Job Results:
Ligand
Structure
Job ID
b66da62373d29d285a275497218b65f3
Job name
NA
Time
2024-11-27 22:32:54
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Glucagon-like peptide 1 receptor (GLP1R) | 6X1A | 8.32 | |
Target general information Gen name GLP1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GLP-1R; GLP-1-R; GLP-1 receptor Protein family G-protein coupled receptor 2 family Biochemical class GPCR secretin Function Ligand binding triggers activation of a signaling cascade that leads to the activation of adenylyl cyclase and increased intracellular cAMP levels. Plays a role in regulating insulin secretion in response to GLP-1. G-protein coupled receptor for glucagon-like peptide 1 (GLP-1). Related diseases Lynch syndrome 2 (LYNCH2) [MIM:609310]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10323887, ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10413423, ECO:0000269|PubMed:10480359, ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10627141, ECO:0000269|PubMed:10660333, ECO:0000269|PubMed:10671064, ECO:0000269|PubMed:10713887, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:11139242, ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11748856, ECO:0000269|PubMed:11754112, ECO:0000269|PubMed:11781295, ECO:0000269|PubMed:11793442, ECO:0000269|PubMed:11839723, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:12095971, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655562, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:14961575, ECO:0000269|PubMed:15064764, ECO:0000269|PubMed:15139004, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15365996, ECO:0000269|PubMed:16083711, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17301300, ECO:0000269|PubMed:17510385, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:20020535, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22753075, ECO:0000269|PubMed:7757073, ECO:0000269|PubMed:8566964, ECO:0000269|PubMed:8571956, ECO:0000269|PubMed:8574961, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:8993976, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9067757, ECO:0000269|PubMed:9218993, ECO:0000269|PubMed:9272156, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9326924, ECO:0000269|PubMed:9399661, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9833759, ECO:0000269|PubMed:9927034, ECO:0000269|Ref.5}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 1 (MMRCS1) [MIM:276300]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:17440981, ECO:0000269|PubMed:7661930}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:8751876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in MLH1 may contribute to lobular carcinoma in situ (LCIS), a non-invasive neoplastic disease of the breast.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Some epigenetic changes can be transmitted unchanged through the germline (termed 'epigenetic inheritance'). Evidence that this mechanism occurs in humans is provided by the identification of individuals in whom 1 allele of the MLH1 gene is epigenetically silenced throughout the soma (implying a germline event). These individuals are affected by Lynch syndrome but does not have identifiable mutations in MLH1, even though it is silenced, which demonstrates that an epimutation can phenocopy a genetic disease.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15184898, ECO:0000269|PubMed:18033691, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9032648, ECO:0000269|PubMed:9087566, ECO:0000269|PubMed:9611074}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09043; DB09045; DB15650; DB01276; DB00040; DB16697; DB06655; DB09265; DB13928; DB14027; DB15171 Interacts with A8MQ03; Q07627; Q8IUG1; P60409; P60410; P60411; Q9BYP8; P26371; Q7Z3S9; P0DPK4 EC number NA Uniprot keywords 3D-structure; ADP-ribosylation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 45579.6 Length 390 Aromaticity 0.16 Instability index 39.66 Isoelectric point 6.73 Charge (pH=7) -0.68 2D Binding mode Binding energy (Kcal/mol) -9.29 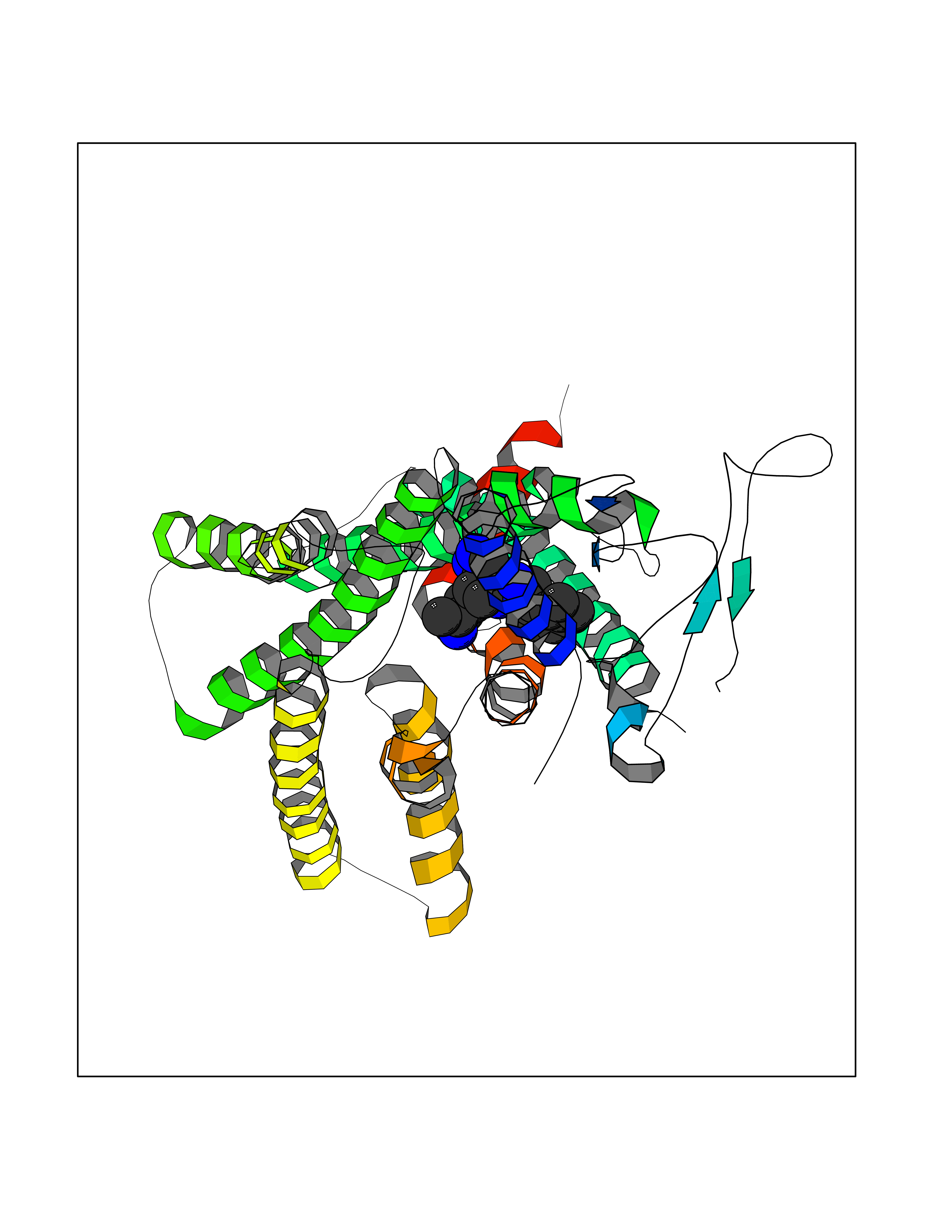 Molscript Map 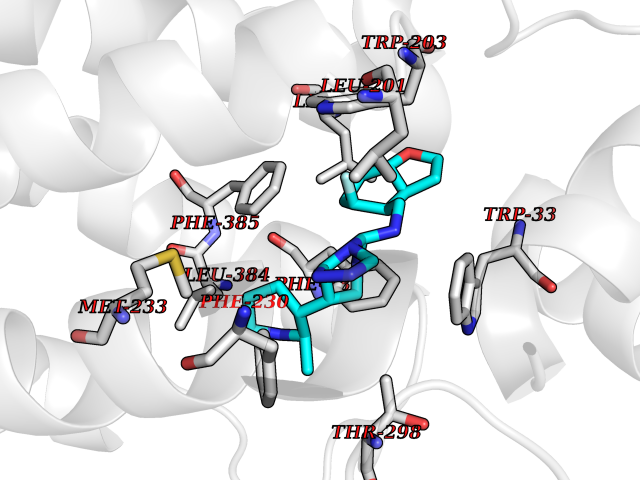 Pymol Map 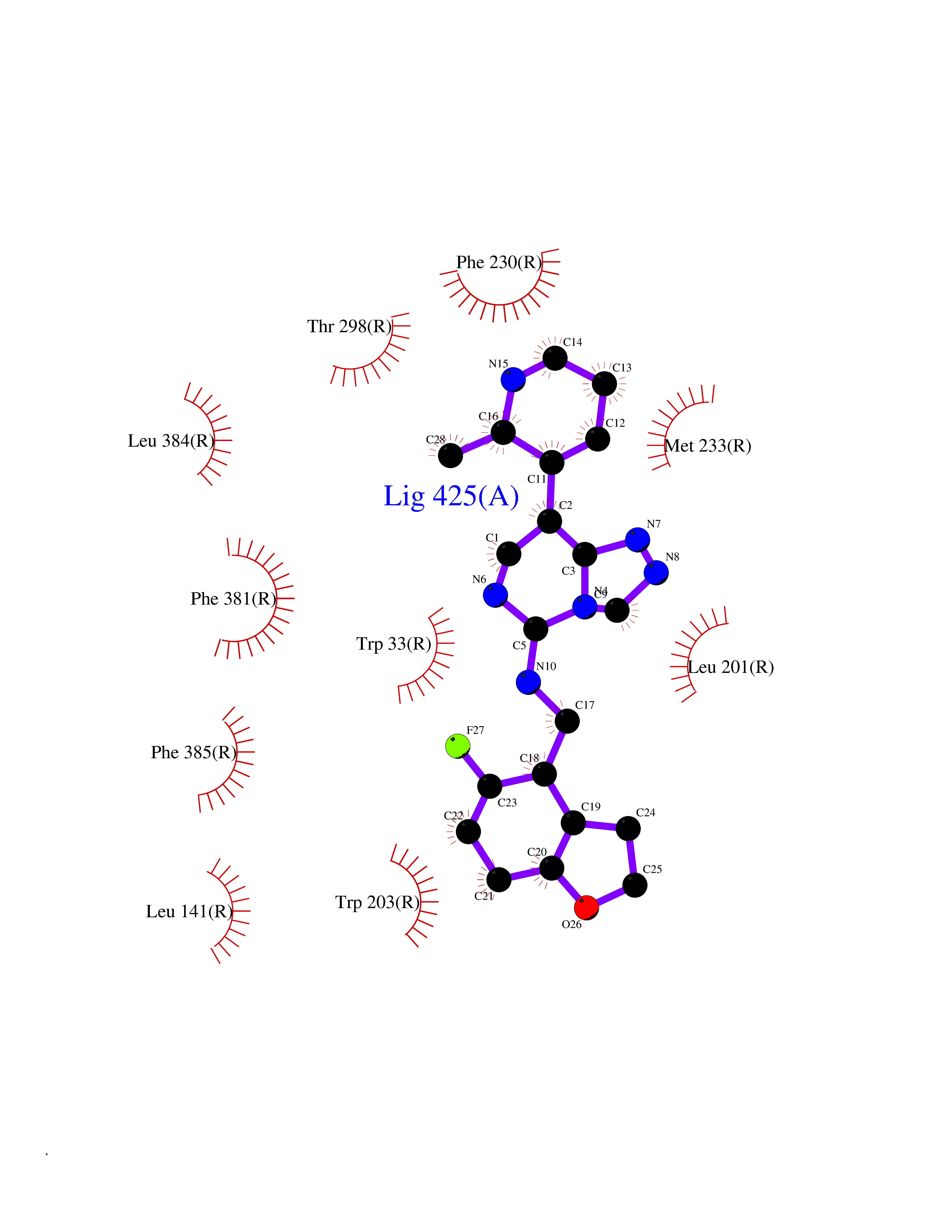 Ligplot Map 3D Binding mode Sequence ATVSLWETVQKWREYRRQCQRSLTEDPPPATDLFCNRTFDEYACWPDGEPGSFVNVSCPWYLPWASSVPQGHVYRFCTAEGLWLQKDNSSLPWRDLSECEESSPEEQLLFLYIIYTVGYALSFSALVIASAILLGFRHLHCTRNYIHLNLFASFILRALSVFIKDAALKWMYSTAAQQHQWDGLLSYQDSLSCRLVFLLMQYCVAANYYWLLVEGVYLYTLLAFSVFSEQWIFRLYVSIGWGVPLLFVVPWGIVKYLYEDEGCWTRNSNMNYWLIIRLPILFAIGVNFLIFVRVICIVVSKLKANLMCKTDIKCRLAKSTLTLIPLLGTHEVIFAFVMDEHARGTLRFIKLFTELSFTSFQGLMVAILYCFVNNEVQLEFRKSWERWRLE Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | T-cell surface glycoprotein CD1a (CD1A) | 6NUX | 8.31 | |
Target general information Gen name CD1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hTa1 thymocyteantigen; hTa1 thymocyte antigen; T-cell surfaceantigen T6/Leu-6; T-cell surface antigen T6/Leu-6; CD1a Protein family NA Biochemical class Immunoglobulin Function Antigen-presenting protein that binds self and non-self lipid and glycolipid antigens and presents them to T-cell receptors on natural killer T-cells. Related diseases Pulmonary hypertension, primary, 1 (PPH1) [MIM:178600]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:10903931, ECO:0000269|PubMed:10973254, ECO:0000269|PubMed:11015450, ECO:0000269|PubMed:11115378, ECO:0000269|PubMed:12045205, ECO:0000269|PubMed:12358323, ECO:0000269|PubMed:15965979, ECO:0000269|PubMed:24936649, ECO:0000269|PubMed:25187962, ECO:0000269|PubMed:28507310}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pulmonary venoocclusive disease 1, autosomal dominant (PVOD1) [MIM:265450]: A disease characterized by widespread fibrous obstruction and intimal thickening of septal veins and preseptal venules, a low diffusing capacity for carbon monoxide, occult alveolar hemorrhage, and nodular ground-glass opacities, septal lines and lymph node enlargement showed by high-resolution computed tomography of the chest. It is frequently associated with pulmonary capillary dilatation and proliferation, and is a rare and devastating cause of pulmonary hypertension. {ECO:0000269|PubMed:12446270, ECO:0000269|PubMed:16429395}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00098 Interacts with P61769 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 30867.3 Length 270 Aromaticity 0.13 Instability index 37.45 Isoelectric point 6.16 Charge (pH=7) -4.5 2D Binding mode Binding energy (Kcal/mol) -9.16 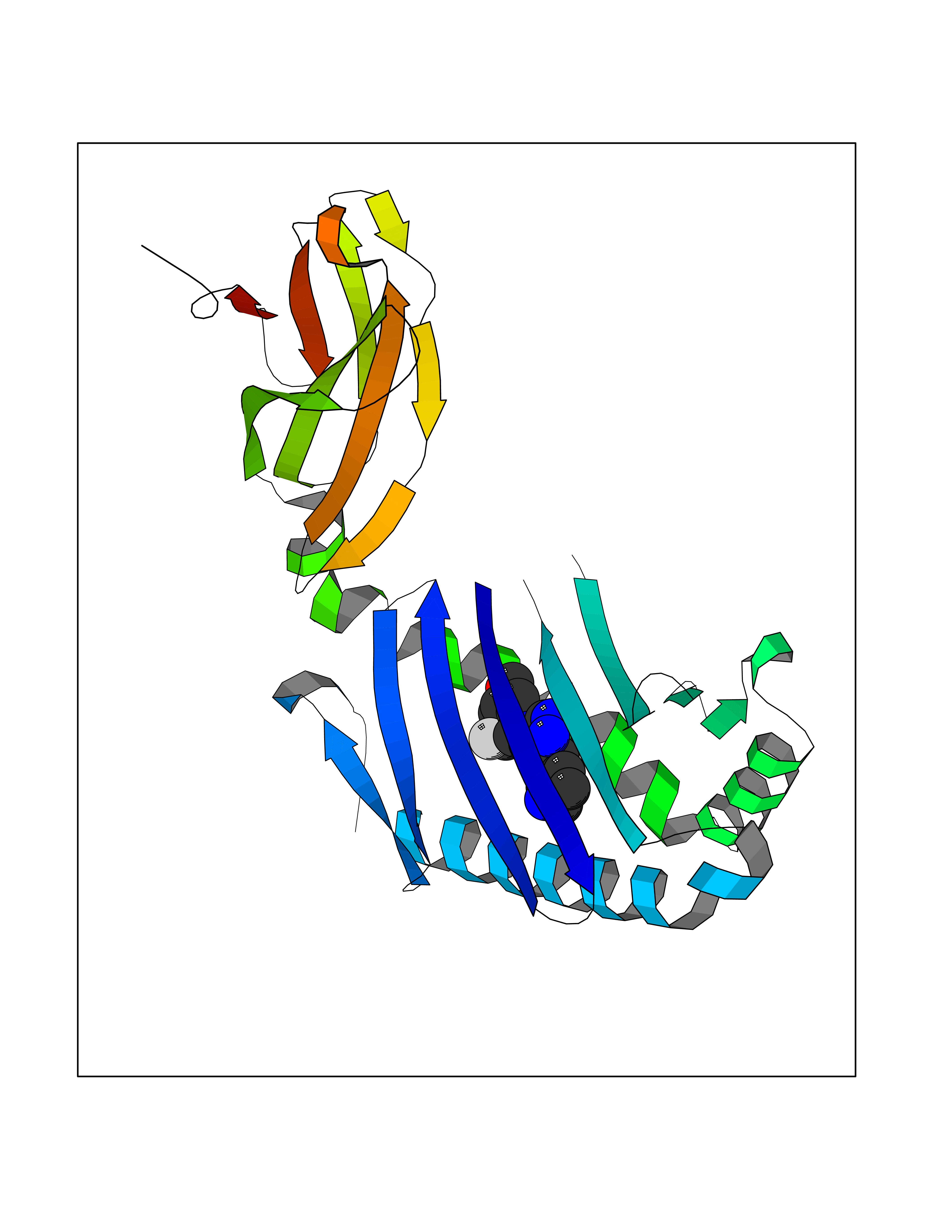 Molscript Map 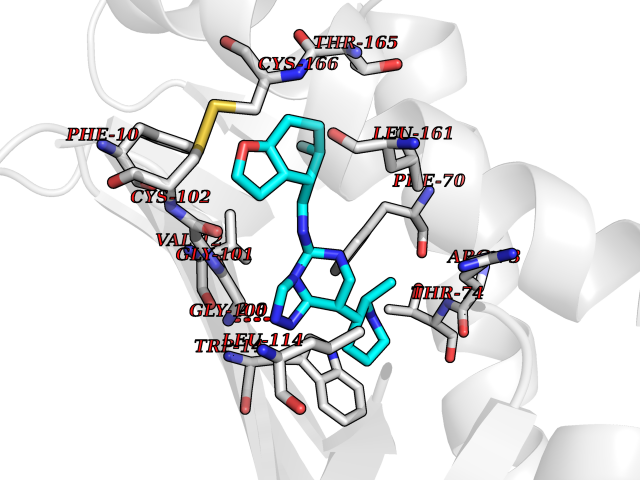 Pymol Map 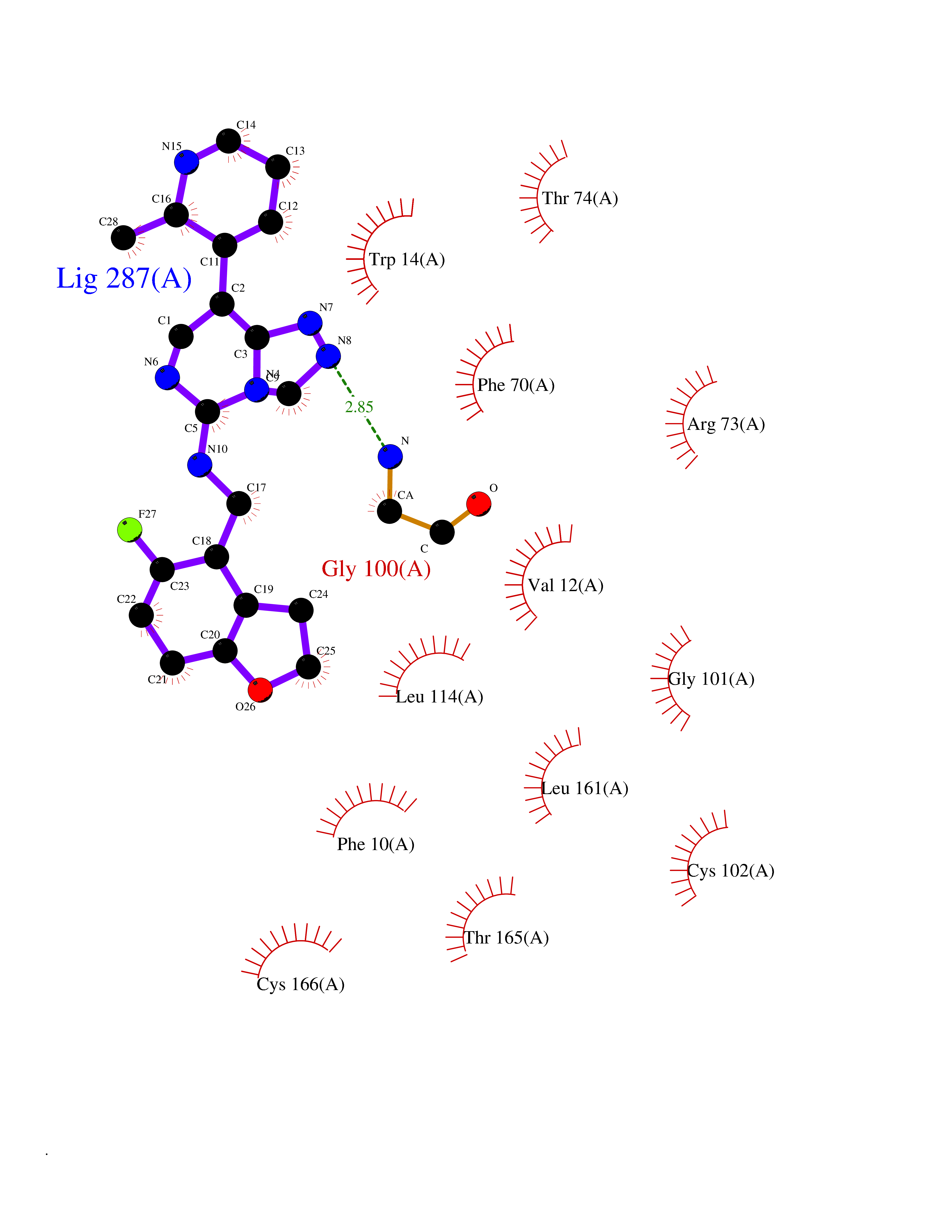 Ligplot Map 3D Binding mode Sequence SFHVIWIASFYNHSWKQNLVSGWLSDLQTHTWDSNSSTIVFLWPWSRGNFSNEWKELETLFRIRTIRSFEGIRRYAHELQFEYPFEIQVTGGCESGSFLQLAYQGSDFVSFQNNSWLPYPVAGNMAKHFCKVLNQNQHENDITHNLLSDTCPRFILGLLDAGKAHLQRQVKPEAWLSHGPSPGPGHLQLVCHVSGFYPKPVWVMWMRGEQEQQGTQRGDILPSADGTWYLRATLEVAAGEAADLSCRVKHSSLEGQDIVLYWEGSLVPRG Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Retinoic acid receptor alpha (RARA) | 3KMR | 8.30 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -9.81 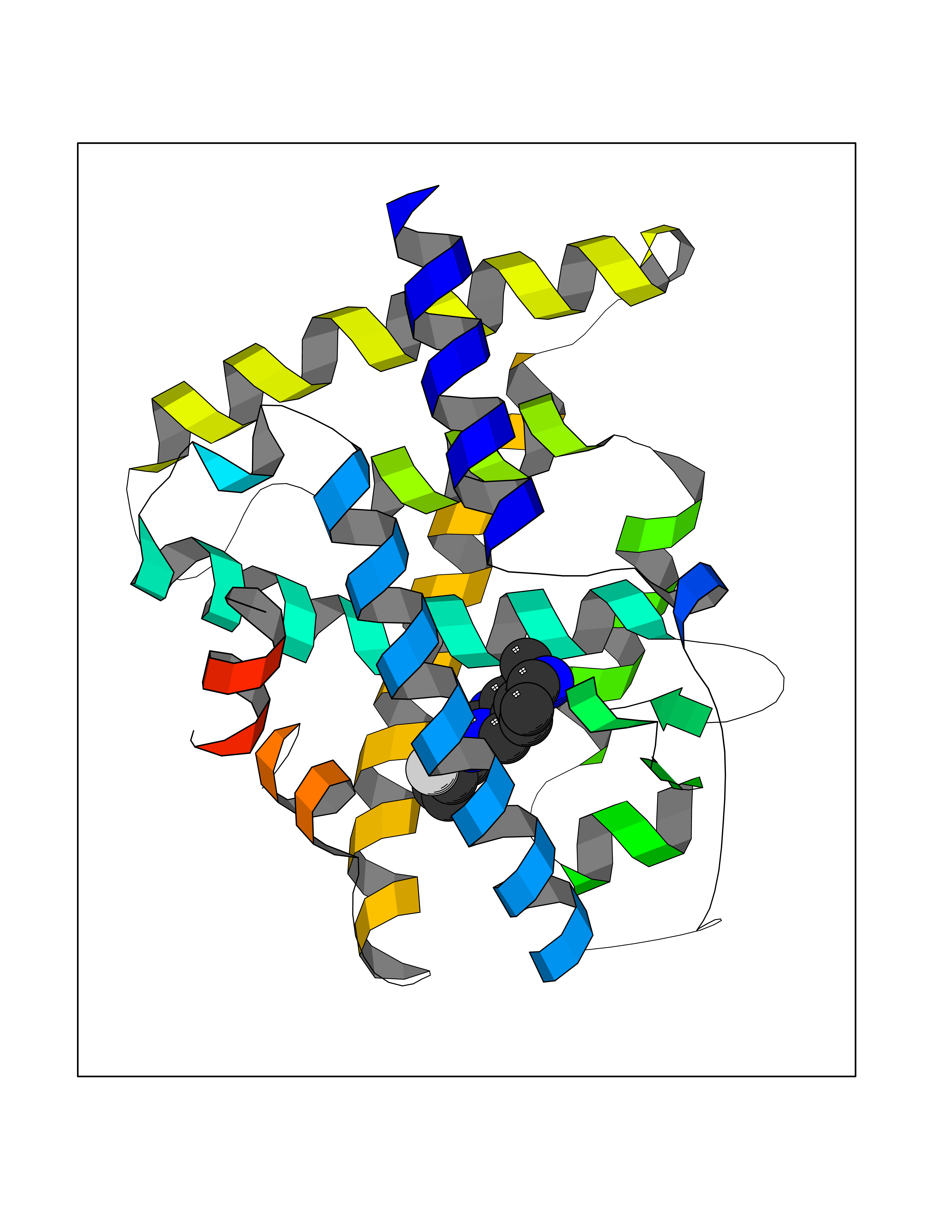 Molscript Map 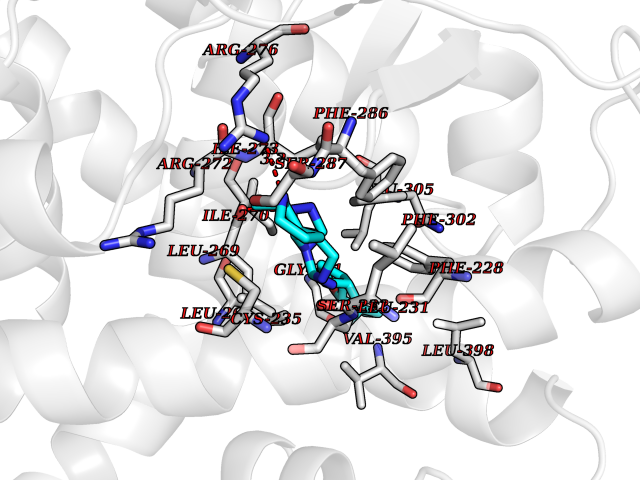 Pymol Map 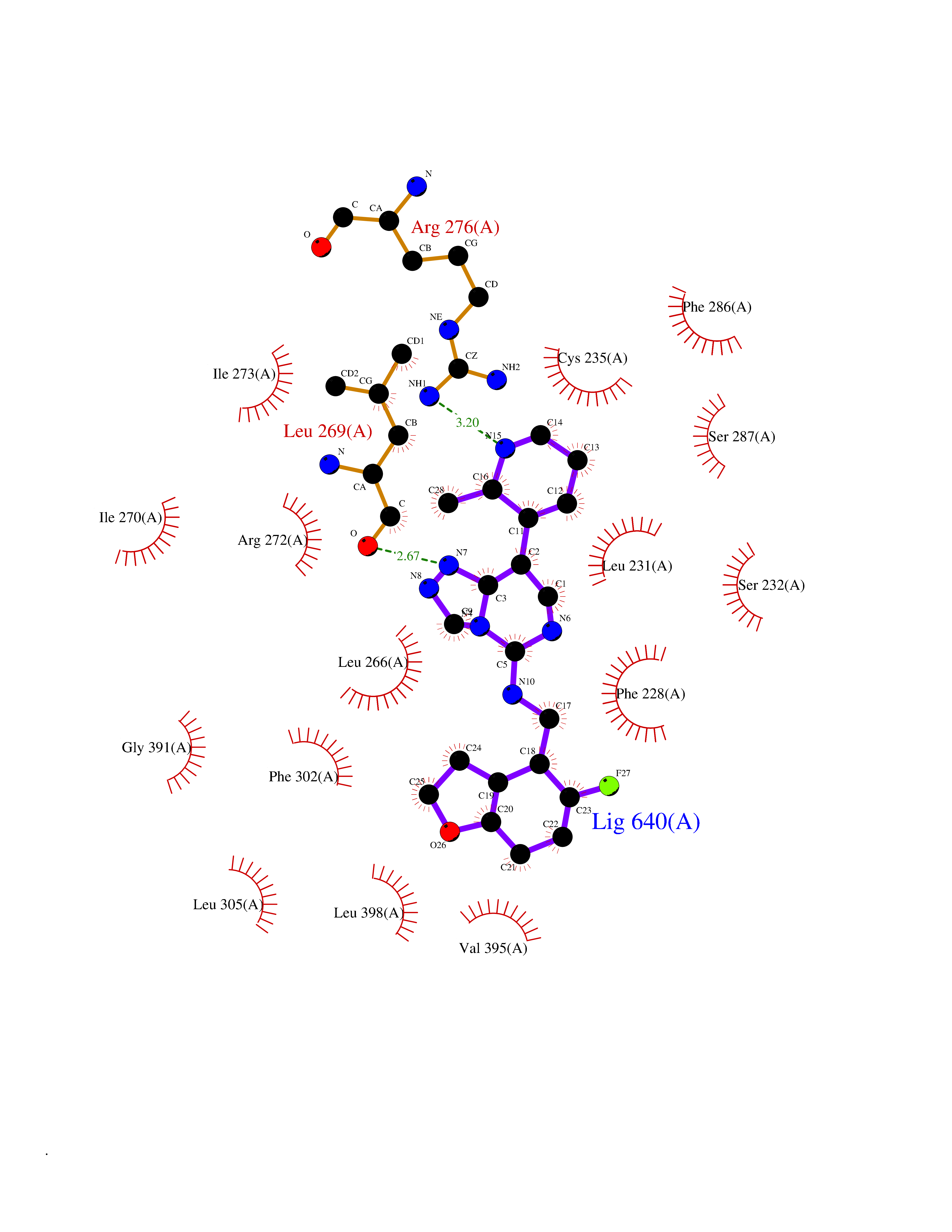 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | MLK-related kinase (MLTK) | 5HES | 8.29 | |
Target general information Gen name MAP3K20 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ZAK; Sterile alpha motif- and leucine zipper-containing kinase AZK; Mixed lineage kinase-related kinase; Mitogen-activated protein kinase kinase kinase MLT; Mitogen-activated protein kinase kinase kin Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class NA Function Stress-activated component of a protein kinase signal transduction cascade. Regulates the JNK and p38 pathways. Part of a signaling cascade that begins with the activation of the adrenergic receptor ADRA1B and leads to the activation of MAPK14. Pro-apoptotic. Role in regulation of S and G2 cell cycle checkpoint by direct phosphorylation of CHEK2. Involved in limb development. Related diseases Split-foot malformation with mesoaxial polydactyly (SFMMP) [MIM:616890]: An autosomal recessive disorder characterized by a split-foot defect, mesoaxial polydactyly, nail abnormalities of the hands, and sensorineural hearing loss. {ECO:0000269|PubMed:26755636, ECO:0000269|PubMed:32266845}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, centronuclear, 6, with fiber-type disproportion (CNM6) [MIM:617760]: A form of centronuclear myopathy, a congenital muscle disorder characterized by progressive muscular weakness and wasting involving mainly limb girdle, trunk, and neck muscles. It may also affect distal muscles. Weakness may be present during childhood or adolescence or may not become evident until the third decade of life. Ptosis is a frequent clinical feature. The most prominent histopathologic features include high frequency of centrally located nuclei in muscle fibers not secondary to regeneration, radial arrangement of sarcoplasmic strands around the central nuclei, and predominance and hypotrophy of type 1 fibers. CNM6 is an autosomal recessive, slowly progressive form with onset in infancy or early childhood. {ECO:0000269|PubMed:27816943, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01254; DB12010 Interacts with O75582; P31947; P63104; Q8N184; Q16512; Q6P2D0; Q6ZN57; P13682; Q8N184; Q6AZW8; Q9NQZ8 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cytoplasm; Disease variant; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding; rRNA-binding; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 64591.5 Length 566 Aromaticity 0.1 Instability index 52.08 Isoelectric point 5.69 Charge (pH=7) -12.83 2D Binding mode Binding energy (Kcal/mol) -9.3 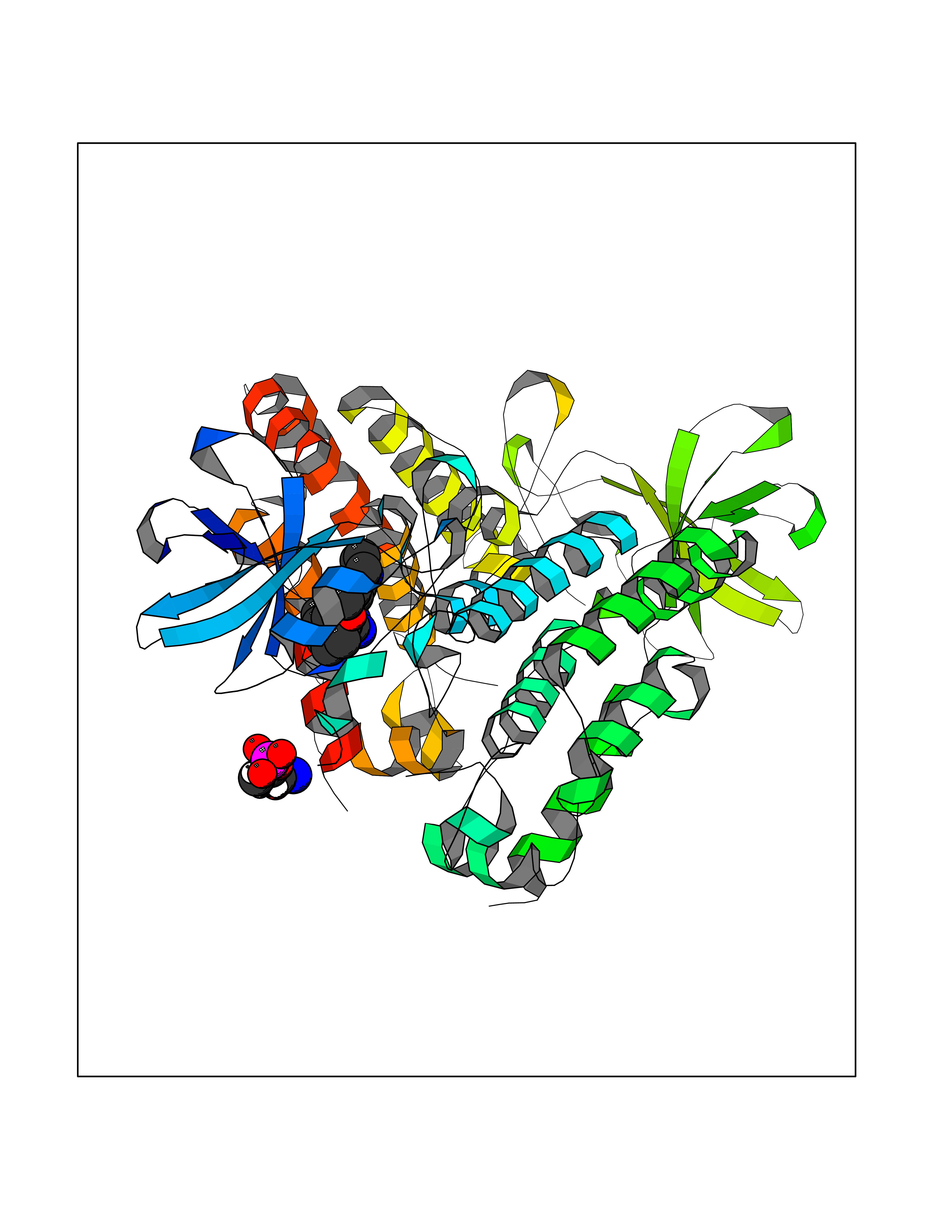 Molscript Map 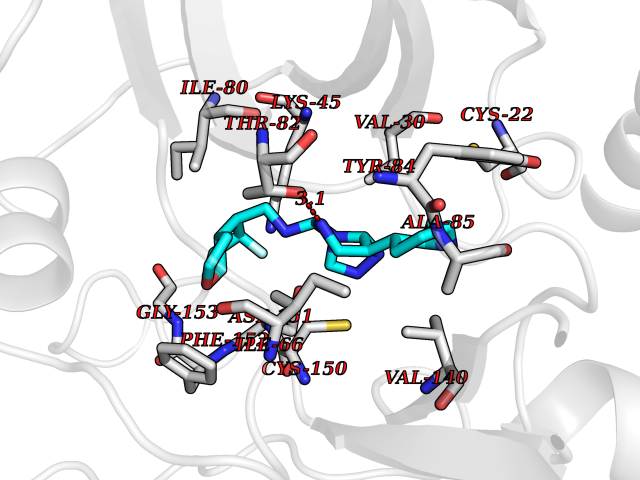 Pymol Map 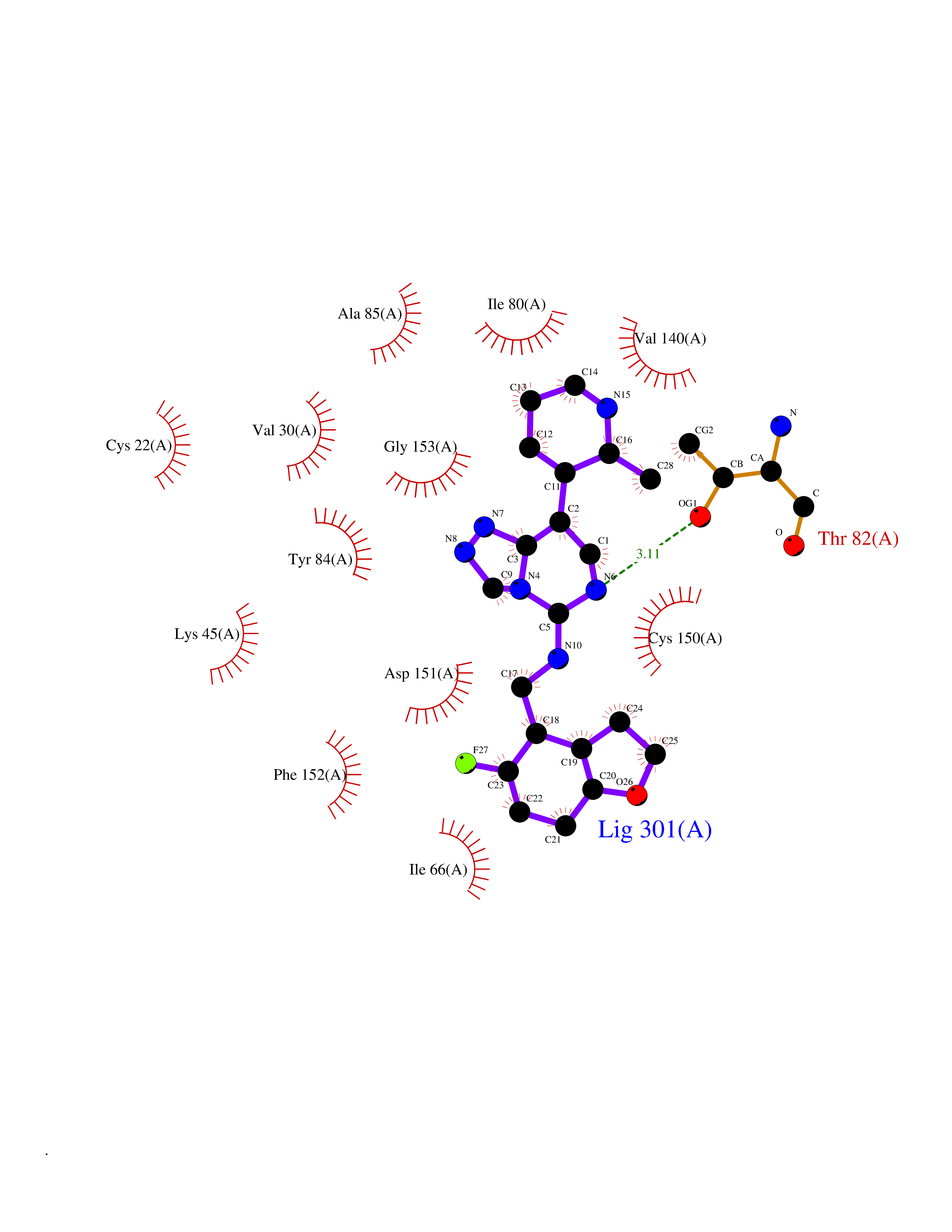 Ligplot Map 3D Binding mode Sequence ASFVQIKFDDLQFFENCGGGSFGSVYRAKWISQDKEVAVKKLLKIEKEAEILSVLSHRNIIQFYGVILEPPNYGIVTEYASLGSLYDYINSNRSEEMDMDHIMTWATDVAKGMHYLHMEAPVKVIHRDLKSRNVVIAADGVLKICDFGASRFHNHXGTFPWMAPEVIQSLPVSETCDTYSYGVVLWEMLTREVPFKGLEGLQVAWLVVEKNERLTIPSSCPRSFAELLHQCWEADAKKRPSFKQIISILESMSNDTSLPDKCNSFLHNKAEWRCEIEATLERLKKLERSFVQIKFDDLQFFENCGGGSFGSVYRAKWISQDKEVAVKKLLKIEKEAEILSVLSHRNIIQFYGVILEPPNYGIVTEYASLGSLYDYINSNRSEEMDMDHIMTWATDVAKGMHYLHMEAPVKVIHRDLKSRNVVIAADGVLKICDFGGTFPWMAPEVIQSLPVSETCDTYSYGVVLWEMLTREVPFKGLEGLQVAWLVVEKNERLTIPSSCPRSFAELLHQCWEADAKKRPSFKQIISILESMSNDTSLPDKCNSFLHNKAEWRCEIEATLERLKKLE Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | 1-deoxy-D-xylulose-5-phosphate synthase (EC 2.2.1.7) (1-deoxyxylulose-5-phosphate synthase) (DXP synthase) (DXPS) | 2O1X | 8.26 | |
Target general information Gen name dxs Organism Deinococcus radiodurans (strain ATCC 13939 / DSM 20539 / JCM 16871 / CCUG 27074 / LMG 4051 / NBRC 15346 / NCIMB 9279 / VKM B-1422 / R1) Uniprot ID TTD ID NA Synonyms DR_1475 Protein family Transketolase family, DXPS subfamily Biochemical class NA Function Catalyzes the acyloin condensation reaction between C atoms 2 and 3 of pyruvate and glyceraldehyde 3-phosphate to yield 1-deoxy-D-xylulose-5-phosphate (DXP). {ECO:0000250}. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 2.2.1.7 Uniprot keywords 3D-structure; Isoprene biosynthesis; Magnesium; Metal-binding; Reference proteome; Thiamine biosynthesis; Thiamine pyrophosphate; Transferase Protein physicochemical properties Chain ID C,D Molecular weight (Da) 114550 Length 1068 Aromaticity 0.06 Instability index 28.21 Isoelectric point 5.51 Charge (pH=7) -32.76 2D Binding mode Binding energy (Kcal/mol) -9.39 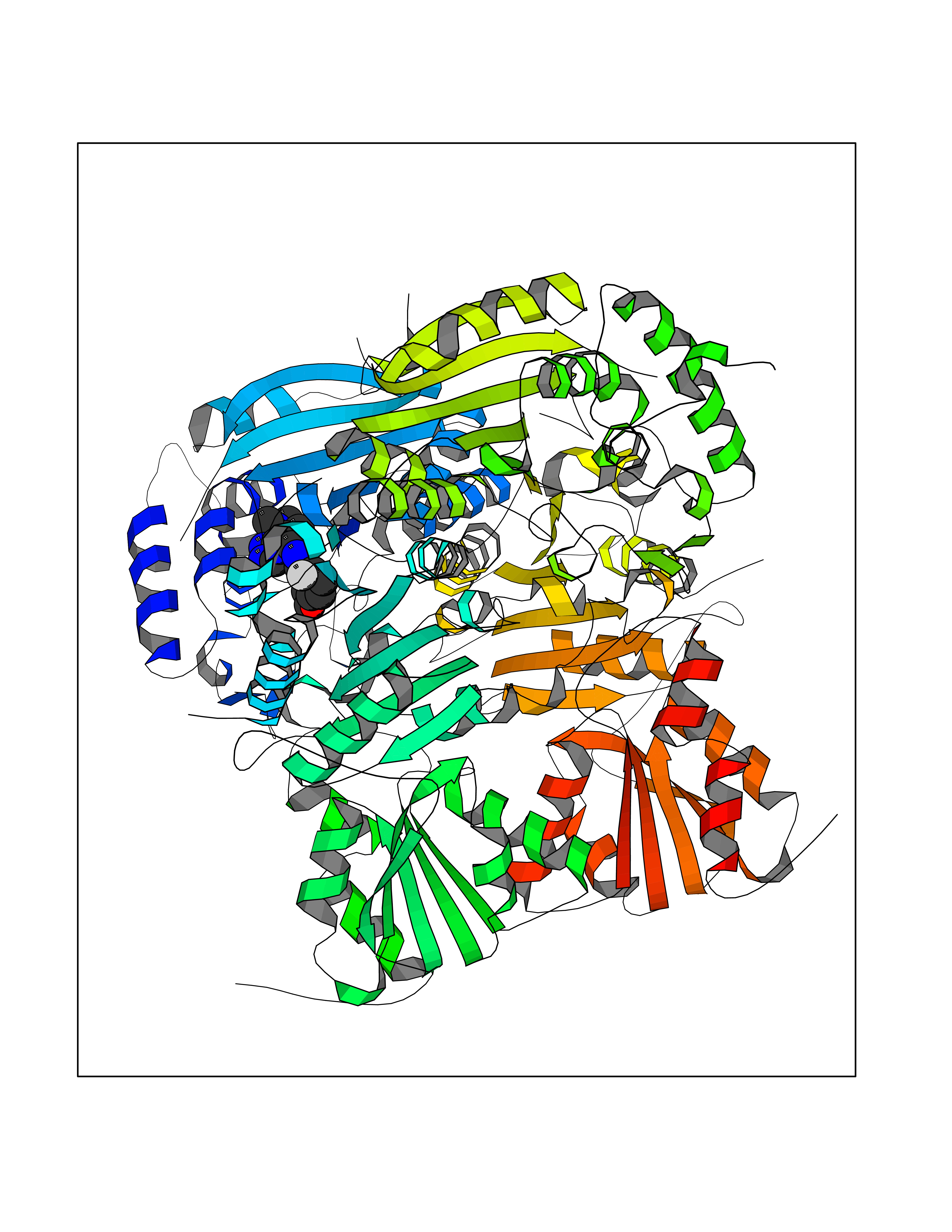 Molscript Map 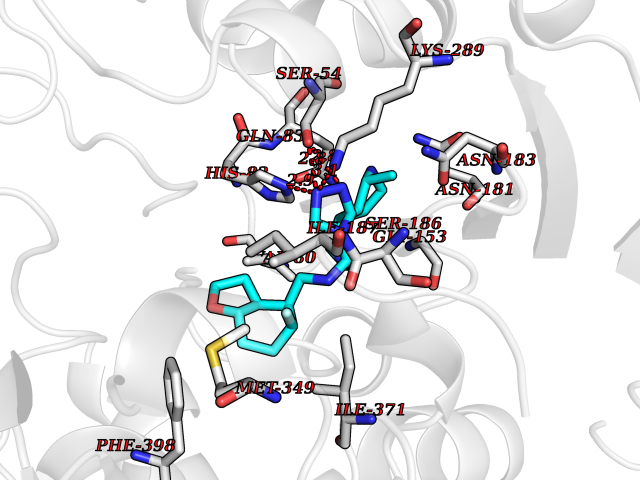 Pymol Map 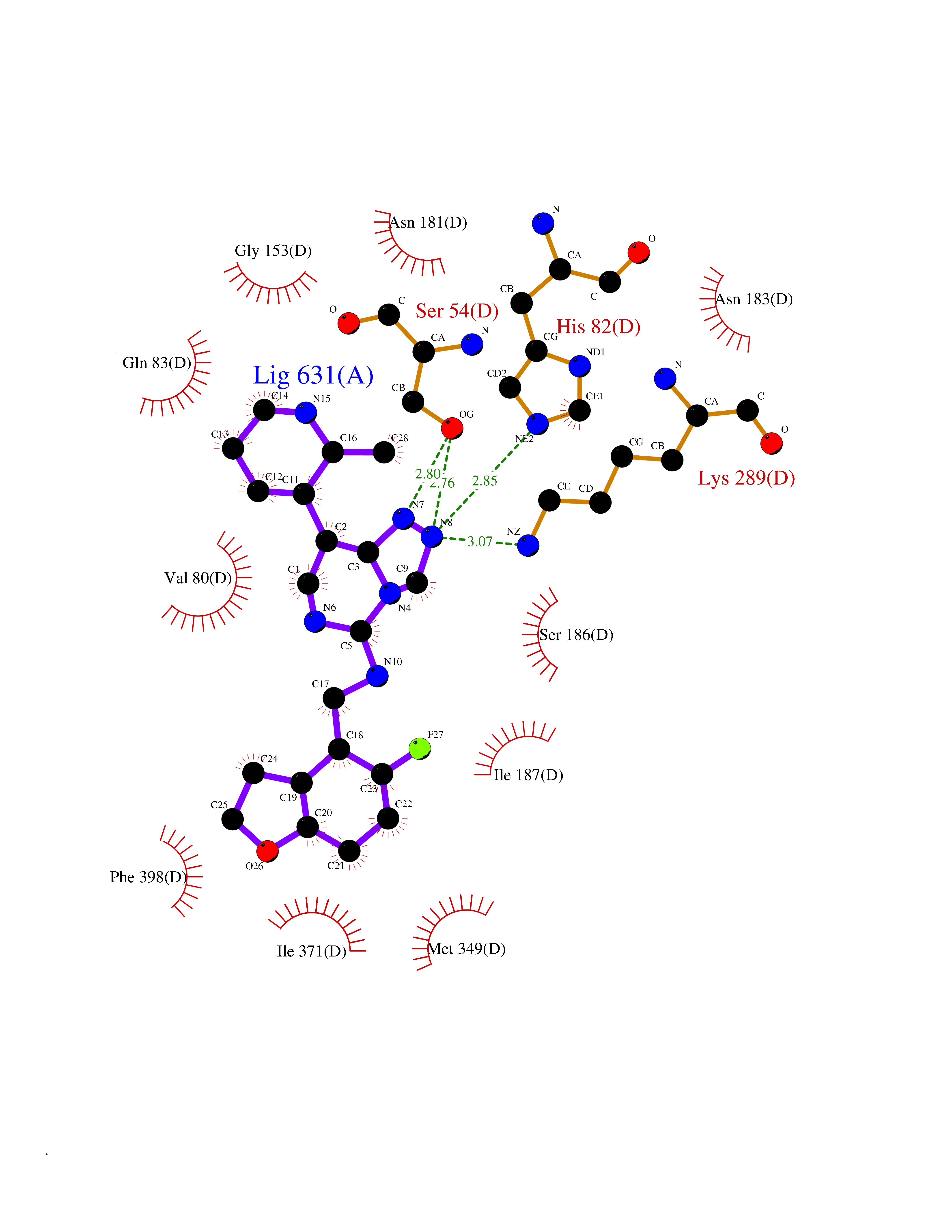 Ligplot Map 3D Binding mode Sequence HGPKDLKRLSREQLPALTEELRGEIVRVCSRGGLHLASSLGAVDIITALHYVLDSPRDRILFDVGHQAYAHKILTGRRDQMADIKKEGGISGFTKVSESEHDAITVGHASTSLTNALGMALARDAQGKDFHVAAVIGDGSLTGGMALAALNTIGDMGRKMLIVLNDNEMSISENVVNPFAAMGVRYVGPVDGHNVQELVWLLERLVDLDGPTILHIVTTKGAYSWSAAFGEAVTEWAKTDPRTFVVTPAMREGSGLVEFSRVHPHRYLDVGIAEEVAVTTAAGMALQGMRPVVAIYSTFLQRAYDQVLHDVAIEHLNVTFCIDRAGIVGADGATHNGVFDLSFLRSIPGVRIGLPKDAAELRGMLKYAQTHDGPFAIRYPRGNTAQVPAGTWPDLKWGEWERLKGGDDVVILAGGKALDYALKAAEDLPGVGVVNARFVKPLDEEMLREVGGRARALITVEDNTVVGGFGGAVLEALNSMNLHPTVRVLGIPDEFQEHATAESVHARAGIDAPAIRTVLAELGVDVPIEVTSDTPLLDQIHGPKDLKRLSREQLPALTEELRGEIVRVCSRGGLHLASSLGAVDIITALHYVLDSPRDRILFDVGHQAYAHKILTGRRDQMADIKKEGGISGFTKVSESEHDAITVGHASTSLTNALGMALARDAQGKDFHVAAVIGDGSLTGGMALAALNTIGDMGRKMLIVLNDNEMSISENVNPFAAMGVRYVGPVDGHNVQELVWLLERLVDLDGPTILHIVTTKGAYSWSAAFGEAVTEWAKTDPRTFVVTPAMREGSGLVEFSRVHPHRYLDVGIAEEVAVTTAAGMALQGMRPVVAIYSTFLQRAYDQVLHDVAIEHLNVTFCIDRAGIVGADGATHNGVFDLSFLRSIPGVRIGLPKDAAELRGMLKYAQTHDGPFAIRYPRGNTAQVPAGTWPDLKWGEWERLKGGDDVVILAGGKALDYALKAAEDLPGVGVVNARFVKPLDEEMLREVGGRARALITVEDNTVVGGFGGAVLEALNSMNLHPTVRVLGIPDEFQEHATAESVHARAGIDAPAIRTVLAELGVDVPIE Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Metabotropic glutamate receptor 2 (mGluR2) | 7E9G | 8.26 | |
Target general information Gen name GRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR2; Group II metabotropic glutamate receptor; Glutamate receptor mGLU2; GPRC1B Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling inhibits adenylate cyclase activity. May mediate suppression of neurotransmission or may be involved in synaptogenesis or synaptic stabilization. G-protein coupled receptor for glutamate. Related diseases Oocyte/zygote/embryo maturation arrest 21 (OZEMA21) [MIM:620610]: An autosomal dominant, female infertility disorder characterized by zygote development arrest due to failure of pronuclei fusion. {ECO:0000269|PubMed:33948904, ECO:0000269|PubMed:33953335}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with Q5T8D3-2; Q9BYF1; Q13520; Q13323; Q8WV48; P57739; O95484; Q7Z7G2; P00387; P27487; P28223-1; Q5SR56; O14880; Q8N4V1; Q58DX5; Q13113; Q9NR31; Q8IWU4; Q9H2H9; P27105; Q8N3G9; Q96Q45-2; Q9NWD8; Q8WUV1; Q9UMX0-2; P0DTC2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 85146.2 Length 769 Aromaticity 0.11 Instability index 36.84 Isoelectric point 8.48 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -9.16 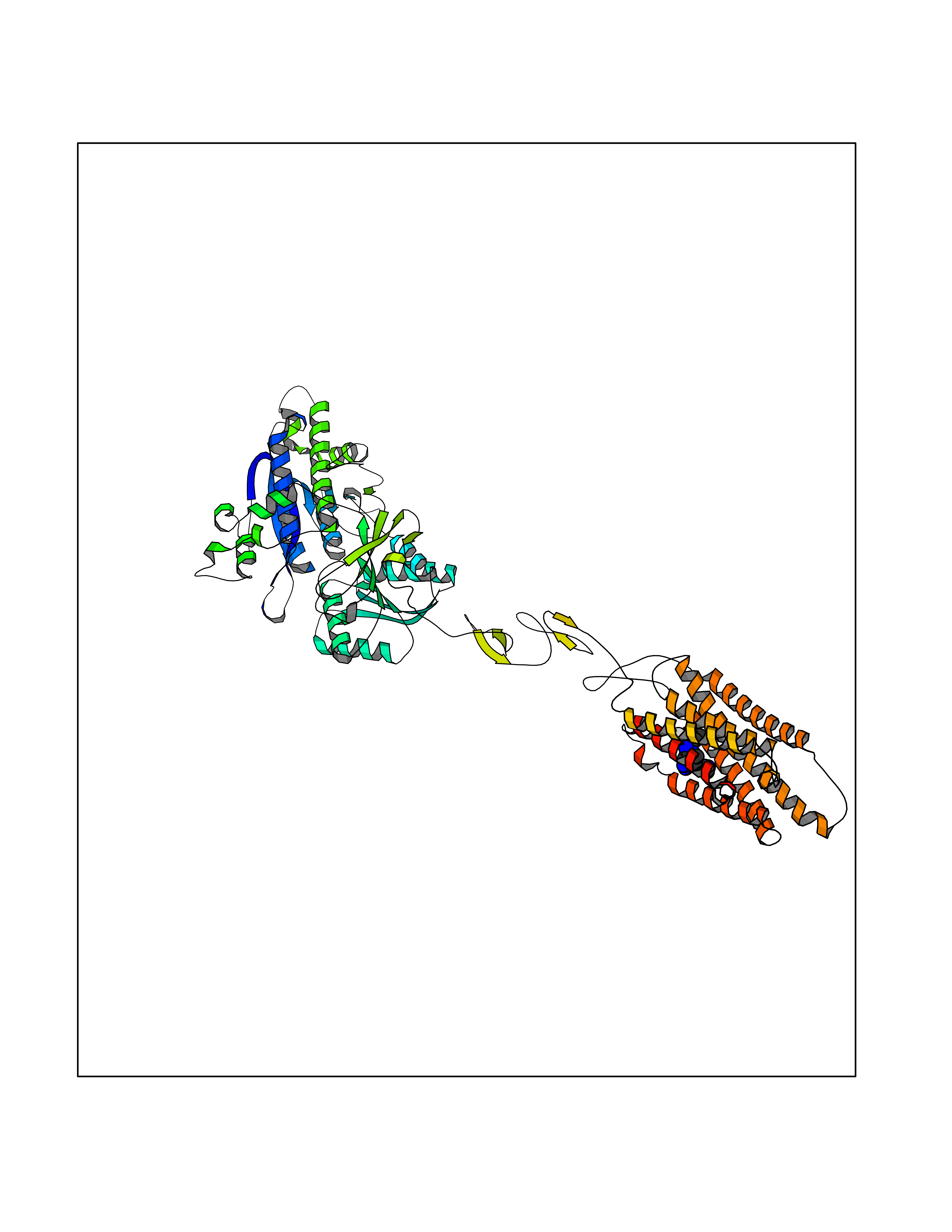 Molscript Map 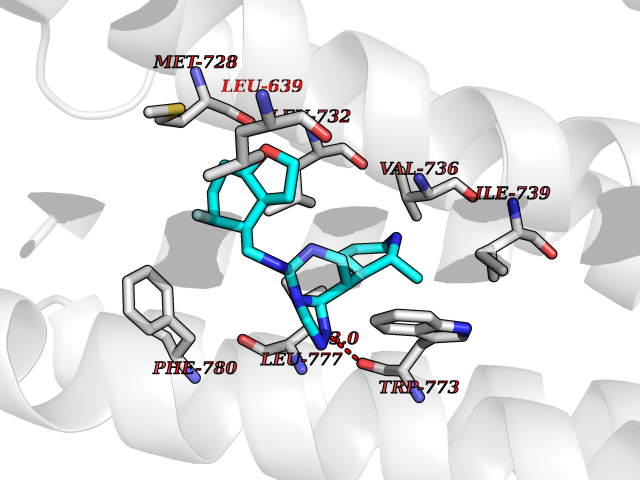 Pymol Map 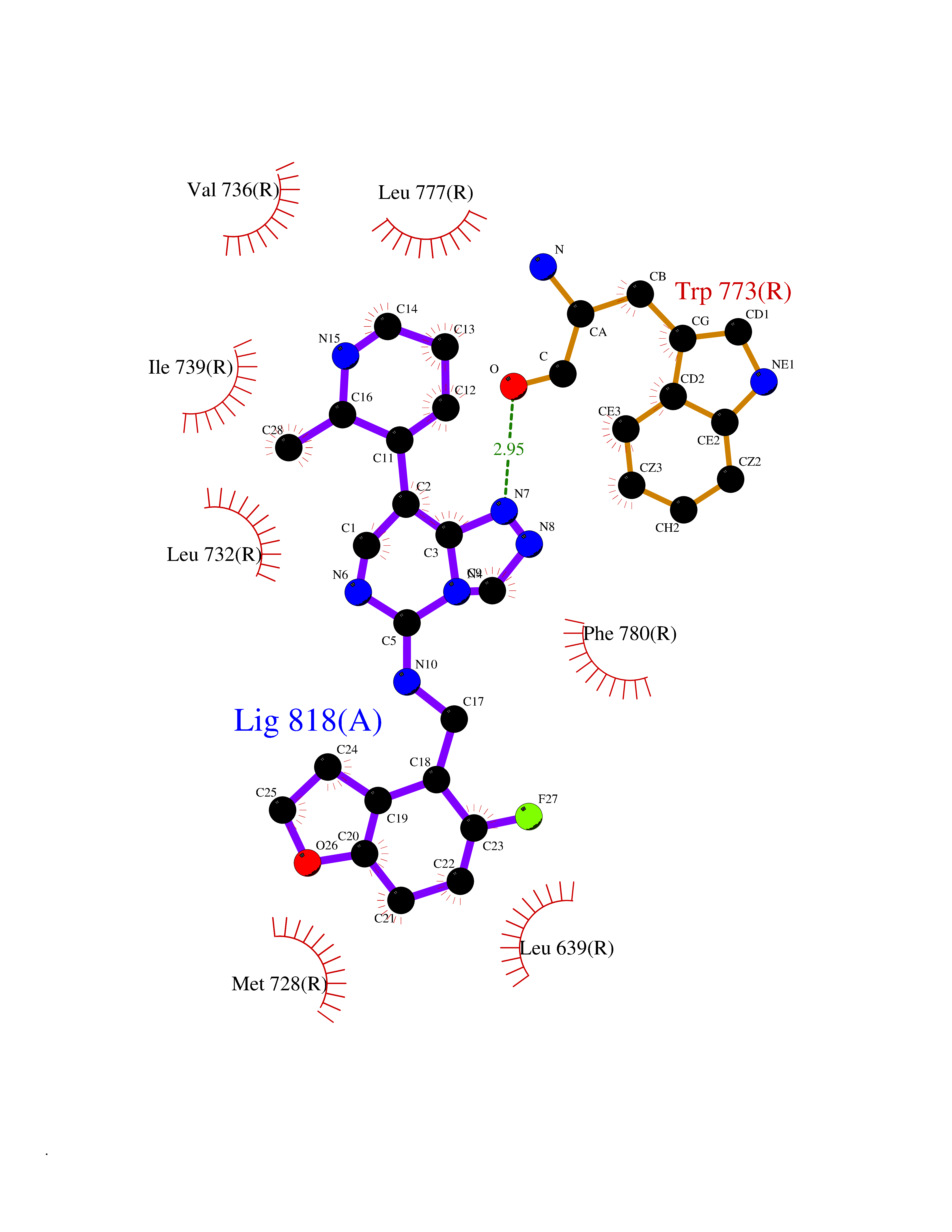 Ligplot Map 3D Binding mode Sequence KKVLTLEGDLVLGGLFPVHQKGGPAEDCGPVNEHRGIQRLEAMLFALDRINRDPHLLPGVRLGAHILDSCSKDTHALEQALDFVRASLITGVIGGSYSDVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFFQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFELEARARNICVATSEKVGRAMSRAAFEGVVRALLQKPSARVAVLFTRSEDARELLAASQRLNASFTWVASDGWGALESVVAGSEGAAEGAITIELASYPISDFASYFQSLDPWNNSRNPWFREFWEQRFRCSFRQRDCAAHSLRAVPFEQESKIMFVVNAVYAMAHALHNMHRALCPNTTRLCDAMRPVNGRRLYKDFVLNVKFDAPFRPADTHNEVRFDRFGDGIGRYNIFTYLRAGSGRYRYQKVGYWAEGLTLDTSLIPWASPSAGPLPASRCSEPCLQNEVKSVQPGEVCCWLCIPCQPYEYRLDEFTCADCGLGYWPNASLTGCFELPQEYIRWGDAWAVGPVTIACLGALATLFVLGVFVRHNATPVVKAAGRELCYILLGGVFLCYCMTFIFIAKPSTAVCTLRRLGLGTAFSVCYSALLTKTNRIARIFGGAREGAQRPRFISPASQVAICLALISGQLLIVVAWLVVEAPGTGKETAPERREVVTLRCNHRDASMLGSLAYNVLLIALCTLYAFKTRKCPENFNEAKFIGFTMYTTCIIWLAFLPIFYVTSSDYRVQTTTMCVSVSLSGSVVLGCLFAPKLHI Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | SET domain containing 8 (KMT5A) | 5TEG | 8.24 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -9.2 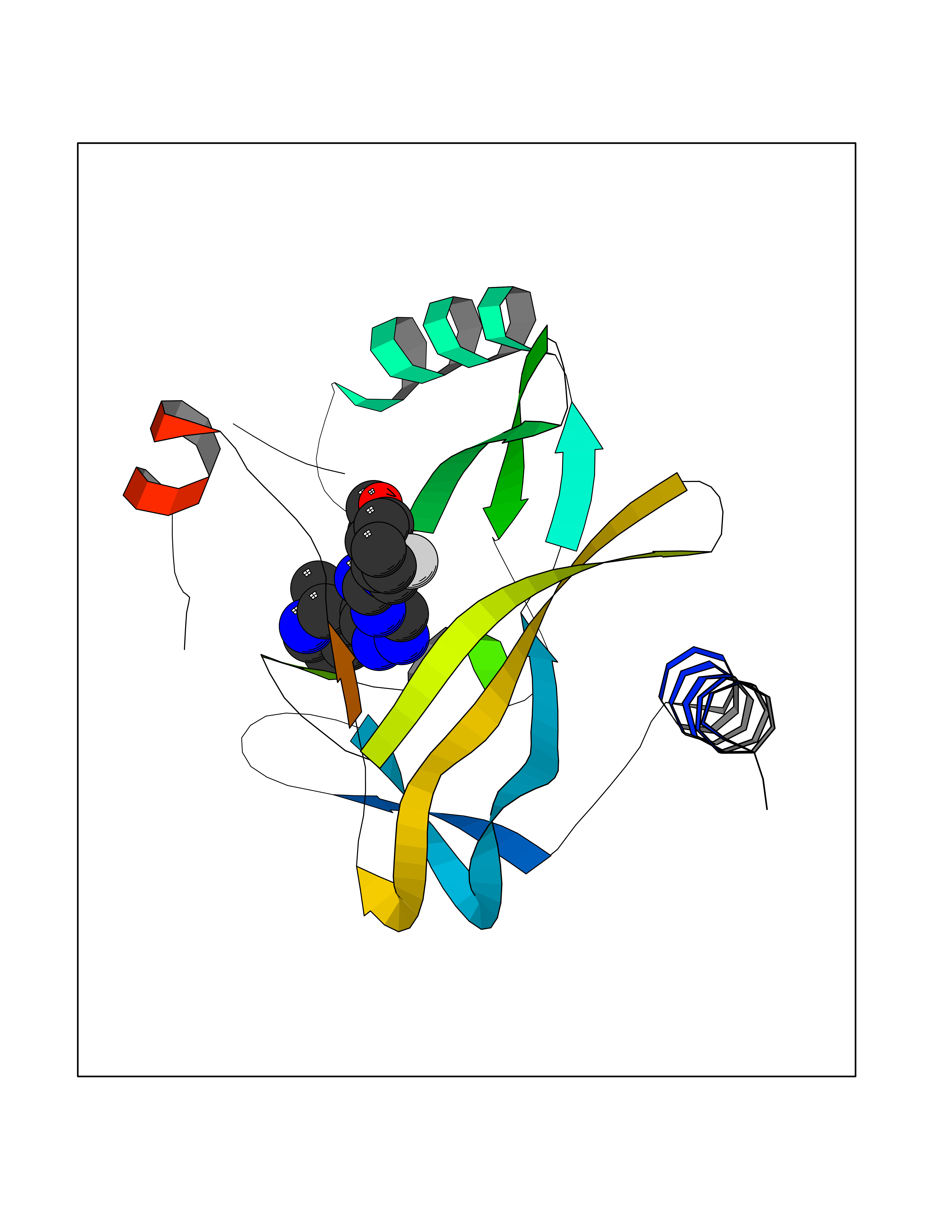 Molscript Map 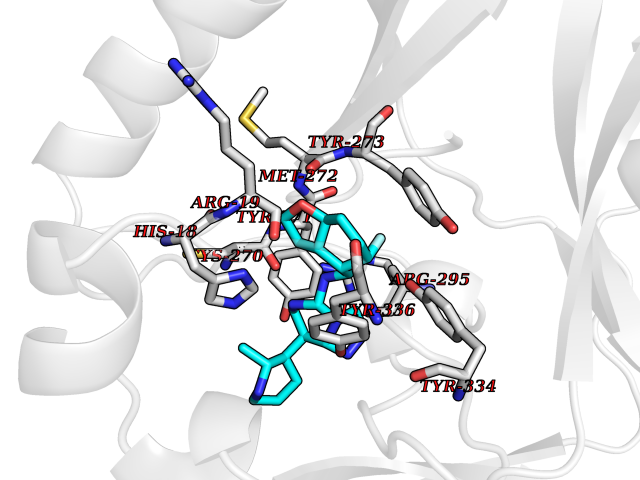 Pymol Map 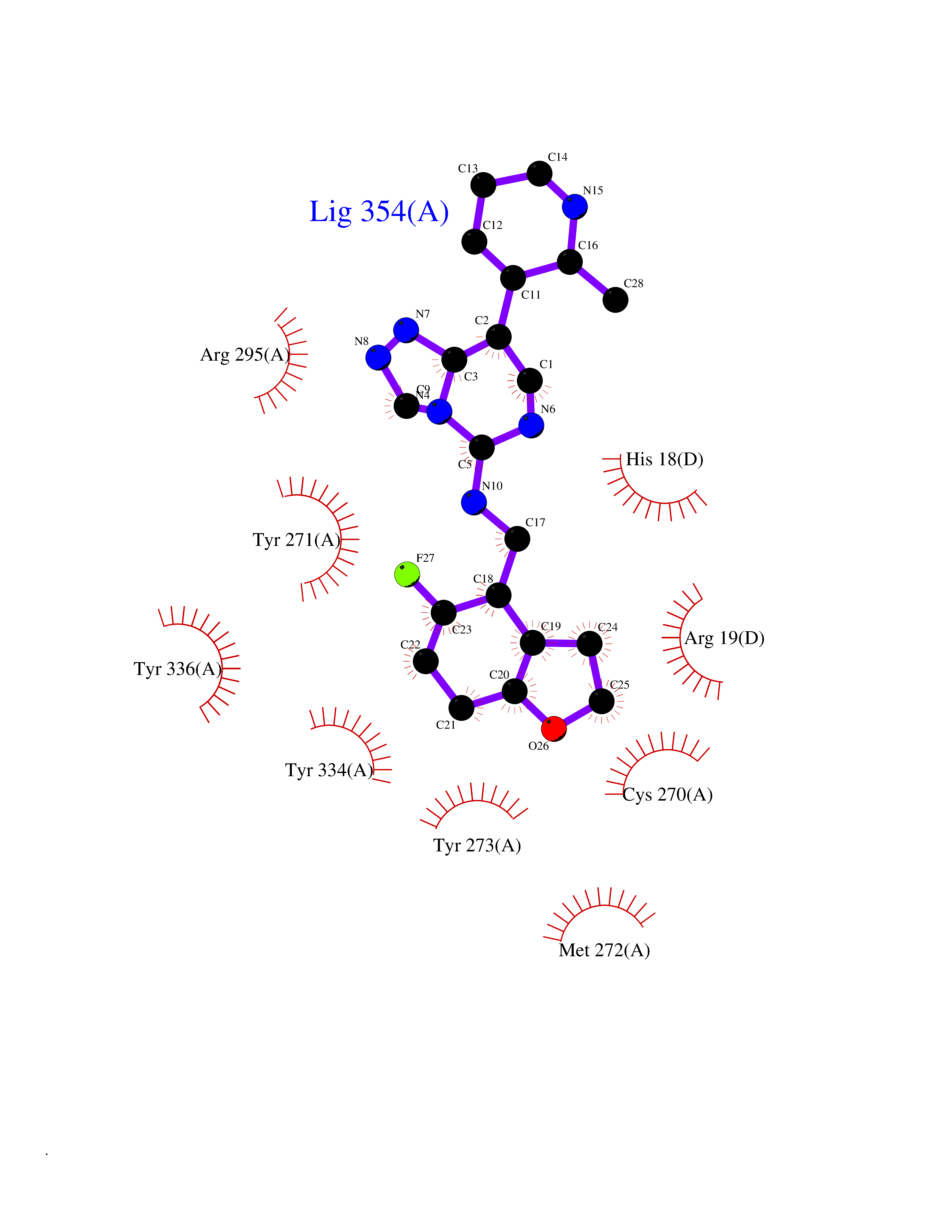 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Clostridium histolyticum Collagenase (CH colG) | 7Z5U | 8.23 | |
Target general information Gen name CH colG Organism Hathewaya histolytica (Clostridium histolyticum) Uniprot ID TTD ID Synonyms Microbial collagenase; Gelatinase ColG; Collagenase ColG; Class I collagenase Protein family Peptidase M9B family, Collagenase subfamily Biochemical class NA Function Clostridial collagenases are among the most efficient degraders of eukaryotic collagen known; saprophytes use collagen as a carbon source while pathogens additionally digest collagen to aid in host colonization. Has both tripeptidylcarboxypeptidase on Gly-X-Y and endopeptidase activities; the endopeptidase cuts within the triple helix region of collagen while tripeptidylcarboxypeptidase successively digests the exposed ends, thus clostridial collagenases can digest large sections of collagen. Active on soluble type I collagen, insoluble collagen, azocoll, soluble PZ-peptide (all collagenase substrates) and gelatin. The full-length protein has collagenase activity, while the in vivo derived C-terminally truncated shorter versions only act on gelatin. In vitro digestion of soluble calf skin collagen fibrils requires both ColG and ColH; ColG forms missing the second collagen-binding domain are also synergistic with ColH, although their overall efficiency is decreased. The activator domain (residues 119-388) and catalytic subdomain (389-670) open and close around substrate using a Gly-rich hinge (387-397), allowing digestion when the protein is closed. Binding of collagen requires Ca(2+) and is inhibited by EGTA; the collagen-binding domain (CBD, S3a plus S3b) specifically recognizes the triple-helical conformation made by 3 collagen protein chains in the triple-helical region. Isolated CBD (S3a plus S3b) binds collagen fibrils and sheets of many tissues. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.24.3 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Hydrolase; Metal-binding; Metalloprotease; Pharmaceutical; Protease; Repeat; Secreted; Signal; Virulence; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44751.2 Length 386 Aromaticity 0.15 Instability index 27.33 Isoelectric point 5.77 Charge (pH=7) -7.33 2D Binding mode Binding energy (Kcal/mol) -8.89 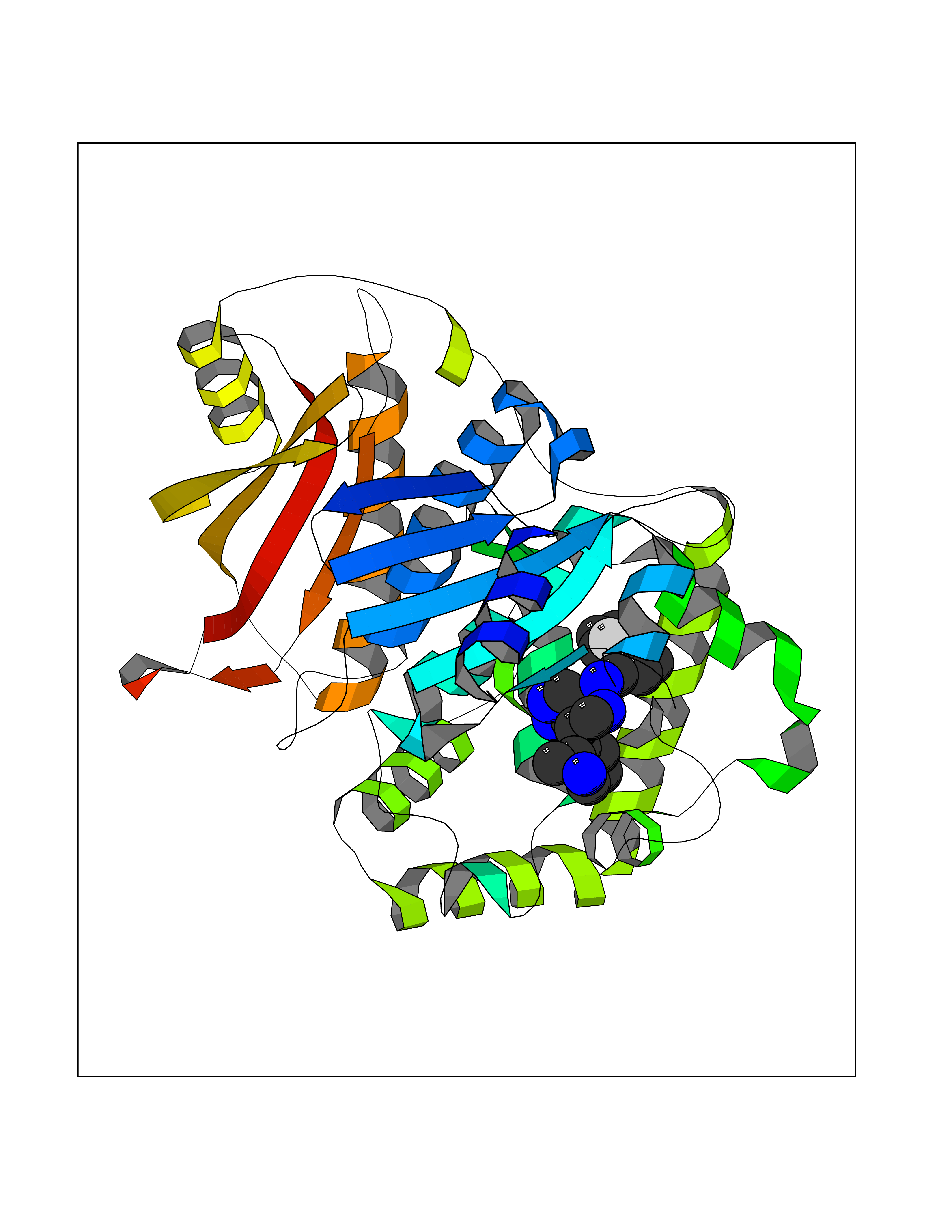 Molscript Map 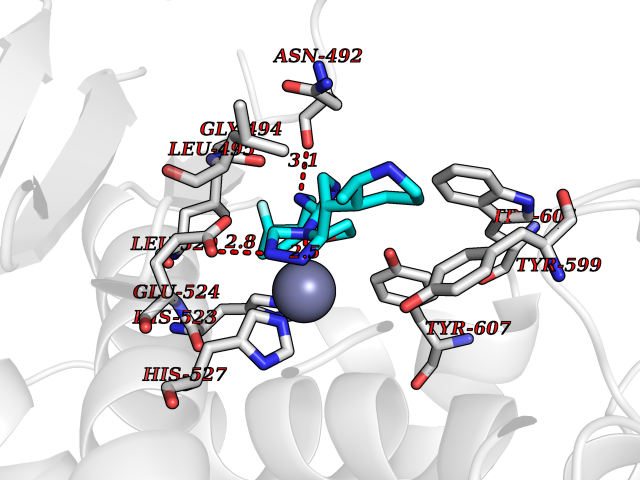 Pymol Map 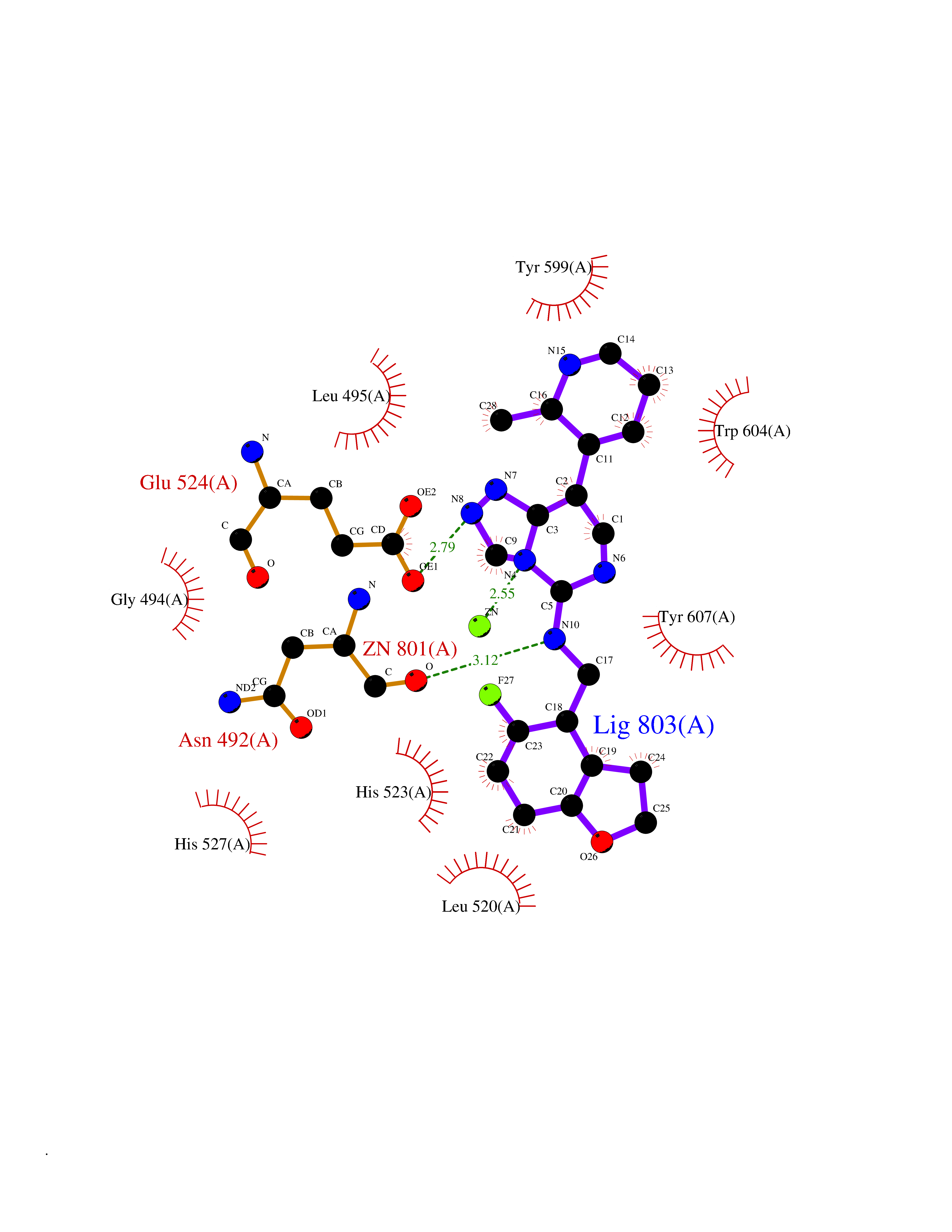 Ligplot Map 3D Binding mode Sequence DHDKFLDDAEKHYLPKTYTFDNGTFIIRAGDKVSEEKIKRLYWASREVKSQFHRVVGNDKALEVGNADDVLTMKIFNSPEEYKFNTTDNGGLYIEPRGTFYTYERTPQQSIFSLEELFRHEYTHYLQARYLVDGLWGQGPFYEKNRLTWFDEGTAEFFAGSTRTSGVLPRKLILGYLAKDKVDHRYSLKKTLNSGYDDSDWMFYNYGFAVAHYLYEKDMPTFIKMNKAILNTDVKSYDEIIKKLSDDANKNTEYQNHIQELVDKYQGAGIPLVSDDYLKDHGYKKASEVYSEISKAASLTNTSVTAEKSQYFNTFTLRGTYTGETSKGEFKDWDEMSKKLDGTLESLAKNSWSGYKTLTAYFTNYRVTSDNKVQYDVVFHGVLTDN Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 8.21 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -8.63  Molscript Map 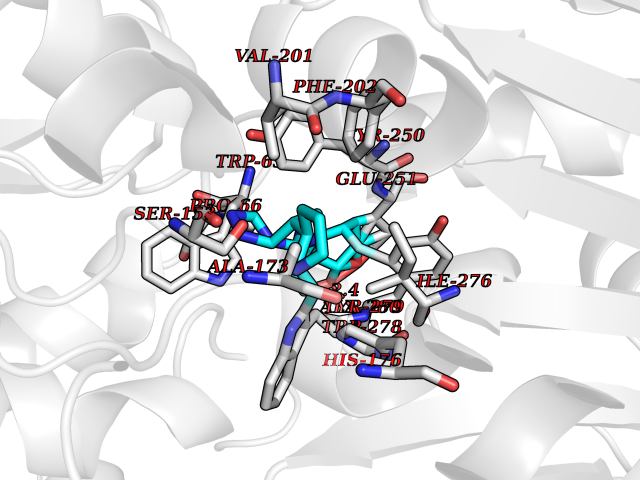 Pymol Map 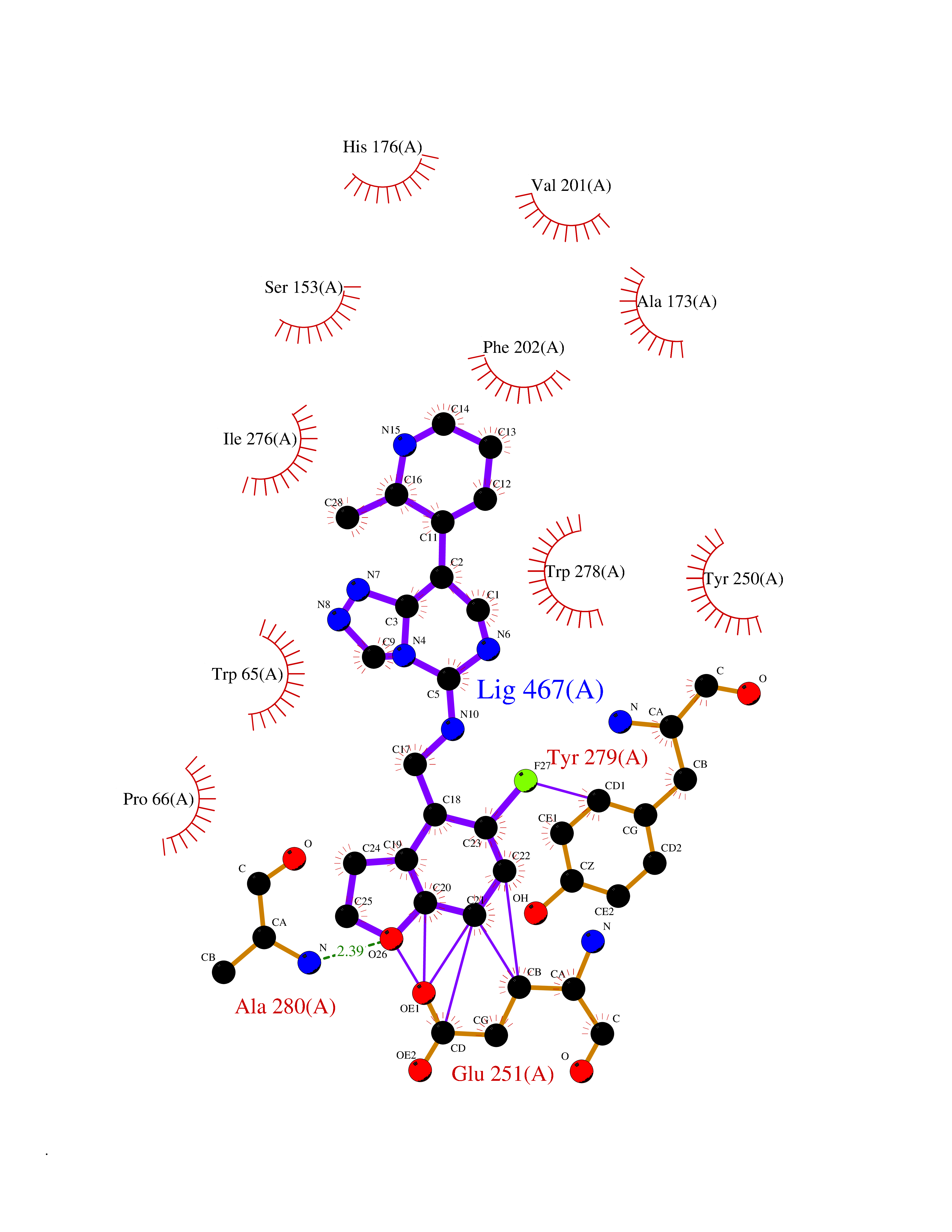 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Extracellular calcium-sensing receptor (CASR) | 5FBK | 8.21 | |
Target general information Gen name CASR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCasR; Parathyroid cell calciumreceptor; Parathyroid cell calcium-sensing receptor 1; Parathyroid calcium receptor; Parathyroid Cell calcium-sensing receptor; PCaR1; GPRC2A; CaSR Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Senses fluctuations in the circulating calcium concentration and modulates the production of parathyroid hormone (PTH) in parathyroid glands. The activity of this receptor is mediated by a G-protein that activates a phosphatidylinositol-calcium second messenger system. The G-protein-coupled receptor activity is activated by a co-agonist mechanism: aromatic amino acids, such as Trp or Phe, act concertedly with divalent cations, such as calcium or magnesium, to achieve full receptor activation. G-protein-coupled receptor that senses changes in the extracellular concentration of calcium ions and plays a key role in maintaining calcium homeostasis. Related diseases Hypocalciuric hypercalcemia, familial 1 (HHC1) [MIM:145980]: A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults. {ECO:0000269|PubMed:11762699, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:15579740, ECO:0000269|PubMed:15879434, ECO:0000269|PubMed:16598859, ECO:0000269|PubMed:16740594, ECO:0000269|PubMed:17473068, ECO:0000269|PubMed:17698911, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:19789209, ECO:0000269|PubMed:21566075, ECO:0000269|PubMed:21643651, ECO:0000269|PubMed:22114145, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25104082, ECO:0000269|PubMed:25292184, ECO:0000269|PubMed:26386835, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:7673400, ECO:0000269|PubMed:7726161, ECO:0000269|PubMed:7916660, ECO:0000269|PubMed:8636323, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9298824}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperparathyroidism, neonatal severe (NSHPT) [MIM:239200]: A disorder characterized by severe hypercalcemia, bone demineralization, and failure to thrive usually manifesting in the first 6 months of life. If untreated, NSHPT can be a devastating neurodevelopmental disorder, which in some cases is lethal without parathyroidectomy. {ECO:0000269|PubMed:14985373, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:17555508, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:8675635, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypocalcemia, autosomal dominant 1 (HYPOC1) [MIM:601198]: A disorder of mineral homeostasis characterized by blood calcium levels below normal, and low or normal serum parathyroid hormone concentrations. Disease manifestations include mild or asymptomatic hypocalcemia, paresthesias, carpopedal spasm, seizures, hypercalciuria with nephrocalcinosis or kidney stones, and ectopic and basal ganglia calcifications. Few patients manifest hypocalcemia and features of Bartter syndrome, including hypomagnesemia, hypokalemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia. {ECO:0000269|PubMed:10487661, ECO:0000269|PubMed:12050233, ECO:0000269|PubMed:12107202, ECO:0000269|PubMed:12241879, ECO:0000269|PubMed:12574188, ECO:0000269|PubMed:12915654, ECO:0000269|PubMed:15551332, ECO:0000269|PubMed:16608894, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:22789683, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25766501, ECO:0000269|PubMed:7874174, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8733126, ECO:0000269|PubMed:8813042, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253358, ECO:0000269|PubMed:9661634, ECO:0000269|PubMed:9920108}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 8 (EIG8) [MIM:612899]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Seizure types are variable, but include myoclonic seizures, absence seizures, febrile seizures, complex partial seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:18756473}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11093; DB11348; DB14481; DB01012; DB12865; DB00994; DB05695; DB05255; DB00127 Interacts with Q15363; P41180-1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 52438.9 Length 467 Aromaticity 0.12 Instability index 39.62 Isoelectric point 5.63 Charge (pH=7) -10.18 2D Binding mode Binding energy (Kcal/mol) -8.8  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GPDQRAQKKGDIILGGLFPIHFGVAAKDQDLKSRPESVECIRYNFRGFRWLQAMIFAIEEINSSPALLPNLTLGYRIFDTCNTVSKALEATLSFVAQNKIDSTIAVVGATGSGVSTAVANLLGLFYIPQVSYASSSRLLSNKNQFKSFLRTIPNDEHQATAMADIIEYFRWNWVGTIAADDDYGRPGIEKFREEAEERDIXIDFSELISQYSDEEEIQHVVEVIQNSTAKVIVVFSSGPDLEPLIKEIVRRNITGKIWLASEAWASSSLIAMPQYFHVVGGTIGFALKAGQIPGFREFLKKVHPRKSVHNGFAKEFWEETFNCHLQFRPLCTGDENISSVETPYIDYTHLRISYNVYLAVYSIAHALQDIYTCLPGRGLFTNGSCADIKKVEAWQVLKHLRHLNFTNNMGEQVTFDEXGDLVGNYSIINWHLSPEDGSIVFKEVGYYNVYAKKGERLFINEEKILWS Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Myeloperoxidase (MPO) | 4DL1 | 8.20 | |
Target general information Gen name MPO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MPO Protein family Peroxidase family, XPO subfamily Biochemical class Peroxidases Function Part of the host defense system of polymorphonuclear leukocytes. It is responsible for microbicidal activity against a wide range of organisms. In the stimulated PMN, MPO catalyzes the production of hypohalous acids, primarily hypochlorous acidin physiologic situations, and other toxic intermediates that greatly enhance PMN microbicidal activity. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06111; DB00233; DB00006; DB02300; DB06774; DB00958; DB06468; DB00833; DB00535; DB00515; DB00847; DB00250; DB05161; DB01225; DB00583; DB01065; DB00461; DB04821; DB00104; DB00526; DB00550; DB00208; DB06823; DB00500; DB04827 Interacts with P27918; Q9UNE7 EC number EC 1.11.2.2 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Heme; Hydrogen peroxide; Iron; Lysosome; Metal-binding; Oxidation; Oxidoreductase; Peroxidase; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A,B,E,F,I,J,M,N Molecular weight (Da) 53052.6 Length 466 Aromaticity 0.08 Instability index 40.64 Isoelectric point 9.48 Charge (pH=7) 15.12 2D Binding mode Binding energy (Kcal/mol) -7.96  Molscript Map 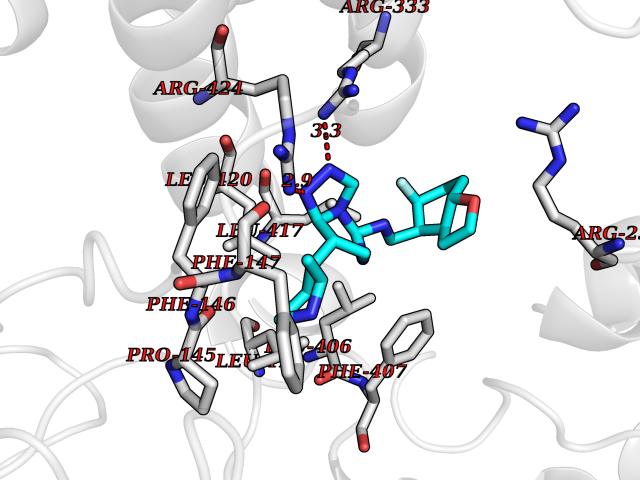 Pymol Map 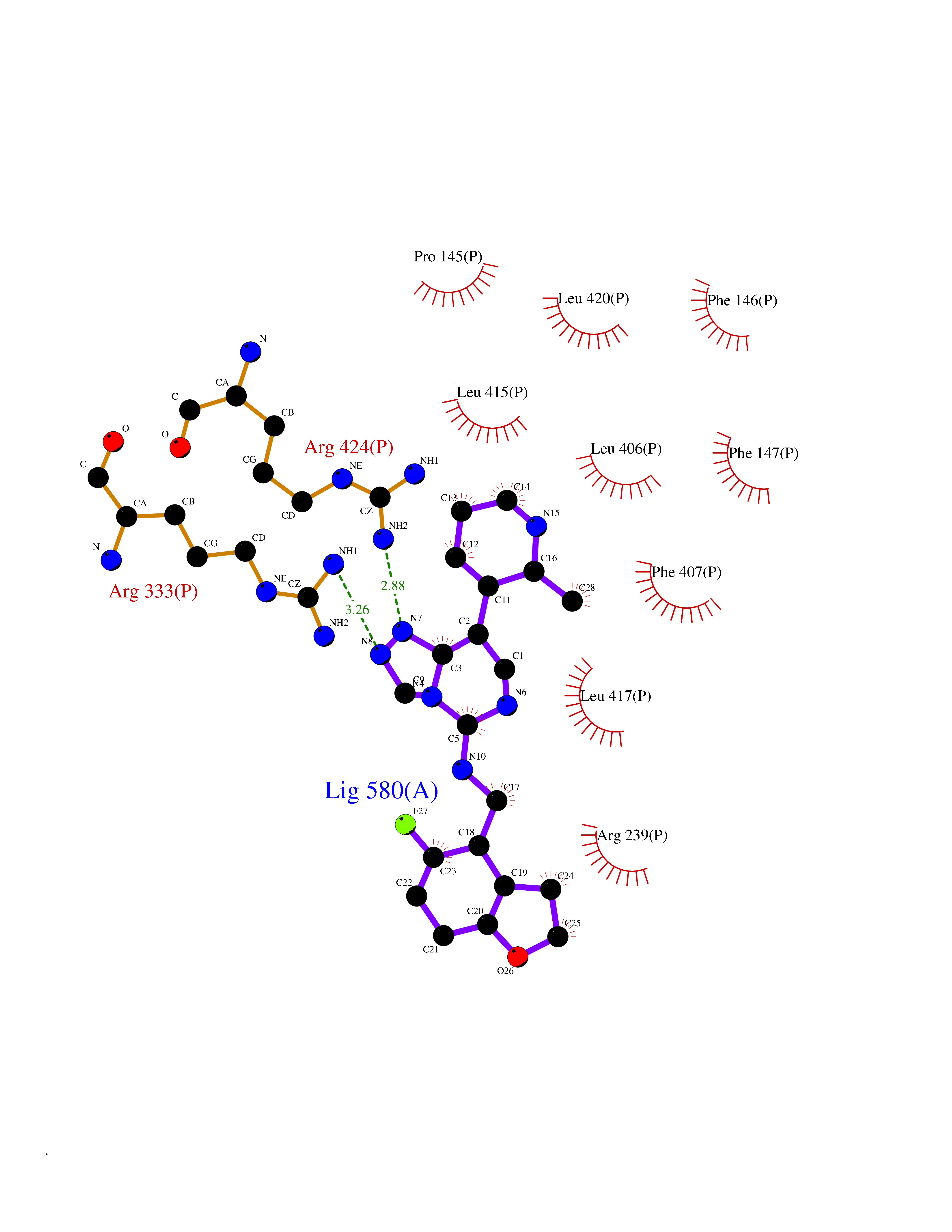 Ligplot Map 3D Binding mode Sequence VNCETSCVQQPPCFPLKIPPNDPRIKNQADCIPFFRSXPACPGSNITIRNQINALTSFVDASMVYGSEEPLARNLRNMSNQLGLLAVNQRFQDNGRALLPFDNLHDDPCLLTNRSARIPCFLAGDTRSSEMPELTSMHTLLLREHNRLATELKSLNPRWDGERLYQEARKIVGAMVQIITYRDYLPLVLGPTAMRKYLPTYRSYNDSVDPRIANVFTNAFRYGHTLIQPFMFRLDNRYQPMEPNPRVPLSRVFFASWRVVLEGGIDPILRGLMATPAKLNRQNQIAVDEIRERLFEQVMRIGLDLPALNMQRSRDHGLPGYNAWRRFCGLPQPETVGQLGTVLRNLKLARKLMEQYGTPNNIDIWMGGVSEPLKRKGRVGPLLACIIGTQFRKLRDGDRFWWENEGVFSMQQRQALAQISLPRIICDNTGITTVSKNNIFMSNSYPRDFVNCSTLPALNLASWREA Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Kallikrein-4 (KLK4) | 7JOW | 8.20 | |
Target general information Gen name KLK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine protease 17; Prostase; PSTS; PRSS17; Kallikreinlike protein 1; Kallikrein4; Kallikrein-like protein 1; KLKL1; KLK-L1; Enamel matrix serine proteinase 1; EMSP1 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function Required during the maturation stage of tooth development for clearance of enamel proteins and normal structural patterning of the crystalline matrix. Has a major role in enamel formation. Related diseases Amelogenesis imperfecta, hypomaturation type, 2A1 (AI2A1) [MIM:204700]: A defect of enamel formation. The disorder involves both primary and secondary dentitions. The teeth have a shiny agar jelly appearance and the enamel is softer than normal. Brown pigment is present in middle layers of enamel. {ECO:0000269|PubMed:15235027}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20155; Q06418 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Alternative splicing; Amelogenesis imperfecta; Biomineralization; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Metal-binding; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID E,I Molecular weight (Da) 41763.9 Length 387 Aromaticity 0.07 Instability index 32.75 Isoelectric point 5.38 Charge (pH=7) -11.36 2D Binding mode Binding energy (Kcal/mol) -8.19 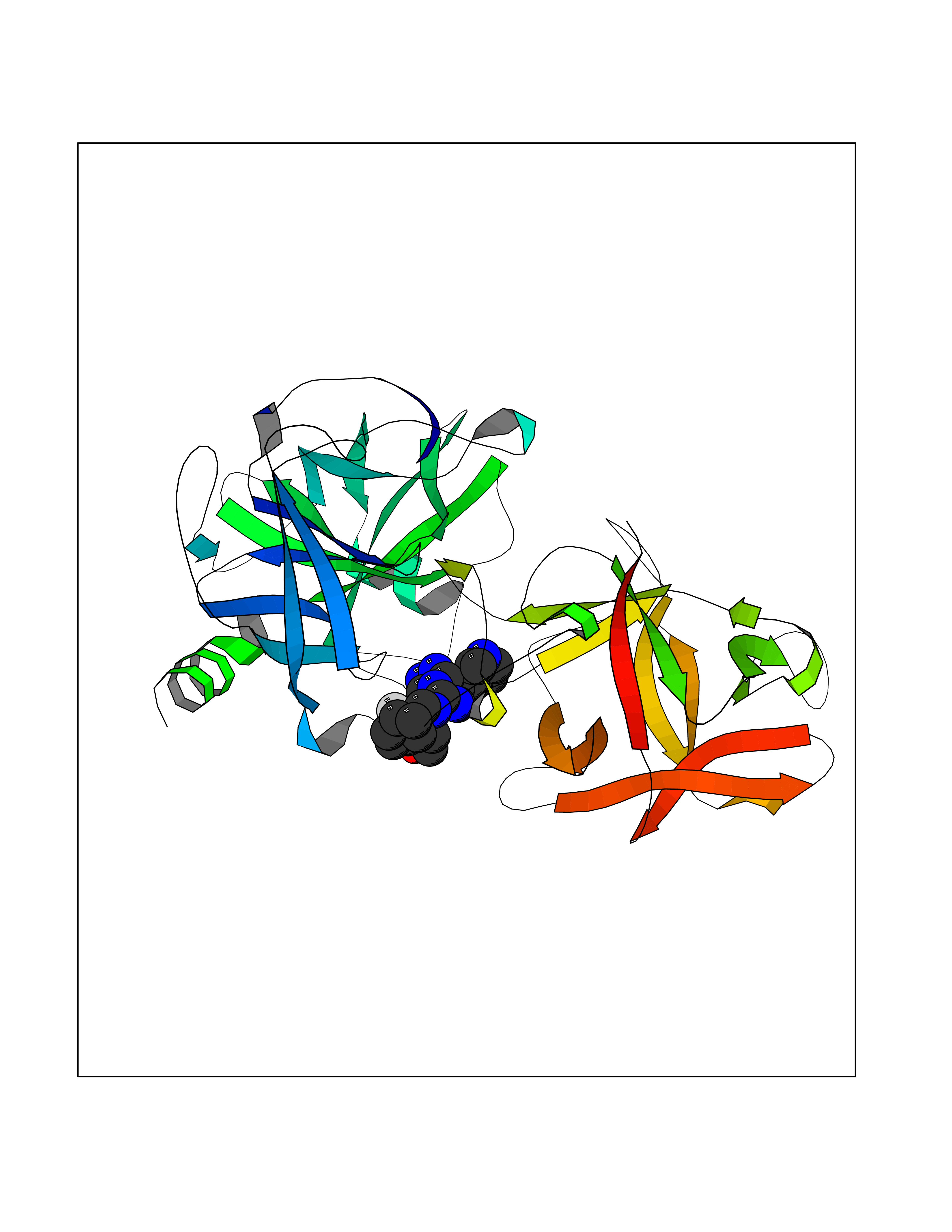 Molscript Map 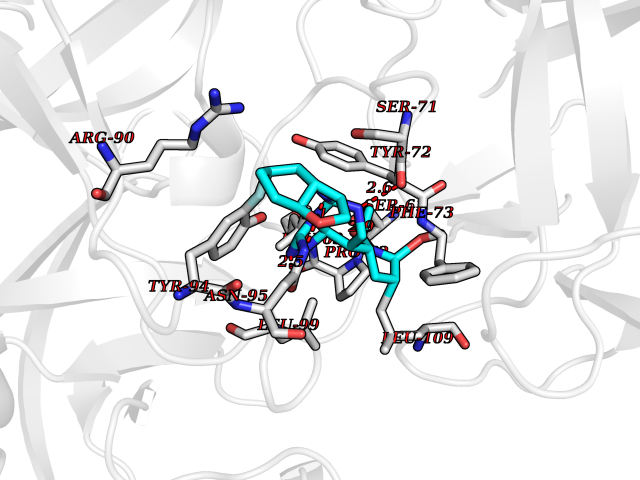 Pymol Map 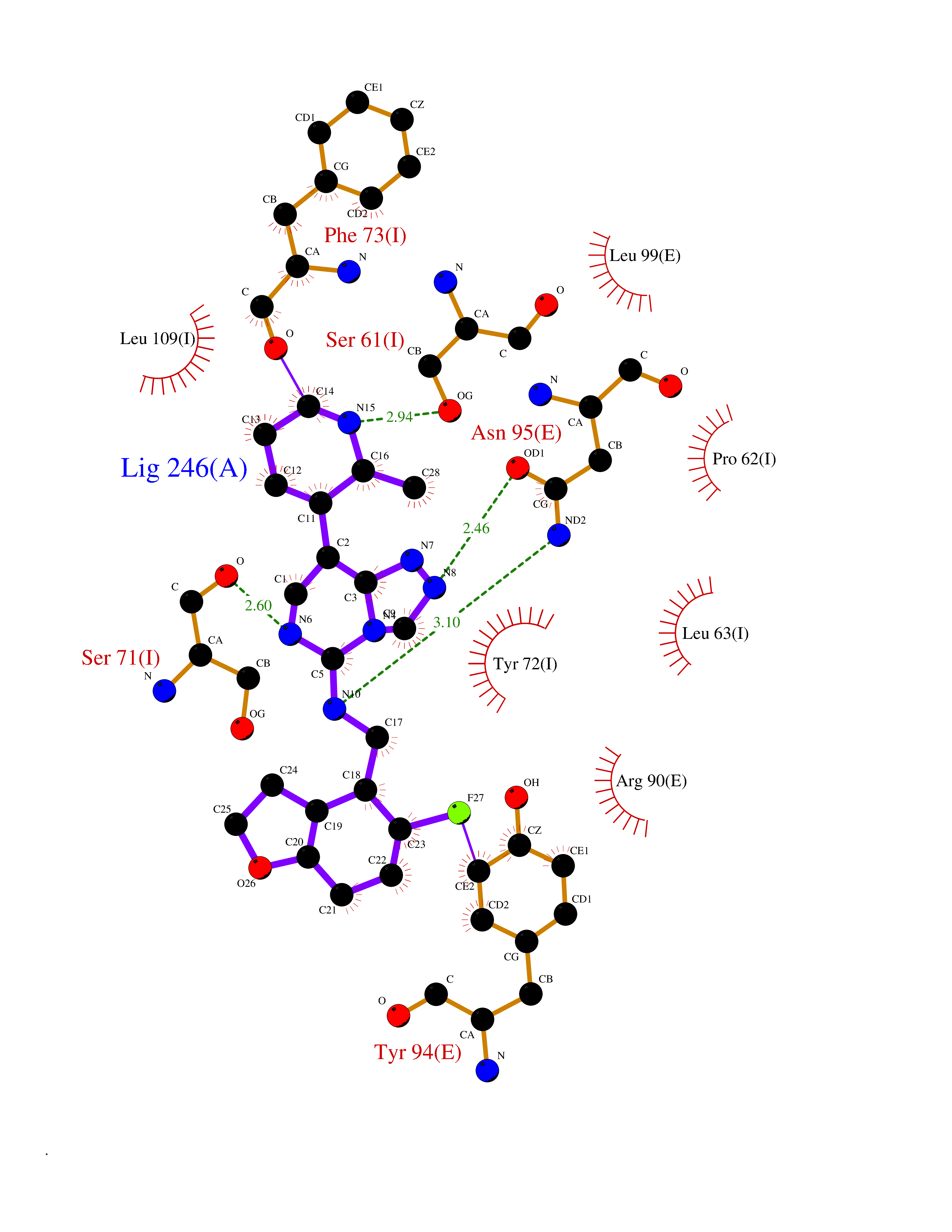 Ligplot Map 3D Binding mode Sequence IINGEDCSPHSQPWQAALVMENELFCSGVLVHPQWVLSAAHCFQNSYTIGLGLHSLEADQEPGSQMVEASLSVRHPEYNRPLLANDLMLIKLDESVSESDTIRSISIASQCPTAGNSCLVSGWGLLANGRMPTVLQCVNVSVVSEEVCSKLYDPLYHPSMFCAGGGQDQKDSCNGDSGGPLICNGYLQGLVSFGKAPCGQVGVPGVYTNLCKFTEWIEKTVQAGSSVVVDTNGQPVSNGADAYYLVPVSHGHAGLALAKIGNEAEPRAVVLDPHHRPGLPVRFESPLRINIIKESYFLNIKFGPSSSDSGVWDVIQQDPIGLAVKVTDTKSLLGPFKVEKEGEGYKIVYYPERGQTGLDIGLVHRNDKYYLAVKDGEPCVFKIRKAT Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Retinoic acid receptor RXR-beta (RXRB) | 5HJP | 8.19 | |
Target general information Gen name RXRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor beta; Nuclear receptor subfamily 2 group B member 2; NR2B2 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE). Receptor for retinoic acid. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB00459; DB00210; DB00523; DB00307; DB01393; DB03756; DB00926; DB01941; DB07929; DB02746; DB00412; DB00799; DB07080; DB00755 Interacts with Q00975; Q9HB07; F1D8P7; Q13133; Q13133-3; Q96RI1-1; P04150; Q9NRD5; P37231; P10276; P10276-2; P10826-2; P13631; Q6IQ16; Q13137; Q96B26; Q08379; Q6A162; Q9UJV3-2; Q13133-3; Q96RI1-1; O43586; P10276; P10826-2; Q8IUQ4-2; O75528; Q12800; Q9UBB9; Q05BL1; P14373; O94972; Q96S82 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 28845.8 Length 251 Aromaticity 0.08 Instability index 54.86 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -9.59 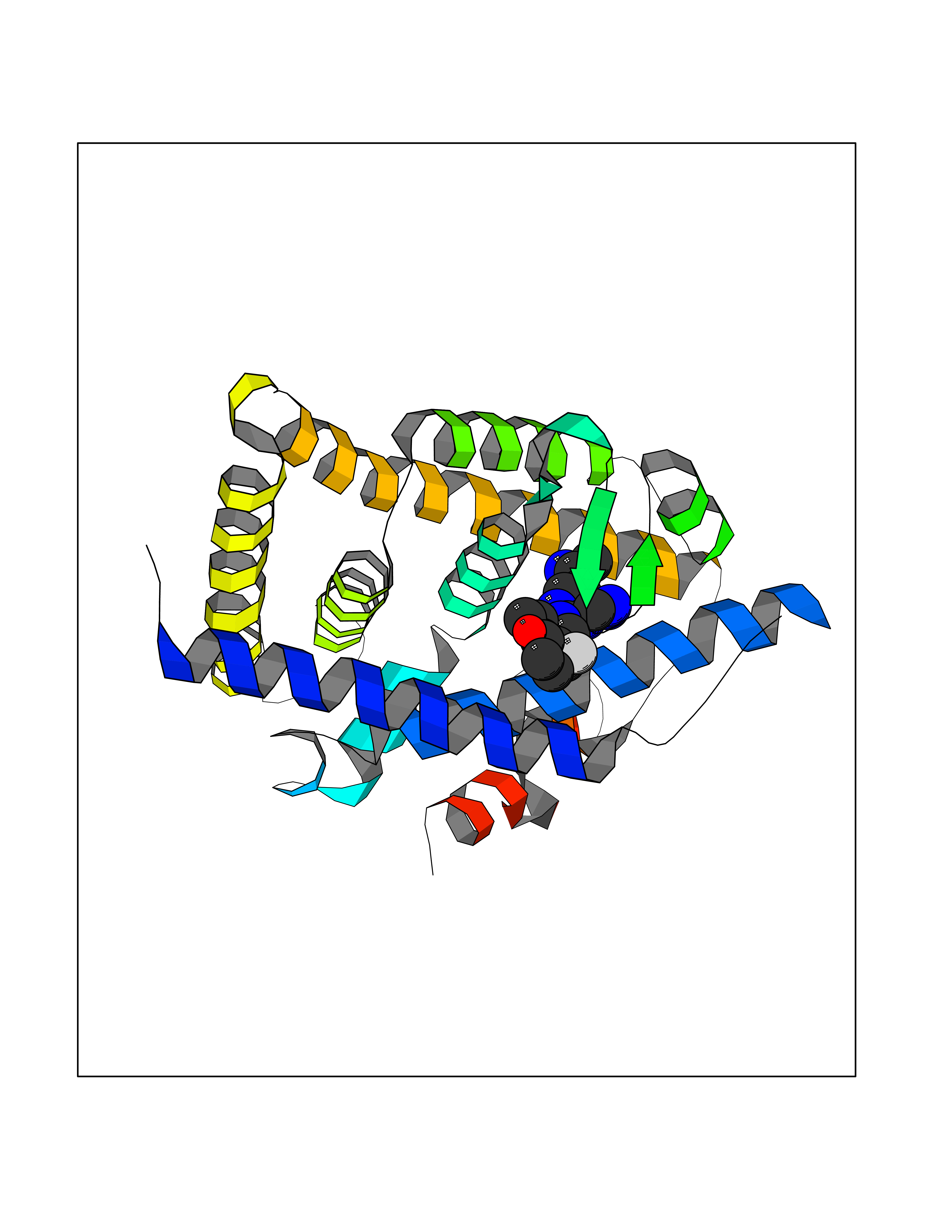 Molscript Map 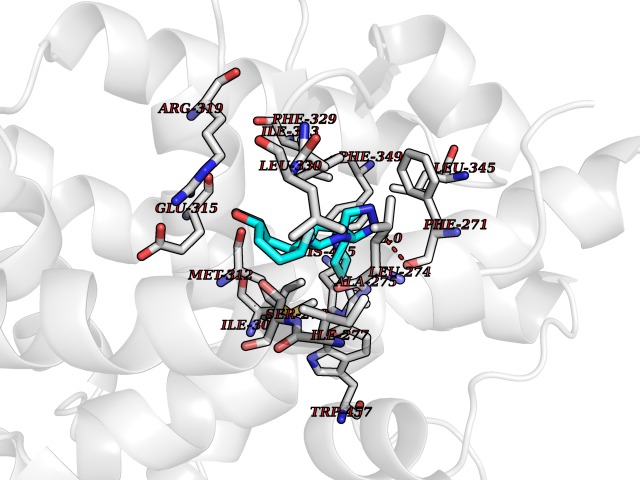 Pymol Map 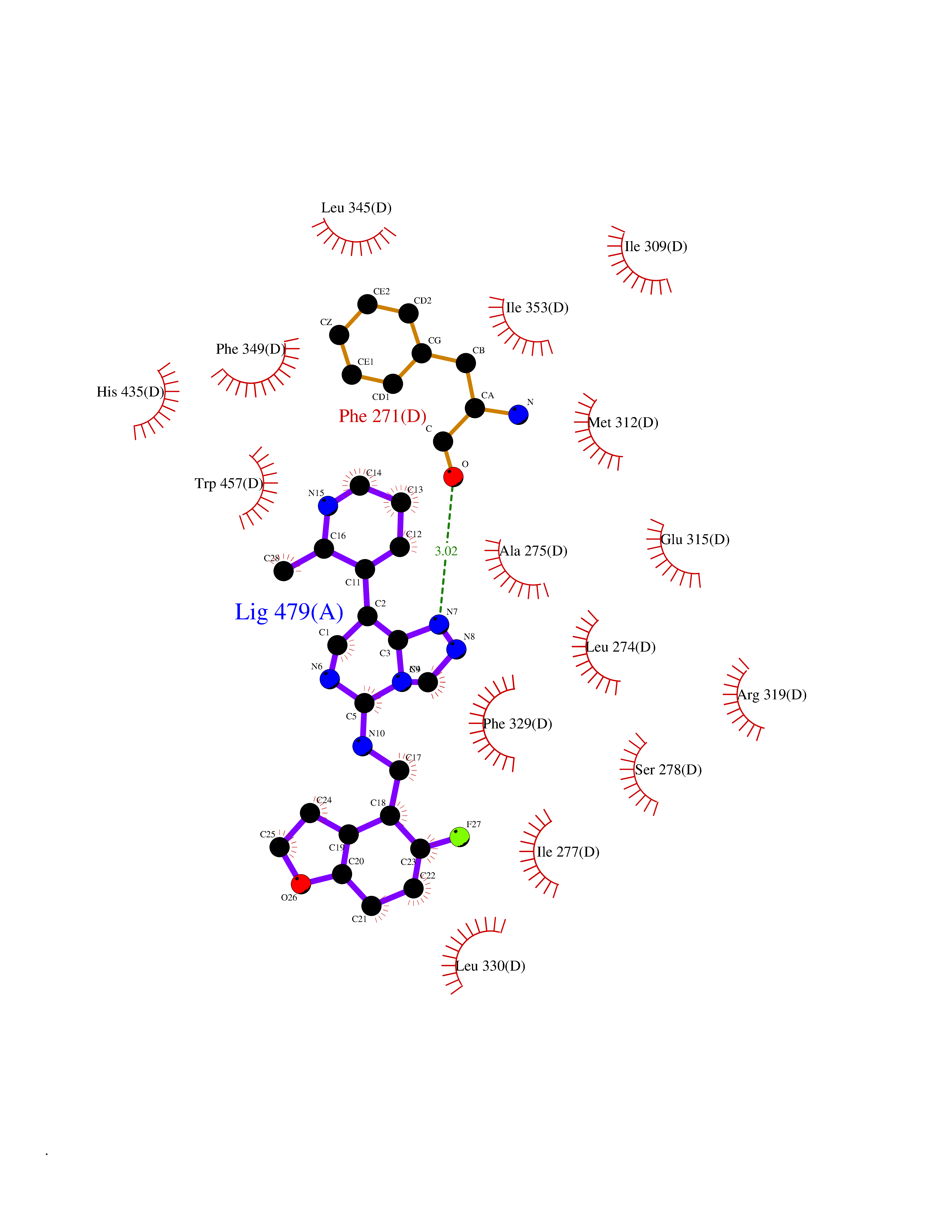 Ligplot Map 3D Binding mode Sequence QLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPSASQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDVHEGSGSGSHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | CAAX farnesyltransferase beta (FNTB) | 1SA4 | 8.17 | |
Target general information Gen name FNTB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAS proteins prenyltransferasebeta; FTase-beta; FNTB; CAAX farnesyltransferase beta subunit Protein family Protein prenyltransferase subunit beta family Biochemical class Alkyl aryl transferase Function Essential subunit of the farnesyltransferase complex. Catalyzes the transfer of a farnesyl moiety from farnesyl diphosphate to a cysteine at the fourth position from the C- terminus of several proteins having the C-terminal sequence Cys- aliphatic-aliphatic-X. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07216; DB08674; DB08676; DB06953; DB07771; DB07895; DB04893; DB07780; DB07841; DB07227; DB06448; DB04960 Interacts with P21549; Q9BWW8; Q8N5M1; Q8N4L8; G5E9A7; P49354; P31273; A5PL33-2; Q9H0W8; Q7Z699; O43711 EC number EC 2.5.1.58 Uniprot keywords 3D-structure; Alternative splicing; Lipid metabolism; Metal-binding; Phosphoprotein; Prenyltransferase; Proteomics identification; Reference proteome; Repeat; Transferase; Zinc Protein physicochemical properties Chain ID B,A Molecular weight (Da) 83751.2 Length 725 Aromaticity 0.11 Instability index 46.44 Isoelectric point 5.61 Charge (pH=7) -20.1 2D Binding mode Binding energy (Kcal/mol) -8.28 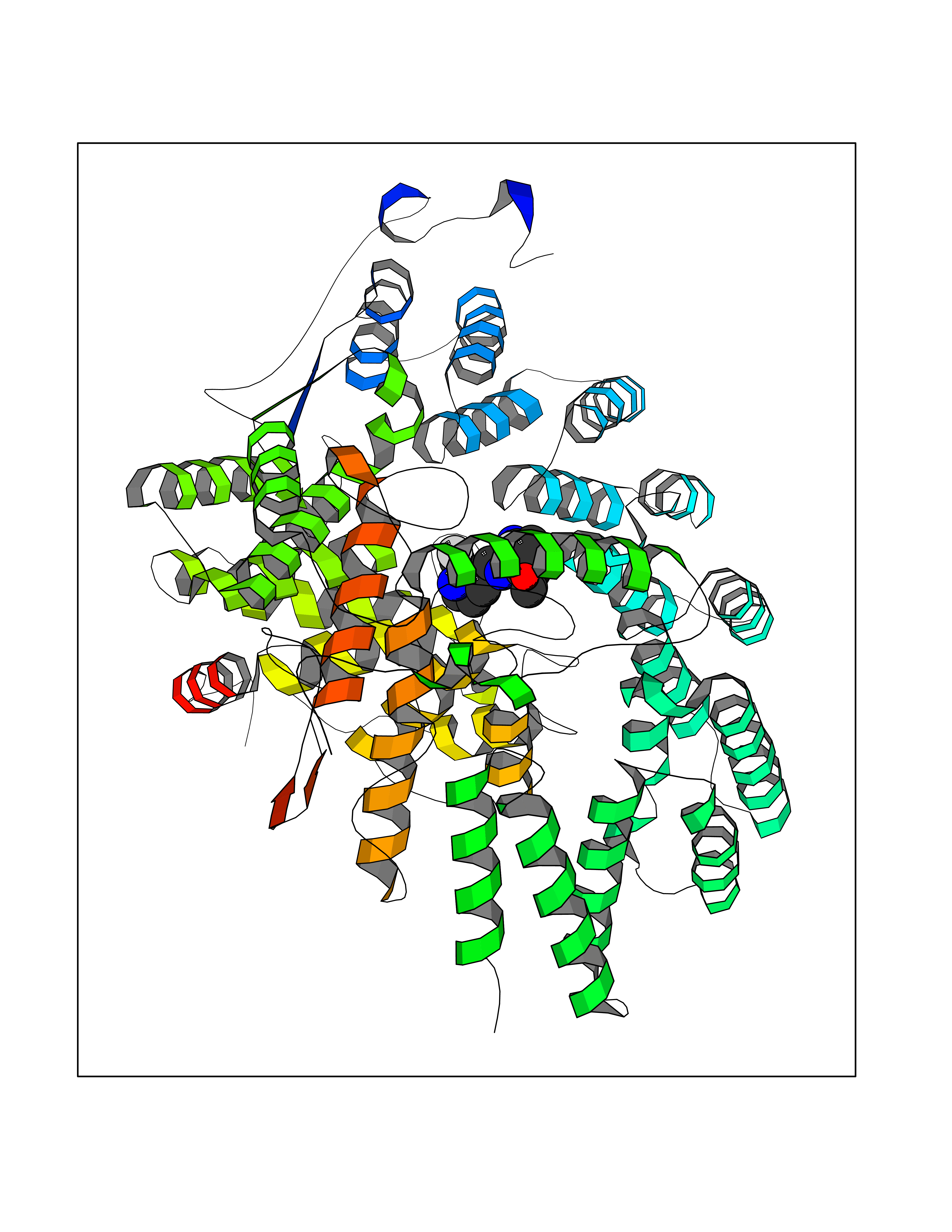 Molscript Map 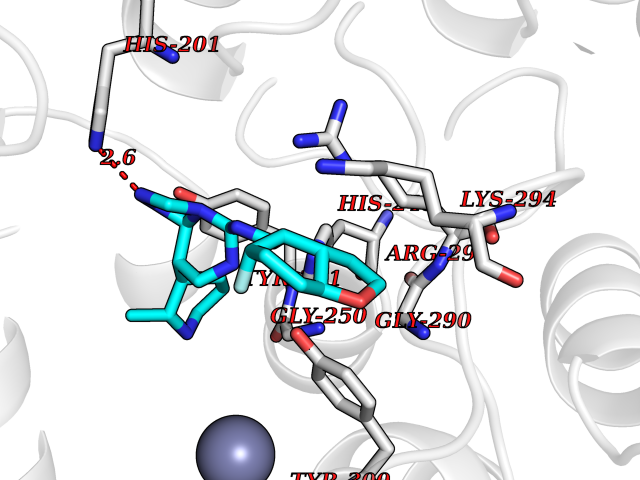 Pymol Map 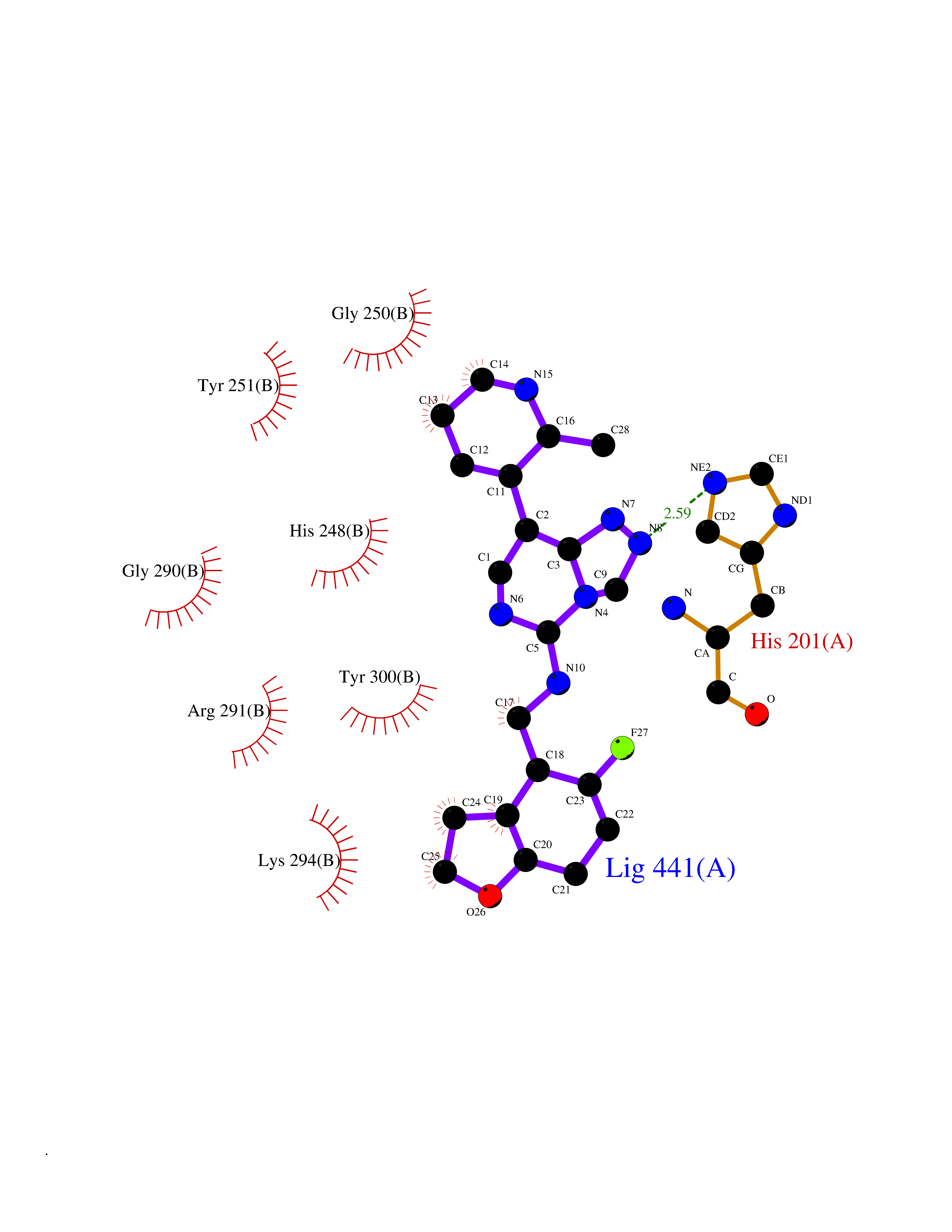 Ligplot Map 3D Binding mode Sequence FVSLDSPSYVLYRDRAEWADIDPVPQNDGPNPVVQIIYSDKFRDVYDYFRAVLQRDERSERAFKLTRDAIELNAANYTVWHFRRVLLKSLQKDLHEEMNYITAIIEEQPKNYQVWHHRRVLVEWLRDPSQELEFIADILNQDAKNYHAWQHRQWVIQEFKLWDNELQYVDQLLKEDVRNNSVWNQRYFVISNTTGYNDRAVLEREVQYTLEMIKLVPHNESAWNYLKGILQDRGLSKYPNLLNQLLDLQPSHSSPYLIAFLVDIYEDMLENQCDNKEDILNKALELCEILAKEKDTIRKEYWRYIGRSLQSKHSTSSPVWSEPLYSLRPEHARERLQDDSVETVTSIEQAKVEEKIQEVFSSYKFNHLVPRLVLQREKHFHYLKRGLRQLTDAYECLDASRPWLCYWILHSLELLDEPIPQIVATDVCQFLELCQSPEGGFGGGPGQYPHLAPTYAAVNALCIIGTEEAYDIINREKLLQYLYSLKQPDGSFLMHVGGEVDVRSAYCAASVASLTNIITPDLFEGTAEWIARCQNWEGGIGGVPGMEAHGGYTFCGLAALVILKRERSLNLKSLLQWVTSRQMRFEGGFQGRCNKLVDGCYSFWQAGLLPLLHRALHAQGDPALSMSHWMFHQQALQEYILMCCQCPAGGLLDKPGKSRDFYHTCYCLSGLSIAQHFGSGAMLHDVVLGVPENALQPTHPVYNIGPDKVIQATTYFLQKPVPGFE Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Kallikrein-7 (KLK7) | 6SHI | 8.17 | |
Target general information Gen name KLK7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hK7; Stratum corneum chymotryptic enzyme; Serine protease 6; SCCE; PRSS6; HSCCE Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function Specific for amino acid residues with aromatic side chains in the P1 position. Cleaves insulin A chain at '14-Tyr-|-Gln-15' and insulin B chain at '6-Leu-|-Cys-7', '16-Tyr-|-Leu-17', '25-Phe-|-Tyr-26' and '26-Tyr-|-Thr-27'. Could play a role in the activation of precursors to inflammatory cytokines. May catalyze the degradation of intercellular cohesive structures in the cornified layer of the skin in the continuous shedding of cells from the skin surface. Related diseases Immunodeficiency 35 (IMD35) [MIM:611521]: A primary immunodeficiency characterized by recurrent skin abscesses, pneumonia, and highly elevated serum IgE. {ECO:0000269|PubMed:17088085}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08038 Interacts with NA EC number EC 3.4.21.117 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48877.7 Length 448 Aromaticity 0.06 Instability index 27.7 Isoelectric point 9.07 Charge (pH=7) 18.76 2D Binding mode Binding energy (Kcal/mol) -8.12 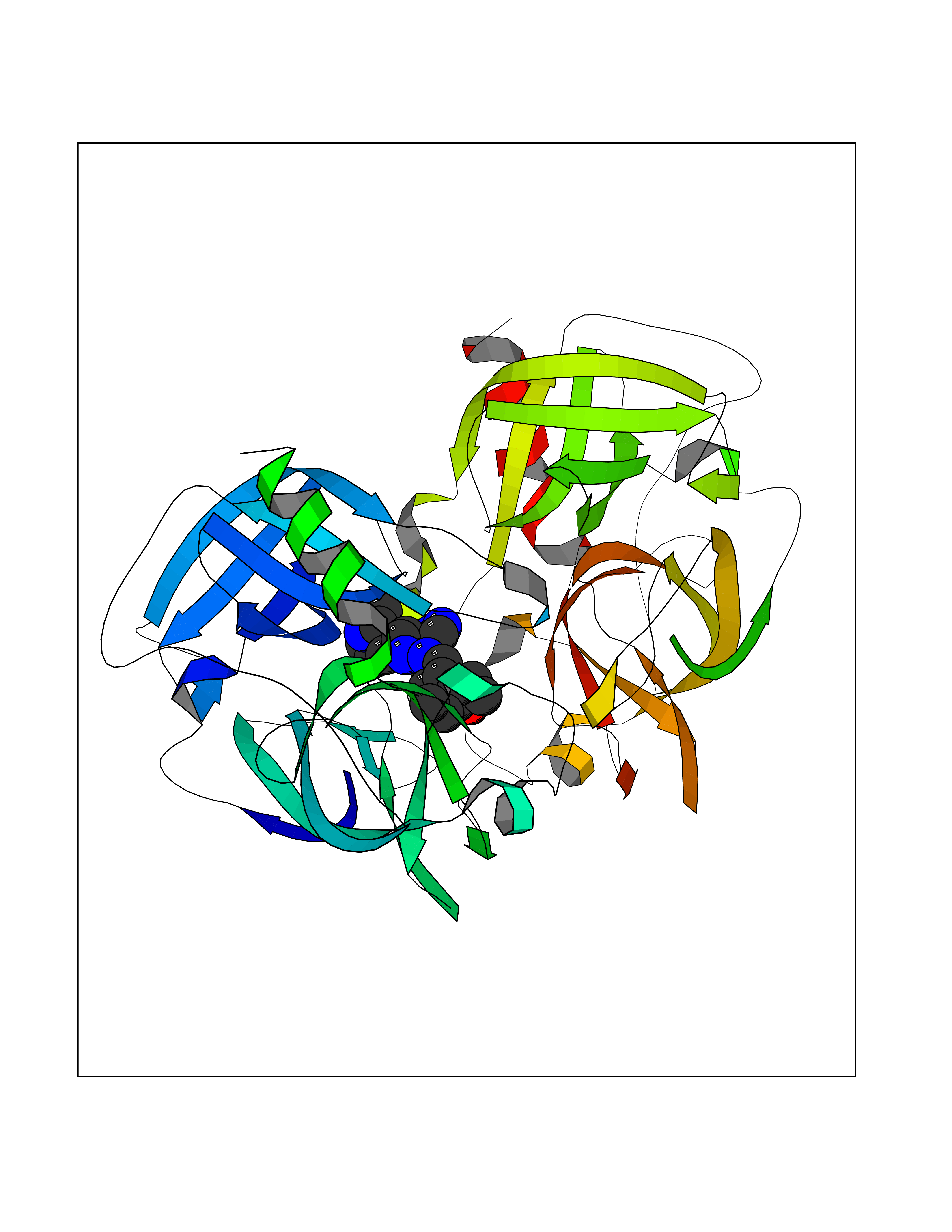 Molscript Map 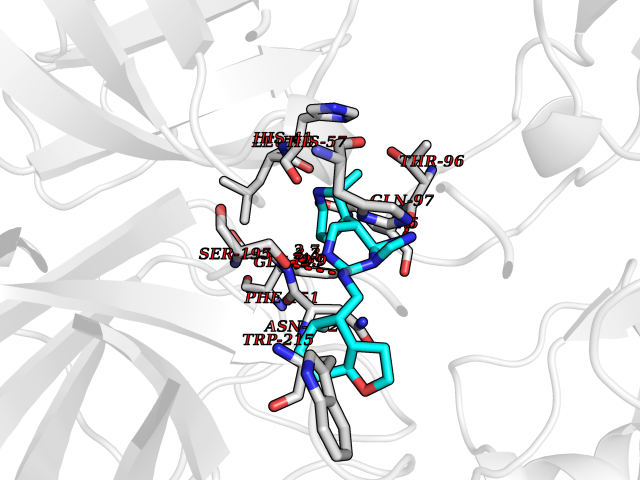 Pymol Map 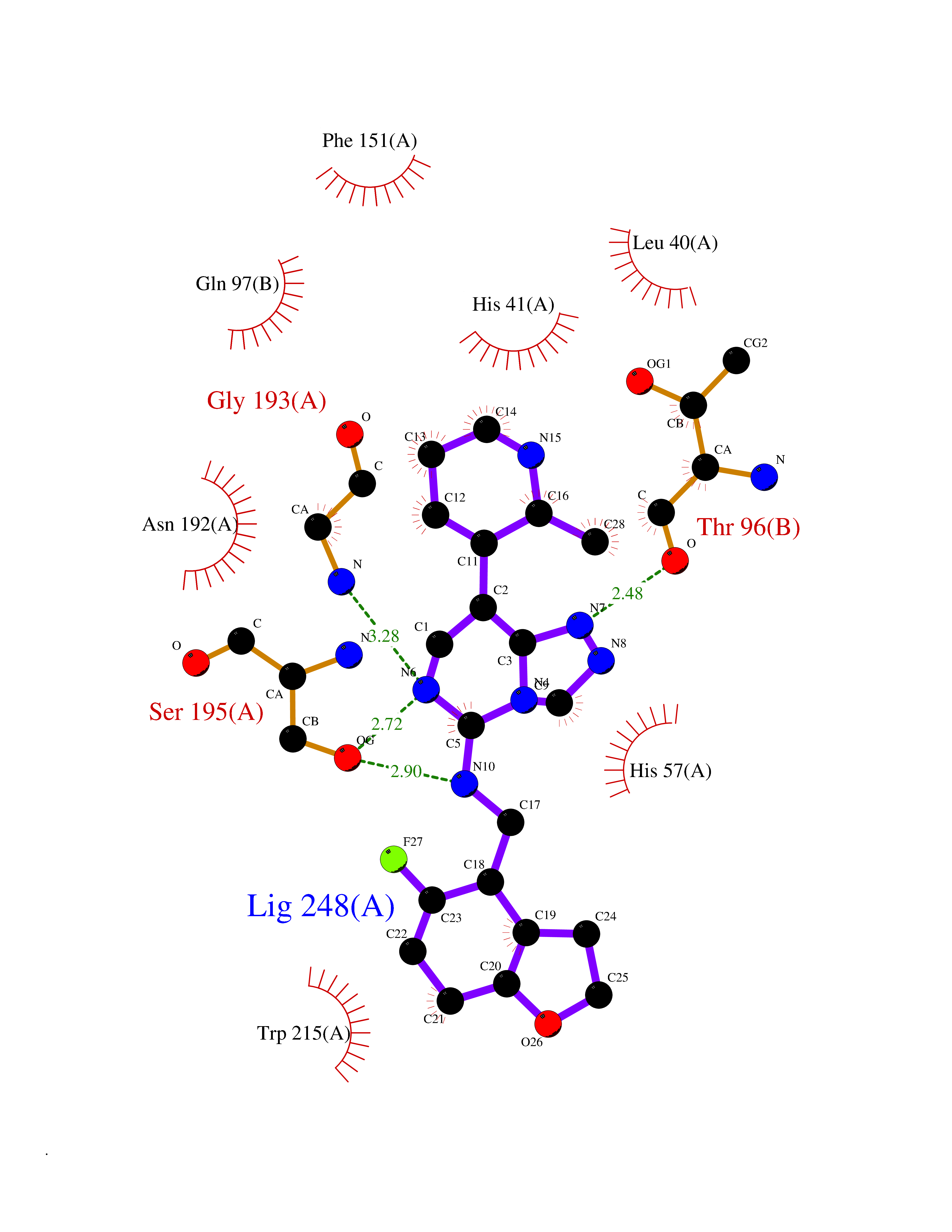 Ligplot Map 3D Binding mode Sequence IIDGAPCARGSHPWQVALLSGNQLHCGGVLVNERWVLTAAHCKMNEYTVHLGSDTLGDRRAQRIKASKSFRHPGYSTQTHVNDLMLVKLNSQARLSSMVKKVRLPSRCEPPGTTCTVSGWGTTTSPDVTFPSDLMCVDVKLISPQDCTKVYKDLLENSMLCAGIPDSKKNACNGDSGGPLVCRGTLQGLVSWGTFPCGQPNDPGVYTQVCKFTKWINDTMKKHRIIDGAPCARGSHPWQVALLSGNQLHCGGVLVNERWVLTAAHCKMNEYTVHLGSDTLGDRRAQRIKASKSFRHPGYSTQTHVNDLMLVKLNSQARLSSMVKKVRLPSRCEPPGTTCTVSGWGTTTSPDVTFPSDLMCVDVKLISPQDCTKVYKDLLENSMLCAGIPDSKKNACNGDSGGPLVCRGTLQGLVSWGTFPCGQPNDPGVYTQVCKFTKWINDTMKKHR Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Vascular endothelial growth factor receptor 2 (KDR) | 2XIR | 8.17 | |
Target general information Gen name KDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms VEGFR2; VEGFR-2; VEGF-2 receptor; Protein-tyrosine kinase receptor flk-1; Kinase insert domain receptor; Fetal liver kinase 1; FLK1; FLK-1; CD309 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Plays an essential role in the regulation of angiogenesis, vascular development, vascular permeability, and embryonic hematopoiesis. Promotes proliferation, survival, migration and differentiation of endothelial cells. Promotes reorganization of the actin cytoskeleton. Isoforms lacking a transmembrane domain, such as isoform 2 and isoform 3, may function as decoy receptors for VEGFA, VEGFC and/or VEGFD. Isoform 2 plays an important role as negative regulator of VEGFA- and VEGFC-mediated lymphangiogenesis by limiting the amount of free VEGFA and/or VEGFC and preventing their binding to FLT4. Modulates FLT1 and FLT4 signaling by forming heterodimers. Binding of vascular growth factors to isoform 1 leads to the activation of several signaling cascades. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate and the activation of protein kinase C. Mediates activation of MAPK1/ERK2, MAPK3/ERK1 and the MAP kinase signaling pathway, as well as of the AKT1 signaling pathway. Mediates phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, reorganization of the actin cytoskeleton and activation of PTK2/FAK1. Required for VEGFA-mediated induction of NOS2 and NOS3, leading to the production of the signaling molecule nitric oxide (NO) by endothelial cells. Phosphorylates PLCG1. Promotes phosphorylation of FYN, NCK1, NOS3, PIK3R1, PTK2/FAK1 and SRC. Tyrosine-protein kinase that acts as a cell-surface receptor for VEGFA, VEGFC and VEGFD. Related diseases Hemangioma, capillary infantile (HCI) [MIM:602089]: A condition characterized by dull red, firm, dome-shaped hemangiomas, sharply demarcated from surrounding skin, usually presenting at birth or occurring within the first two or three months of life. They result from highly proliferative, localized growth of capillary endothelium and generally undergo regression and involution without scarring. {ECO:0000269|PubMed:11807987, ECO:0000269|PubMed:18931684}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Plays a major role in tumor angiogenesis. In case of HIV-1 infection, the interaction with extracellular viral Tat protein seems to enhance angiogenesis in Kaposi's sarcoma lesions. Drugs (DrugBank ID) DB04727; DB07514; DB07528; DB06938; DB07326; DB06626; DB08875; DB04849; DB05198; DB12147; DB12307; DB12010; DB11679; DB06101; DB09078; DB06080; DB06595; DB07537; DB07183; DB07333; DB07334; DB07274; DB09079; DB08519; DB08042; DB16265; DB06589; DB05931; DB08901; DB15822; DB05984; DB05578; DB08896; DB14840; DB06436; DB00398; DB01268; DB05075; DB11800; DB04879; DB05146; DB05014 Interacts with P35916; O60565; P98160; PRO_0000391621 [P98160]; PRO_0000391622 [P98160]; P17301; P35968; P09382; P08581; P16333; O14786; O75340; P09619; P29350; Q12913; P12931; P15692; P15692-4; P49767; Q9MYV3-3 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell junction; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Developmental protein; Differentiation; Disulfide bond; Endoplasmic reticulum; Endosome; Glycoprotein; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33863.9 Length 296 Aromaticity 0.1 Instability index 40.01 Isoelectric point 8.5 Charge (pH=7) 4.59 2D Binding mode Binding energy (Kcal/mol) -9.34  Molscript Map 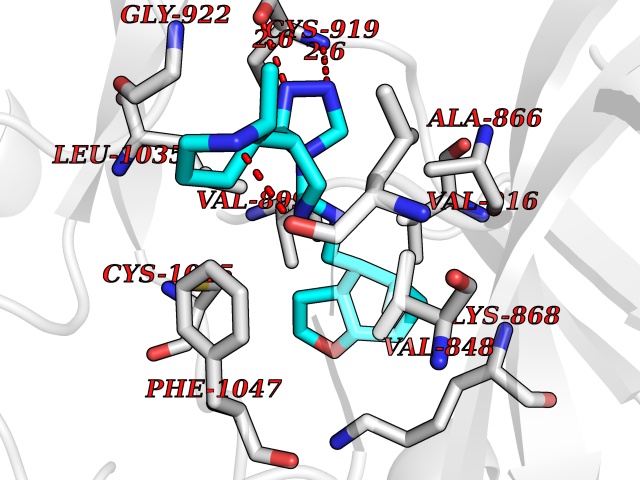 Pymol Map 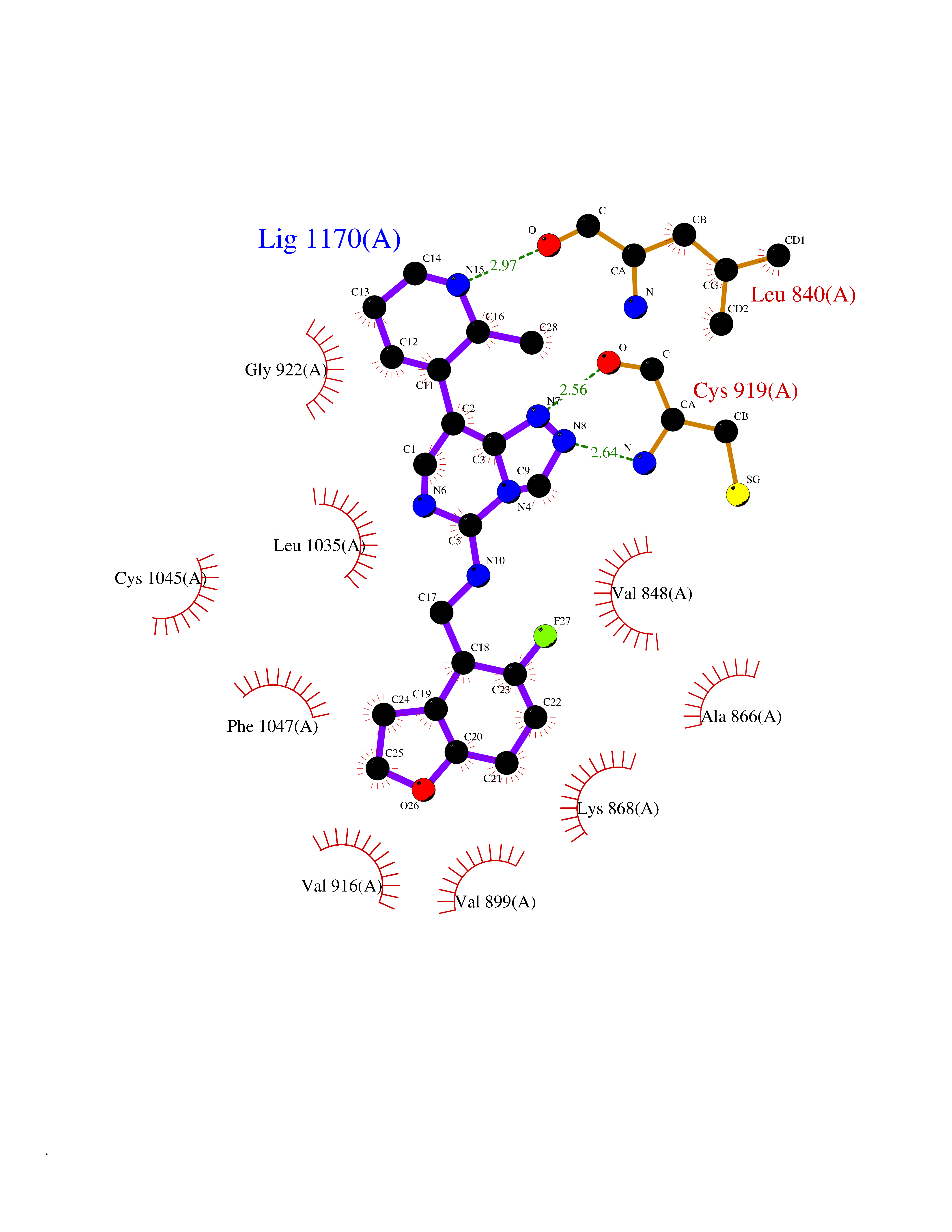 Ligplot Map 3D Binding mode Sequence HCERLPYDASKWEFPRDRLKLGKPLGRGAFGQVIEADAFGIDKTATCRTVAVKMLKEGATHSEHRALMSELKILIHIGHHLNVVNLLGACTKPGGPLMVIVEFCKFGNLSTYLRSKRNEFVPYYKDFLTLEHLICYSFQVAKGMEFLASRKCIHRDLAARNILLSEKNVVKICDFGLARDIYKDPDYVRKGDARLPLKWMAPETIFDRVYTIQSDVWSFGVLLWEIFSLGASPYPGVKIDEEFCRRLKEGTRMRAPDYTTPEMYQTMLDCWHGEPSQRPTFSELVEHLGNLLQANA Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Dopamine beta-hydroxylase | 4ZEL | 8.16 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -8.44 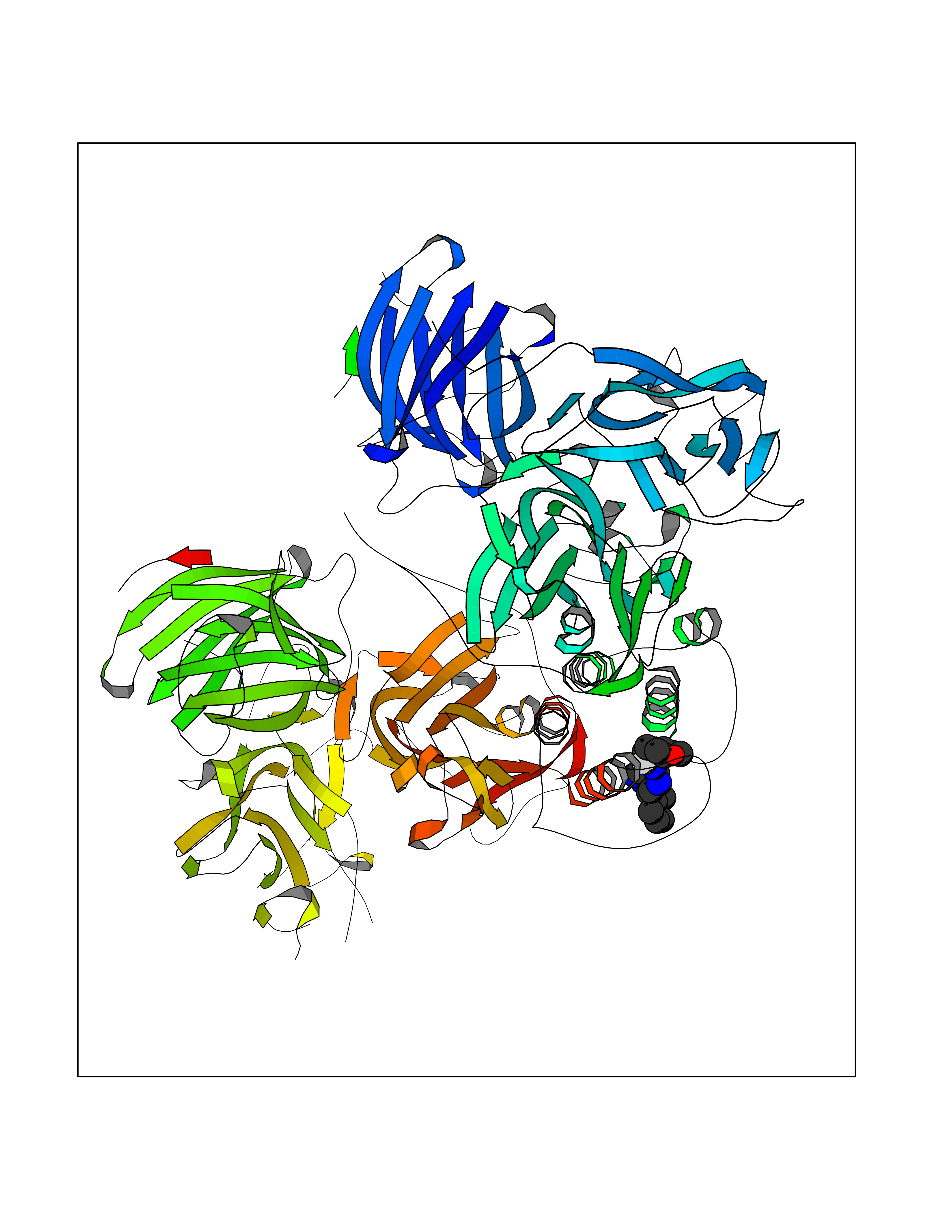 Molscript Map 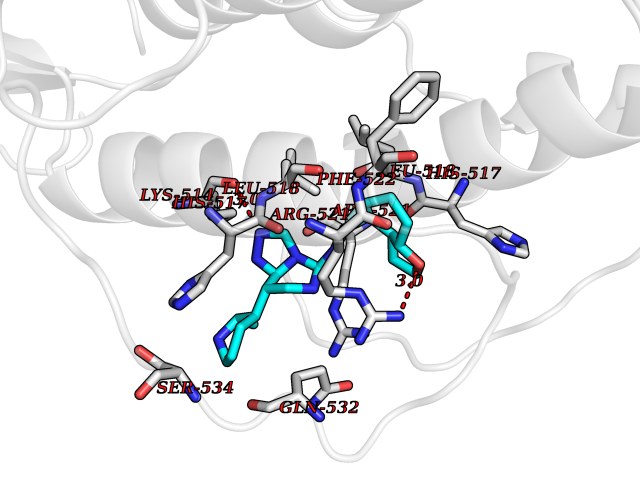 Pymol Map 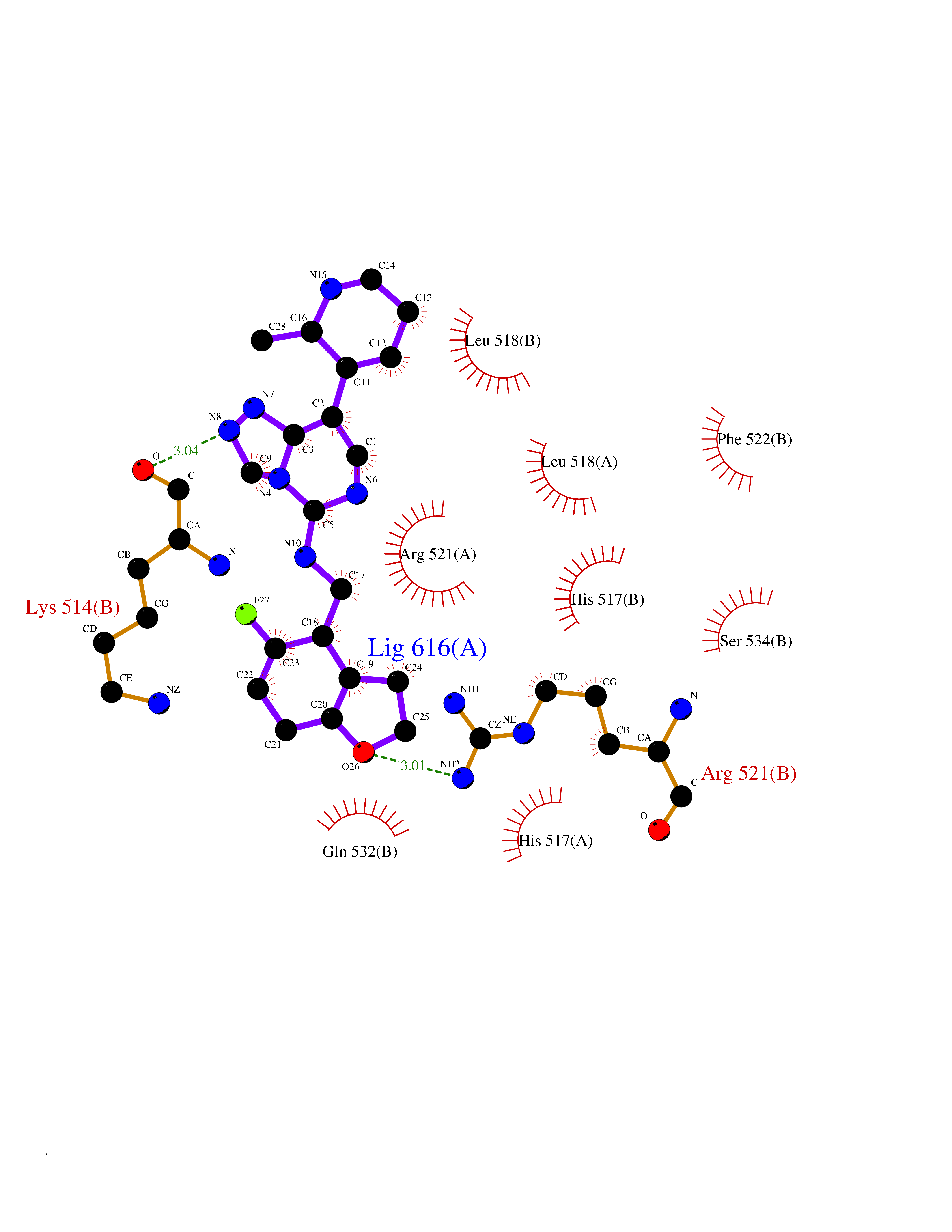 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Folate receptor beta (FOLR2) | 4KN0 | 8.16 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -9.17 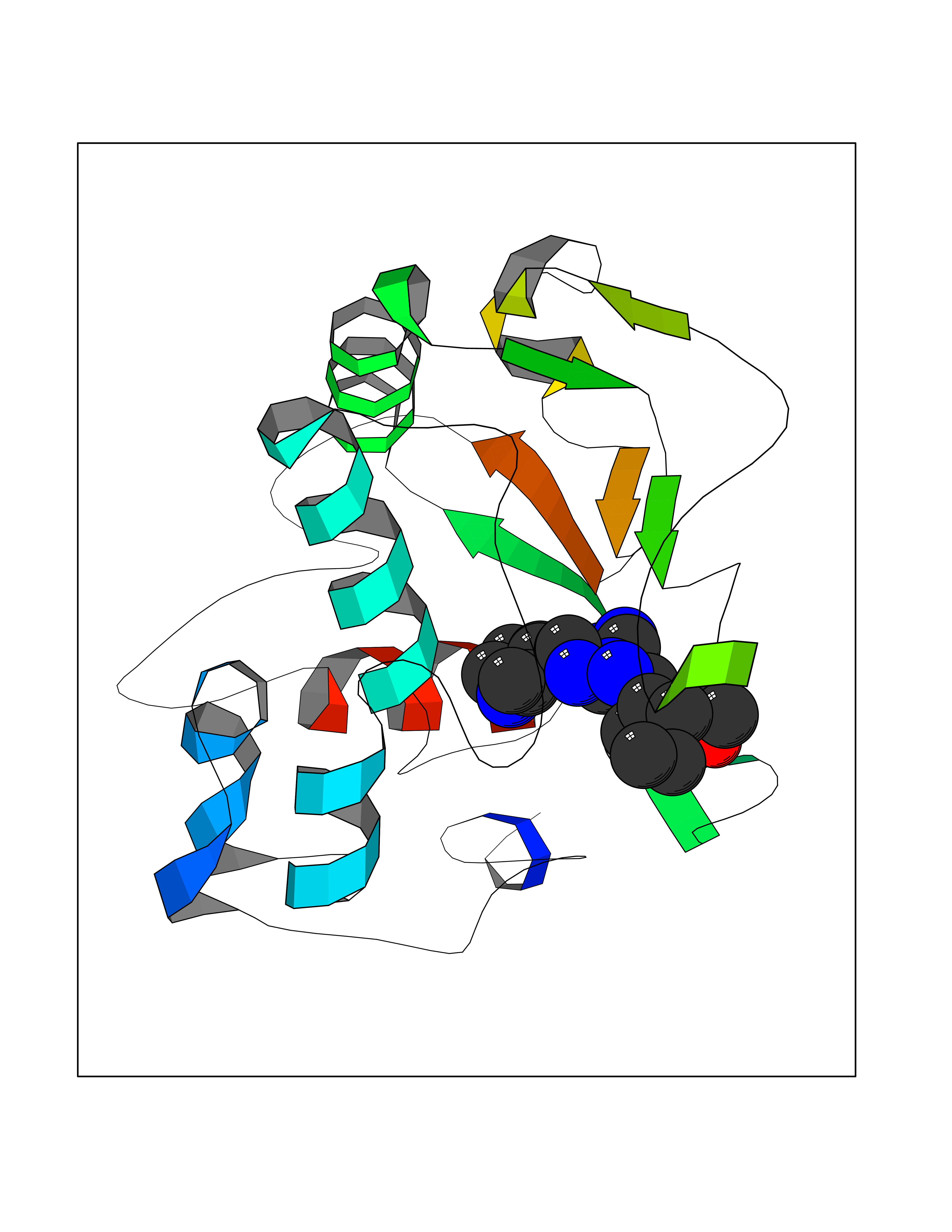 Molscript Map 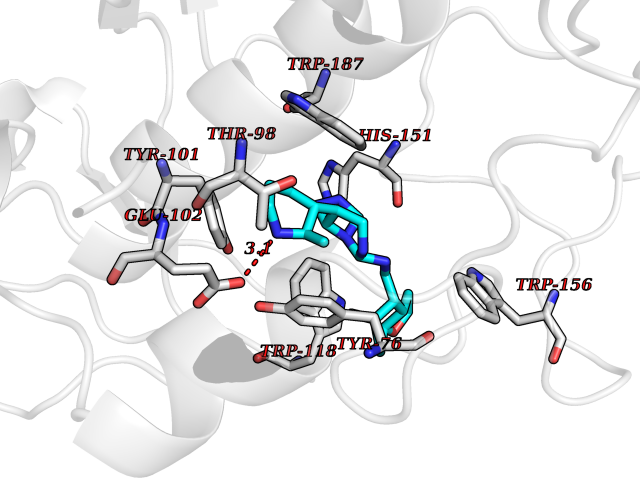 Pymol Map 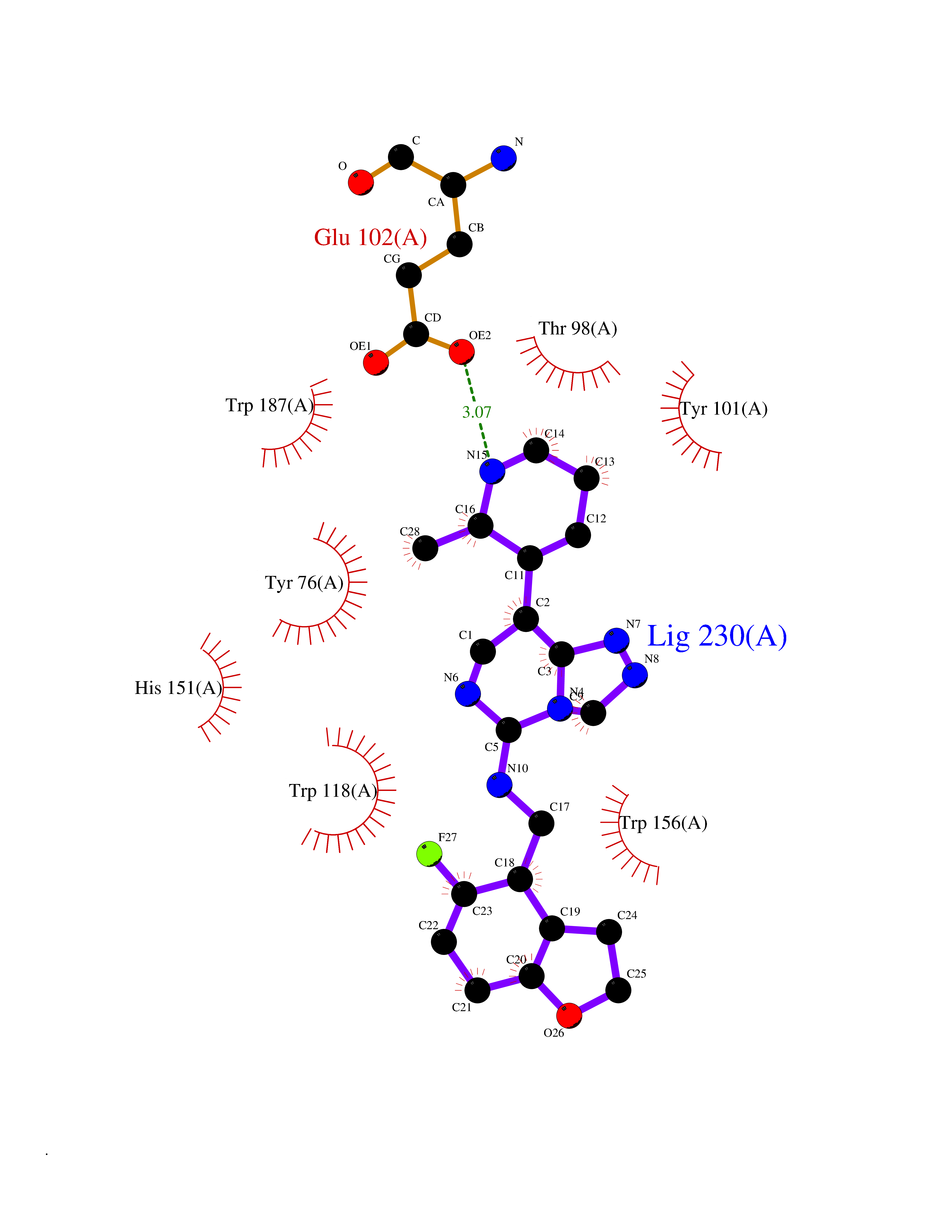 Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Steroid 5-alpha-reductase 2 (SRD5A2) | 7BW1 | 8.14 | |
Target general information Gen name SRD5A2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Type 2 steroid 5alpha-reductase (5alpha-R); SRD5A2; SR type 2; 5-alpha reductase 2; 5 alpha-SR2 Protein family Steroid 5-alpha reductase family Biochemical class CH-CH donor oxidoreductase Function Converts testosterone (T) into 5-alpha- dihydrotestosterone (DHT) and progesterone or corticosterone into their corresponding 5-alpha-3-oxosteroids. It plays a central role in sexual differentiation and androgen physiology. Related diseases Pseudovaginal perineoscrotal hypospadias (PPSH) [MIM:264600]: A form of male pseudohermaphroditism in which 46,XY males show ambiguous genitalia at birth, including perineal hypospadias and a blind perineal pouch, and develop masculinization at puberty. The name of the disorder stems from the finding of a blind-ending perineal opening resembling a vagina and a severely hypospadiac penis with the urethra opening onto the perineum. {ECO:0000269|PubMed:10718838, ECO:0000269|PubMed:10898110, ECO:0000269|PubMed:10999800, ECO:0000269|PubMed:12843198, ECO:0000269|PubMed:15064320, ECO:0000269|PubMed:1522235, ECO:0000269|PubMed:15528927, ECO:0000269|PubMed:15770495, ECO:0000269|PubMed:16098368, ECO:0000269|PubMed:16181229, ECO:0000269|PubMed:7554313, ECO:0000269|PubMed:8626825, ECO:0000269|PubMed:8768837, ECO:0000269|PubMed:9208814, ECO:0000269|PubMed:9745434, ECO:0000269|PubMed:9843052}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00548; DB01126; DB01216; DB00741; DB00717; DB13943; DB13944; DB01420; DB13946 Interacts with O96005; A8MQ03; Q14749 EC number EC 1.3.1.22 Uniprot keywords 3D-structure; Differentiation; Disease variant; Endoplasmic reticulum; Lipid metabolism; Membrane; Microsome; NADP; Oxidoreductase; Proteomics identification; Pseudohermaphroditism; Reference proteome; Sexual differentiation; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 27409.8 Length 245 Aromaticity 0.17 Instability index 45.26 Isoelectric point 9.4 Charge (pH=7) 9.32 2D Binding mode Binding energy (Kcal/mol) -9.16 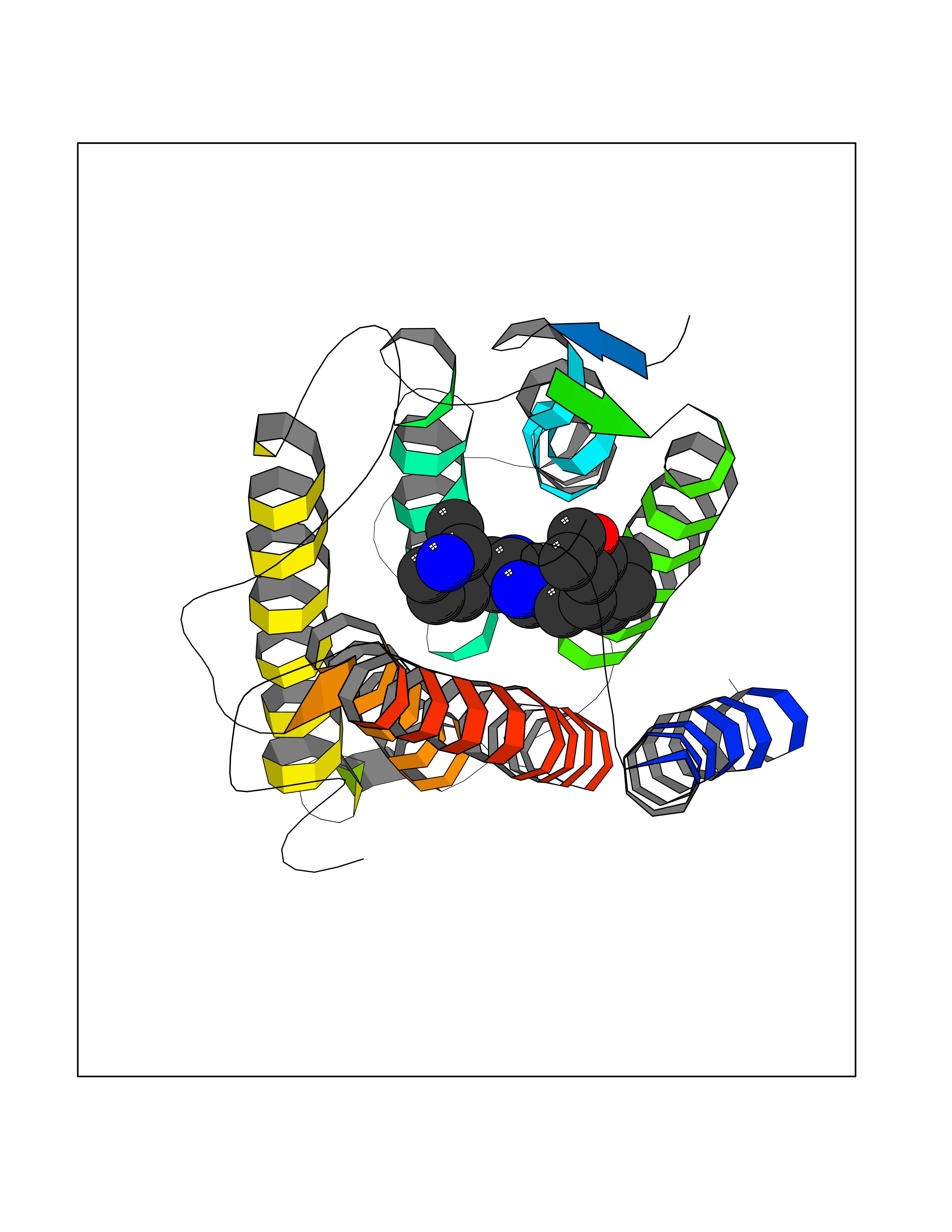 Molscript Map  Pymol Map 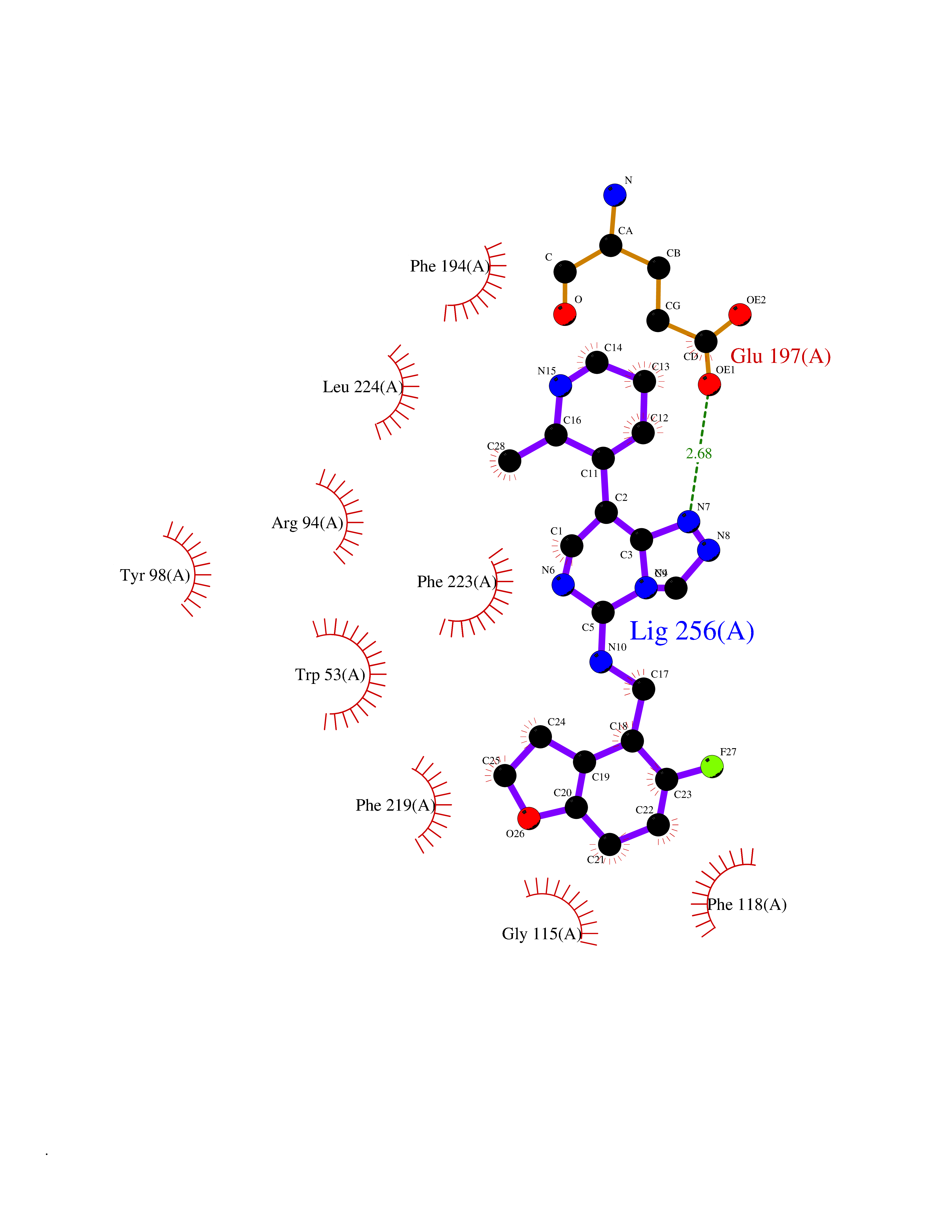 Ligplot Map 3D Binding mode Sequence CQQSPVLAGSATLVALGALALYVAKPSGYGKHTEATRLPARAAWFLQELPSFAVPAGILARQPLSLFGPPGTVLLGLFCVHYFHRTFVYSLLNRGRPYPAILILRGTAFCTGNGVLQGYYLIYCAEYPDGWYTDIRFSLGVFLFILGMGINIHSDYILRQLRKPGEISYRIPQGGLFTYVSGANFLGEIIEWIGYALATWSLPALAFAFFSLCFLGLRAFHHHRFYLKMFEDYPKSRKALIPFIF Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Phosphodiesterase 4A (PDE4A) | 2QYK | 8.13 | |
Target general information Gen name PDE4A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4A; Type 4A cAMP phosphodiesterase; PDE46; DPDE2 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB08299; DB01427; DB00201; DB05219; DB00975; DB06751; DB00651; DB00824; DB05266; DB01088; DB01303; DB01791; DB06479; DB01656; DB01954; DB00277; DB08811; DB09283 Interacts with P55212; O14569; P13473-2; Q9UJX0; P16118; O75400-2; Q9Y371 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cell projection; Cytoplasm; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 38579.6 Length 335 Aromaticity 0.08 Instability index 37.43 Isoelectric point 5.01 Charge (pH=7) -19.92 2D Binding mode Binding energy (Kcal/mol) -8.96 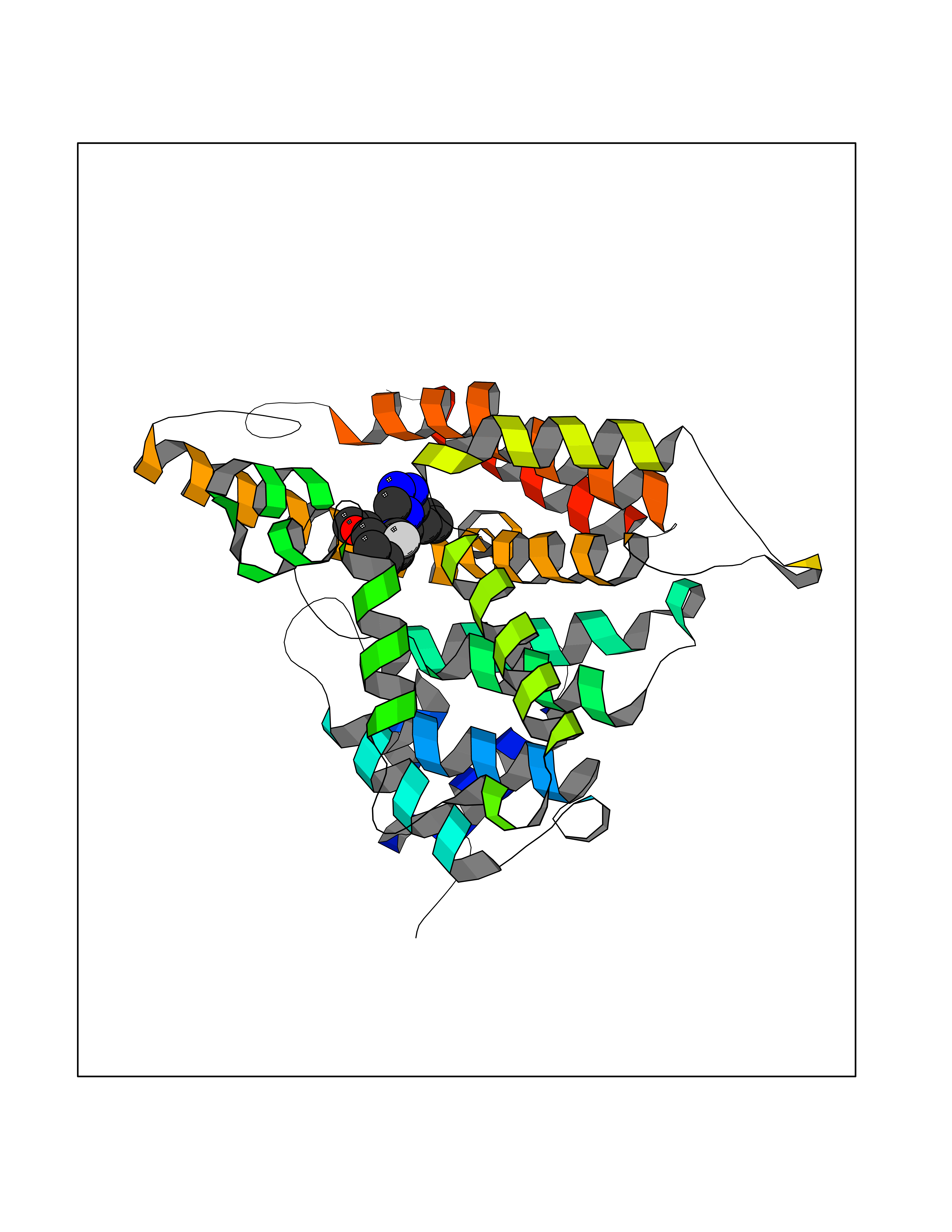 Molscript Map 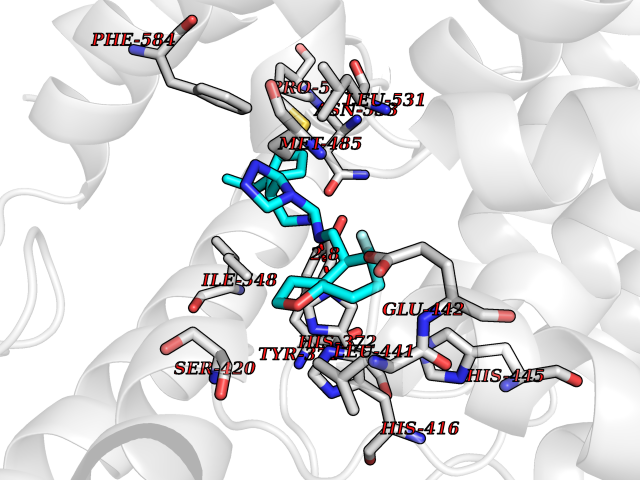 Pymol Map 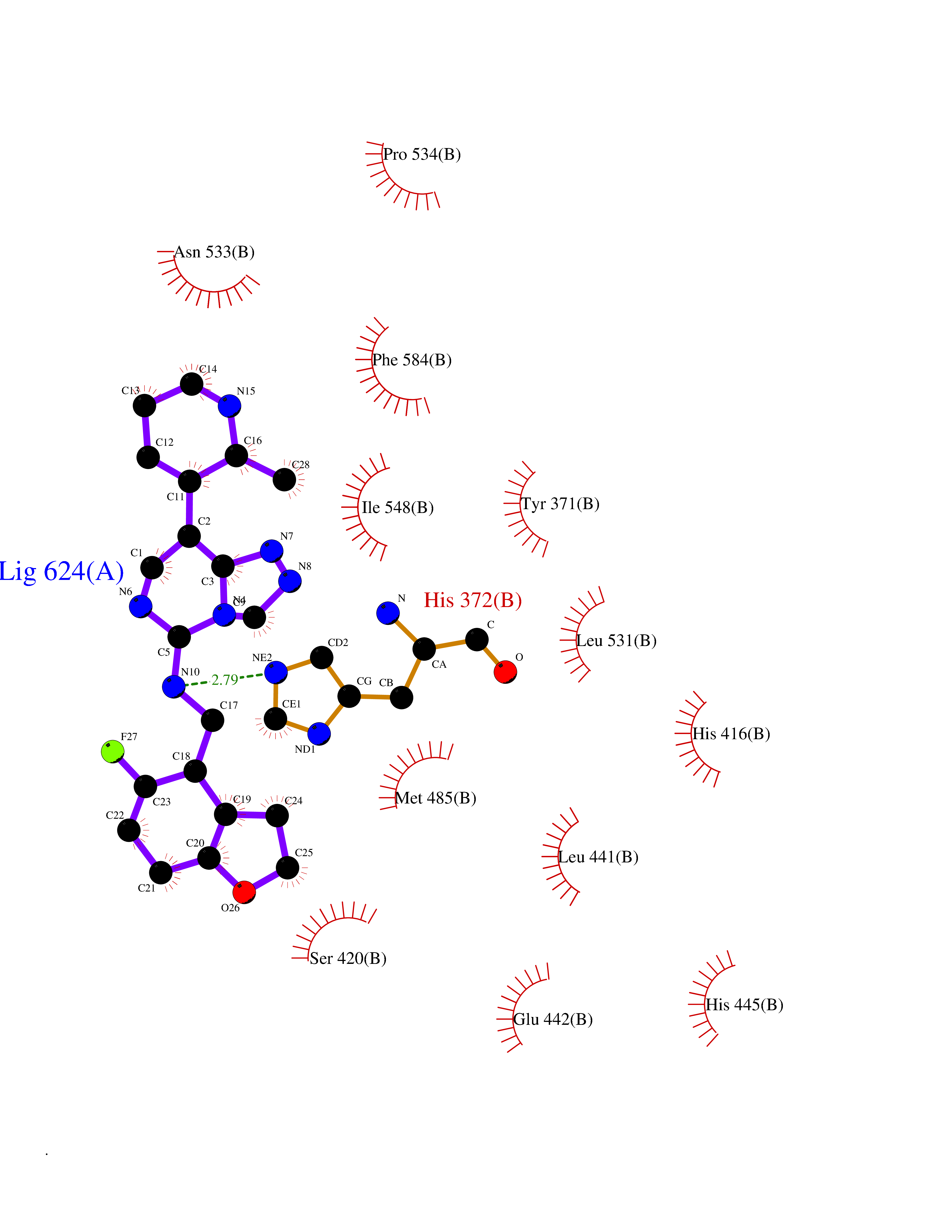 Ligplot Map 3D Binding mode Sequence HMNIPRFGVKTDQEELLAQELENLNKWGLNIFCVSDYAGGRSLTCIMYMIFQERDLLKKFRIPVDTMVTYMLTLEDHYHADVAYHNSLHAADVLQSTHVLLATPALDAVFTDLEILAALFAAAIHDVDHPGVSNQFLINTNSELALMYNDESVLENHHLAVGFKLLQEDNCDIFQNLSKRQRQSLRKMVIDMVLATDMSKHMTLLADLKTMVETKKVTSSGVLLLDNYSDRIQVLRNMVHCADLSNPTKPLELYRQWTDRIMAEFFQQGDRERERGMEISPMCDKHTASVEKSQVGFIDYIVHPLWETWADLVHPDAQEILDTLEDNRDWYYSAI Hydrogen bonds contact Hydrophobic contact | ||||