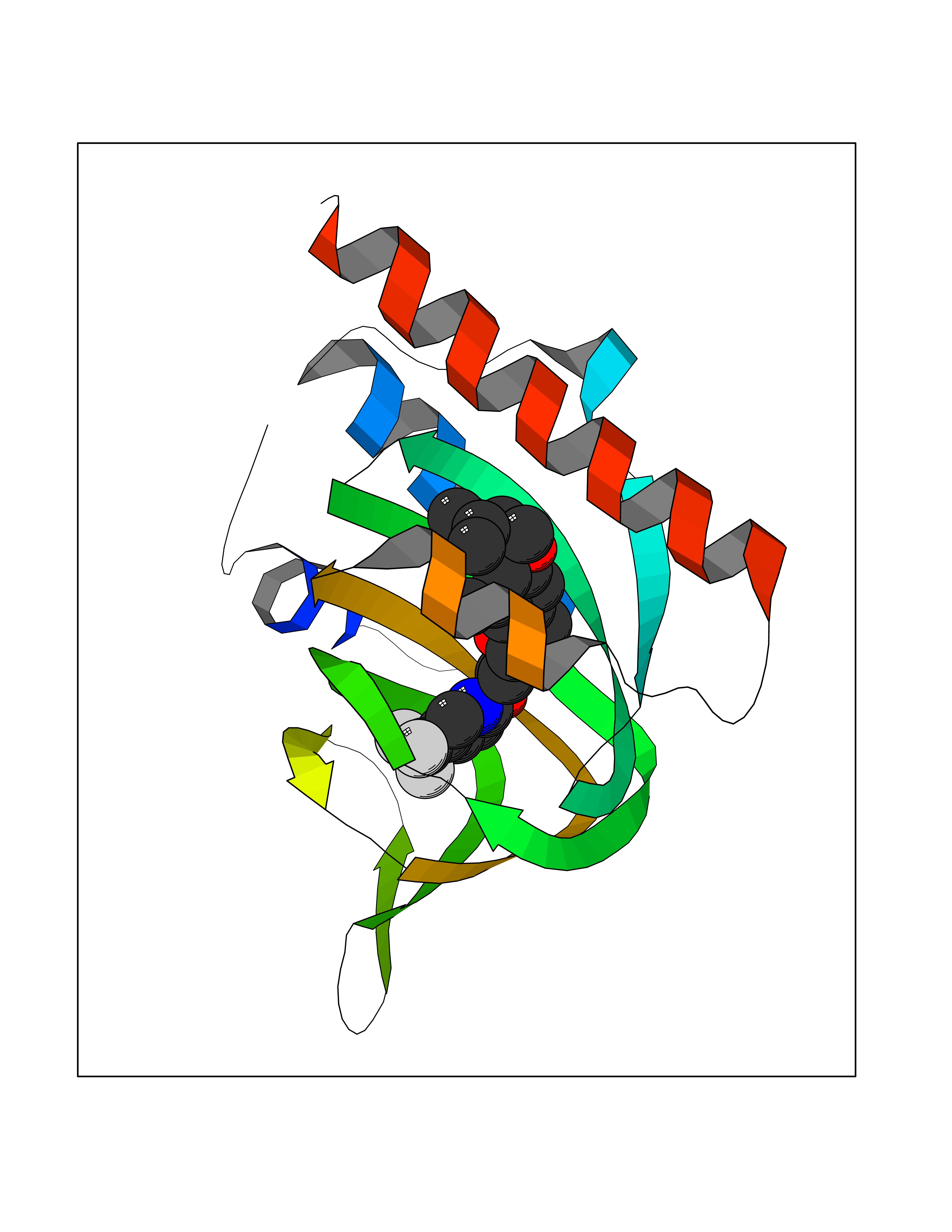 Molscript Map 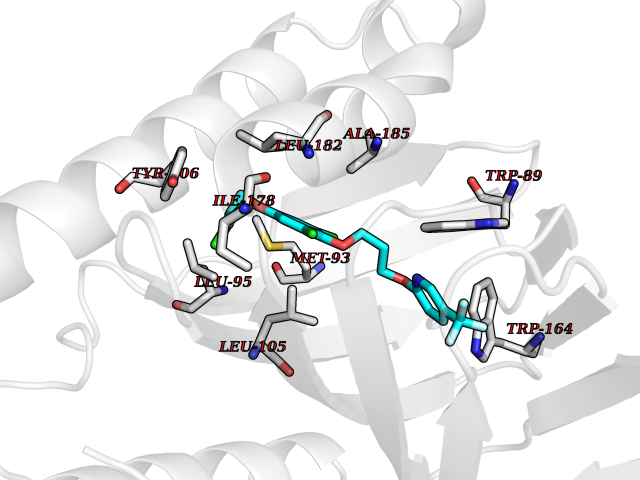 Pymol Map 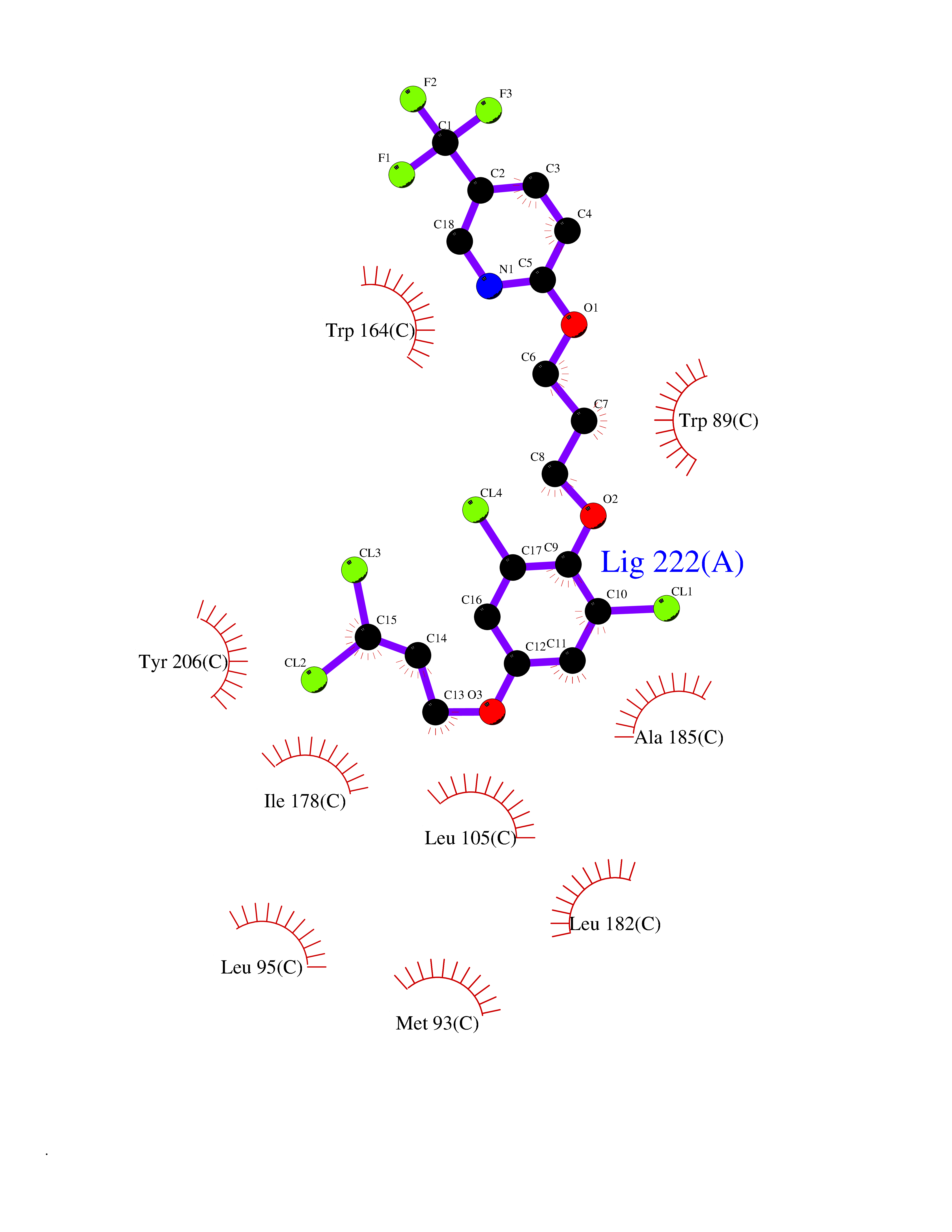 Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Thyroid hormone receptor alpha (THRA) | 3ILZ | 7.21 | |
Target general information Gen name THRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms V-erbA-related protein 7; THRA2; THRA1; Nuclear receptor subfamily 1 group A member 1; NR1A1; ERBA1; EAR7; EAR-7; C-erbA-alpha; C-erbA-1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Isoform Alpha-1: Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Hypothyroidism, congenital, non-goitrous, 6 (CHNG6) [MIM:614450]: A disease characterized by growth retardation, developmental retardation, skeletal dysplasia, borderline low thyroxine levels and high triiodothyronine levels. There is differential sensitivity to thyroid hormone action, with retention of hormone responsiveness in the hypothalamic pituitary axis and liver but skeletal, gastrointestinal, and myocardial resistance. {ECO:0000269|PubMed:22168587, ECO:0000269|PubMed:24969835, ECO:0000269|PubMed:25670821, ECO:0000269|PubMed:26037512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01118; DB00509; DB04855; DB05035; DB03176; DB00451; DB00279; DB01583; DB05235; DB09100 Interacts with Q9Y2J4; Q9Y2J4-4; O95971; Q8TAP6; Q96JM7; Q15648; Q6FHY5; P31321; Q96A49; O75410-7; Q9JLI4 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Congenital hypothyroidism; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29910.1 Length 267 Aromaticity 0.07 Instability index 52.75 Isoelectric point 5.31 Charge (pH=7) -11.32 2D Binding mode Binding energy (Kcal/mol) -9.83 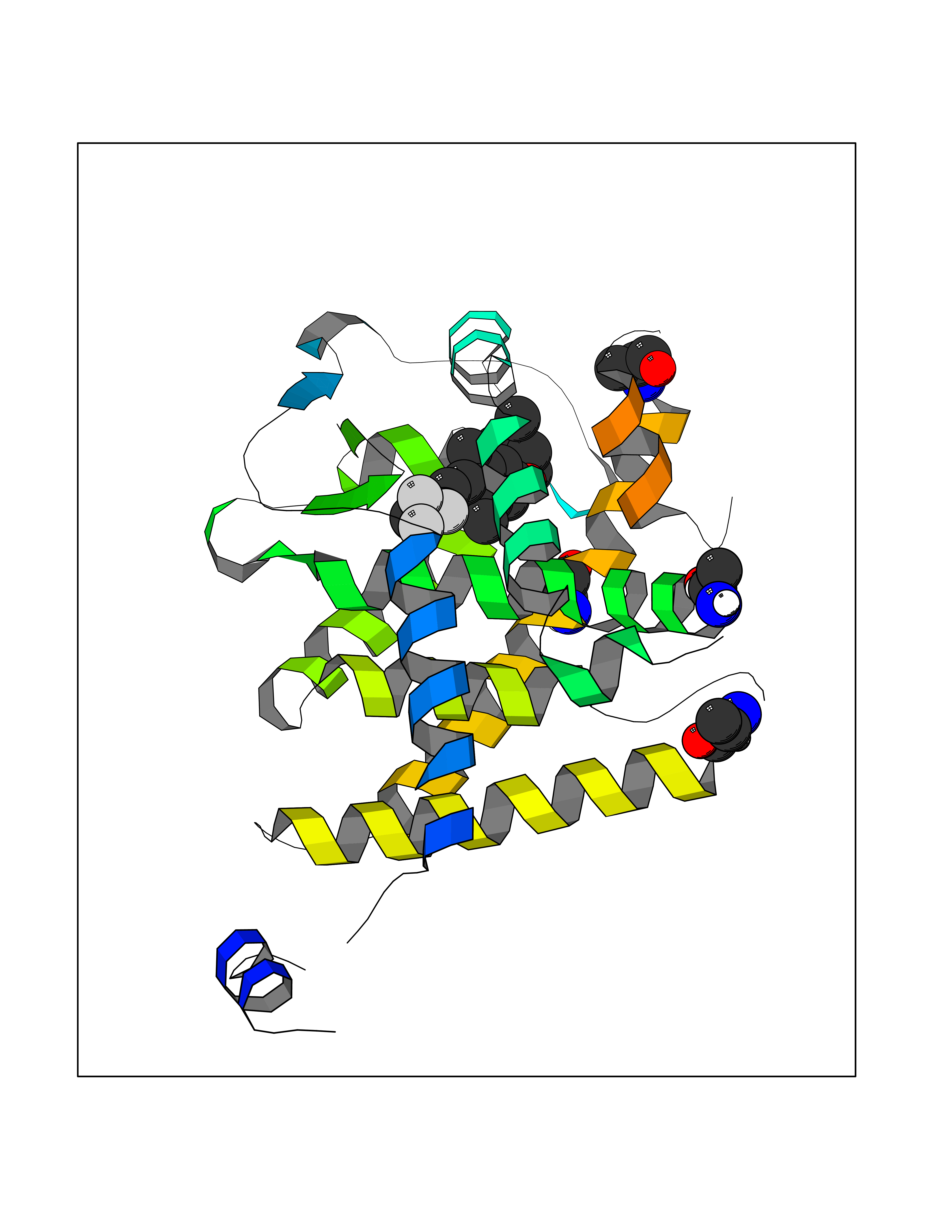 Molscript Map 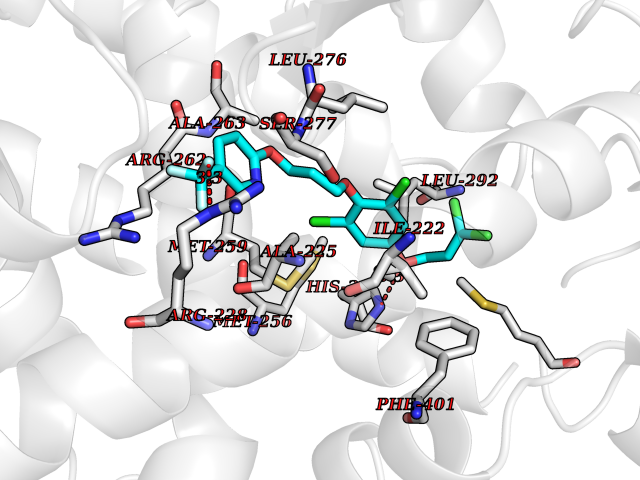 Pymol Map 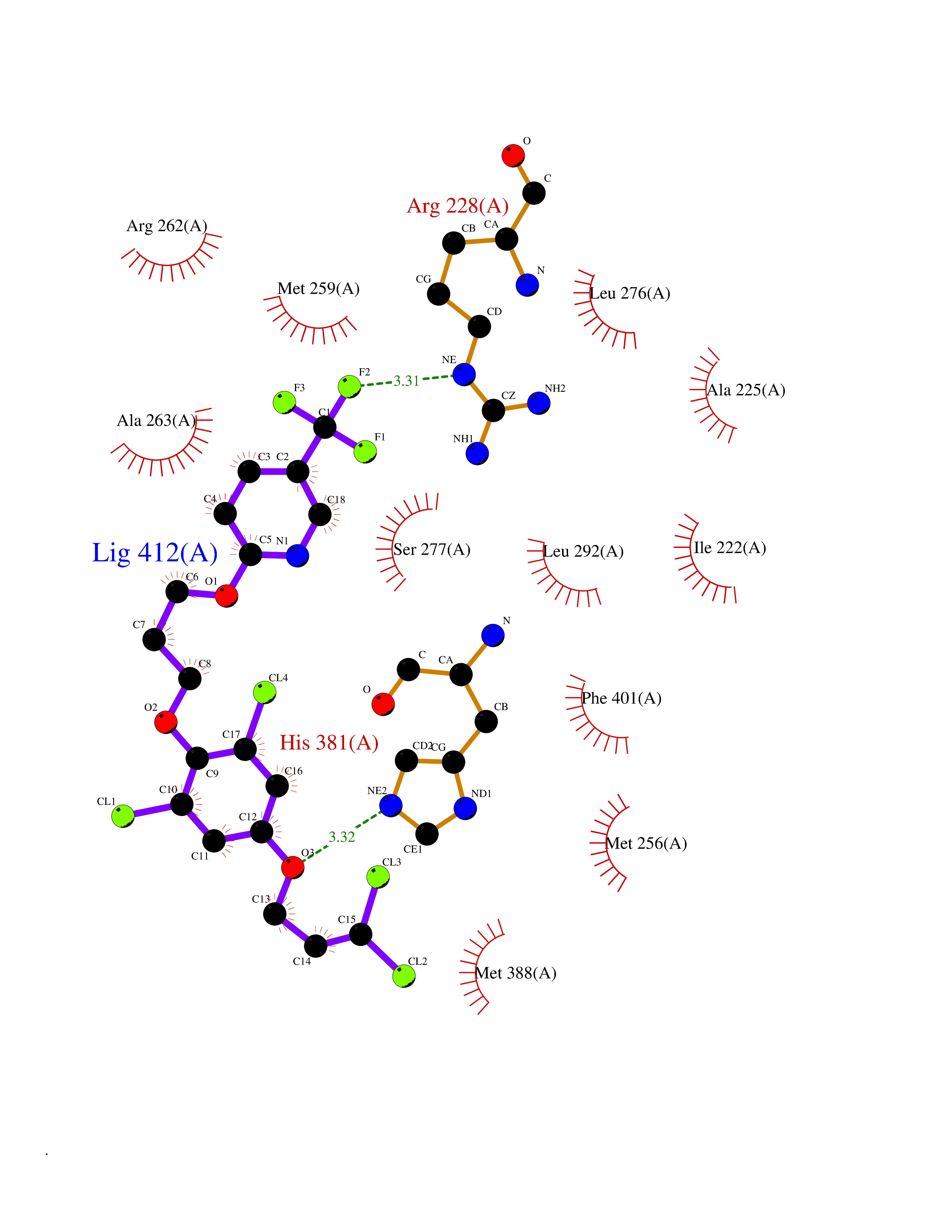 Ligplot Map 3D Binding mode Sequence GSHMEEMIRSLQQRPEPTPEEWDLIHIATEAHRSTNAQGSHWKQRRKFLPDDIGQSPIVSMPDGDKVDLEAFSEFTKIITPAITRVVDFAKKLPMFSELPXEDQIILLKGCCMEIMSLRAAVRYDPESDTLTLSGEMAVKREQLKNGGLGVVSDAIFELGKSLSAFNLDDTEVALLQAVLLMSTDRSGLLXVDKIEKSQEAYLLAFEHYVNHRKHNIPHFWPKLLMKVTDLRMIGAXHASRFLHMKVEXPTELFPPLFLEVFEDQEV Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Aldehyde oxidoreductase | 4USA | 7.21 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -9.84  Molscript Map  Pymol Map 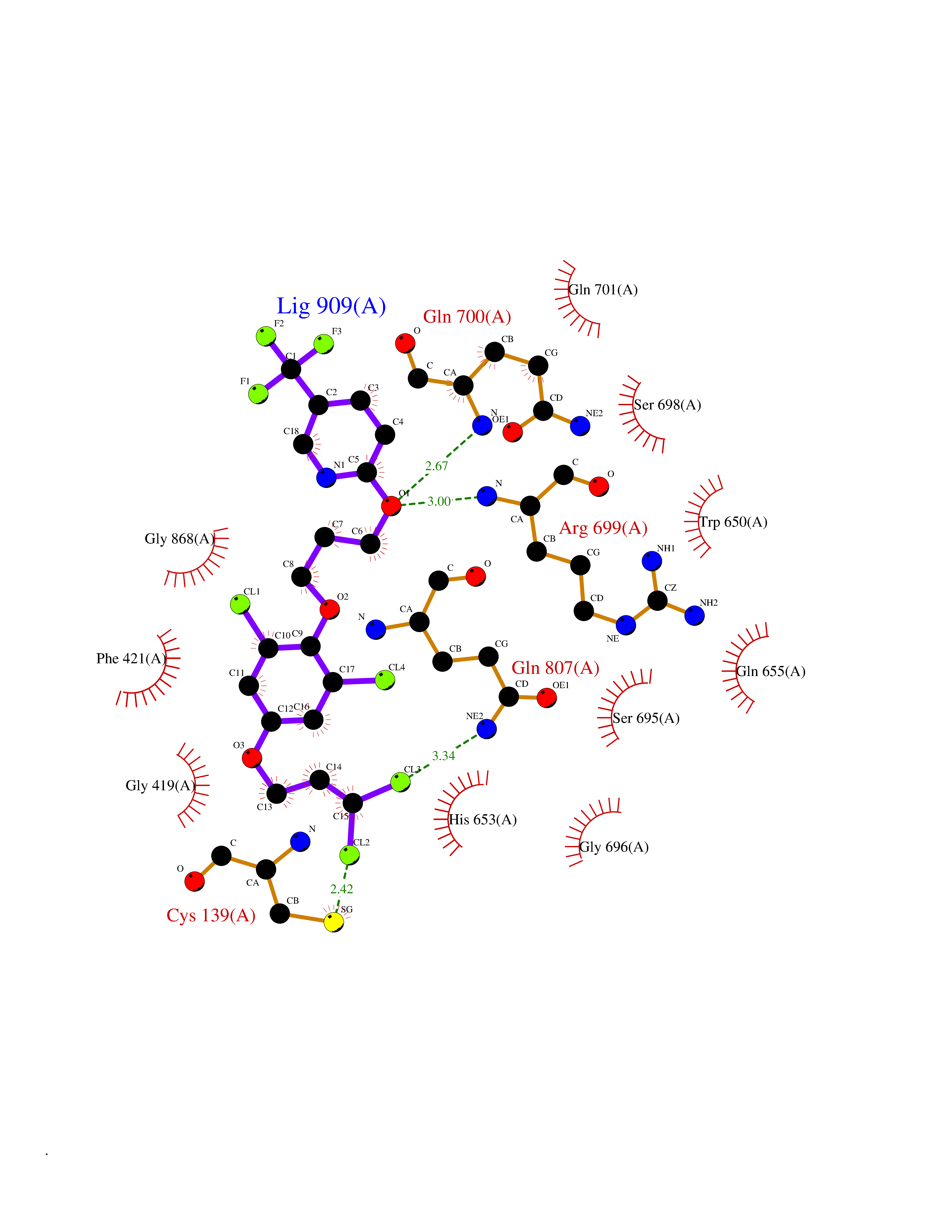 Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Guanidinoacetate N-methyltransferase | 3ORH | 7.19 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -9.81  Molscript Map  Pymol Map 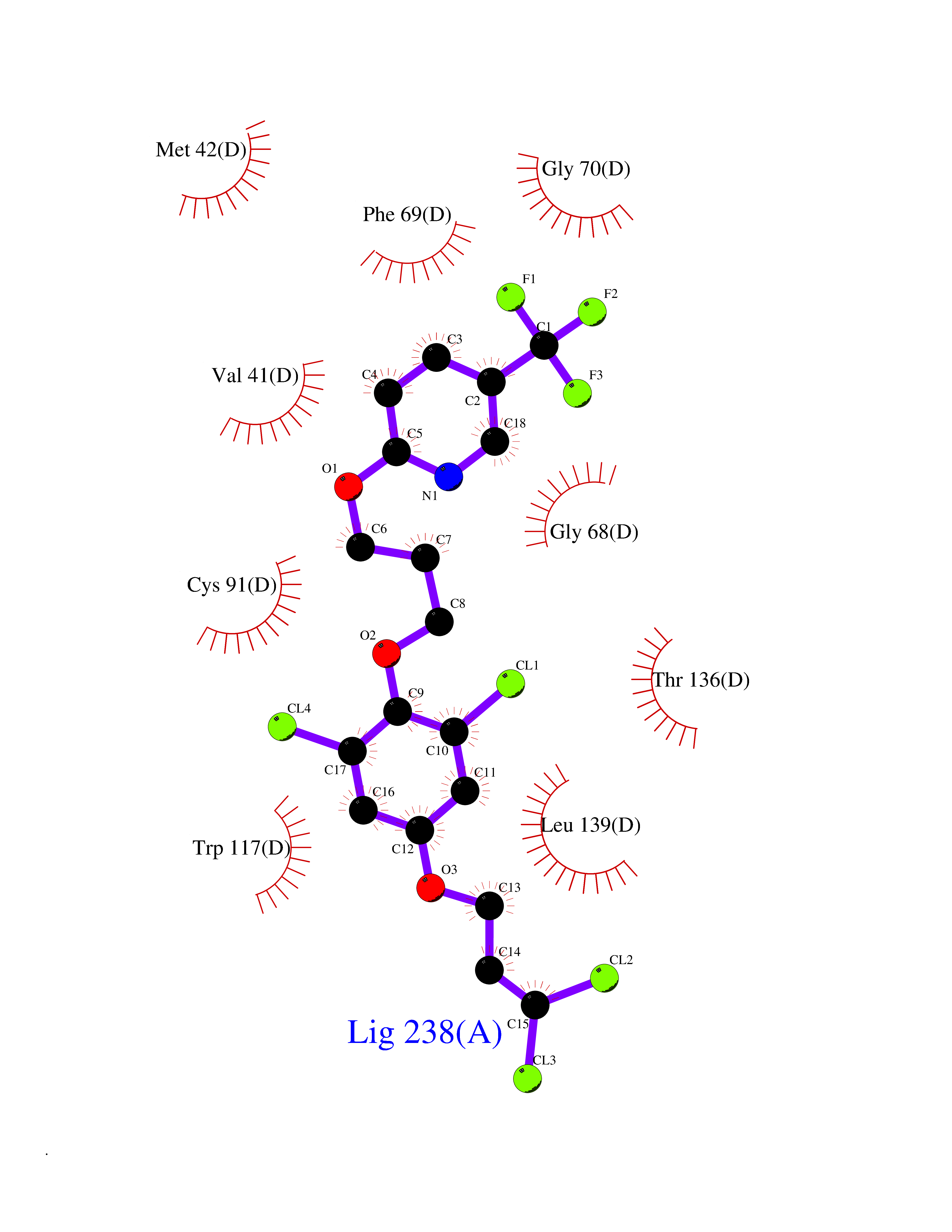 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | PRKR-like endoplasmic reticulum kinase (PERK) | 4G31 | 7.19 | |
Target general information Gen name EIF2AK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PEK Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class Kinase Function Converts phosphorylated eIF-2-alpha/EIF2S1 either in a global protein synthesis inhibitor, leading to a reduced overall utilization of amino acids, or to a translation initiation activator of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Serves as a critical effector of unfolded protein response (UPR)-induced G1 growth arrest due to the loss of cyclin-D1 (CCND1). Involved in control of mitochondrial morphology and function. Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' during the unfolded protein response (UPR) and in response to low amino acid availability. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NZJ5; P11021 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ADP-ribosylation; ATP-binding; Diabetes mellitus; Disease variant; Endoplasmic reticulum; Glycoprotein; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Signal; Stress response; Transferase; Translation regulation; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Unfolded protein response Protein physicochemical properties Chain ID A Molecular weight (Da) 29033.5 Length 248 Aromaticity 0.1 Instability index 43.71 Isoelectric point 7.75 Charge (pH=7) 1.27 2D Binding mode Binding energy (Kcal/mol) -9.81  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GRYLTDFEPIQCLGRGGVVFEAKNKVDDCNYAIKRIRLPNRELAREKVMREVKALAKLEHPGIVRYFNAWLEKNKVYLYIQMQLCRKENLKDWMNGRCTIEERERSVCLHIFLQIAEAVEFLHSKGLMHRDLKPSNIFFTMDDVVKVGDFGLVGTKLYMSPEQIHGNSYSHKVDIFSLGLILFELLYPFSTQMERVRTLTDVRNLKFPPLFTQKYPCEYVMVQDMLSPSPMERPEAINIIENAVFEDL Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Vitamin K-dependent protein C (PROC) | 1LQV | 7.18 | |
Target general information Gen name PROC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K-dependent protein C light chain; Vitamin K-dependent protein C heavy chain; PROC; Blood coagulation factor XIV; Autoprothrombin IIA; Anticoagulant protein C; Activation peptide Protein family Peptidase S1 family Biochemical class Peptidase Function Protein C is avitamin K-dependent serine protease that regulates blood coagulation by inactivating factors Va and VIIIa in the presence of calcium ions and phospholipids. Related diseases Thrombophilia due to protein C deficiency, autosomal dominant (THPH3) [MIM:176860]: A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. Individuals with decreased amounts of protein C are classically referred to as having type I protein C deficiency and those with normal amounts of a functionally defective protein as having type II deficiency. {ECO:0000269|PubMed:1301959, ECO:0000269|PubMed:1347706, ECO:0000269|PubMed:1511989, ECO:0000269|PubMed:1868249, ECO:0000269|PubMed:2437584, ECO:0000269|PubMed:25618265, ECO:0000269|PubMed:25748729, ECO:0000269|PubMed:2602169, ECO:0000269|PubMed:7792728, ECO:0000269|PubMed:7865674, ECO:0000269|PubMed:8292730, ECO:0000269|PubMed:8398832, ECO:0000269|PubMed:8499568, ECO:0000269|PubMed:8560401, ECO:0000269|PubMed:8829639, ECO:0000269|PubMed:9798967}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to protein C deficiency, autosomal recessive (THPH4) [MIM:612304]: A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. It results in a thrombotic condition that can manifest as a severe neonatal disorder or as a milder disorder with late-onset thrombophilia. The severe form leads to neonatal death through massive neonatal venous thrombosis. Often associated with ecchymotic skin lesions which can turn necrotic called purpura fulminans, this disorder is very rare. {ECO:0000269|PubMed:1511988, ECO:0000269|PubMed:1593215, ECO:0000269|PubMed:1611081, ECO:0000269|PubMed:25618265, ECO:0000269|PubMed:7841323, ECO:0000269|PubMed:7841324, ECO:0000269|PubMed:7878626}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13192; DB00025; DB09131; DB09332; DB13998; DB00170; DB13999; DB13149; DB00464; DB14738 Interacts with A8MQ03; P51511 EC number EC 3.4.21.69 Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endoplasmic reticulum; Gamma-carboxyglutamic acid; Glycoprotein; Golgi apparatus; Hemostasis; Hydrolase; Hydroxylation; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID C,D Molecular weight (Da) 45326 Length 411 Aromaticity 0.12 Instability index 40.68 Isoelectric point 7.07 Charge (pH=7) 0.29 2D Binding mode Binding energy (Kcal/mol) -9.8  Molscript Map 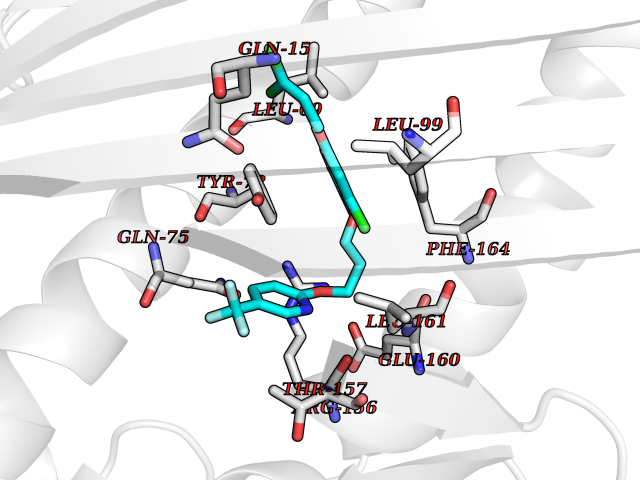 Pymol Map 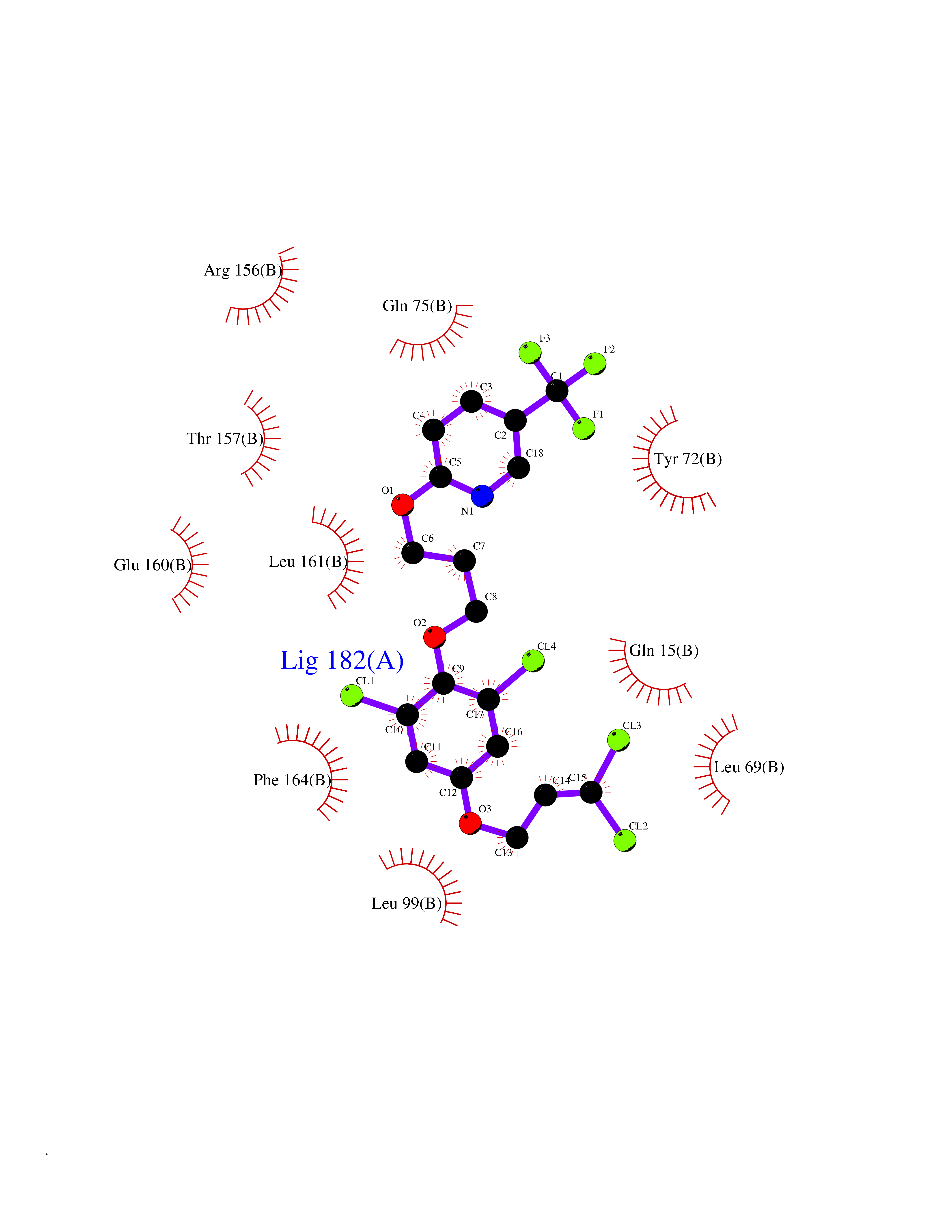 Ligplot Map 3D Binding mode Sequence GLQRLHMLQISYFRDPYHVWYQGNASLGGHLTHVLEGPDTNTTIIQLQPLQEPESWARTQSGLQSYLLQFHGLVRLVHQERTLAFPLTIRCFLGCELPPEGSRAHVFFEVAVNGSSFVSFRPERALWQADTQVTSGVVTFTLQQLNAYNRTRYELREFLEDTCVQYVQKHISANSFLXXLRHSSLXRXCIXXICDFXXAKXIFQNANSFLXXLRHSSLXRXCIXXICDFXXAKXIFQNLQRLHMLQISYFRDPYHVWYQGNASLGGHLTHVLEGPDTNTTIIQLQPLQEPESWARTQSGLQSYLLQFHGLVRLVHQERTLAFPLTIRCFLGCELPPEGSRAHVFFEVAVNGSSFVSFRPERALWQADTQVTSGVVTFTLQQLNAYNRTRYELREFLEDTCVQYVQKHISAE Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 7.18 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -9.8 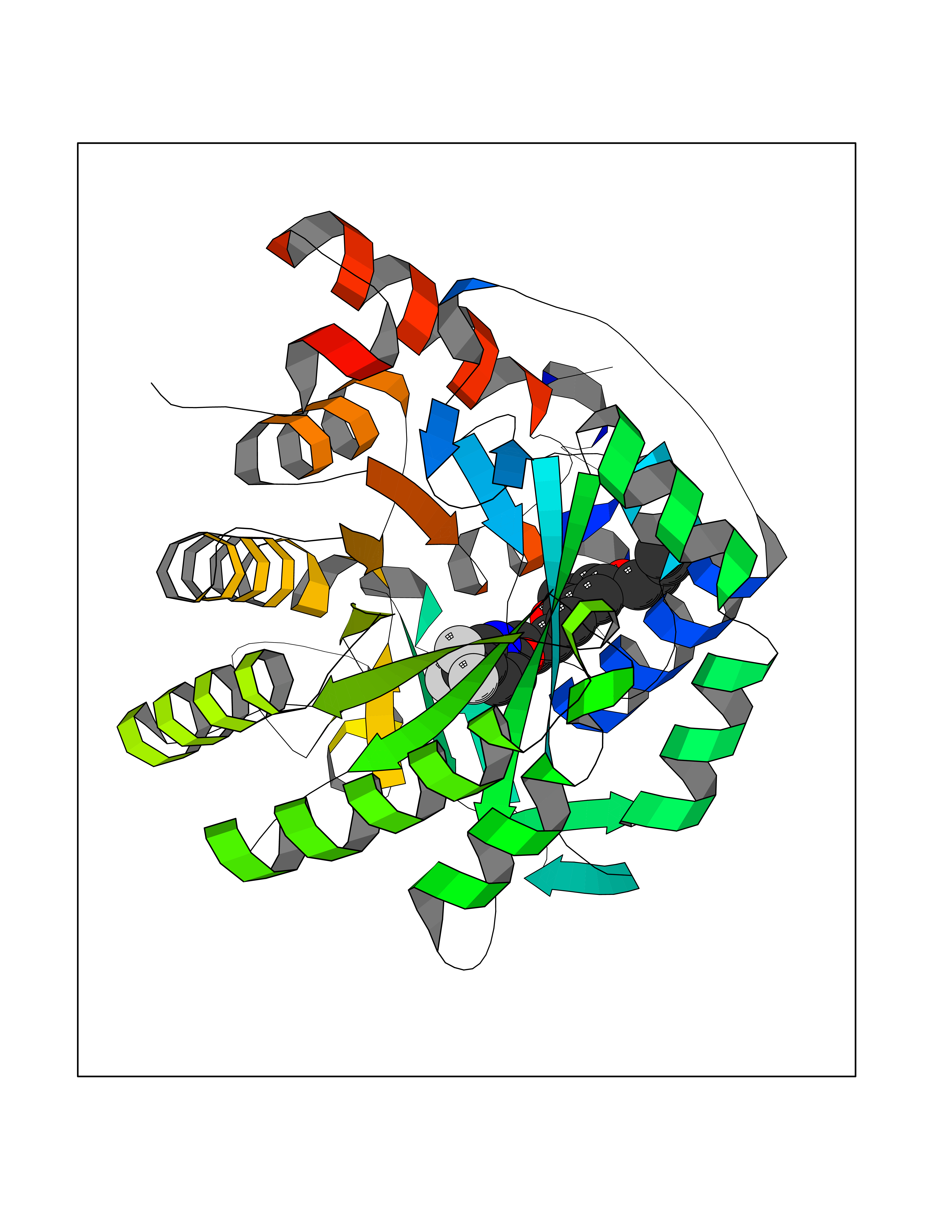 Molscript Map 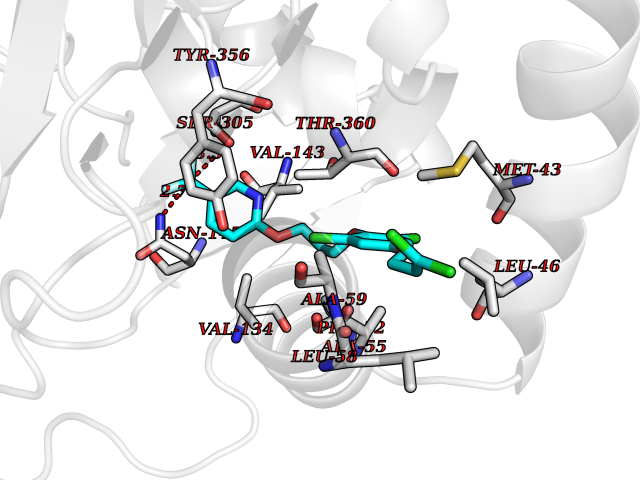 Pymol Map 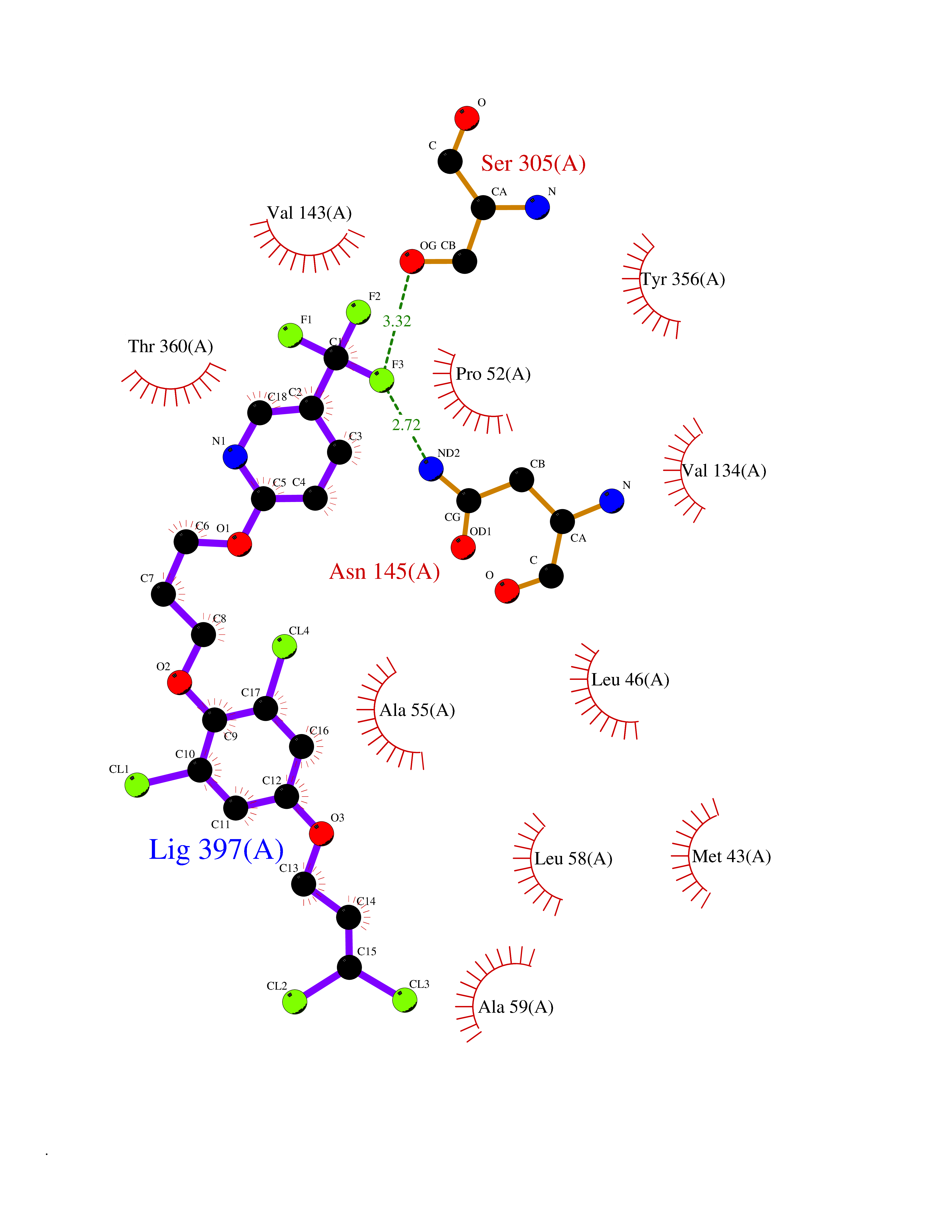 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Melatonin receptor type 1B (MTNR1B) | 6ME9 | 7.18 | |
Target general information Gen name MTNR1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1b receptor; Mel1b melatonin receptor; Mel-1B-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071; DB15133 Interacts with P28335; P48039; O76081; Q14669 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50184.9 Length 448 Aromaticity 0.11 Instability index 37.2 Isoelectric point 5.72 Charge (pH=7) -5.68 2D Binding mode Binding energy (Kcal/mol) -9.79 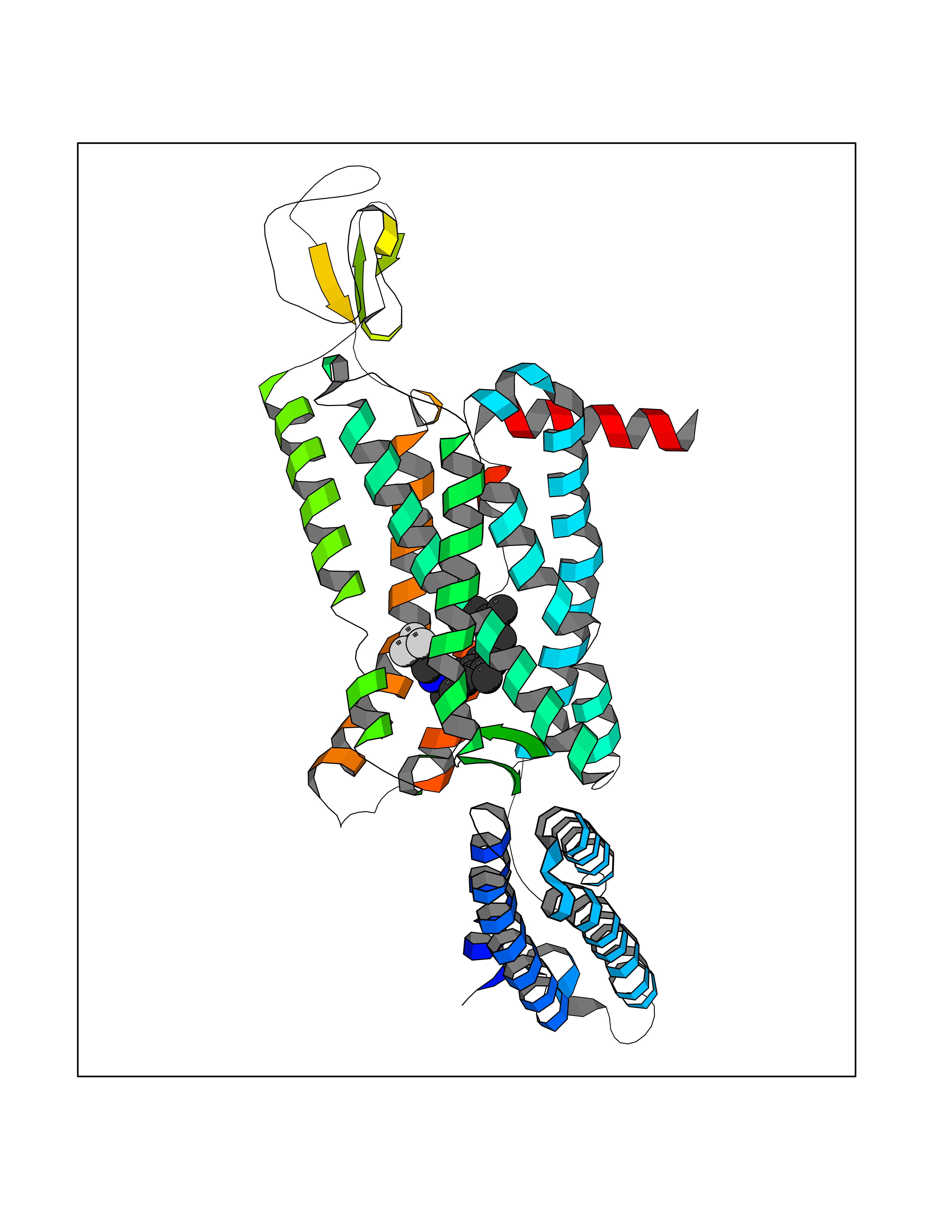 Molscript Map 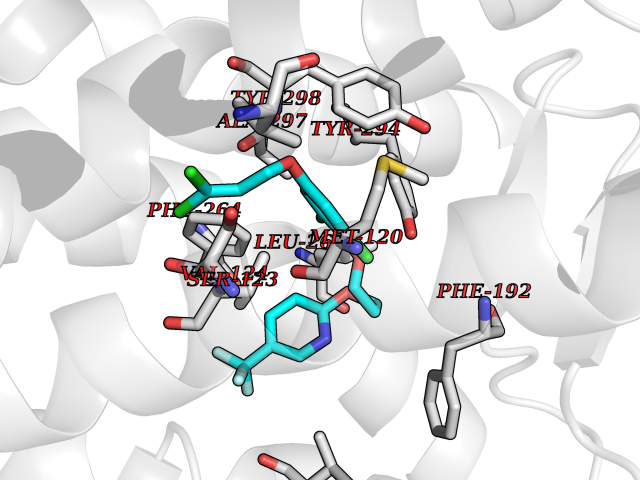 Pymol Map 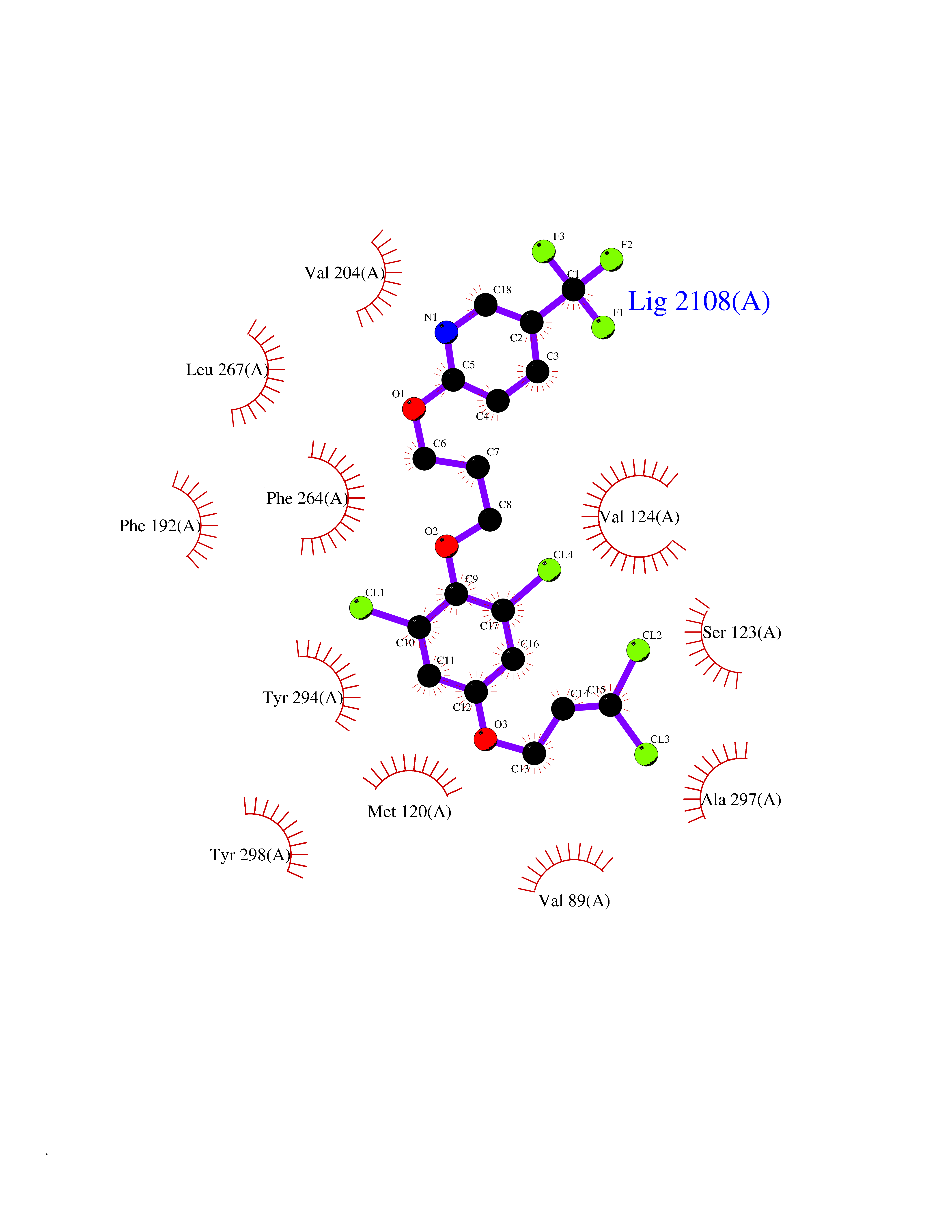 Ligplot Map 3D Binding mode Sequence ADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLGDGARPSWVAPALSAVLIVTTAVDVVGNLLVILSVLRNRKLRNAGNLFLVSLALANLVVAFYPYPLILVAIFYDGWAFGEEHCKASAFVMGLSVIGSVWNITAIAIDRYLYICHSMAYHRIYRRWHTPLHICLIWLLTVVALLPNFFVGSLEYDPRIYSCTFIQTASTQYTAAVVVIHFLLPIAVVSFCYLRIWVLVLQARMKKYTCTVCGYIYNPEDGDPDNGVNPGTDFKDIPDDWVCPLCGVGKDQFEEVECLKPSDLRSFLTMFVVFVIFAICFAPLNCIGLAVAINPQEMAPQIPEGLFVTSYLLAYFNSCLNPIVYGLLDQNFRREYKRILLALWN Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | SEC14-like protein 2 | 4OMJ | 7.17 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -9.79  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 7.17 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -9.78  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 7.15 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -9.75  Molscript Map  Pymol Map 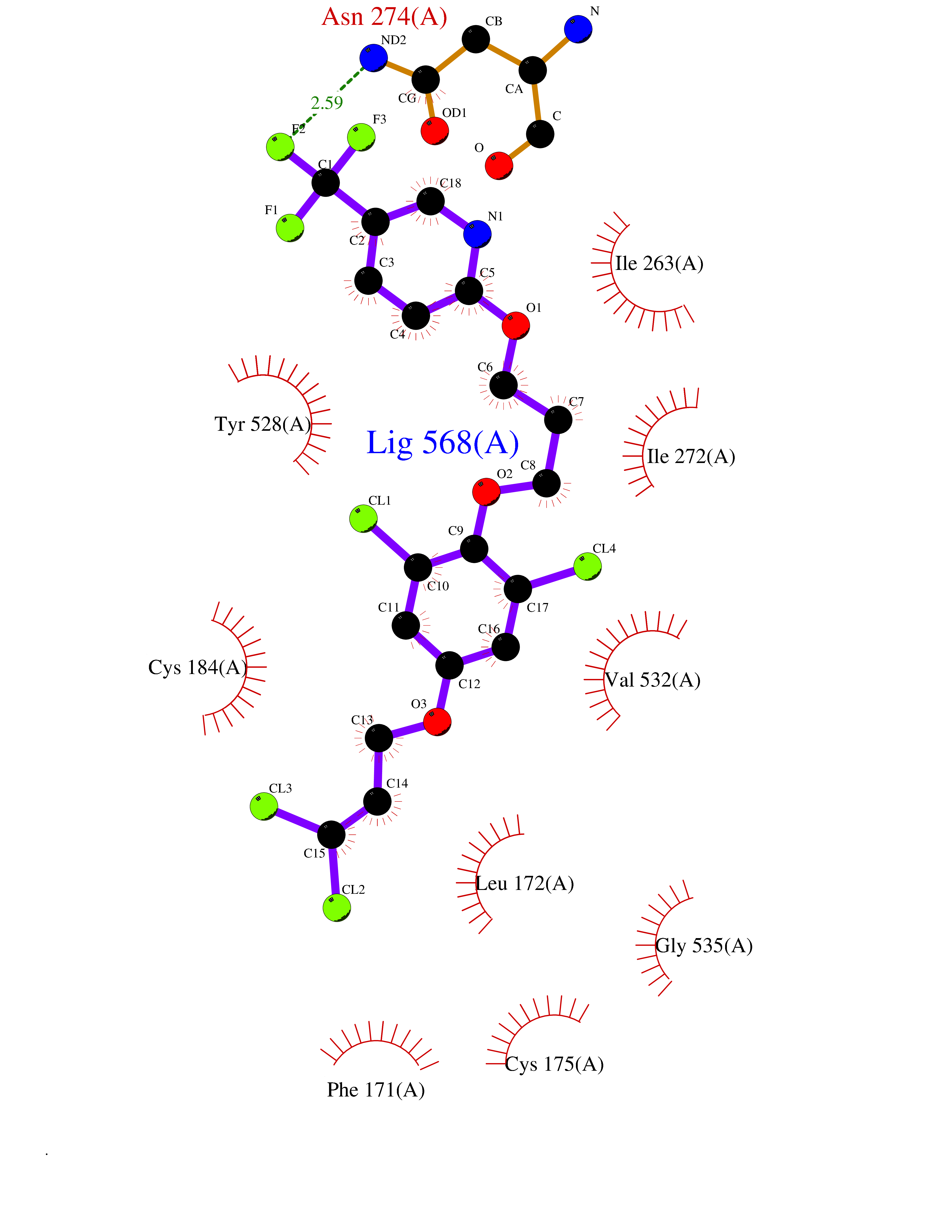 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 7.14 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -9.74  Molscript Map 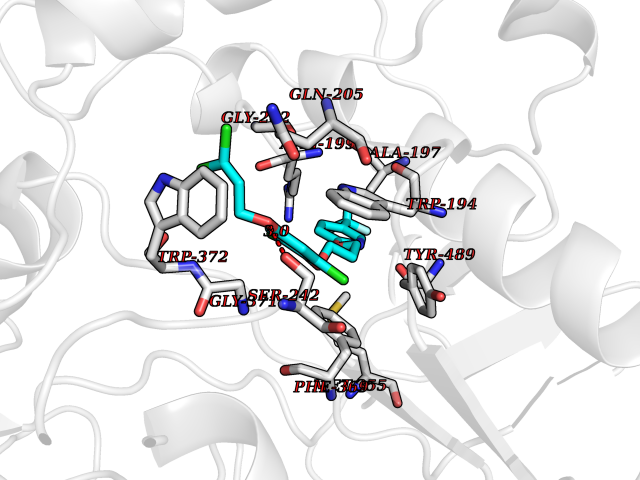 Pymol Map 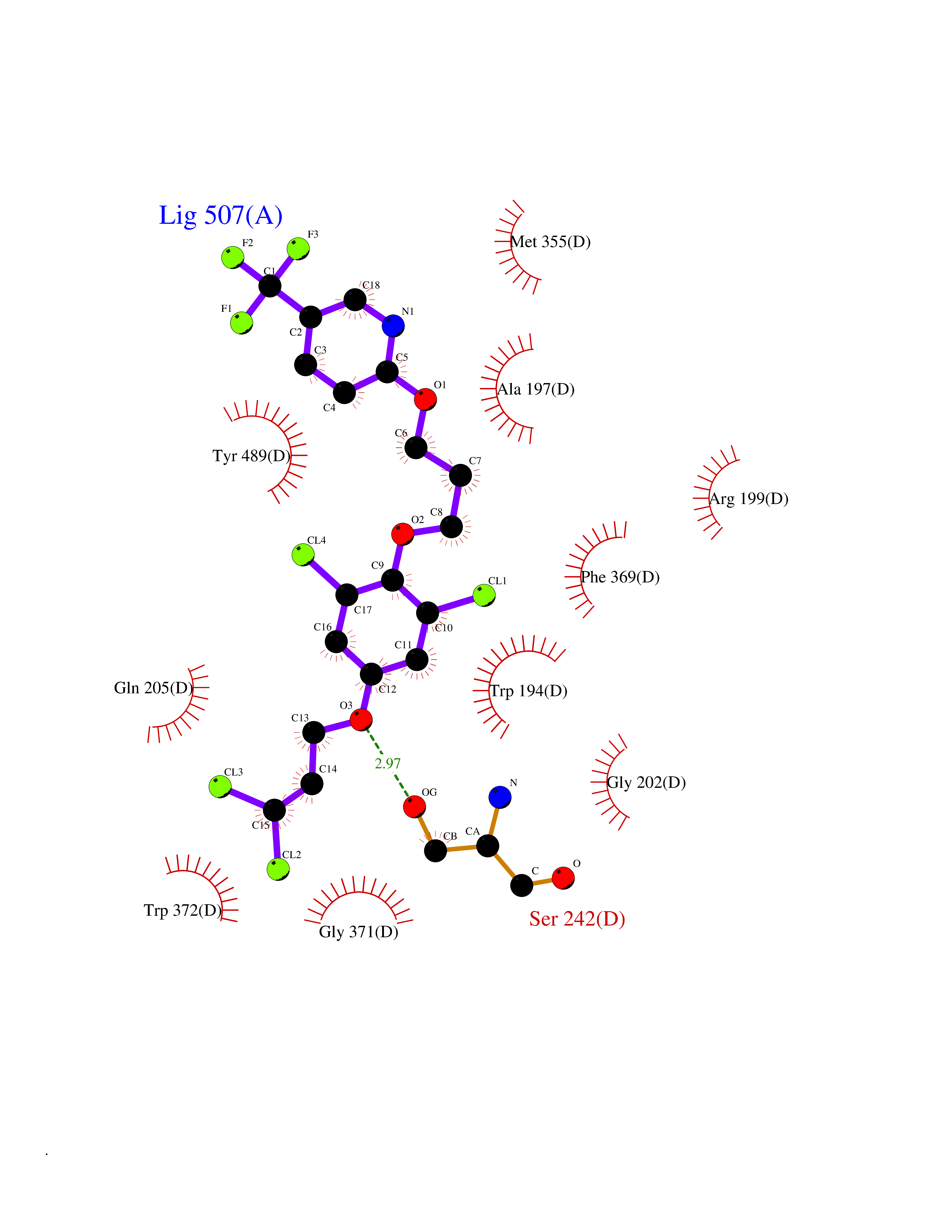 Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5H8Q | 7.13 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 63014.6 Length 557 Aromaticity 0.1 Instability index 28.81 Isoelectric point 8.59 Charge (pH=7) 7.66 2D Binding mode Binding energy (Kcal/mol) -9.72 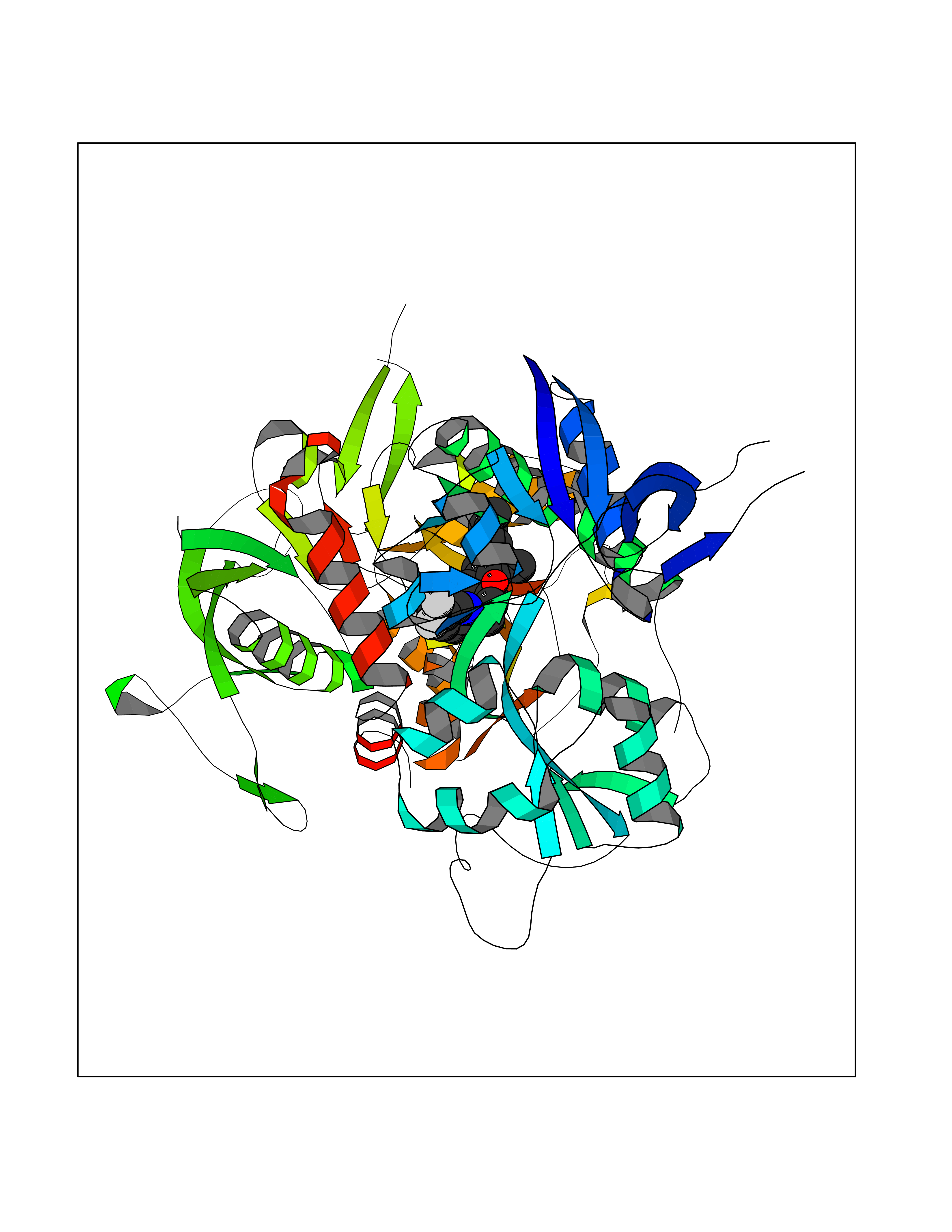 Molscript Map 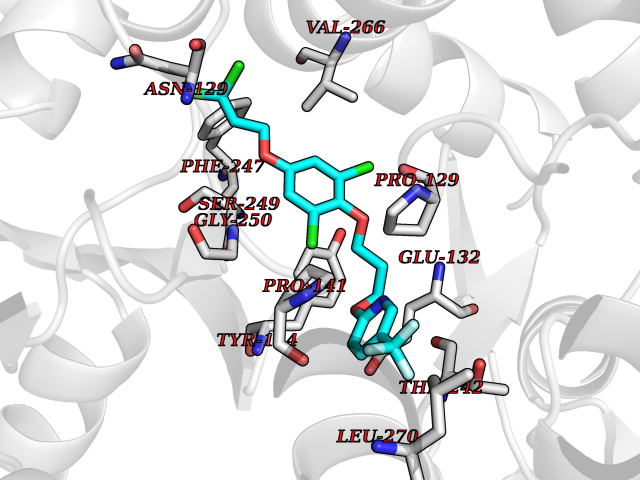 Pymol Map 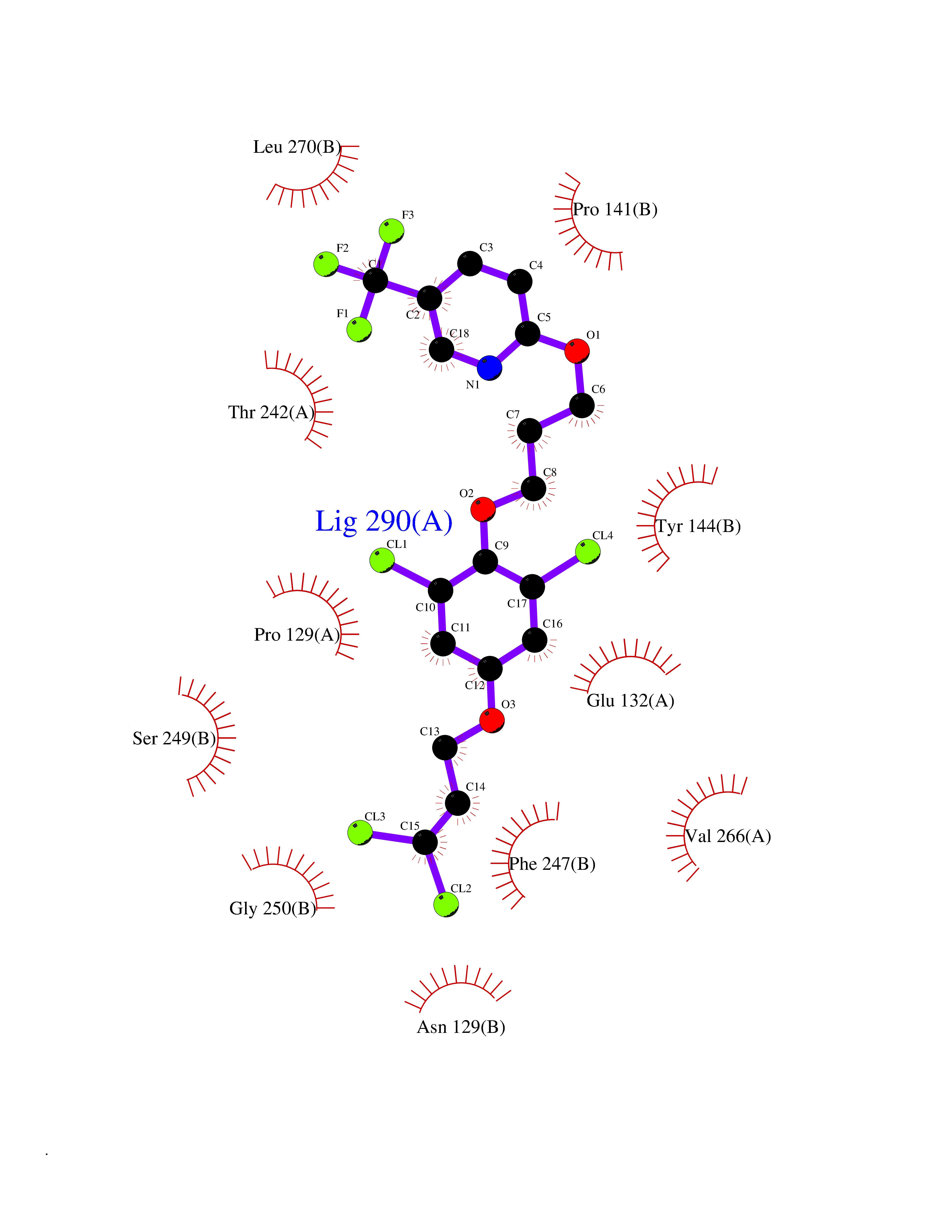 Ligplot Map 3D Binding mode Sequence NHLSIVTLEEAPFVIVEDIDPETCVRNTVPCRKFVKINNSTNEGMNVKKCCKGFCIDILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYVKPTLSDGTCKEEFTVNGDPVKKVICTGPNDTSPGSPRHTVPQCCYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVRY Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5I2N | 7.13 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 63184.8 Length 559 Aromaticity 0.1 Instability index 29.67 Isoelectric point 8.51 Charge (pH=7) 6.66 2D Binding mode Binding energy (Kcal/mol) -9.72 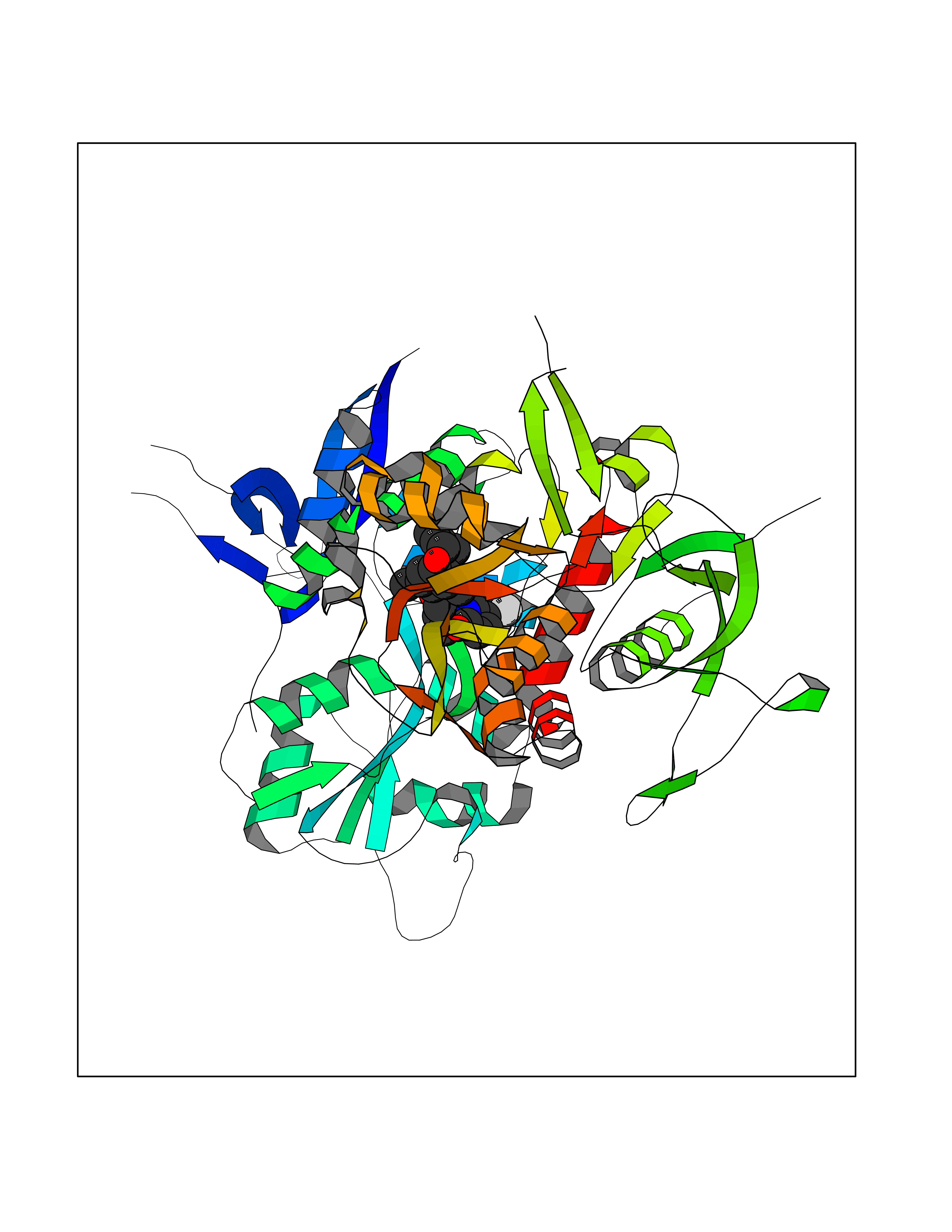 Molscript Map 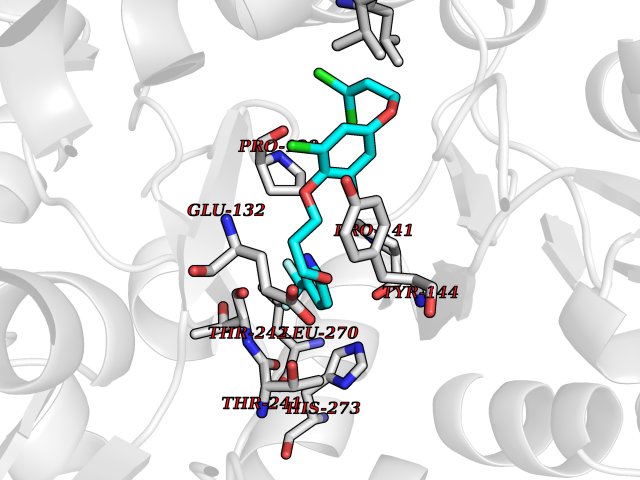 Pymol Map 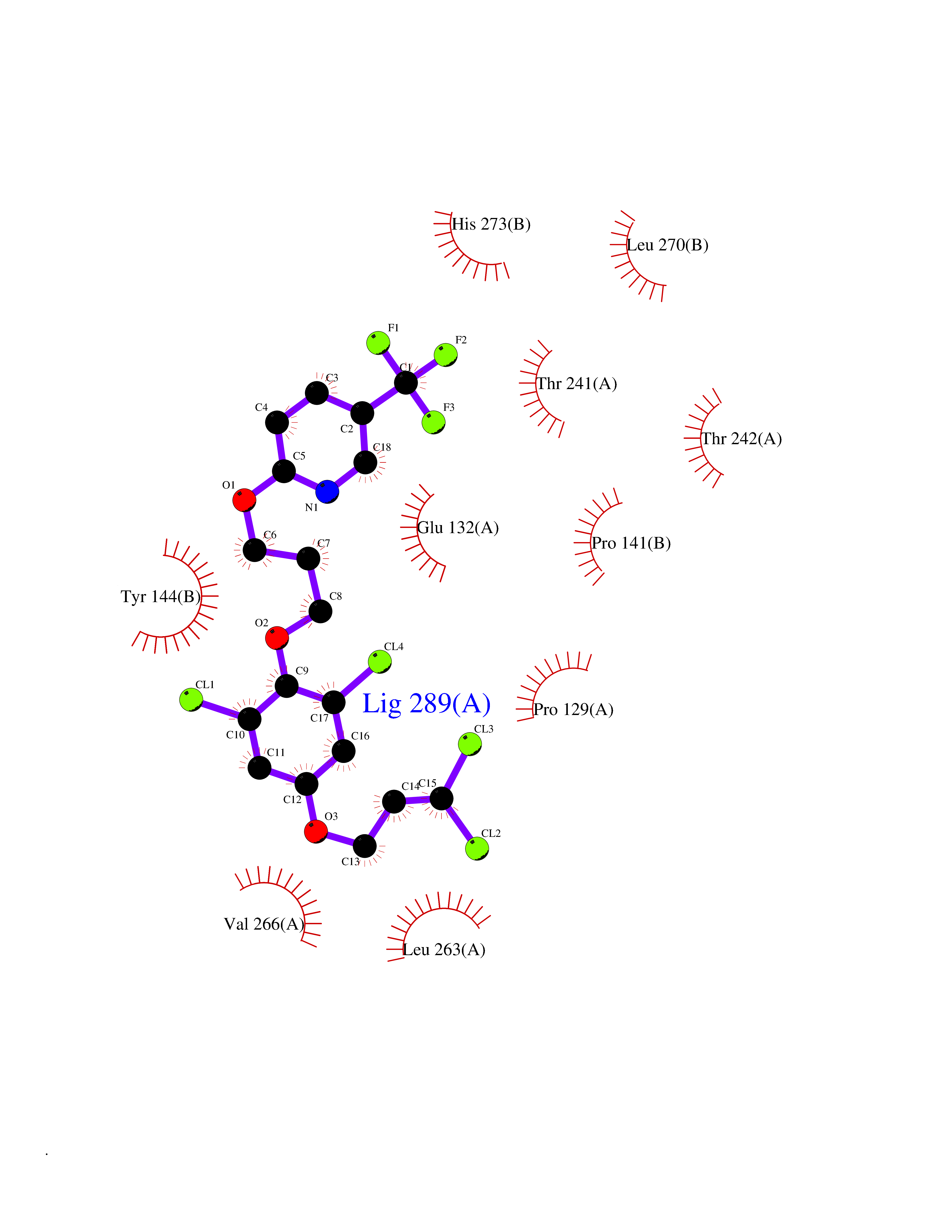 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVIVEDIDPETCVRNTVPCRKFVKINNSTNEGMNVKKCCKGFCIDILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICMSTRLKIVTIHQEPFVYVKPTLSDGTCKEEFTVNGDPVKKVICTGPNDTSPGSPRHTVPQCCYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Leucine carboxyl methyltransferase 1 | 3IEI | 7.13 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -9.72  Molscript Map 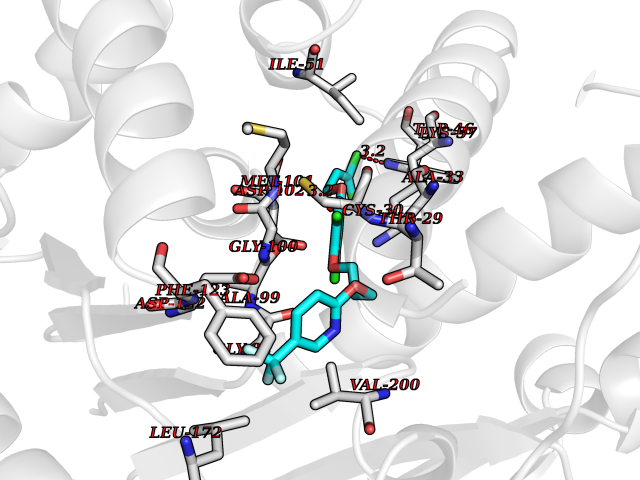 Pymol Map 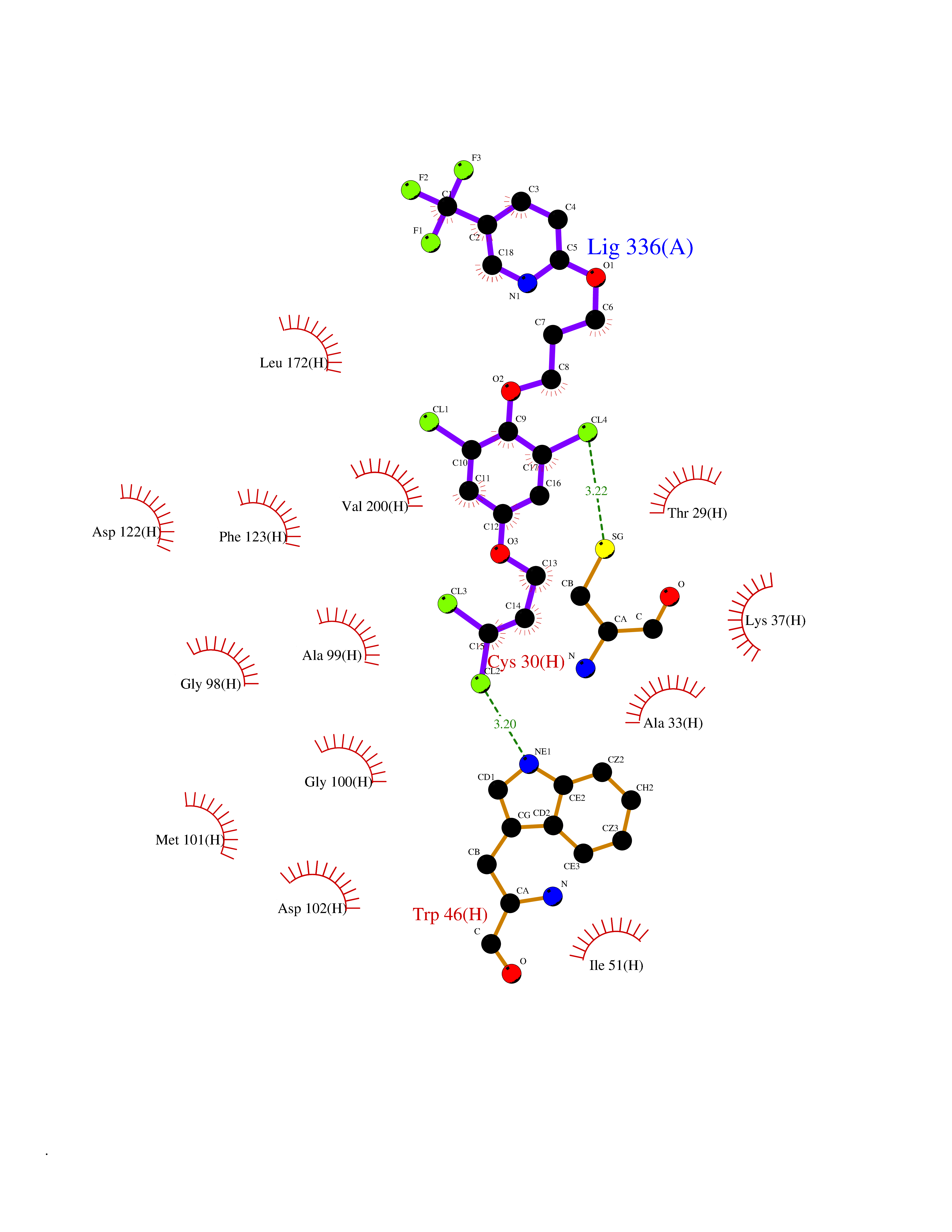 Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Estrogen-related receptor-beta (ESRRB) | 6LIT | 7.13 | |
Target general information Gen name ESRRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid hormone receptor ERR2; Nuclear receptor subfamily 3 group B member 2; NR3B2; Estrogen-related receptor beta; Estrogen receptor-like 2; ESRL2; ERRB2; ERR-beta; ERR beta-2 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Isoform 3: Transcription factor that binds a canonical ESRRB recognition (ERRE) sequence 5'TCAAGGTCA-3' localized on promoter and enhancer of targets genes regulating their expression or their transcription activity. Plays a role, in a LIF-independent manner, in maintainance of self-renewal and pluripotency of embryonic and trophoblast stem cells through different signaling pathways including FGF signaling pathway and Wnt signaling pathways. Upon FGF signaling pathway activation, interacts with KDM1A by directly binding to enhancer site of ELF5 and EOMES and activating their transcription leading to self-renewal of trophoblast stem cells. Also regulates expression of multiple rod-specific genes and is required for survival of this cell type (By similarity). Plays a role as transcription factor activator of GATA6, NR0B1, POU5F1 and PERM1. Plays a role as transcription factor repressor of NFE2L2 transcriptional activity and ESR1 transcriptional activity. During mitosis remains bound to a subset of interphase target genes, including pluripotency regulators, through the canonical ESRRB recognition (ERRE) sequence, leading to their transcriptional activation in early G1 phase. Can coassemble on structured DNA elements with other transcription factors like SOX2, POU5F1, KDM1A and NCOA3 to trigger ESRRB-dependent gene activation. This mechanism, in the case of SOX2 corecruitment prevents the embryonic stem cells (ESCs) to epiblast stem cells (EpiSC) transition through positive regulation of NR0B1 that inhibits the EpiSC transcriptional program. Also plays a role inner ear development by controlling expression of ion channels and transporters and in early placentation (By similarity). Related diseases Deafness, autosomal recessive, 35 (DFNB35) [MIM:608565]: A form of non-syndromic deafness characterized by non-progressive, prelingual hearing loss. {ECO:0000269|PubMed:18179891}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00255; DB07776; DB01645 Interacts with P62508-3 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromosome; Cytoplasm; Deafness; Disease variant; DNA-binding; Metal-binding; Non-syndromic deafness; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 22767.3 Length 198 Aromaticity 0.08 Instability index 56.41 Isoelectric point 5.76 Charge (pH=7) -5.51 2D Binding mode Binding energy (Kcal/mol) -9.73  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GDIKALTTLCDLADRELVVIIGWAKHIPGFSSLSLGDQMSLLQSAWMEILILGIVYRSLPYDDKLVYAEDYIMDEEHSRLAGLLELYRAILQLVRRYKKLKVEKEEFVTLKALALANSDSMHIEDLEAVQKLQDLLHEALQDYELSQHHEEPWRTGKLLLTLPLLRQTAAKAVQHFYSVKLQGKVPMHKLFLEMLEAK Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Monoamine oxidase type A (MAO-A) | 2Z5Y | 7.12 | |
Target general information Gen name MAOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoamine oxidase A; Amine oxidase [flavin-containing] A Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function MAOA preferentially oxidizes biogenic amines such as 5-hydroxytryptamine (5-HT), norepinephrine and epinephrine. Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. Related diseases Brunner syndrome (BRNRS) [MIM:300615]: A form of X-linked non-dysmorphic mild intellectual disability. Male patients are affected by borderline intellectual deficit and exhibit abnormal behavior, including disturbed regulation of impulsive aggression. Obligate female carriers have normal intelligence and behavior. {ECO:0000269|PubMed:8211186}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00918; DB00182; DB06698; DB04889; DB13876; DB01445; DB06774; DB00215; DB04017; DB09130; DB05205; DB07641; DB00988; DB01363; DB00668; DB12329; DB01175; DB03147; DB14914; DB00614; DB01381; DB07919; DB04818; DB01247; DB00601; DB01577; DB00805; DB01442; DB01171; DB08804; DB00952; DB04820; DB00184; DB04821; DB06412; DB01626; DB00780; DB00191; DB00388; DB00397; DB09244; DB04850; DB00721; DB01168; DB00571; DB00852; DB09363; DB00140; DB00953; DB06654; DB01037; DB01104; DB00669; DB14569; DB09042; DB00624; DB13943; DB13944; DB13946; DB09245; DB00752; DB15328; DB09185; DB04832; DB00315; DB00909 Interacts with P27338 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine metabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Intellectual disability; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurotransmitter degradation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 58195.3 Length 513 Aromaticity 0.11 Instability index 34.97 Isoelectric point 7.98 Charge (pH=7) 2.87 2D Binding mode Binding energy (Kcal/mol) -9.71  Molscript Map 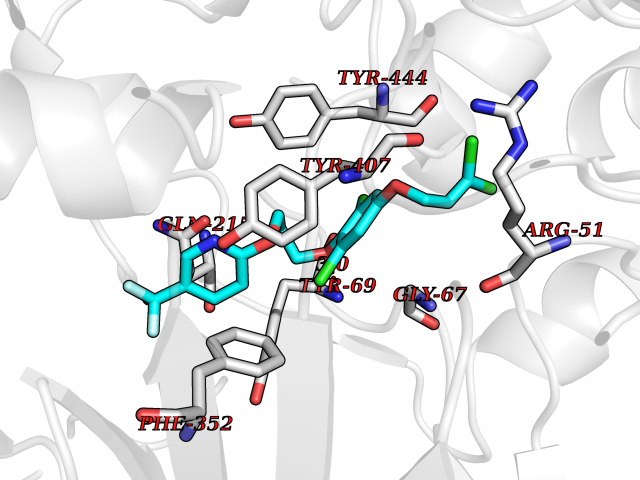 Pymol Map 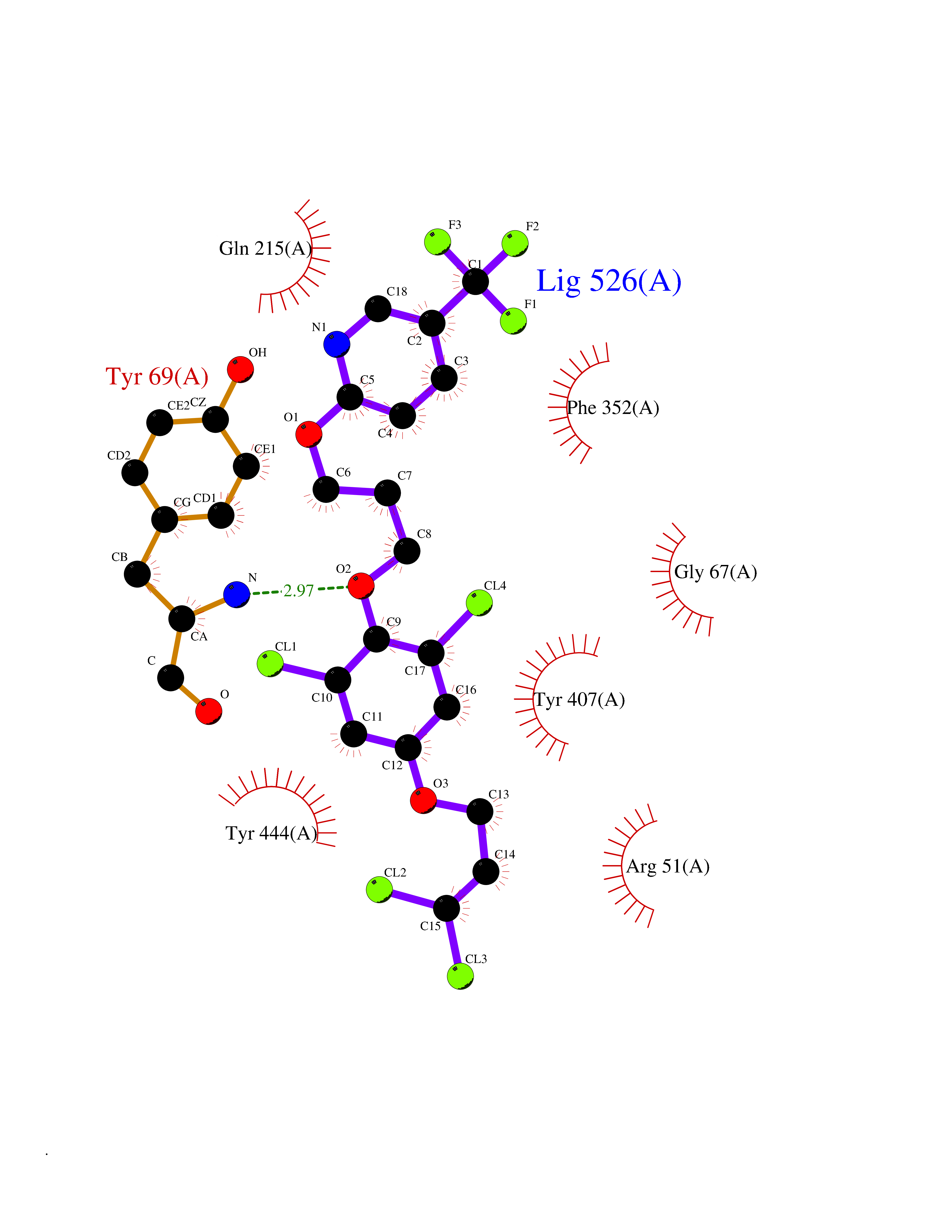 Ligplot Map 3D Binding mode Sequence HMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYVGPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRAAFPPVWNPIAYLDYNNLWRTIDNMGKEIPTDAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSVTNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAKIHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIMGFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYGRVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTFWERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLL Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Proteinase-activated receptor 1 | 3VW7 | 7.12 | |
Target general information Gen name F2R Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TR;PAR1;CF2R Protein family G-protein coupled receptor 1 family Biochemical class Signaling protein / antagonist Function G-protein alpha-subunit binding.G-protein beta-subunit binding.G-protein coupled receptor activity.Receptor binding.Thrombin-activated receptor activity. Related diseases 3-ketothiolase deficiency (3KTD) [MIM:203750]: An autosomal recessive inborn error of isoleucine catabolism characterized by intermittent ketoacidotic attacks associated with unconsciousness. Some patients die during an attack or are mentally retarded. Urinary excretion of 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid, triglylglycine, butanone is increased. It seems likely that the severity of this disease correlates better with the environmental or acquired factors than with the ACAT1 genotype. {ECO:0000269|PubMed:1346617, ECO:0000269|PubMed:1715688, ECO:0000269|PubMed:7728148, ECO:0000269|PubMed:9744475}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05361; DB00086; DB11300; DB09030 Interacts with Q03135; Q9UNN8 EC number NA Uniprot keywords 3D-structure; Blood coagulation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Hemostasis; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 31193.7 Length 282 Aromaticity 0.16 Instability index 36.7 Isoelectric point 8.2 Charge (pH=7) 2.98 2D Binding mode Binding energy (Kcal/mol) -9.71 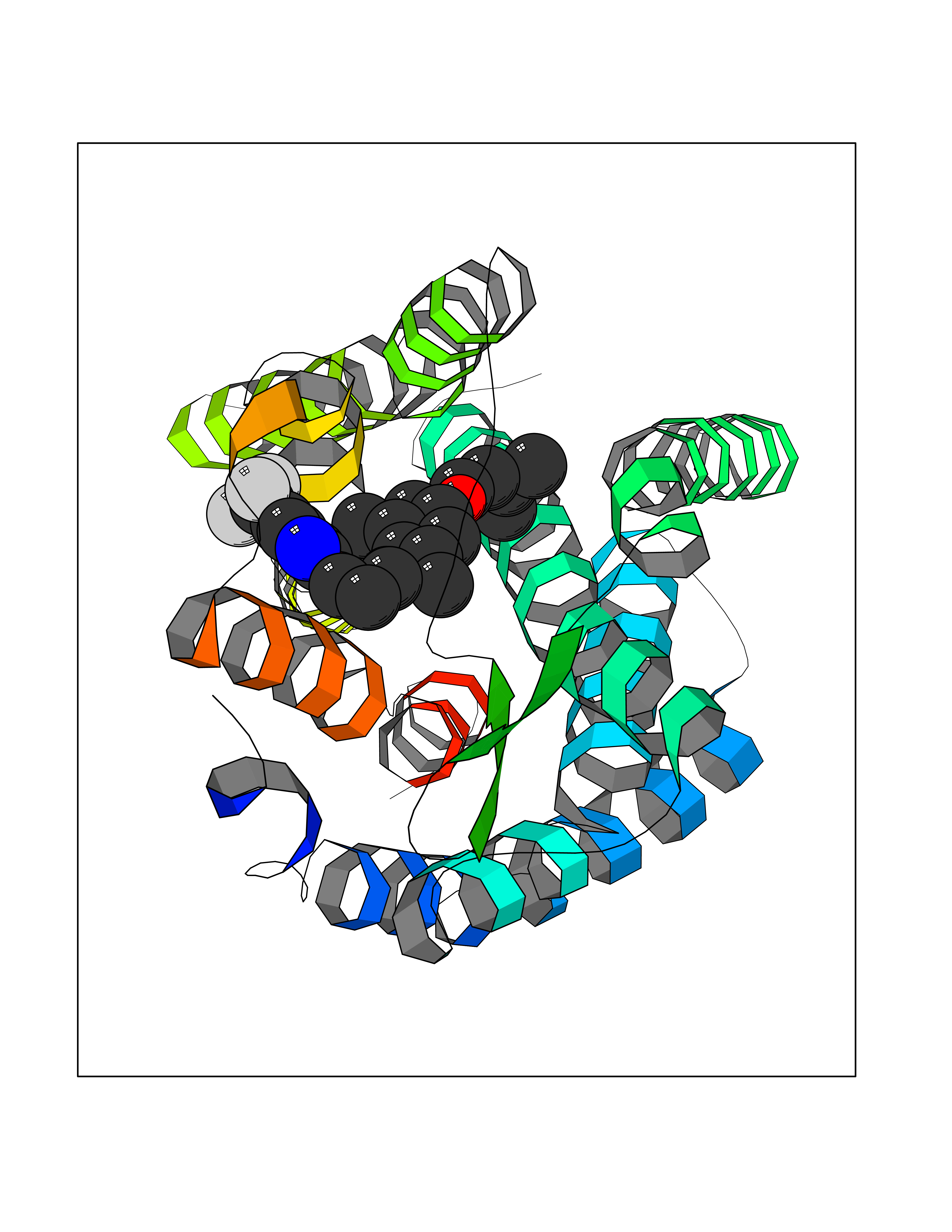 Molscript Map 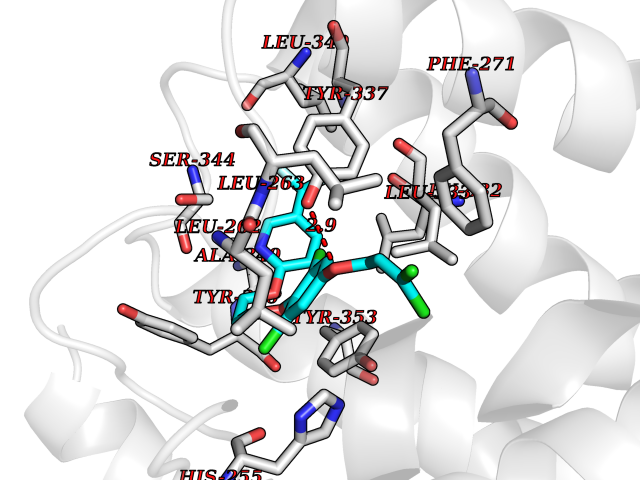 Pymol Map 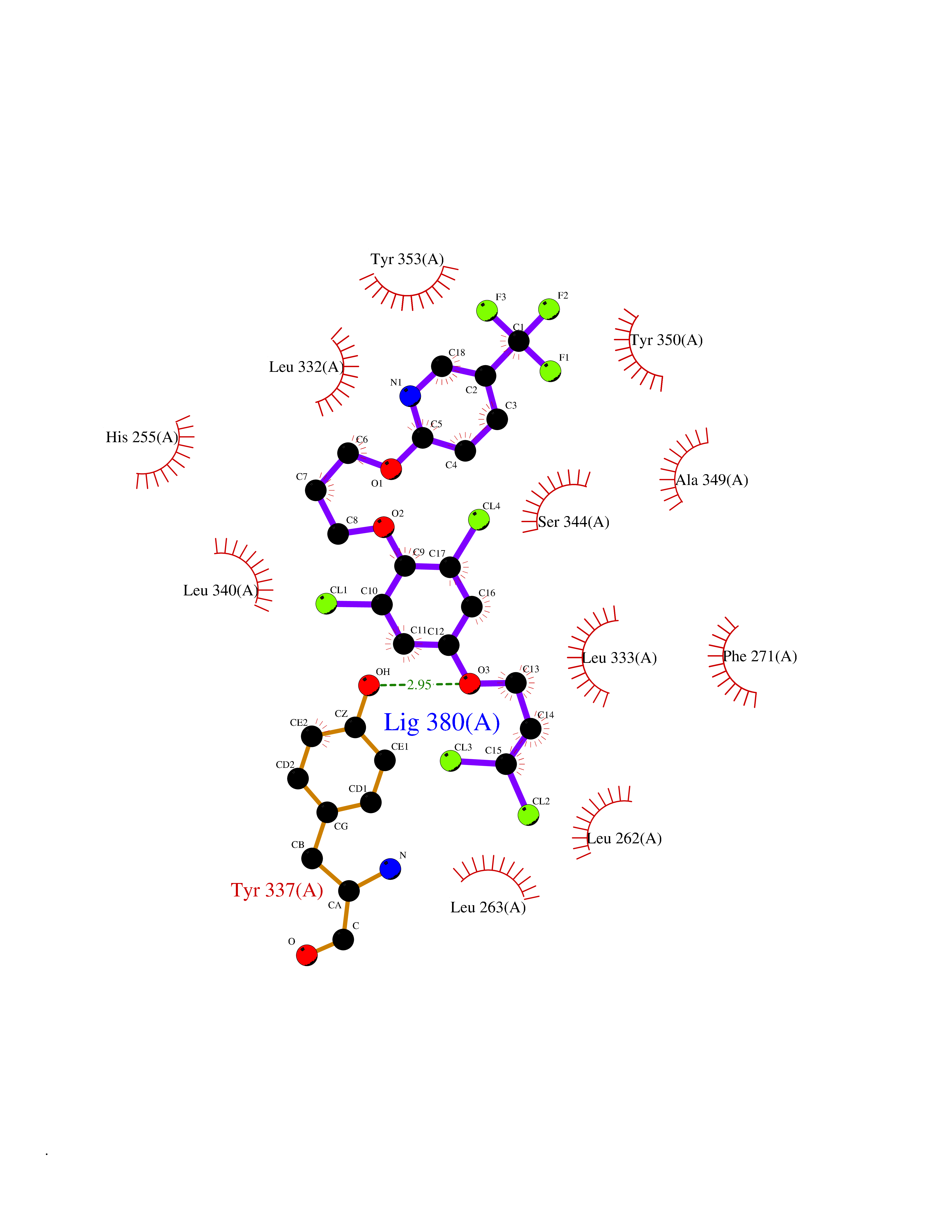 Ligplot Map 3D Binding mode Sequence DASGYLTSSWLTLFVPSVYTGVFVVSLPLNIMAIVVFILKMKVKKPAVVYMLHLATADVLFVSVLPFKISYYFSGSDWQFGSELCRFVTAAFYCNMYASILLMTVISIDRFLAVVYPMRTLGRASFTCLAIWALAIAGVVPLLLKEQTIQVPGLGITTCHDVLSETLLEGYYAYYFSAFSAVFFFVPLIISTVCYVSIIRCLSSSAANRSKKSRALFLSAAVFCIFIICFGPTNVLLIAHYSFLSHTSTTEAAYFAYLLCVCVSSISCCIDPLIYYYASSEC Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Androgen receptor (AR) | 2AM9 | 7.12 | |
Target general information Gen name AR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Testosterone receptor; Nuclear receptor subfamily 3 group C member 4; NR3C4; Dihydrotestosterone receptor; DHTR Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Transcription factor activity is modulated by bound coactivator and corepressor proteins like ZBTB7A that recruits NCOR1 and NCOR2 to the androgen response elements/ARE on target genes, negatively regulating androgen receptor signaling and androgen-induced cell proliferation. Transcription activation is also down-regulated by NR0B2. Activated, but not phosphorylated, by HIPK3 and ZIPK/DAPK3. Steroid hormone receptors are ligand-activated transcription factors that regulate eukaryotic gene expression and affect cellular proliferation and differentiation in target tissues. Related diseases Androgen insensitivity syndrome (AIS) [MIM:300068]: An X-linked recessive form of pseudohermaphroditism due end-organ resistance to androgen. Affected males have female external genitalia, female breast development, blind vagina, absent uterus and female adnexa, and abdominal or inguinal testes, despite a normal 46,XY karyotype. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10221770, ECO:0000269|PubMed:10404311, ECO:0000269|PubMed:10458483, ECO:0000269|PubMed:10571951, ECO:0000269|PubMed:10590024, ECO:0000269|PubMed:10690872, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:11744994, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1426313, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:1464650, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:1480178, ECO:0000269|PubMed:1487249, ECO:0000269|PubMed:1569163, ECO:0000269|PubMed:1609793, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:16595706, ECO:0000269|PubMed:1775137, ECO:0000269|PubMed:1999491, ECO:0000269|PubMed:2082179, ECO:0000269|PubMed:2594783, ECO:0000269|PubMed:7537149, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7633398, ECO:0000269|PubMed:7641413, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7962294, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:7981689, ECO:0000269|PubMed:7993455, ECO:0000269|PubMed:8040309, ECO:0000269|PubMed:8096390, ECO:0000269|PubMed:8103398, ECO:0000269|PubMed:8162033, ECO:0000269|PubMed:8224266, ECO:0000269|PubMed:8281140, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8339746, ECO:0000269|PubMed:8413310, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8626869, ECO:0000269|PubMed:8647313, ECO:0000269|PubMed:8683794, ECO:0000269|PubMed:8723113, ECO:0000269|PubMed:8768864, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8830623, ECO:0000269|PubMed:8918984, ECO:0000269|PubMed:8990010, ECO:0000269|PubMed:9001799, ECO:0000269|PubMed:9007482, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9106550, ECO:0000269|PubMed:9160185, ECO:0000269|PubMed:9252933, ECO:0000269|PubMed:9255042, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9328206, ECO:0000269|PubMed:9544375, ECO:0000269|PubMed:9554754, ECO:0000269|PubMed:9610419, ECO:0000269|PubMed:9627582, ECO:0000269|PubMed:9698822, ECO:0000269|PubMed:9851768, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.116, ECO:0000269|Ref.182}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal and bulbar muscular atrophy X-linked 1 (SMAX1) [MIM:313200]: An X-linked recessive form of spinal muscular atrophy. Spinal muscular atrophy refers to a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAX1 occurs only in men. Age at onset is usually in the third to fifth decade of life, but earlier involvement has been reported. It is characterized by slowly progressive limb and bulbar muscle weakness with fasciculations, muscle atrophy, and gynecomastia. The disorder is clinically similar to classic forms of autosomal spinal muscular atrophy. {ECO:0000269|PubMed:15851746}. The disease is caused by variants affecting the gene represented in this entry. Caused by trinucleotide CAG repeat expansion. In SMAX1 patients the number of Gln ranges from 38 to 62. Longer expansions result in earlier onset and more severe clinical manifestations of the disease.; DISEASE: Prostate cancer, hereditary, X-linked 3 (HPCX3) [MIM:301120]: A condition associated with familial predisposition to cancer of the prostate. Most prostate cancers are adenocarcinomas that develop in the acini of the prostatic ducts. Other rare histopathologic types of prostate cancer that occur in approximately 5% of patients include small cell carcinoma, mucinous carcinoma, prostatic ductal carcinoma, transitional cell carcinoma, squamous cell carcinoma, basal cell carcinoma, adenoid cystic carcinoma (basaloid), signet-ring cell carcinoma and neuroendocrine carcinoma. {ECO:0000269|PubMed:8530589}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Defects in AR may play a role in metastatic prostate cancer. The mutated receptor stimulates prostate growth and metastases development despite of androgen ablation. This treatment can reduce primary and metastatic lesions probably by inducing apoptosis of tumor cells when they express the wild-type receptor. {ECO:0000269|PubMed:10363963, ECO:0000269|PubMed:10569618, ECO:0000269|PubMed:1562539, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:17311914, ECO:0000269|PubMed:2260966, ECO:0000269|PubMed:25091737, ECO:0000269|PubMed:8187068, ECO:0000269|PubMed:8274409, ECO:0000269|PubMed:8827083}.; DISEASE: Androgen insensitivity, partial (PAIS) [MIM:312300]: A disorder that is characterized by hypospadias, hypogonadism, gynecomastia, genital ambiguity, normal XY karyotype, and a pedigree pattern consistent with X-linked recessive inheritance. Some patients present azoospermia or severe oligospermia without other clinical manifestations. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10470409, ECO:0000269|PubMed:10502786, ECO:0000269|PubMed:10543676, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:1303262, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1424203, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:2010552, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7649358, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7909256, ECO:0000269|PubMed:7910529, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:8033918, ECO:0000269|PubMed:8097257, ECO:0000269|PubMed:8126121, ECO:0000269|PubMed:8205256, ECO:0000269|PubMed:8281139, ECO:0000269|PubMed:8325932, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8550758, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8823308, ECO:0000269|PubMed:8824883, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9196614, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9329414, ECO:0000269|PubMed:9543136, ECO:0000269|PubMed:9607727, ECO:0000269|PubMed:9768671, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.124}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypospadias 1, X-linked (HYSP1) [MIM:300633]: A common malformation in which the urethra opens on the ventral side of the penis, due to developmental arrest of urethral fusion. The opening can be located glandular, penile, or even more posterior in the scrotum or perineum. Hypospadias is a feature of several syndromic disorders, including the androgen insensitivity syndrome and Opitz syndrome. {ECO:0000269|PubMed:8097257}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07422; DB07039; DB04709; DB07717; DB07454; DB02932; DB08035; DB01481; DB08088; DB08461; DB08087; DB07421; DB01063; DB07423; DB11901; DB01128; DB07286; DB01541; DB14639; DB01564; DB12499; DB04839; DB01406; DB12941; DB09123; DB00255; DB06133; DB01395; DB00858; DB15488; DB11219; DB08899; DB13155; DB00655; DB09086; DB02266; DB01185; DB00623; DB00499; DB11619; DB11064; DB01026; DB15647; DB00367; DB08089; DB05234; DB13934; DB11425; DB06710; DB02998; DB11429; DB00648; DB08804; DB00984; DB00665; DB06713; DB00717; DB09371; DB00957; DB09389; DB00621; DB01428; DB06412; DB01608; DB11447; DB01708; DB00396; DB07419; DB07769; DB14583; DB00421; DB02901; DB13951; DB06718; DB00675; DB00624; DB13943; DB13944; DB01420; DB13946; DB06870; DB08604; DB08867 Interacts with P00519; Q9UBL3; P51451; Q8WV28; O60885-1; P78543; Q14790; P24385; Q92793; O14595; P35222; Q9UER7; P20711; P11308; P07332; P09769; Q02790; P55317; O75593; Q14451; P06396; P56524; Q16665; Q16666; O15357; Q15652; O95251; Q9BY66; Q9BY66-3; Q03164; O14686; P06239; P07948; P20794; P42679; Q00987; Q15596; Q14686; O96028; Q99497; P27986; O00459; Q92569; P19174; P16885; Q06830; P78527; Q06124; P20936; Q9UBS8; Q9Y252; O14796; Q9NP31; P29353; Q6S5L8; Q5VZ18; Q15797; O14544; P12931; Q9ULZ2; P63165; Q9HBL0; P07947; Q9R1E0; Q06986 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Isopeptide bond; Lipid-binding; Lipoprotein; Metal-binding; Neurodegeneration; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Pseudohermaphroditism; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Triplet repeat expansion; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29137.9 Length 250 Aromaticity 0.11 Instability index 42.11 Isoelectric point 8.94 Charge (pH=7) 5.43 2D Binding mode Binding energy (Kcal/mol) -9.71  Molscript Map 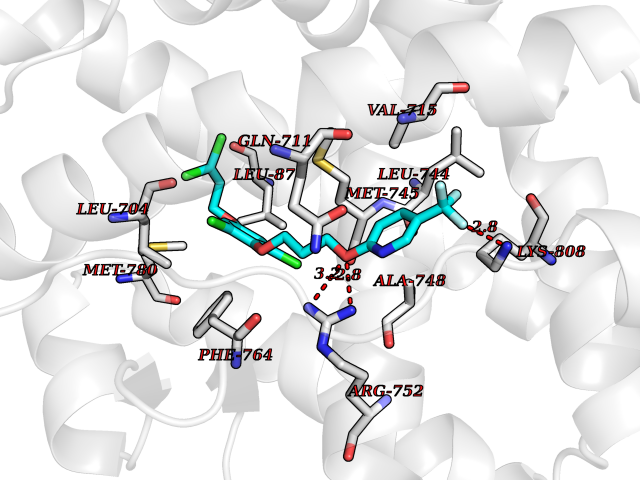 Pymol Map 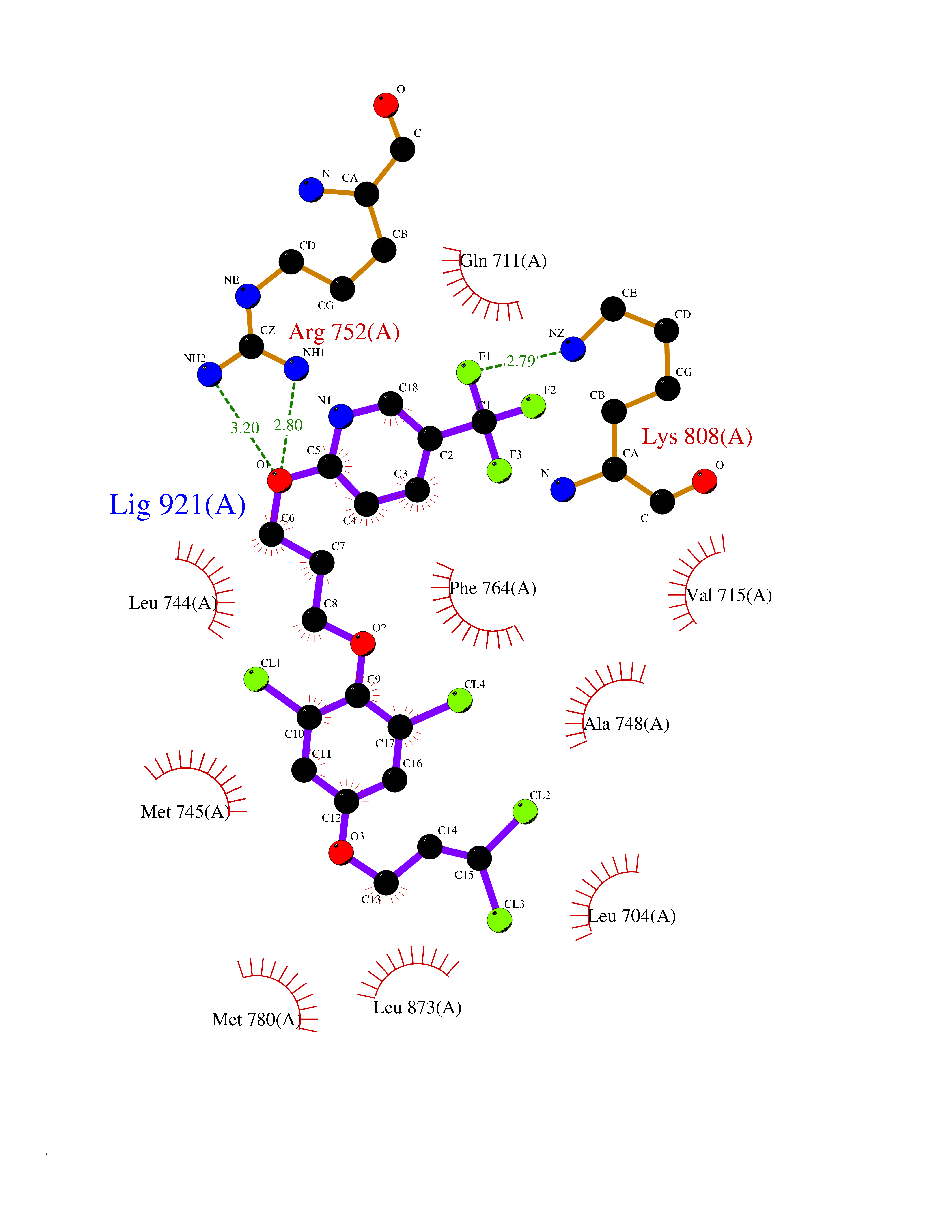 Ligplot Map 3D Binding mode Sequence QPIFLNVLEAIEPGVVCAGHDNNQPDSFAALLSSLNELGERQLVHVVKWAKALPGFRNLHVDDQMAVIQYSWMGLMVFAMGWRSFTNVNSRMLYFAPDLVFNEYRMHKSRMYSQCVRMRHLSQEFGWLQITPQEFLCMKALLLFSIIPVDGLKNQKFFDELRMNYIKELDRIIACKRKNPTSCSRRFYQLTKLLDSVQPIARELHQFTFDLLIKSHMVSVDFPEMMAEIISVQVPKILSGKVKPIYFHTQ Hydrogen bonds contact Hydrophobic contact | ||||