Job Results:
Ligand
Structure
Job ID
e848ab91d8ae28235d6e66c6c8c71011
Job name
NA
Time
2024-09-30 12:19:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit gamma | 3JWR | 6.78 | |
Target general information Gen name PDE6G Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PDEG Protein family Rod/cone cGMP-PDE gamma subunit family Biochemical class Hydrolase Function 3',5'-cyclic-GMP phosphodiesterase activity.CGMP binding.Enzyme inhibitor activity.Spectrin binding. Related diseases Retinitis pigmentosa 57 (RP57) [MIM:613582]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:20655036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB00203; DB00820; DB00862 Interacts with O14503; Q96JM7; A0A6Q8PF08; O43741; Q8R511; P62994; Q9QY17; Q63787 EC number 3.1.4.35 Uniprot keywords 3D-structure; Acetylation; cGMP; Hydrolase; Reference proteome; Retinitis pigmentosa; Sensory transduction; Vision Protein physicochemical properties Chain ID C,D Molecular weight (Da) 40027.7 Length 345 Aromaticity 0.09 Instability index 37.44 Isoelectric point 6.02 Charge (pH=7) -6.88 2D Binding mode Binding energy (Kcal/mol) -7.38  Molscript Map 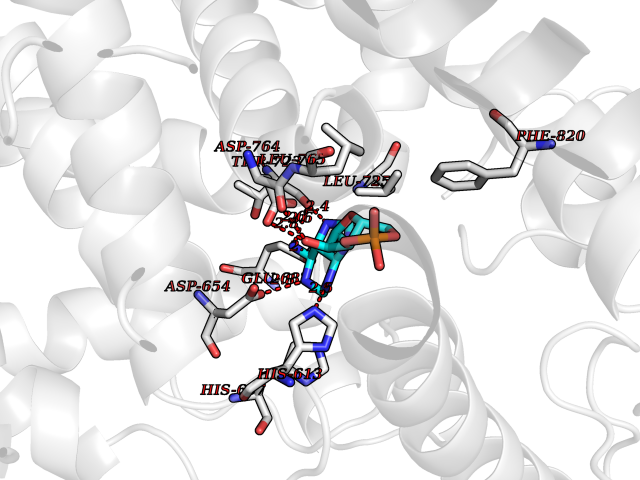 Pymol Map 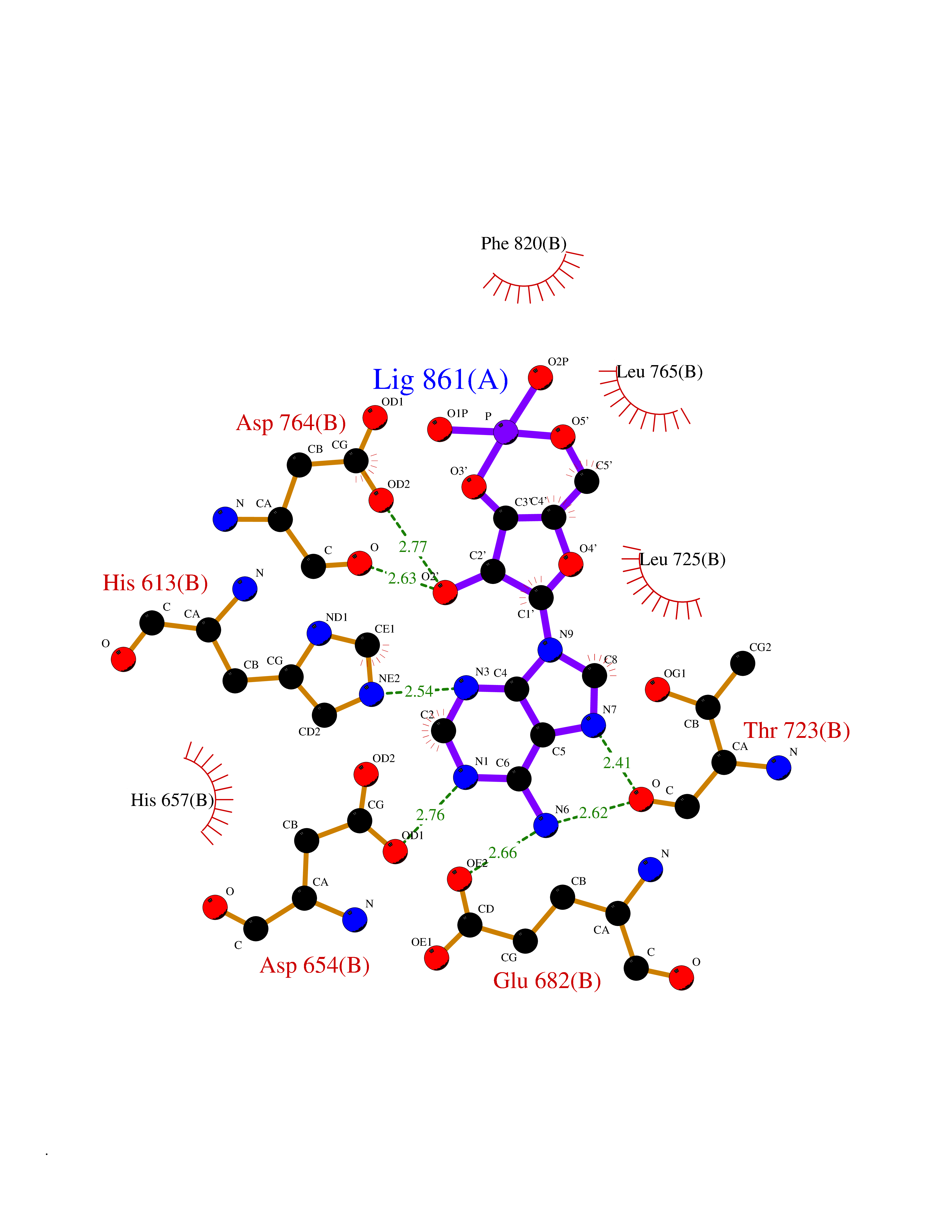 Ligplot Map 3D Binding mode Sequence EAFNHLELHELAQYGIISHMEETRELQSLAAAVVPSAQTLKITDFSFSDFELSDLETALCTIRMFTDLNLVQNFQMKHEVLCRWILSVKKNYRKNVAYHNWRHAFNTAQCMFAALKAGKIQNKLTDLEILALLIAALSHDLDHRGVNNSYIQRSEHPLAQLYCHSIMEHHHFDQCLMILNSPGNQILSGLSIEEYKTTLKIIKQAILATDLALYIKRRGEFFELIRKNQFNLEDPHQKELFLAMLMTACDLSAITKPWPIQQRIAELVATEFWEQGDLERTVLQQQPIPMMDRNKRDELPKLQVGFIDFVCTQLYEALTHVSEDCFPLLDGCRKNRQKWQALAEQ Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Carbonic anhydrase IX (CA-IX) | 5FL4 | 6.77 | |
Target general information Gen name CA9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Renal cell carcinoma-associated antigen G250; RCC-associated antigen G250; PMW1; P54/58N; Membrane antigen MN; MN; G250 antigen (MN/CA IX/G250); G250; Carbonic anhydrase 9; Carbonate dehydratase IX; C Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Participates in pH regulation. May be involved in the control of cell proliferation and transformation. Appears to be a novel specific biomarker for a cervical neoplasia. Reversible hydration of carbon dioxide. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00562; DB00606; DB12741; DB08846; DB05304; DB00774; DB09460; DB00909 Interacts with P21291; O76003 EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Cell membrane; Cell projection; Direct protein sequencing; Disulfide bond; Glycoprotein; Lyase; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27522.8 Length 251 Aromaticity 0.08 Instability index 48.97 Isoelectric point 5.48 Charge (pH=7) -7.5 2D Binding mode Binding energy (Kcal/mol) -7.29 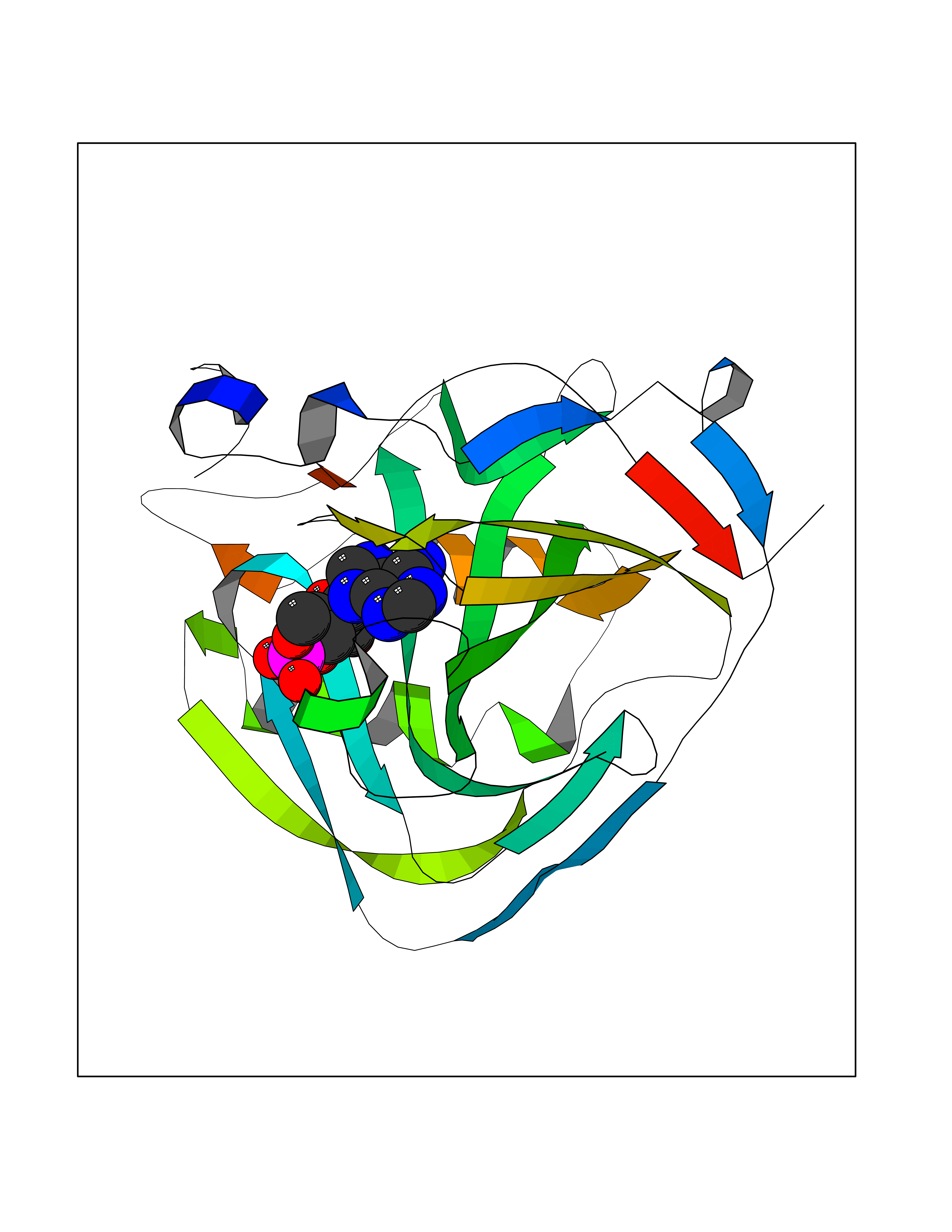 Molscript Map 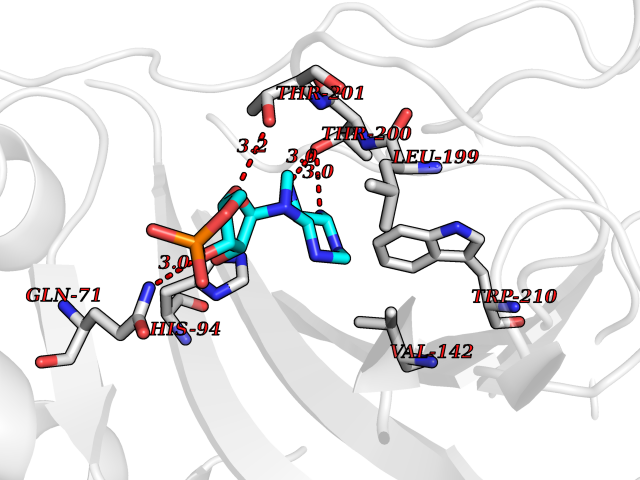 Pymol Map 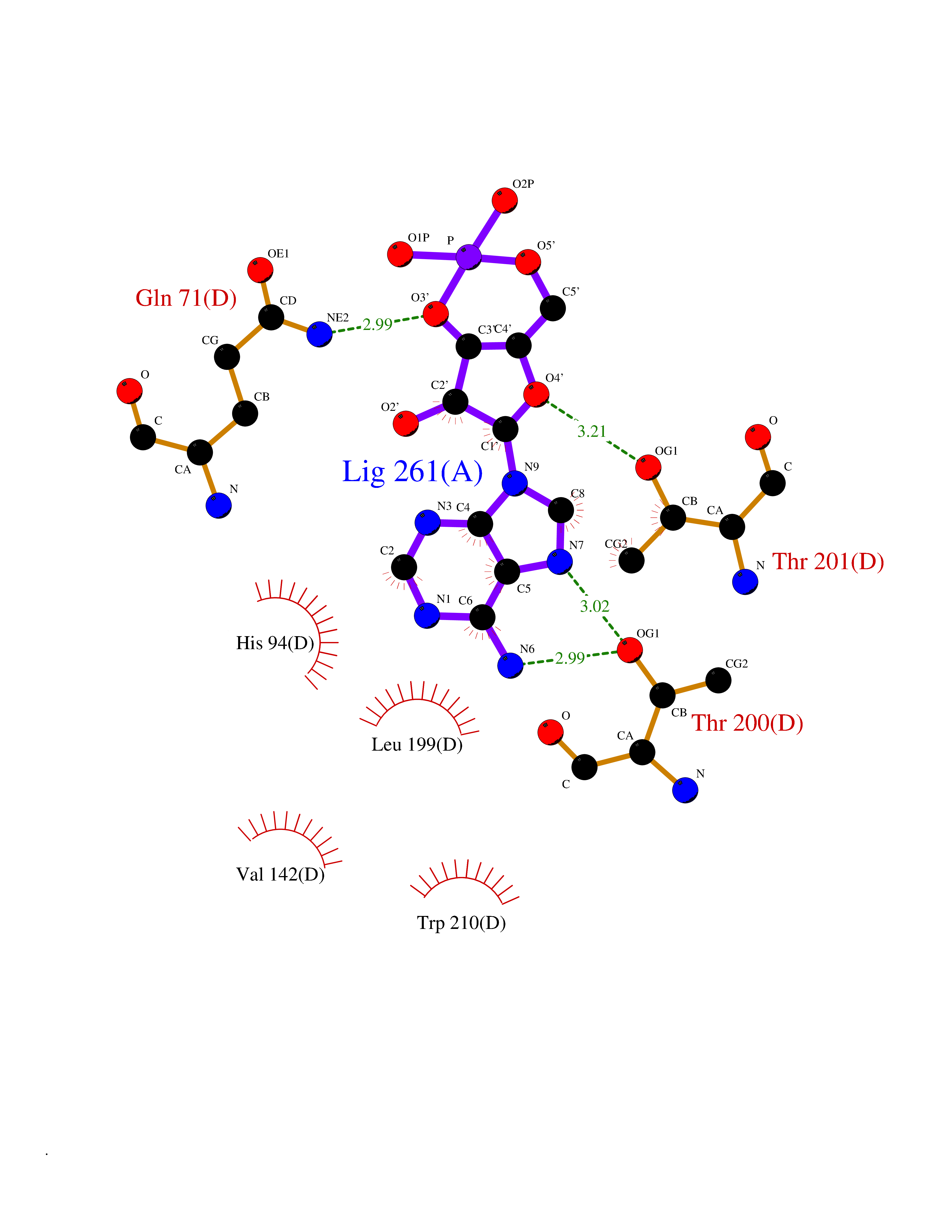 Ligplot Map 3D Binding mode Sequence WRYGGDPPWPRVSPACAGRFQSPVDIRPQLAAFSPALRPLELLGFQLPPLPELRLRNNGHSVQLTLPPGLEMALGPGREYRALQLHLHWGAAGRPGSEHTVEGHRFPAEIHVVHLSTAFARVDEALGRPGGLAVLAAFLEEGPEENSAYEQLLSRLEEIAEEGSETQVPGLDISALLPSDFSRYFQYEGSLTTPPCAQGVIWTVFNQTVMLSAKQLHTLSDTLWGPGDSRLQLNFRATQPLNGRVIEASFP Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Mucin-1 (MUC1) | 6KX1 | 6.77 | |
Target general information Gen name MUC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumour-associated antigen mucin 1; Tumor-associated mucin; Tumor-associated epithelial membraneantigen; Tumor-associated epithelial membrane antigen; Polymorphic epithelial mucin; Peanut-reactive urin Protein family NA Biochemical class NA Function Can act both as an adhesion and an anti-adhesion protein. May provide a protective layer on epithelial cells against bacterial and enzyme attack. The alpha subunit has cell adhesive properties. Related diseases MUC1/CA 15-3 is used as a serological clinical marker of breast cancer to monitor response to breast cancer treatment and disease recurrence (PubMed:20816948). Decreased levels over time may be indicative of a positive response to treatment. Conversely, increased levels may indicate disease progression. At an early stage disease, only 21% of patients exhibit high MUC1/CA 15-3 levels, that is why CA 15-3 is not a useful screening test. Most antibodies target the highly immunodominant core peptide domain of 20 amino acid (APDTRPAPGSTAPPAHGVTS) tandem repeats. Some antibodies recognize glycosylated epitopes. {ECO:0000269|PubMed:20816948}.; DISEASE: Tubulointerstitial kidney disease, autosomal dominant, 2 (ADTKD2) [MIM:174000]: A form of autosomal dominant tubulointerstitial kidney disease, a genetically heterogeneous disorder characterized by slowly progressive loss of kidney function, bland urinary sediment, hyperuricemia, absent or mildly increased albuminuria, lack of severe hypertension during the early stages, and normal or small kidneys on ultrasound. Renal histology shows variable abnormalities including interstitial fibrosis with tubular atrophy, microcystic dilatation of the tubules, thickening of tubular basement membranes, medullary cysts, and secondary glomerulosclerotic or glomerulocystic changes with abnormal glomerular tufting. There is significant variability, as well as incomplete penetrance. {ECO:0000269|PubMed:23396133}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11090; DB06584 Interacts with P00519; P00533; P08581; P15941-7; Q08AM2; O60242; Q15848; Q86W74-2; P02652; P05067-2; P29972; P41181; Q92482; Q9H2C2; Q92843; Q6PL45-2; Q8WVV5; P06681; O14523; Q06432; Q9P0B6; Q08722-3; P19397; P34810; Q8N6F1-2; P56747; Q8NHS1; Q96FZ5; Q4VAQ0; Q8N6G5; Q07325; O43169; P78329; P56851; Q9BV81; P54852; O75355-2; Q9UKR5; P01350; P39905-3; Q9Y3E0; Q9NPR9; Q9HCP6; O60725; Q9Y5U4; P11215; Q969L2; Q13021; Q9P0N8; Q6N075; P30301; Q96S97; O95167; Q99519; Q92982; Q9NZG7; Q16617; Q8N912; Q8NH19; Q6TCH4; P26678; P60201-2; Q8IY26; P54315; Q59EV6; P30405; Q96AA3; Q02161-2; Q8TAC9; Q9Y6D0; Q8N6R1; P11686; Q8IWU4; Q969S0; Q6ICL7; Q9NVC3; Q9NRQ5; B2RUZ4; Q9NZ01; P07204; Q9BZW4; P17152; A0PK00; Q9BTD3; Q5BJH2-2; Q9BVK8; Q9Y6G1; Q9P0S9; Q14656; Q8NBD8; Q9BU79; Q8N2M4; Q8N661; Q5BJF2; Q9Y2Y6; O14763; Q8N609; Q5BVD1; Q53HI1; O95183; Q9BQB6; Q8IVQ6; P00519; P17676 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Cell membrane; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Lipoprotein; Membrane; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Tumor suppressor Protein physicochemical properties Chain ID B,C Molecular weight (Da) 25132.6 Length 230 Aromaticity 0.09 Instability index 44.8 Isoelectric point 7.12 Charge (pH=7) 0.18 2D Binding mode Binding energy (Kcal/mol) -6.84 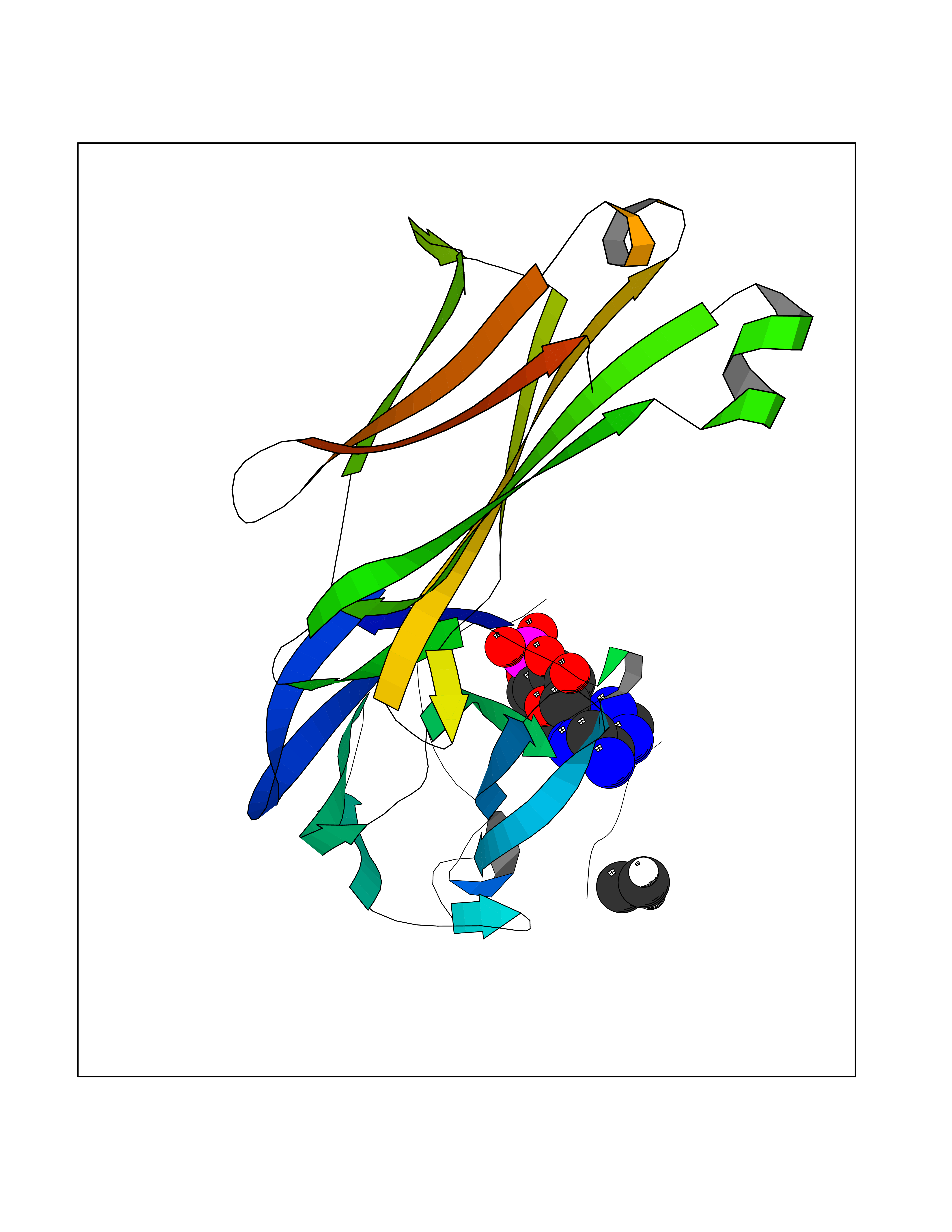 Molscript Map 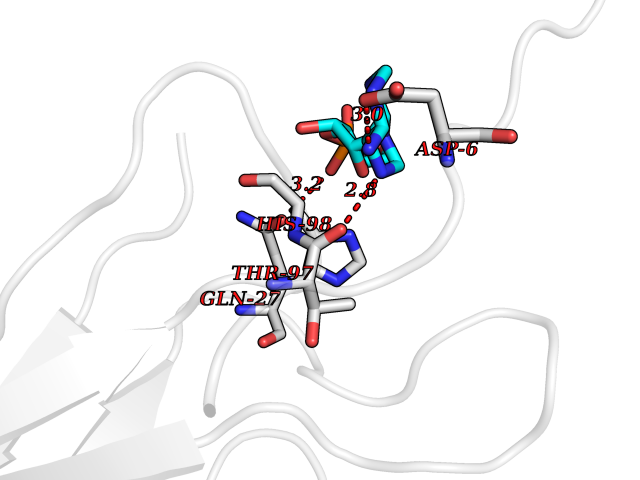 Pymol Map 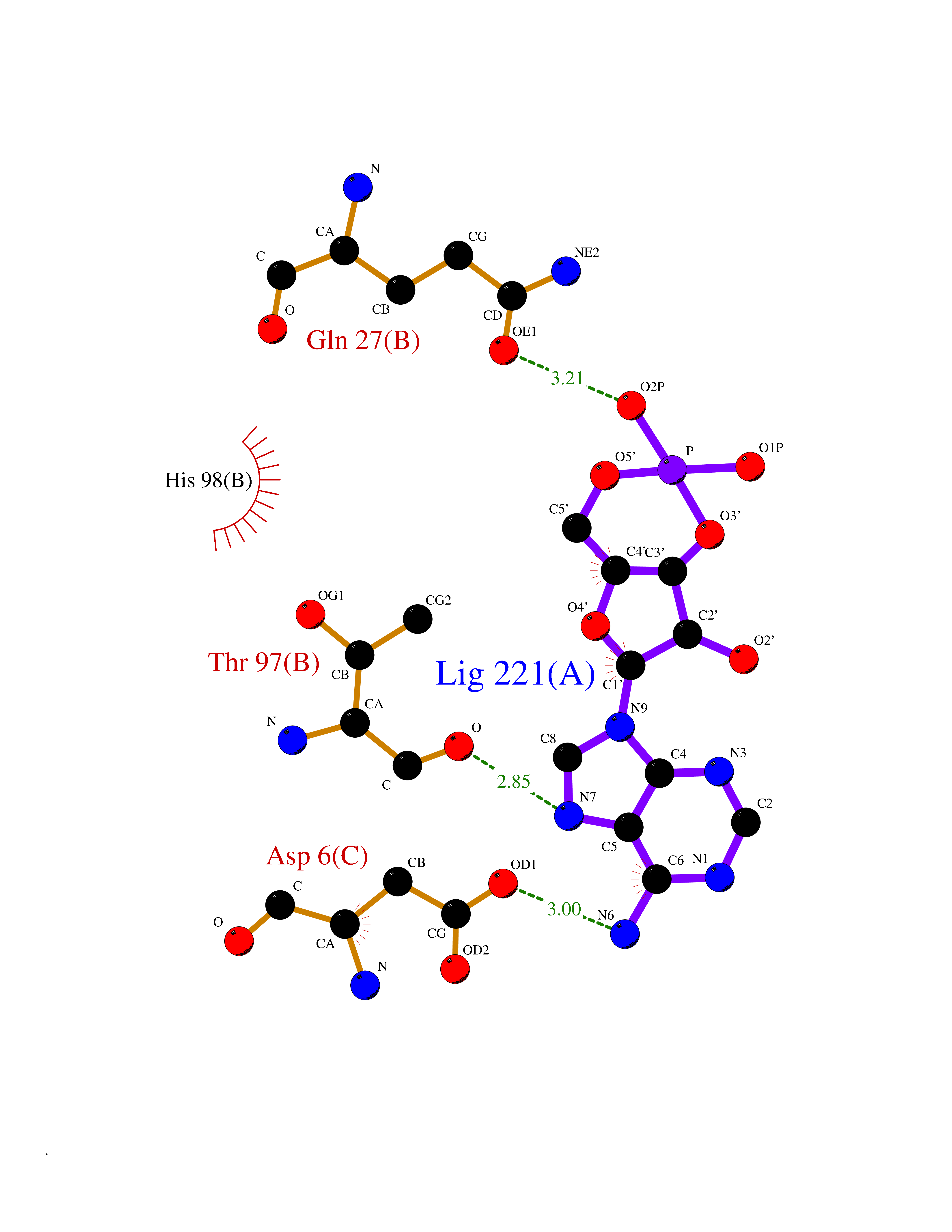 Ligplot Map 3D Binding mode Sequence DVVMTQTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLKISRVEAEDLGVYFCSQSTHVPPWTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNEXVTSAPDTRPA Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Dopamine beta-hydroxylase | 4ZEL | 6.74 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.66 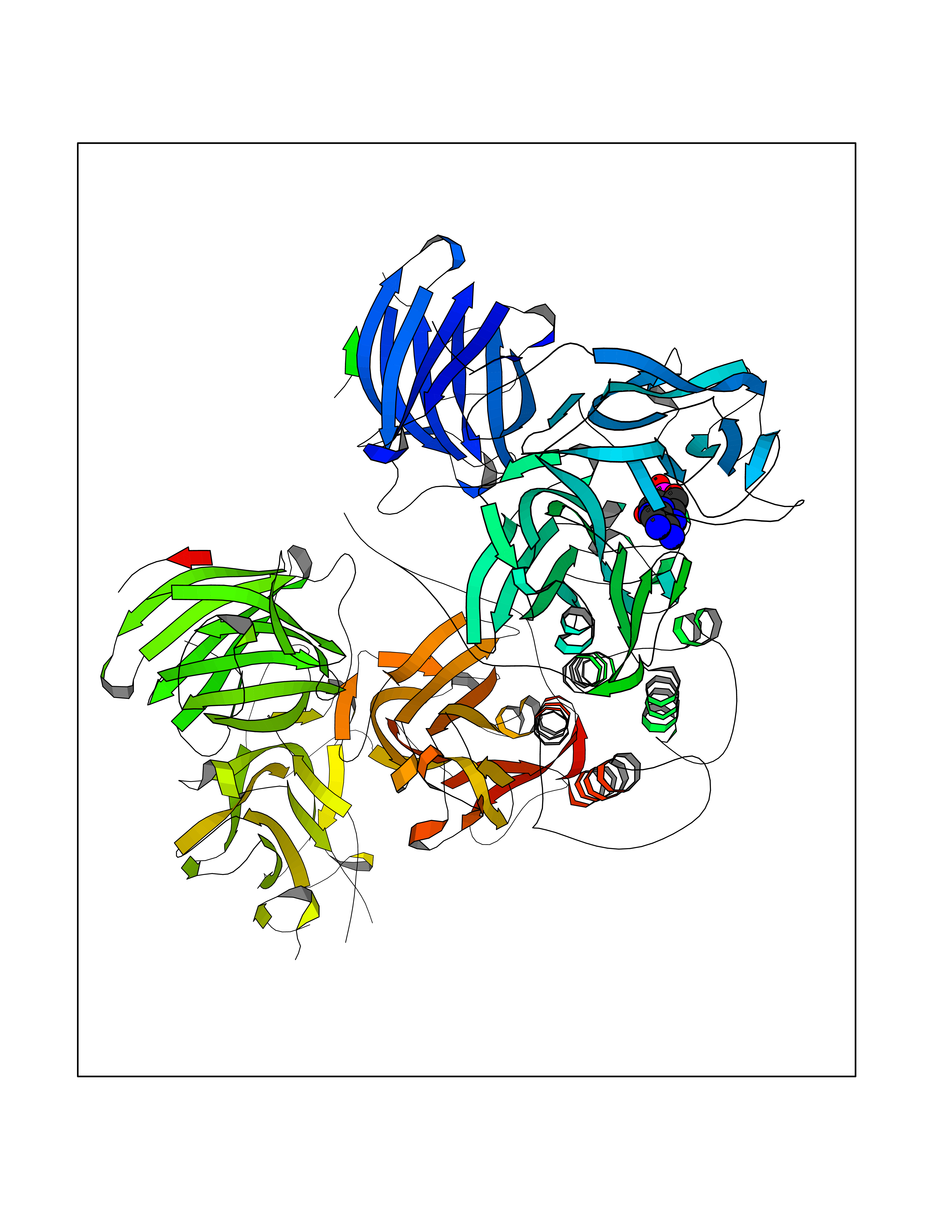 Molscript Map 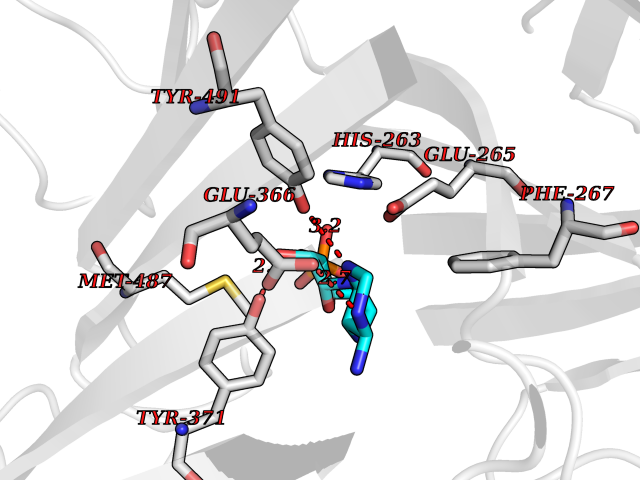 Pymol Map 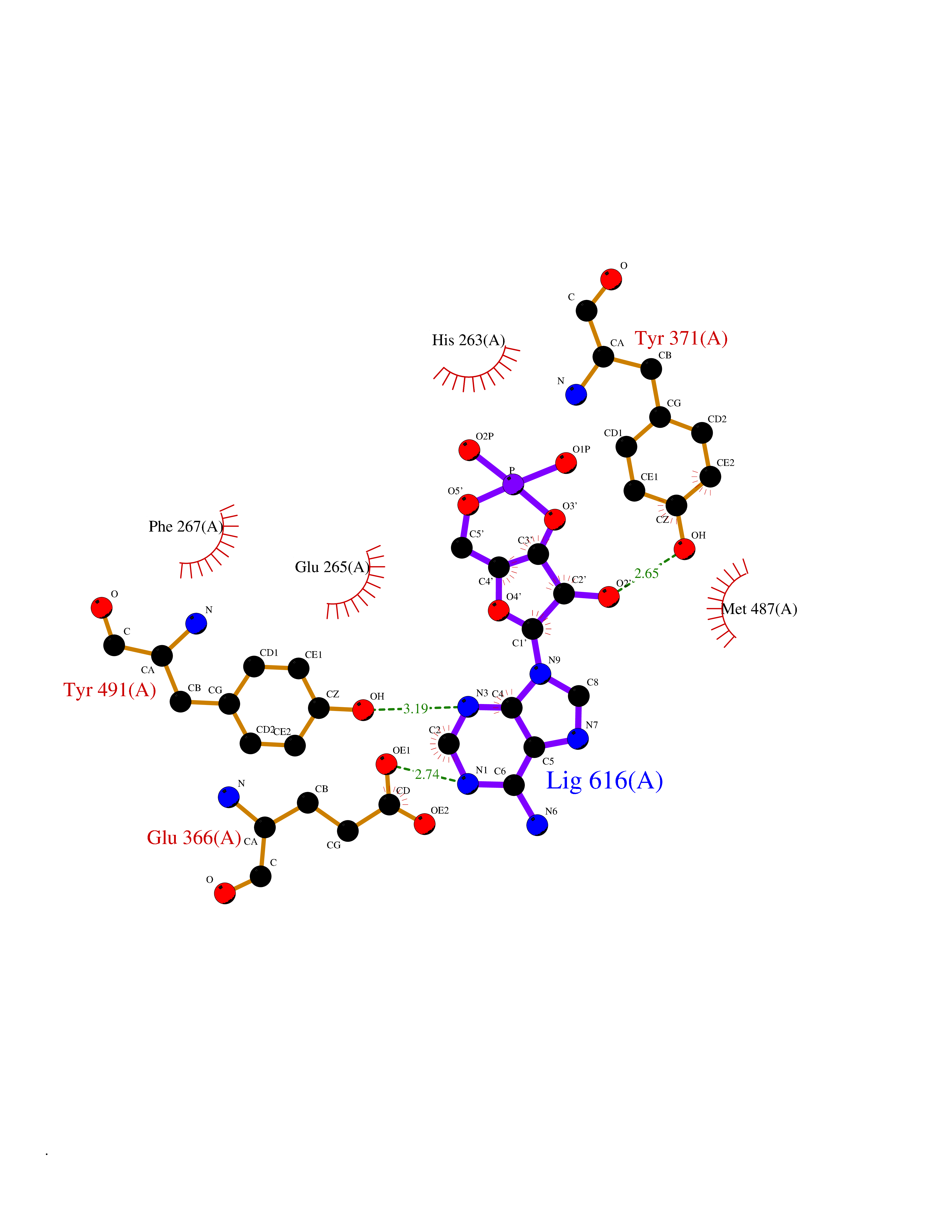 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Solute carrier family 19 member 1 (SLC19A1) | 8GOF | 6.73 | |
Target general information Gen name SLC19A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Reduced folate carrier protein; RFC1; RFC; Placental folate transporter; Intestinal folate carrier 1; IFC-1; Folate transporter 1; FOLT; FLOT1 Protein family Reduced folate carrier (RFC) transporter (TC 2.A.48) family Biochemical class NA Function Transporter for the intake of folate. Uptake of folate in human placental choriocarcinoma cells occurs by a novel mechanism called potocytosis which functionally couples three components, namely the folate receptor, the folate transporter, and a V-type H(+)-pump. Related diseases Megaloblastic anemia, folate-responsive (MEGAF) [MIM:601775]: An autosomal recessive metabolic disorder characterized by megaloblastic anemia resulting from decreased folate transport into erythrocytes. Disease manifestations include hemolytic anemia, hyperhomocysteinemia, and low vitamin B12. Serum folate levels are normal, but erythrocyte folate levels are decreased. Treatment with oral folate corrects the anemia and normalizes homocysteine. {ECO:0000269|PubMed:32276275}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 114, folate-responsive (IMD114) [MIM:620603]: An autosomal recessive immunologic disorder manifesting in early infancy and characterized by recurrent skin and respiratory infections, mucosal bleeding, oral ulcers, chronic diarrhea, and poor overall growth. Affected individuals have lymphopenia, low serum immunoglobulins, and impaired T cell proliferation. Some patients have global developmental delay. {ECO:0000269|PubMed:36517554, ECO:0000269|PubMed:36745868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11256; DB00563; DB00642; DB06813; DB01157 Interacts with Q7Z3Y9 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Antiport; Cell membrane; Disease variant; Folate-binding; Glycoprotein; Hereditary hemolytic anemia; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46087.7 Length 407 Aromaticity 0.15 Instability index 34.62 Isoelectric point 9.82 Charge (pH=7) 17.33 2D Binding mode Binding energy (Kcal/mol) -8.06  Molscript Map  Pymol Map 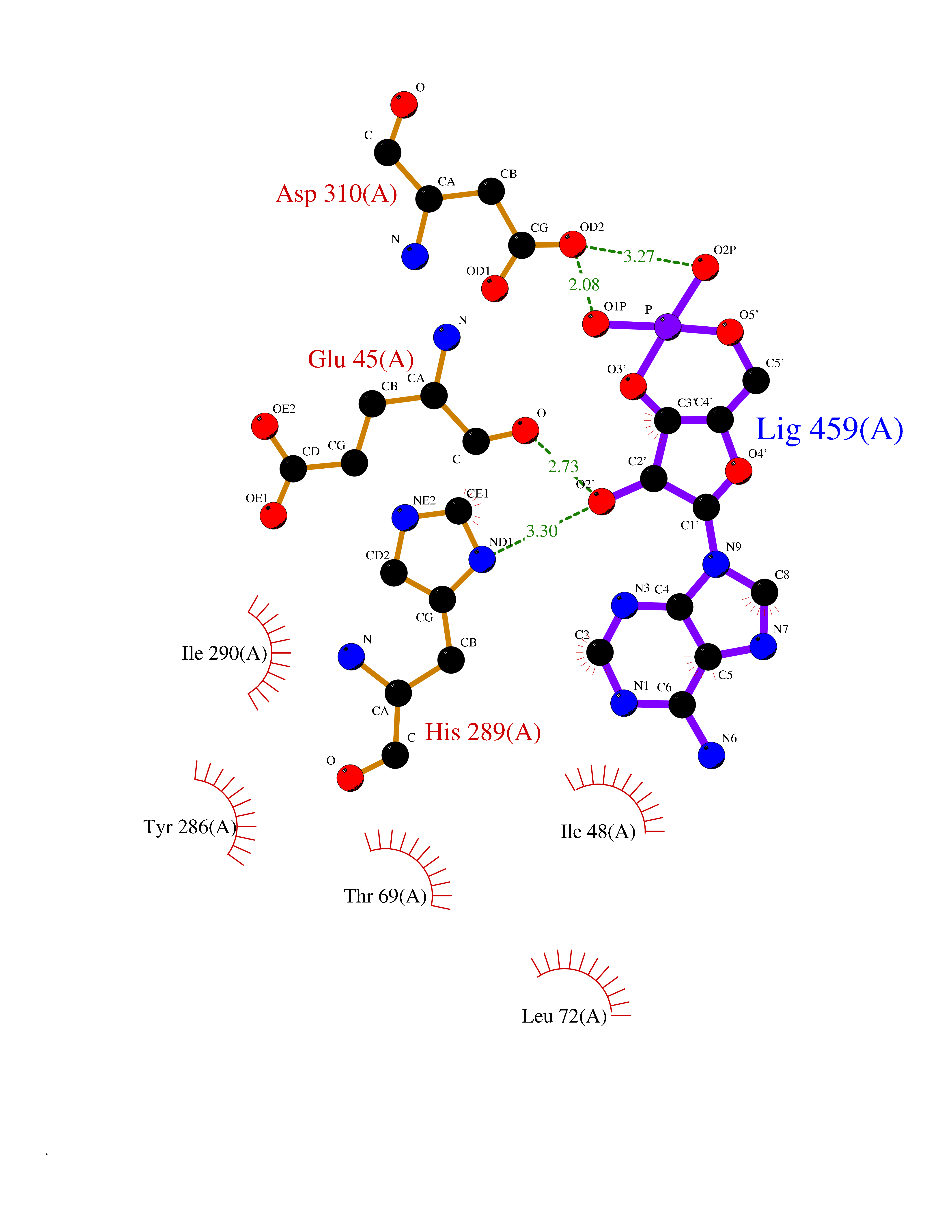 Ligplot Map 3D Binding mode Sequence DPELRSWRHLVCYLCFYGFMAQIRPGESFITPYLLGPDKNFTREQVTNEITPVLSYSYLAVLVPVFLLTDYLRYTPVLLLQGLSFVSVWLLLLLGHSVAHMQLMELFYSVTMAARIAYSSYIFSLVRPARYQRVAGYSRAAVLLGVFTSSVLGQLLVTVGRVSFSTLNYISLAFLTFSVVLALFLKRPKRSLFFNRDDSVLARMLRELGDSLRRPQLRLWSLWWVFNSAGYYLVVYYVHILWNEVDPTTNSARVYNGAADAASTLLGAITSFAAGFVKIRWARWSKLLIAGVTATQAGLVFLLAHTRHPSSIWLCYAAFVLFRGSYQFLVPIATFQIASSLSKELCALVFGVNTFFATIVKTIITFIVSDVRGLGLPVRKQFQLYSVYFLILSIIYFLGAMLDGLRH Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Prostaglandin D2 receptor 2 (PTGDR2) | 6D26 | 6.72 | |
Target general information Gen name PTGDR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PTGDR2; Chemoattractant receptor-homologous molecule expressed on Th2 cells; CD294 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for prostaglandin D2 (PGD2). Coupled to the G(i)-protein. Receptor activation may result in pertussis toxin- sensitive decreases in cAMP levels and Ca(2+) mobilization. PI3K signaling is also implicated in mediating PTGDR2 effects. PGD2 induced receptor internalization. CRTH2 internalization can be regulated by diverse kinases such as, PKC, PKA, ADRBK1/GRK2, GPRK5/GRK5 and GRK6. Receptoractivation is responsible, at least in part, in immune regulation and allergic/inflammation responses. Related diseases Neurodevelopmental disorder with seizures, hypotonia, and brain imaging abnormalities (NEDSHBA) [MIM:618922]: An autosomal recessive neurodevelopmental disorder characterized by global developmental delay, hypotonia, severe to profound intellectual disability, early-onset epilepsy, and microcephaly. Neuroimaging shows cerebral atrophy, thin corpus callosum and hypomyelination in a majority of cases. Death in childhood may occur. {ECO:0000269|PubMed:27435318, ECO:0000269|PubMed:28097321, ECO:0000269|PubMed:32286009, ECO:0000269|PubMed:33476302, ECO:0000269|PubMed:33500274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00770; DB12789; DB00917; DB01088; DB00328; DB02056; DB13036; DB00605; DB04828 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 49740.6 Length 447 Aromaticity 0.1 Instability index 37.89 Isoelectric point 10.13 Charge (pH=7) 21.88 2D Binding mode Binding energy (Kcal/mol) -7.52  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ATLKPLCPILEQMSRLQSHSATSIRYIDHAAVLLHGLASLLGLVENGVILFVVGCRMRQTVVTTWVLHLALSDLLASASLPFFTYFLAVGHSWELGTTFCKLHSSIFFLNMFASGFLLSAISLDRCLQVVRPVWAQNHRTVAAAHKVCLVLWALAVLNTVPYFVFRDTISRLDGRIMCYYNVLLLNPGPDRDATCNSRQAALAVSKFLLAFLVPLAIIASSHAAVSLRLQHRADLGLQHRNIFEMLRIDEGGGSGGDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRAALINMVFQMGETGVAGFTNSLRMLQQKRWDEAAVNLAKSRWYNQTPNRAKRVITTFRTGTWDAYRRRPGRFVRLVAAVVAAFALCWGPYHVFSLLEARAHANPGLRPLVWRGLPFVTSLAFFNSVANPVLYVLTXPDMLRKLRRSLRTVLESVL Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Myeloperoxidase (MPO) | 4DL1 | 6.70 | |
Target general information Gen name MPO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MPO Protein family Peroxidase family, XPO subfamily Biochemical class Peroxidases Function Part of the host defense system of polymorphonuclear leukocytes. It is responsible for microbicidal activity against a wide range of organisms. In the stimulated PMN, MPO catalyzes the production of hypohalous acids, primarily hypochlorous acidin physiologic situations, and other toxic intermediates that greatly enhance PMN microbicidal activity. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06111; DB00233; DB00006; DB02300; DB06774; DB00958; DB06468; DB00833; DB00535; DB00515; DB00847; DB00250; DB05161; DB01225; DB00583; DB01065; DB00461; DB04821; DB00104; DB00526; DB00550; DB00208; DB06823; DB00500; DB04827 Interacts with P27918; Q9UNE7 EC number EC 1.11.2.2 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Heme; Hydrogen peroxide; Iron; Lysosome; Metal-binding; Oxidation; Oxidoreductase; Peroxidase; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A,B,E,F,I,J,M,N Molecular weight (Da) 53052.6 Length 466 Aromaticity 0.08 Instability index 40.64 Isoelectric point 9.48 Charge (pH=7) 15.12 2D Binding mode Binding energy (Kcal/mol) -6.9  Molscript Map 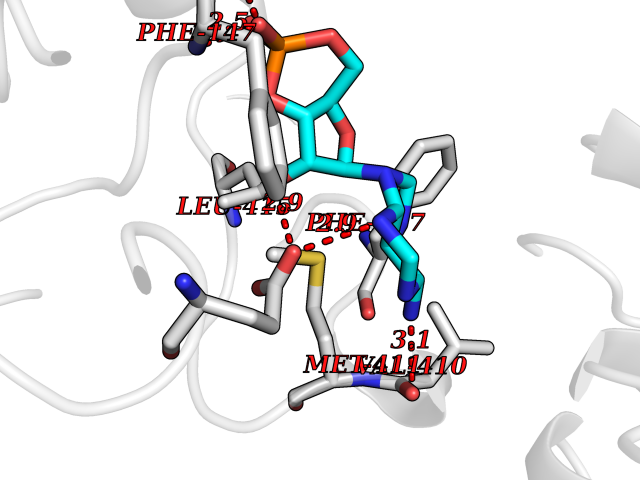 Pymol Map  Ligplot Map 3D Binding mode Sequence VNCETSCVQQPPCFPLKIPPNDPRIKNQADCIPFFRSXPACPGSNITIRNQINALTSFVDASMVYGSEEPLARNLRNMSNQLGLLAVNQRFQDNGRALLPFDNLHDDPCLLTNRSARIPCFLAGDTRSSEMPELTSMHTLLLREHNRLATELKSLNPRWDGERLYQEARKIVGAMVQIITYRDYLPLVLGPTAMRKYLPTYRSYNDSVDPRIANVFTNAFRYGHTLIQPFMFRLDNRYQPMEPNPRVPLSRVFFASWRVVLEGGIDPILRGLMATPAKLNRQNQIAVDEIRERLFEQVMRIGLDLPALNMQRSRDHGLPGYNAWRRFCGLPQPETVGQLGTVLRNLKLARKLMEQYGTPNNIDIWMGGVSEPLKRKGRVGPLLACIIGTQFRKLRDGDRFWWENEGVFSMQQRQALAQISLPRIICDNTGITTVSKNNIFMSNSYPRDFVNCSTLPALNLASWREA Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | MAPK-activated protein kinase 3 (MAPKAPK3) | 3FHR | 6.70 | |
Target general information Gen name MAPKAPK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAPKAPK3 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class NA Function Stress-activated serine/threonine-protein kinase involved in cytokines production, endocytosis, cell migration, chromatin remodeling and transcriptional regulation. Following stress, it is phosphorylated and activated by MAP kinase p38-alpha/MAPK14, leading to phosphorylation of substrates. Phosphorylates serine in the peptide sequence, Hyd-X-R-X2-S, where Hyd is a large hydrophobic residue. MAPKAPK2 and MAPKAPK3, share the same function and substrate specificity, but MAPKAPK3 kinase activity and level in protein expression are lower compared to MAPKAPK2. Phosphorylates HSP27/HSPB1, KRT18, KRT20, RCSD1, RPS6KA3, TAB3 and TTP/ZFP36. Mediates phosphorylation of HSP27/HSPB1 in response to stress, leading to dissociate HSP27/HSPB1 from large small heat-shock protein (sHsps) oligomers and impair their chaperone activities and ability to protect against oxidative stress effectively. Involved in inflammatory response by regulating tumor necrosis factor (TNF) and IL6 production post-transcriptionally: acts by phosphorylating AU-rich elements (AREs)-binding proteins, such as TTP/ZFP36, leading to regulate the stability and translation of TNF and IL6 mRNAs. Phosphorylation of TTP/ZFP36, a major post-transcriptional regulator of TNF, promotes its binding to 14-3-3 proteins and reduces its ARE mRNA affinity leading to inhibition of dependent degradation of ARE-containing transcript. Involved in toll-like receptor signaling pathway (TLR) in dendritic cells: required for acute TLR-induced macropinocytosis by phosphorylating and activating RPS6KA3. Also acts as a modulator of Polycomb-mediated repression. Related diseases Macular dystrophy, patterned, 3 (MDPT3) [MIM:617111]: A form of retinal patterned dystrophy, characterized by retinal pigment epithelium and Bruch's membrane changes resembling a 'dry desert land'. It begins around the age of 30 and progresses to retinitis pigmentosa. MDPT3 inheritance is autosomal dominant. {ECO:0000269|PubMed:26744326}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08358; DB07728 Interacts with P04792; Q15759; Q16539; PRO_0000037566 [P27958] EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cytoplasm; Disease variant; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 31536.8 Length 273 Aromaticity 0.08 Instability index 44.27 Isoelectric point 5.65 Charge (pH=7) -9.13 2D Binding mode Binding energy (Kcal/mol) -7.37  Molscript Map  Pymol Map 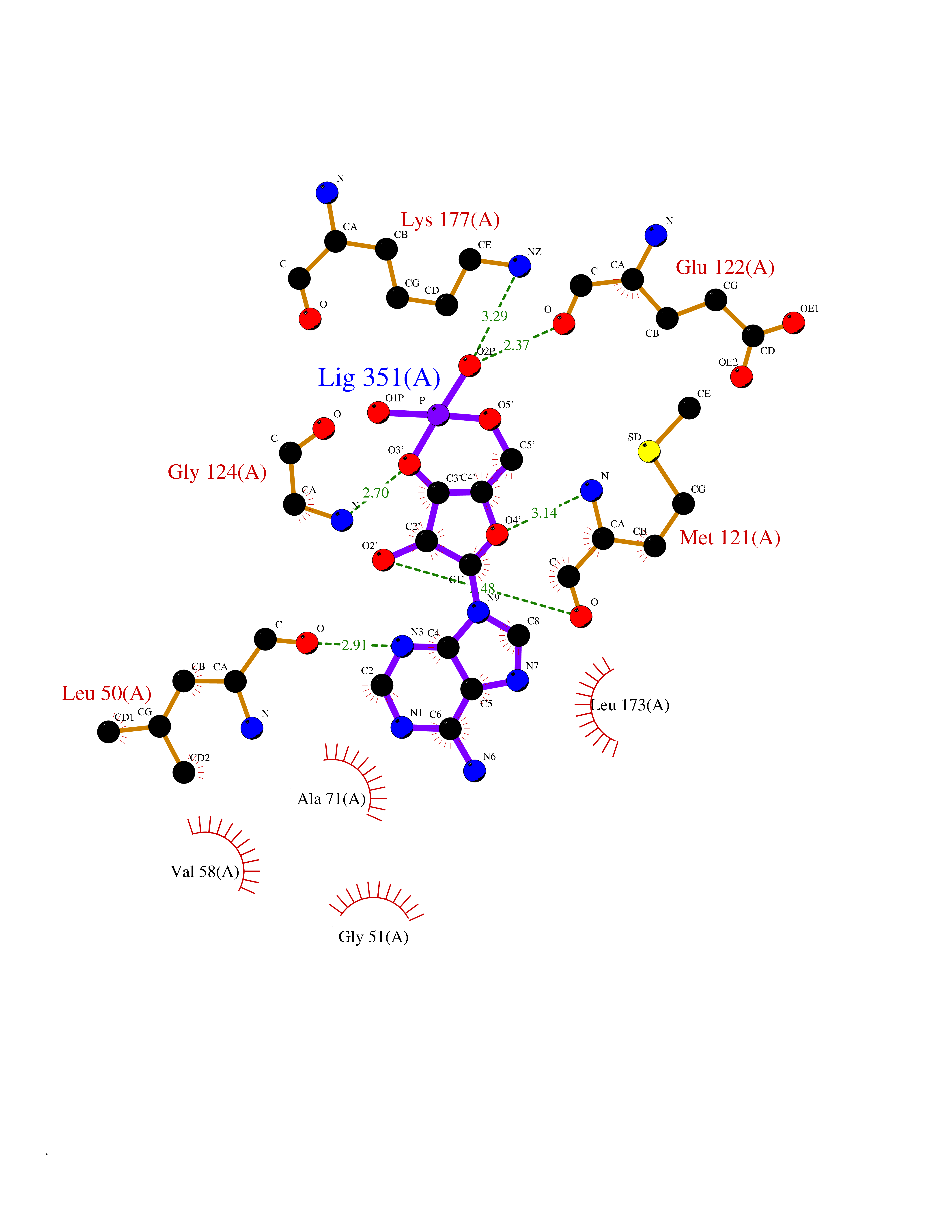 Ligplot Map 3D Binding mode Sequence EPKKYAVTDDYQLSKQVLGLGVNGKVLECFHRRTGQKCALKLLYDSPKARQEVDHHWQASGGPHIVCILDVYENMHHGKRCLLIIMECMEGGELFSRIQERDQAFTEREAAEIMRDIGTAIQFLHSHNIAHRDVKPENLLYTSKEKDAVLKLTDFGFAKETEKYDKSCDMWSLGVIMYILLCGFPPFGFPNPEWSEVSEDAKQLIRLLLKTDPTERLTITQFMNHPWINQSMVVPQTPLHTARVLQEDKDHWDEVKEEMTSALATMRVDYDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 6.70 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -8.02 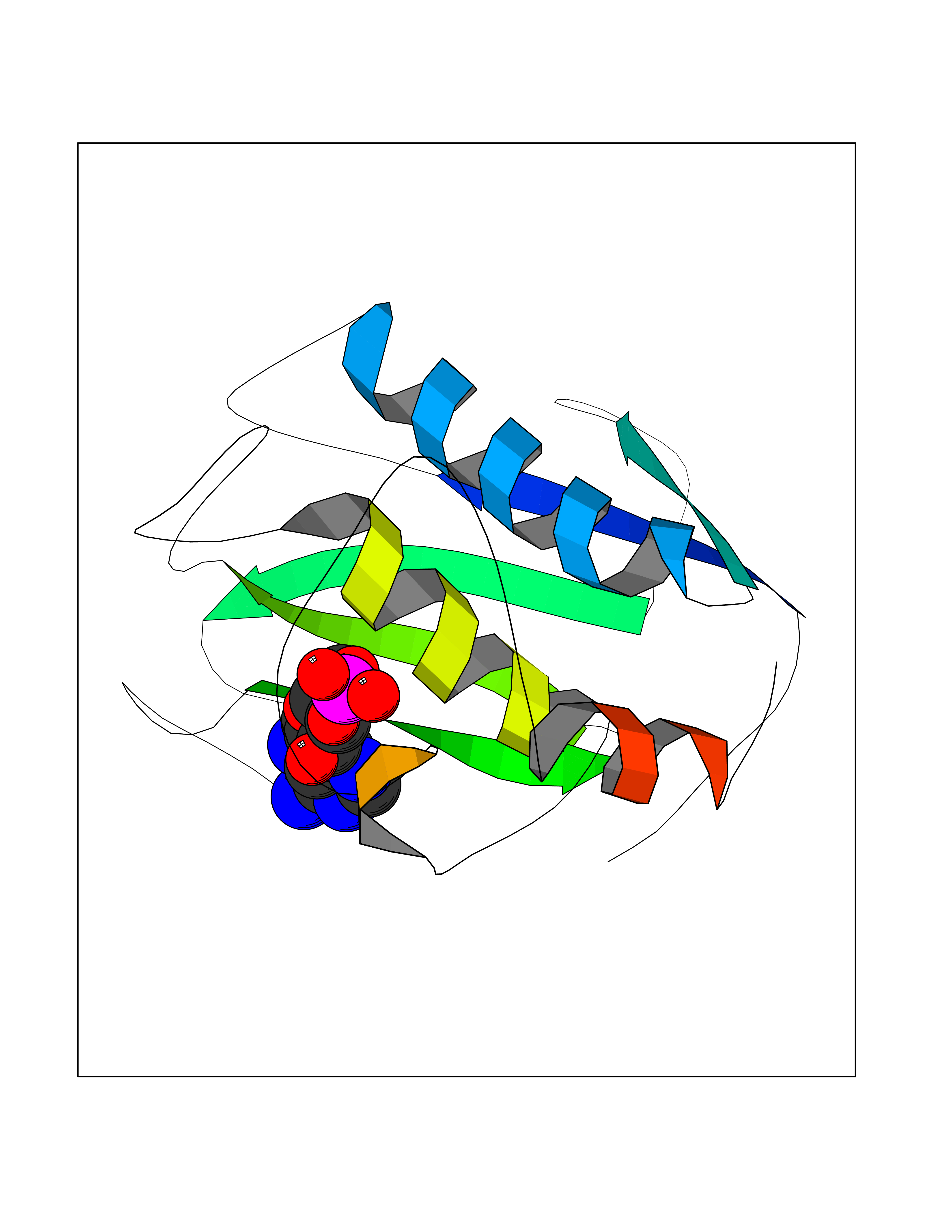 Molscript Map 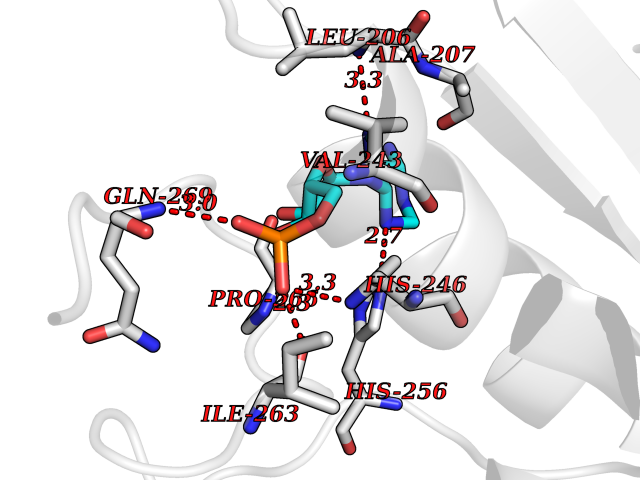 Pymol Map 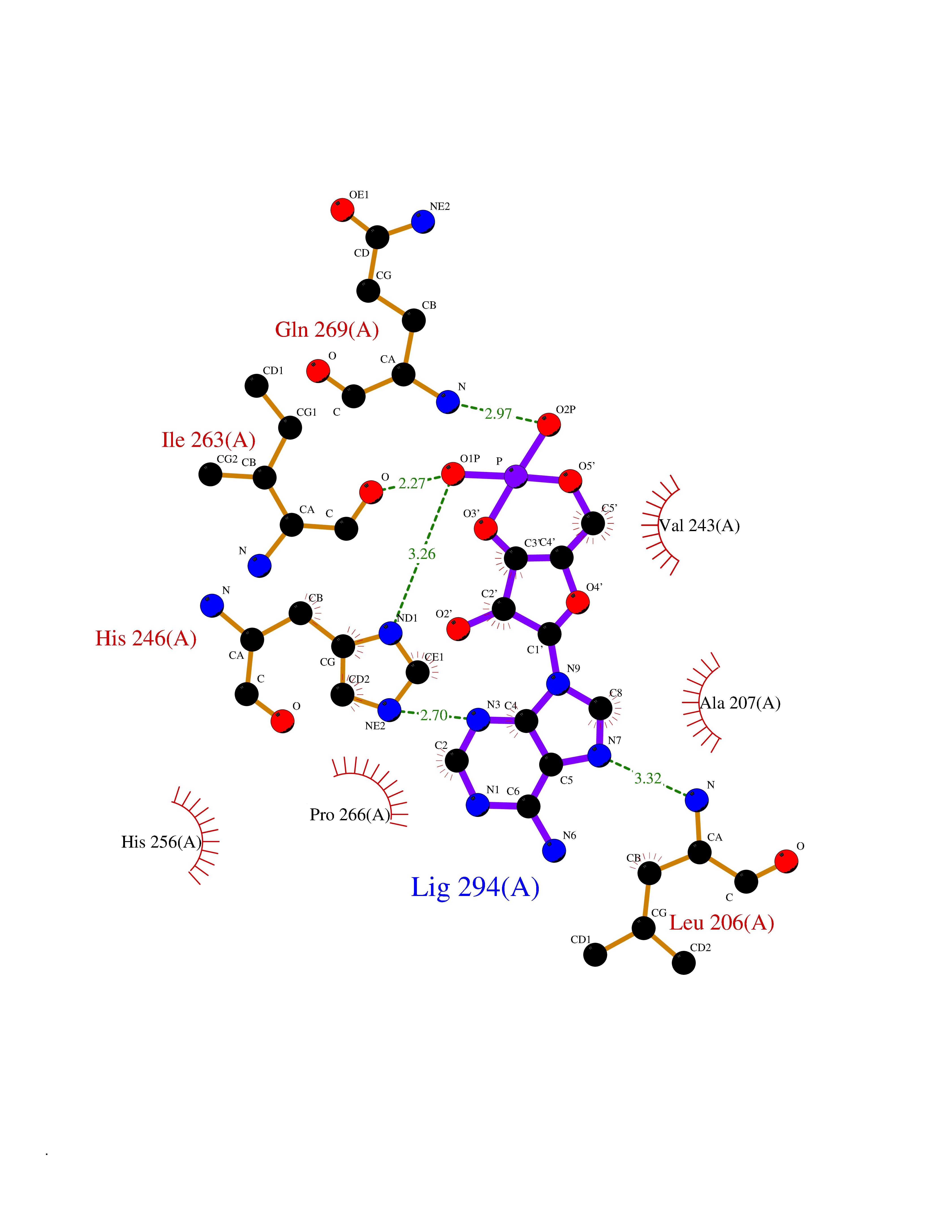 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Carbonic anhydrase II (CA-II) | 3K34 | 6.69 | |
Target general information Gen name CA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase C; Carbonic anhydrase 2; Carbonate dehydratase II; CAC Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Can hydrate cyanamide to urea. Involved in the regulation of fluid secretion into the anterior chamber of the eye. Contributes to intracellular pH regulation in the duodenal upper villous epithelium during proton-coupled peptide absorption. Stimulates the chloride-bicarbonate exchange activity of SLC26A6. Essential for bone resorption and osteoclast differentiation. Related diseases Osteopetrosis, autosomal recessive 3 (OPTB3) [MIM:259730]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability. {ECO:0000269|PubMed:15300855, ECO:0000269|PubMed:1542674, ECO:0000269|PubMed:1928091, ECO:0000269|PubMed:8834238, ECO:0000269|PubMed:9143915}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07596; DB03333; DB08418; DB04081; DB08416; DB02479; DB07467; DB03950; DB03594; DB03294; DB04763; DB03270; DB08083; DB06954; DB08659; DB08046; DB02087; DB08156; DB04203; DB04394; DB08782; DB04549; DB02221; DB04180; DB03039; DB02861; DB02429; DB08202; DB04600; DB04601; DB01784; DB03385; DB03697; DB04002; DB07632; DB07050; DB06891; DB08645; DB08765; DB00819; DB03877; DB03262; DB03598; DB04089; DB01964; DB03526; DB04371; DB02220; DB03221; DB02602; DB02535; DB00436; DB00562; DB01194; DB00482; DB00880; DB02679; DB00606; DB02866; DB01144; DB00869; DB08846; DB01031; DB00311; DB08157; DB01942; DB00695; DB00774; DB08165; DB02292; DB03975; DB00703; DB00232; DB02610; DB07742; DB03844; DB02069; DB02986; DB08301; DB07048; DB03596; DB07476; DB01748; DB08155; DB01671; DB07710; DB01325; DB09460; DB09472; DB02894; DB00391; DB08329; DB07363; DB00273; DB01021; DB03904; DB00580; DB14533; DB14548; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Lyase; Membrane; Metal-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29027.4 Length 258 Aromaticity 0.1 Instability index 20.07 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -6.44 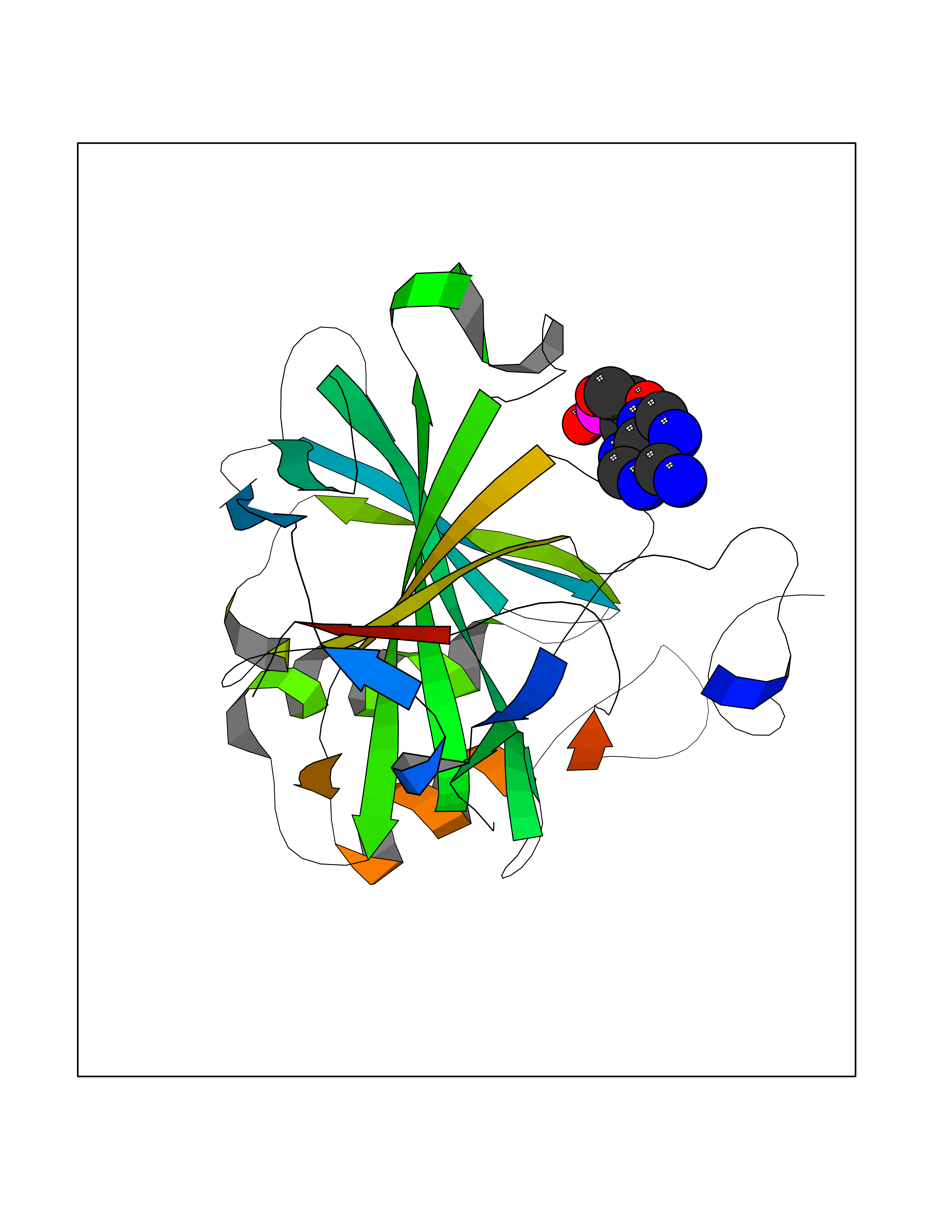 Molscript Map 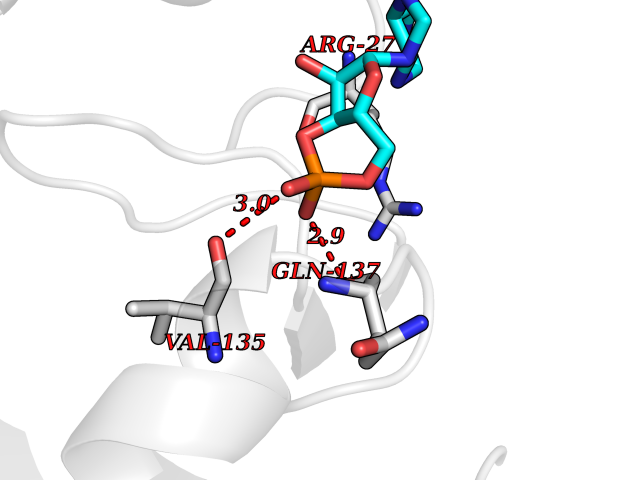 Pymol Map 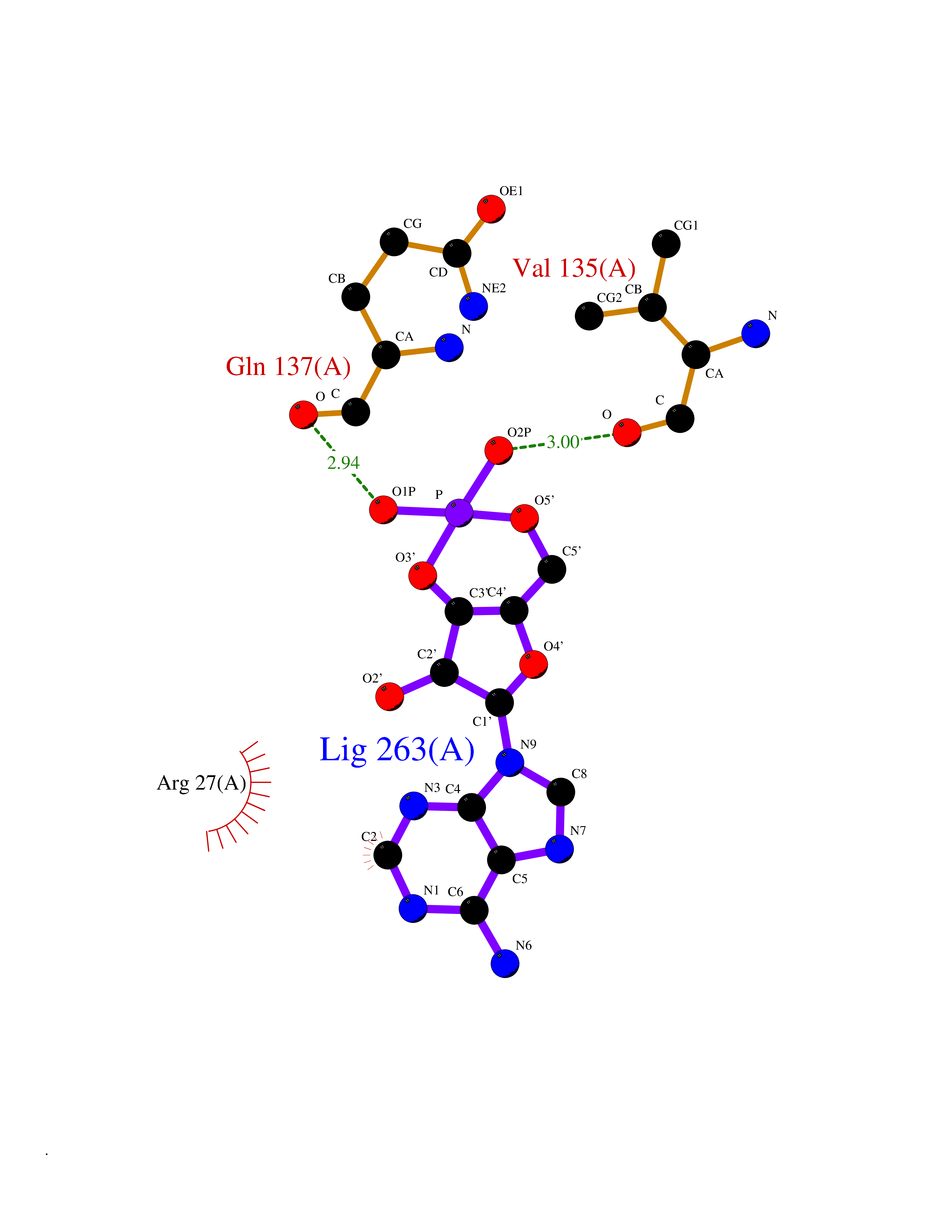 Ligplot Map 3D Binding mode Sequence HHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEPISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Phosphodiesterase 4A (PDE4A) | 2QYK | 6.68 | |
Target general information Gen name PDE4A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4A; Type 4A cAMP phosphodiesterase; PDE46; DPDE2 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06842; DB08299; DB01427; DB00201; DB05219; DB00975; DB06751; DB00651; DB00824; DB05266; DB01088; DB01303; DB01791; DB06479; DB01656; DB01954; DB00277; DB08811; DB09283 Interacts with P55212; O14569; P13473-2; Q9UJX0; P16118; O75400-2; Q9Y371 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cell projection; Cytoplasm; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 38579.6 Length 335 Aromaticity 0.08 Instability index 37.43 Isoelectric point 5.01 Charge (pH=7) -19.92 2D Binding mode Binding energy (Kcal/mol) -7.67 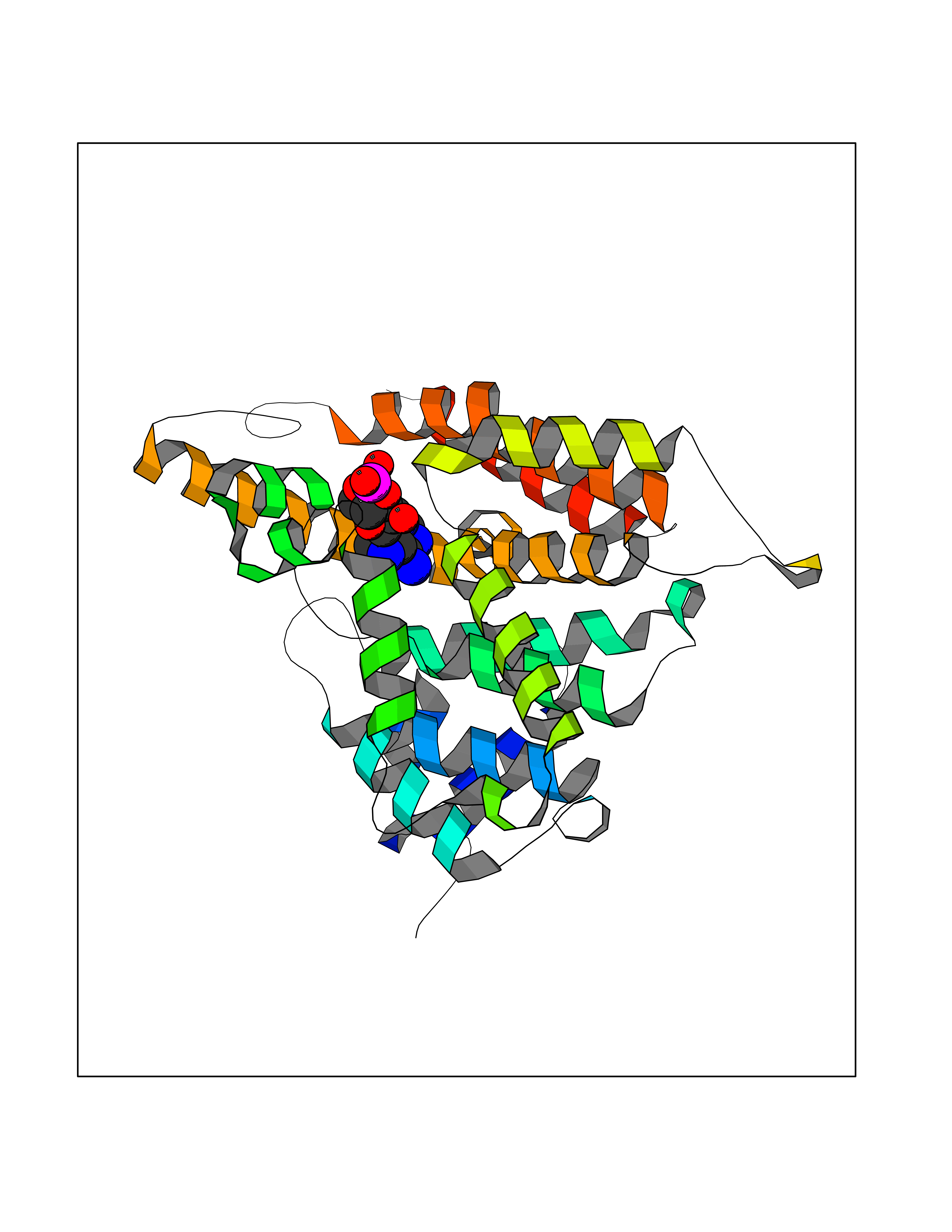 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence HMNIPRFGVKTDQEELLAQELENLNKWGLNIFCVSDYAGGRSLTCIMYMIFQERDLLKKFRIPVDTMVTYMLTLEDHYHADVAYHNSLHAADVLQSTHVLLATPALDAVFTDLEILAALFAAAIHDVDHPGVSNQFLINTNSELALMYNDESVLENHHLAVGFKLLQEDNCDIFQNLSKRQRQSLRKMVIDMVLATDMSKHMTLLADLKTMVETKKVTSSGVLLLDNYSDRIQVLRNMVHCADLSNPTKPLELYRQWTDRIMAEFFQQGDRERERGMEISPMCDKHTASVEKSQVGFIDYIVHPLWETWADLVHPDAQEILDTLEDNRDWYYSAI Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Prolyl endopeptidase (PREP) | 3DDU | 6.67 | |
Target general information Gen name PREP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Post-proline cleaving enzyme; PREP; PE Protein family Peptidase S9A family Biochemical class Peptidase Function Cleaves peptide bonds on the C-terminal side of prolyl residues within peptides that are up to approximately 30 amino acids long. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07148; DB01684; DB08738; DB08739; DB03864; DB00107; DB03382; DB03535 Interacts with P04406 EC number EC 3.4.21.26 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A Molecular weight (Da) 80424.2 Length 707 Aromaticity 0.13 Instability index 35.02 Isoelectric point 5.52 Charge (pH=7) -20.31 2D Binding mode Binding energy (Kcal/mol) -7.03  Molscript Map  Pymol Map 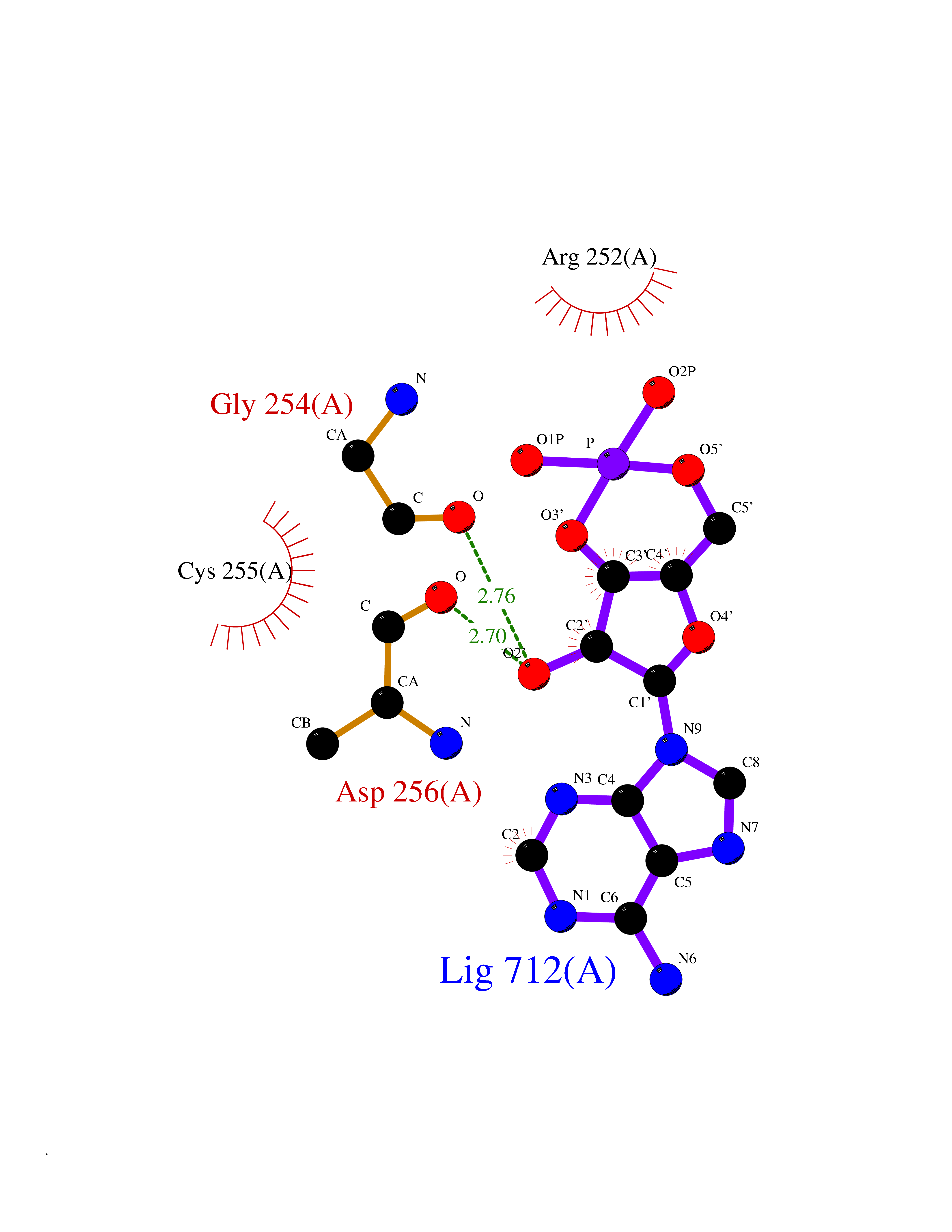 Ligplot Map 3D Binding mode Sequence LSFQYPDVYRDETAVQDYHGHKICDPYAWLEDPDSEQTKAFVEAQNKITVPFLEQCPIRGLYKERMTELYDYPKYSCHFKKGKRYFYFYNTGLQNQRVLYVQDSLEGEARVFLDPNILSDDGTVALRGYAFSEDGEYFAYGLSASGSDWVTIKFMKVDGAKELPDVLERVKFSCMAWTHDGKGMFYNSYPQQDGKSDGTETSTNLHQKLYYHVLGTDQSEDILCAEFPDEPKWMGGAELSDDGRYVLLSIREGCDPVNRLWYCDLQQESSGIAGILKWVKLIDNFEGEYDYVTNEGTVFTFKTNRHSPNYRVINIDFTDPEESKWKVLVPEHEKDVLEWIACVRSNFLVLCYLHDVKNTLQLHDLTTGALLKIFPLDVGSIVGYSGQKKDTEIFYQFTSFLSPGIIYHCDLTKEELEPRVFREVVKIDASDYQTVQIFYPSKDGTKIPMFIVHKKGIKLDGSHPAFLYGYGGFNISITPNYSVSRLIFVRHMGGILAVANIRGGGEYGETWHKGGILANKQNCFDDFQCAAEYLIKEGYTSPKRLTINGGSNGGLLVAACANQRPDLFGCVIAQVGVMDMLKFHKYTIGHAWTTDYGCSDSKQHFEWLVKYSPLHNVKLPEADDIQYPSMLLLTADHDDRVVPLHSLKFIATLQYIVGRSRKQNNPLLIHVDTKAGHGAGKPTAKVIEEVSDMFAFIARCLNVDWIP Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Beta-adrenergic receptor kinase 1 (ADRBK1) | 3V5W | 6.66 | |
Target general information Gen name GRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GRK2; G-protein coupled receptor kinase 2; BetaARK1; Beta-ARK-1; Beta ARK1 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, GPRK subfamily Biochemical class Kinase Function Specifically phosphorylates the agonist-occupied form of the beta-adrenergic and closely related receptors, probably inducing a desensitization of them. Key regulator of LPAR1 signaling. Competes with RALA for binding to LPAR1 thus affecting the signaling properties of the receptor. Desensitizes LPAR1 and LPAR2 in a phosphorylation-independent manner. Related diseases 3-ketothiolase deficiency (3KTD) [MIM:203750]: An autosomal recessive inborn error of isoleucine catabolism characterized by intermittent ketoacidotic attacks associated with unconsciousness. Some patients die during an attack or are mentally retarded. Urinary excretion of 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid, triglylglycine, butanone is increased. It seems likely that the severity of this disease correlates better with the environmental or acquired factors than with the ACAT1 genotype. {ECO:0000269|PubMed:1346617, ECO:0000269|PubMed:1715688, ECO:0000269|PubMed:7728148, ECO:0000269|PubMed:9744475}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00171 Interacts with P05067; P48730-2; P21860; P21462; Q9Y2X7; P35626; Q00987; P13591; P25963; Q13635; P0CG48 EC number EC 2.7.11.15 Uniprot keywords 3D-structure; ATP-binding; Cell membrane; Cell projection; Cytoplasm; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Synapse; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 38433.9 Length 337 Aromaticity 0.09 Instability index 52.17 Isoelectric point 7.36 Charge (pH=7) 1.05 2D Binding mode Binding energy (Kcal/mol) -7.42  Molscript Map 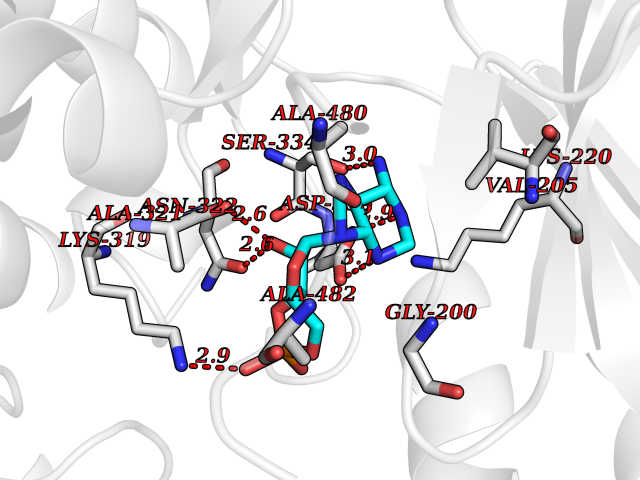 Pymol Map 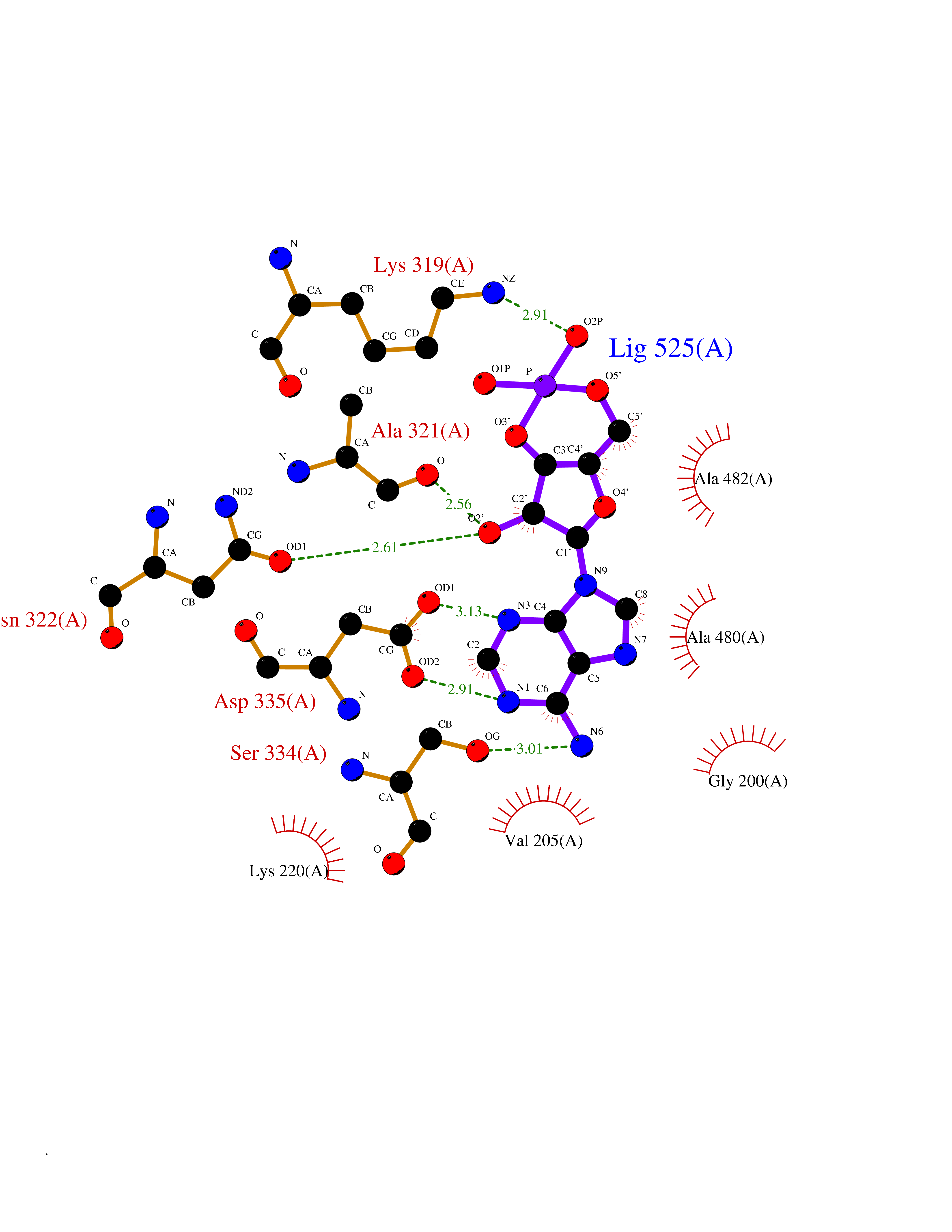 Ligplot Map 3D Binding mode Sequence KNVELNIHLTMNDFSVHRIIGRGGFGEVYGCRKADTGKMYAMKCLDKKRIKMKQGETLALNERIMLSLVSTGDCPFIVCMSYAFHTPDKLSFILDLMNGGDLHYHLSQHGVFSEADMRFYAAEIILGLEHMHNRFVVYRDLKPANILLDEHGHVRISDLGLACDFSKKKPHASVGTHGYMAPEVLQKGVAYDSSADWFSLGCMLFKLLRGHSPFRQHKTKDKHEIDRMTLTMAVELPDSFSPELRSLLEGLLQRDVNRRLGCLGRGAQEVKESPFFRSLDWQMVFLQKYPPPLIPPRGEVNAADAFDKGIKLLDSDQELYRNFPLTISERWQQEVAE Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Plasmodium DOXP reductoisomerase (Malaria DXR) | 3AU9 | 6.66 | |
Target general information Gen name Malaria DXR Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID Synonyms IspC; DXR; DXP reductoisomerase; DOXP reductoisomerase; 2-C-Methyl-d-erythritol 4-phosphate synthase; 1-deoxyxylulose-5-phosphate reductoisomerase Protein family DXR family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADP-dependent rearrangement and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol 4-phosphate (MEP). Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.1.267 Uniprot keywords 3D-structure; Apicoplast; Isoprene biosynthesis; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Plastid; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46644.4 Length 410 Aromaticity 0.09 Instability index 36.77 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -7.78 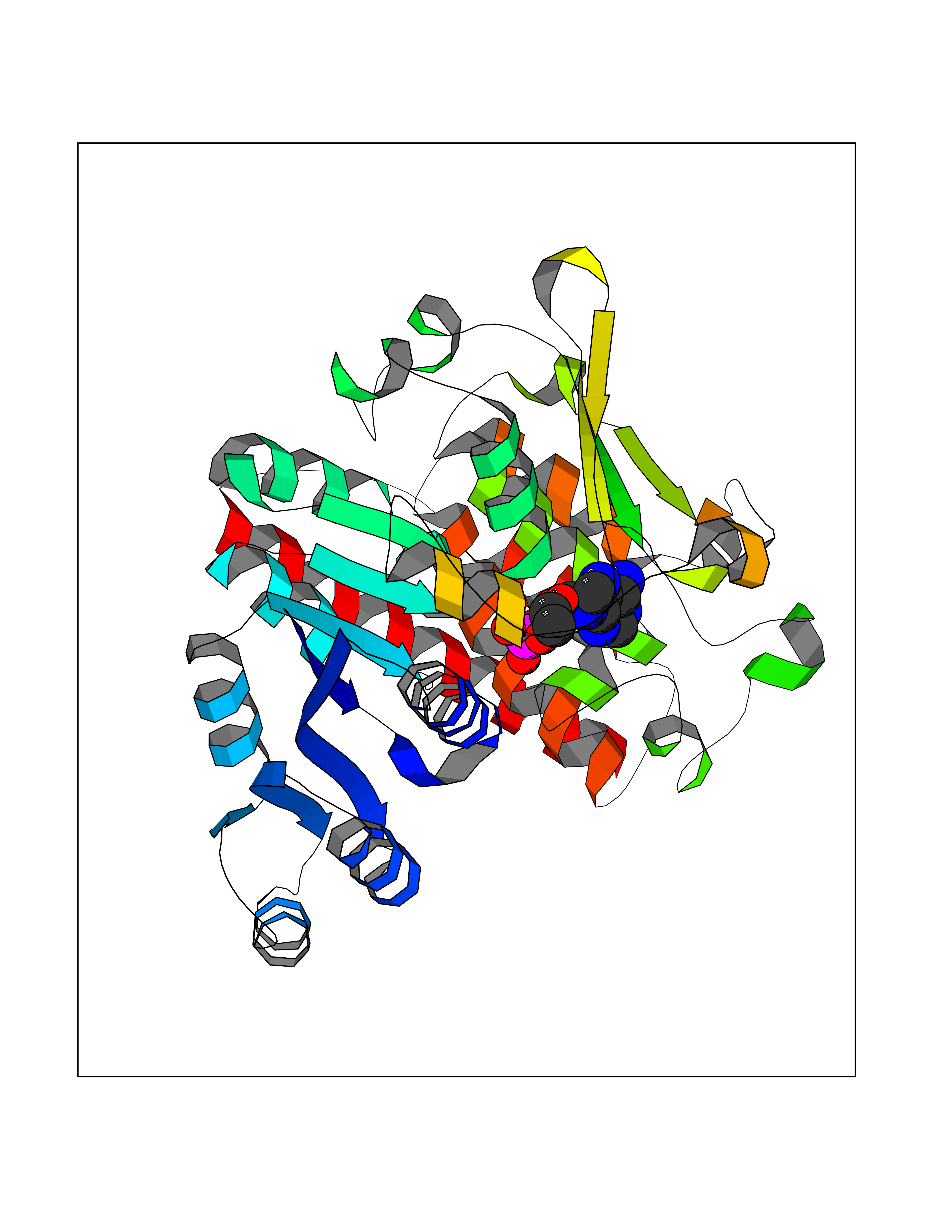 Molscript Map 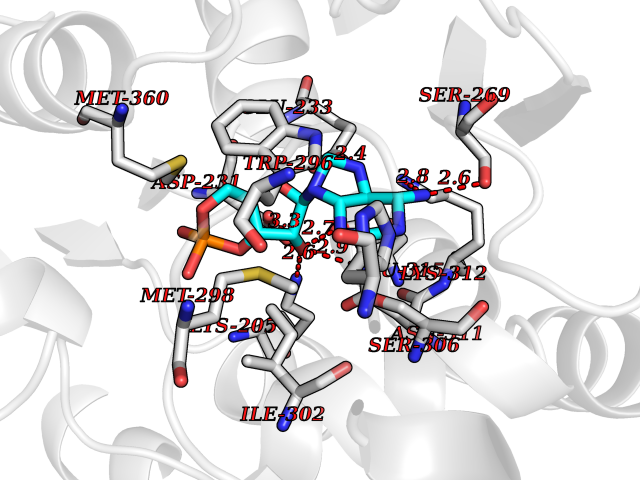 Pymol Map 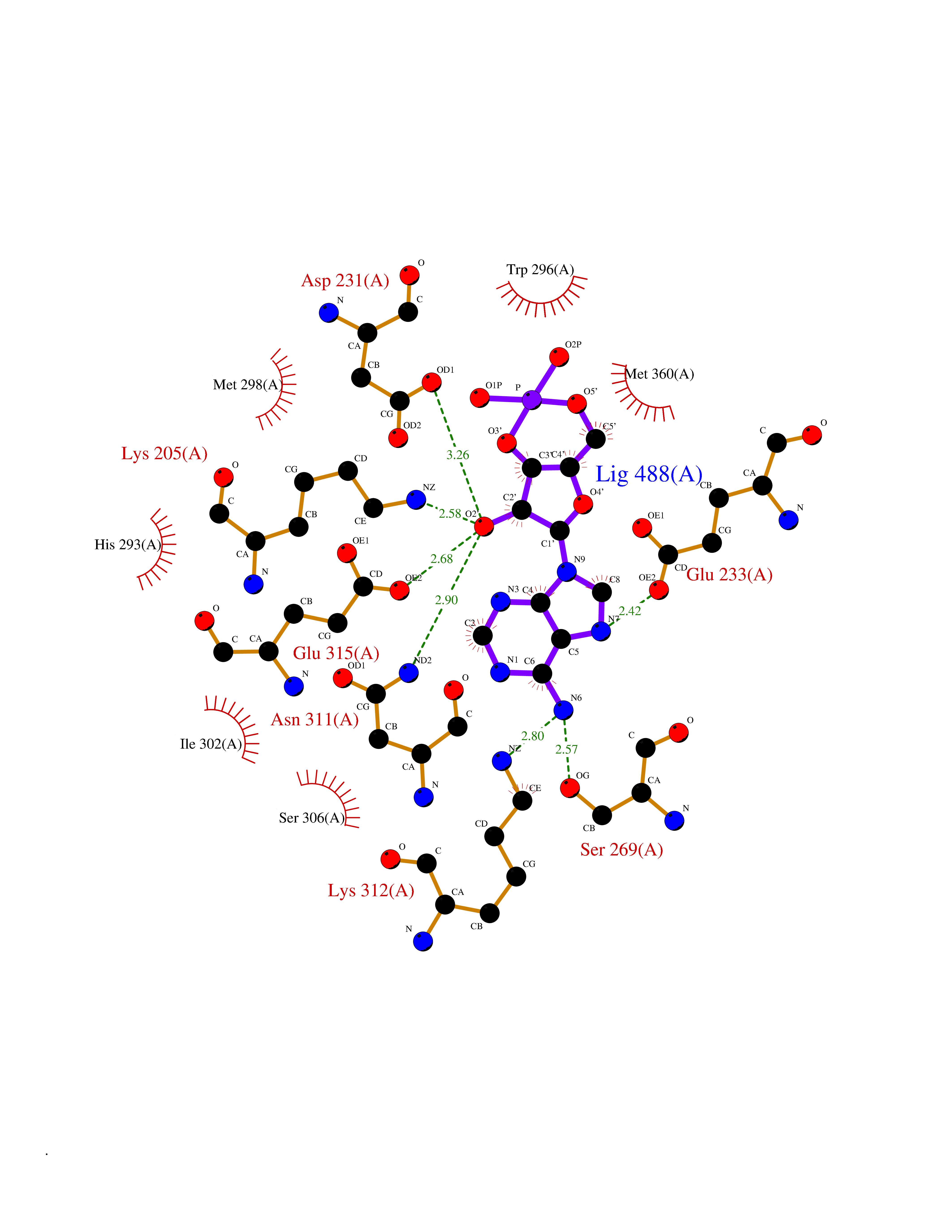 Ligplot Map 3D Binding mode Sequence PINVAIFGSTGSIGTNALNIIRECNKIENVFNVKALYVNKSVNELYEQAREFLPEYLCIHDKSVYEELKELVKNIKDYKPIILCGDEGMKEICSSNSIDKIVIGIDSFQGLYSTMYAIMNNKIVALANKESIVSAGFFLKKLLNIHKNAKIIPVDSEHSAIFQCLDNNKVLKTKCLQDNFSKINNINKIFLCSSGGPFQNLTMDELKNVTSENALKHPKWKMGKKITIDSATMMNKGLEVIETHFLFDVDYNDIEVIVHKECIIHSCVEFIDKSVISQMYYPDMQIPILYSLTWPDRIKTNLKPLDLAQVSTLTFHKPSLEHFPCIKLAYQAGIKGNFYPTVLNASNEIANNLFLNNKIKYFDISSIISQVLESFNSQKVSENSEDLMKQILQIHSWAKDKATDIYNKHN Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Cytochrome P450 1A2 | 2HI4 | 6.65 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -7.39 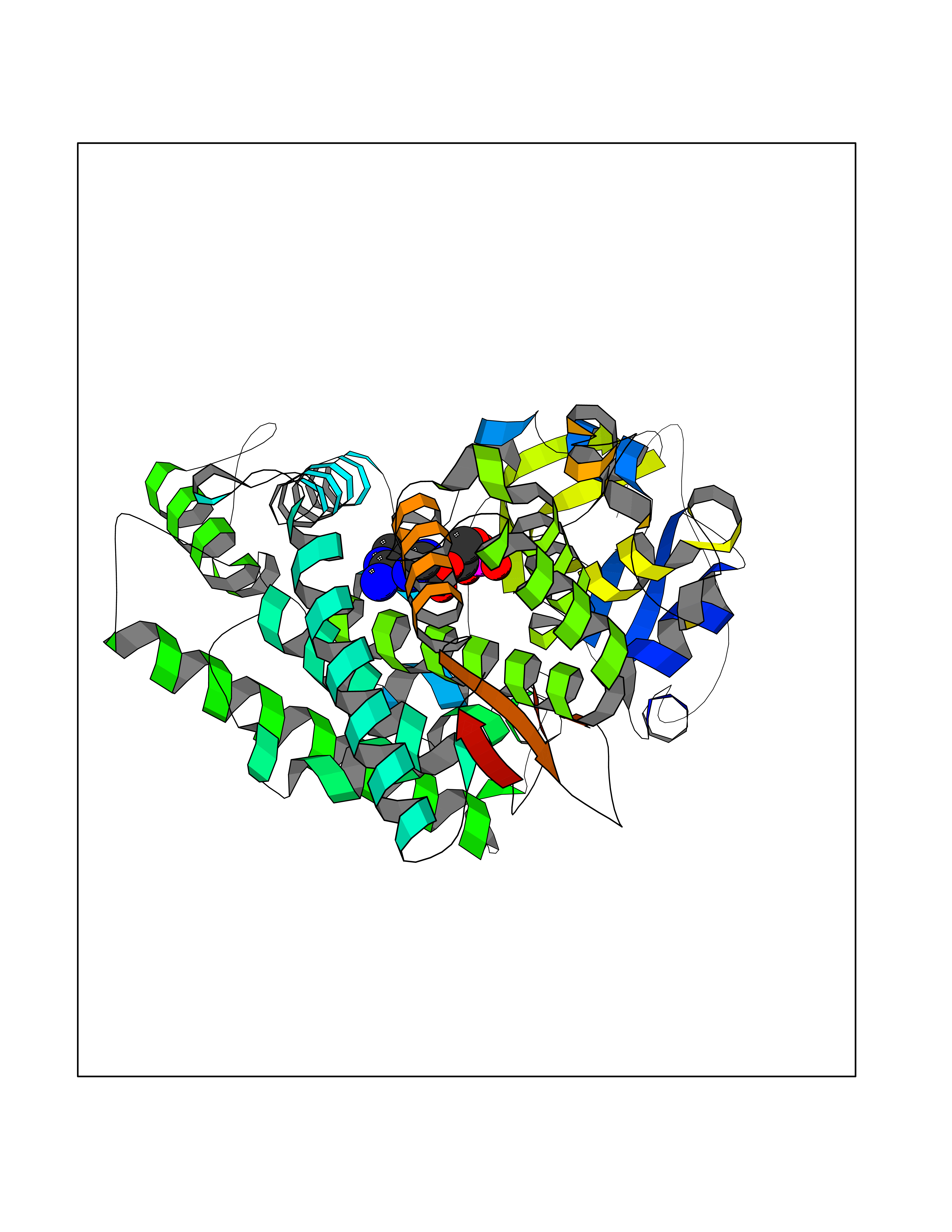 Molscript Map 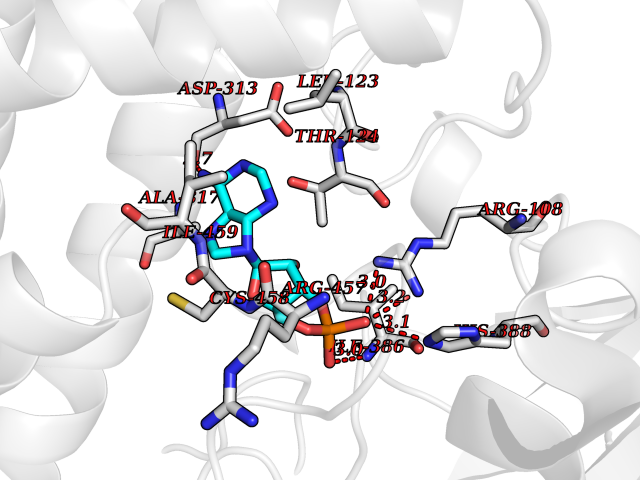 Pymol Map 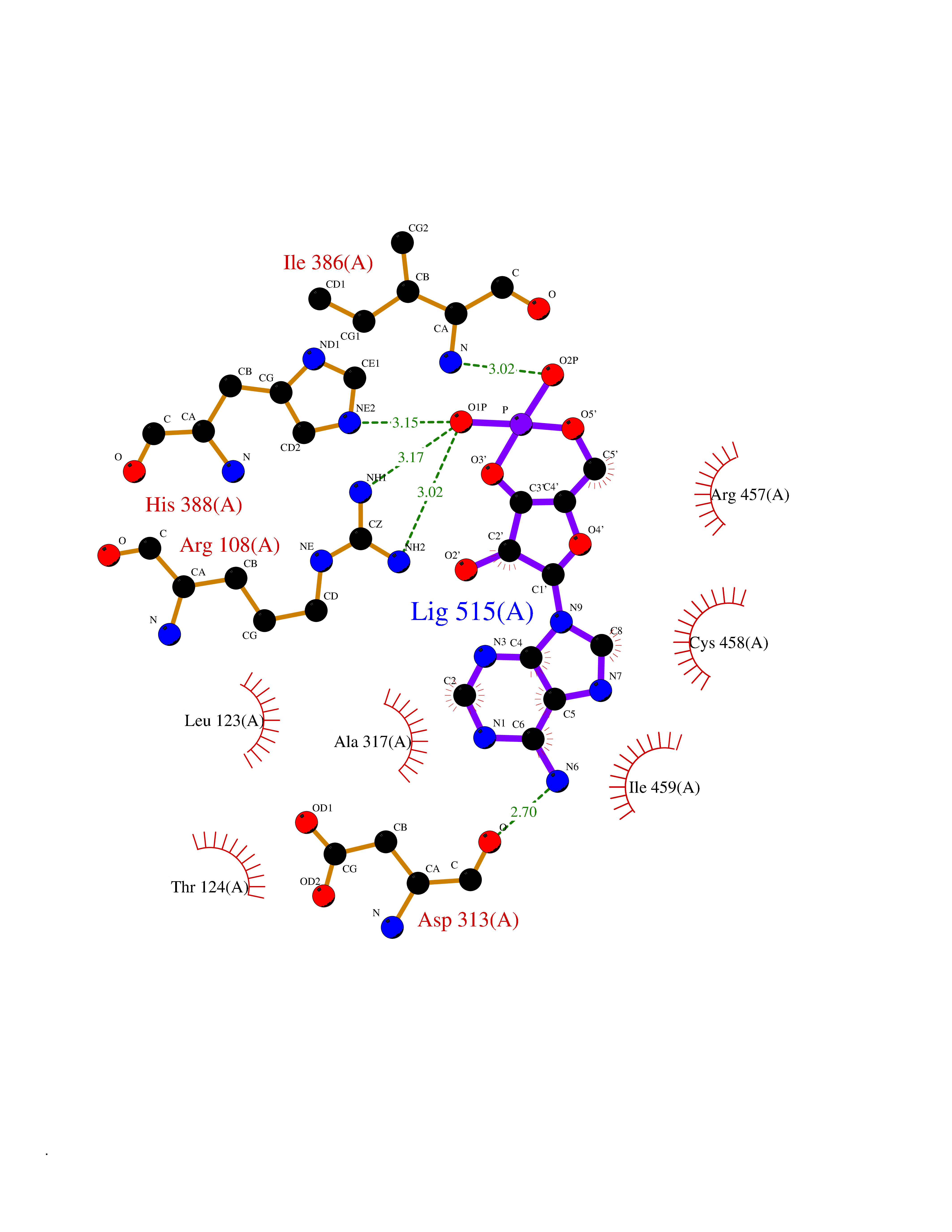 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Retinoic acid receptor RXR-beta (RXRB) | 5HJP | 6.64 | |
Target general information Gen name RXRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoid X receptor beta; Nuclear receptor subfamily 2 group B member 2; NR2B2 Protein family Nuclear hormone receptor family, NR2 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE). Receptor for retinoic acid. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB00459; DB00210; DB00523; DB00307; DB01393; DB03756; DB00926; DB01941; DB07929; DB02746; DB00412; DB00799; DB07080; DB00755 Interacts with Q00975; Q9HB07; F1D8P7; Q13133; Q13133-3; Q96RI1-1; P04150; Q9NRD5; P37231; P10276; P10276-2; P10826-2; P13631; Q6IQ16; Q13137; Q96B26; Q08379; Q6A162; Q9UJV3-2; Q13133-3; Q96RI1-1; O43586; P10276; P10826-2; Q8IUQ4-2; O75528; Q12800; Q9UBB9; Q05BL1; P14373; O94972; Q96S82 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 28845.8 Length 251 Aromaticity 0.08 Instability index 54.86 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -7.91 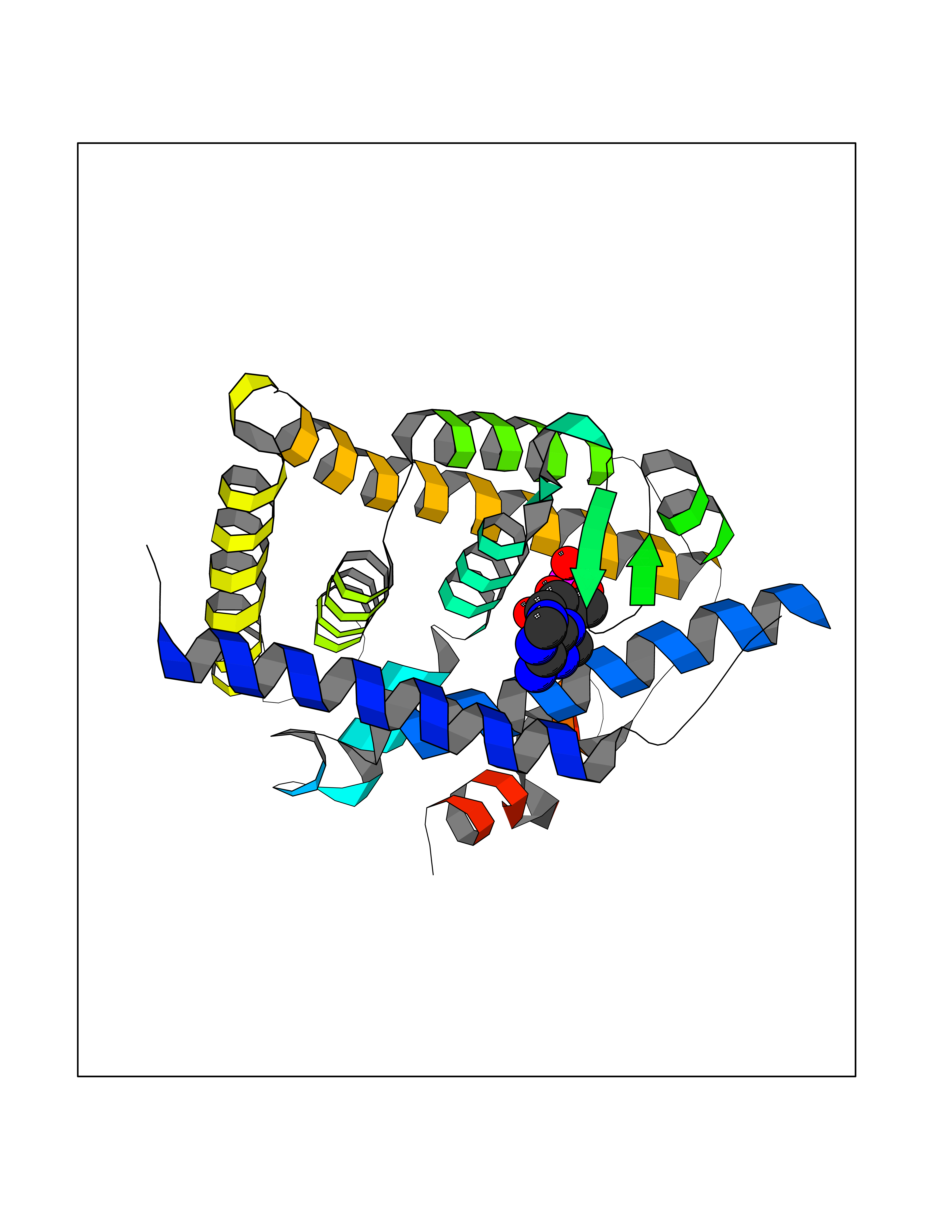 Molscript Map 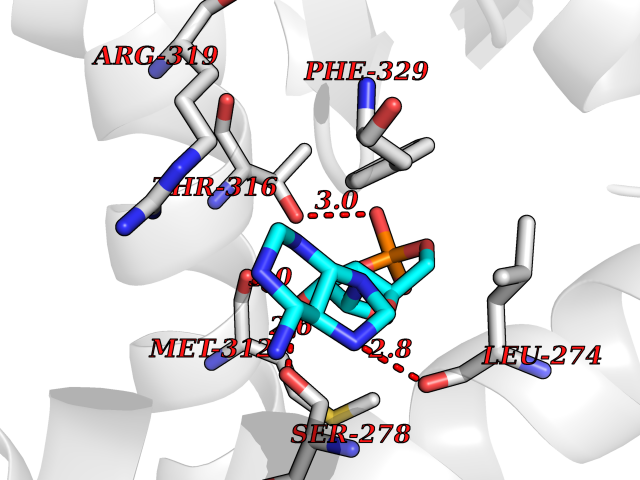 Pymol Map 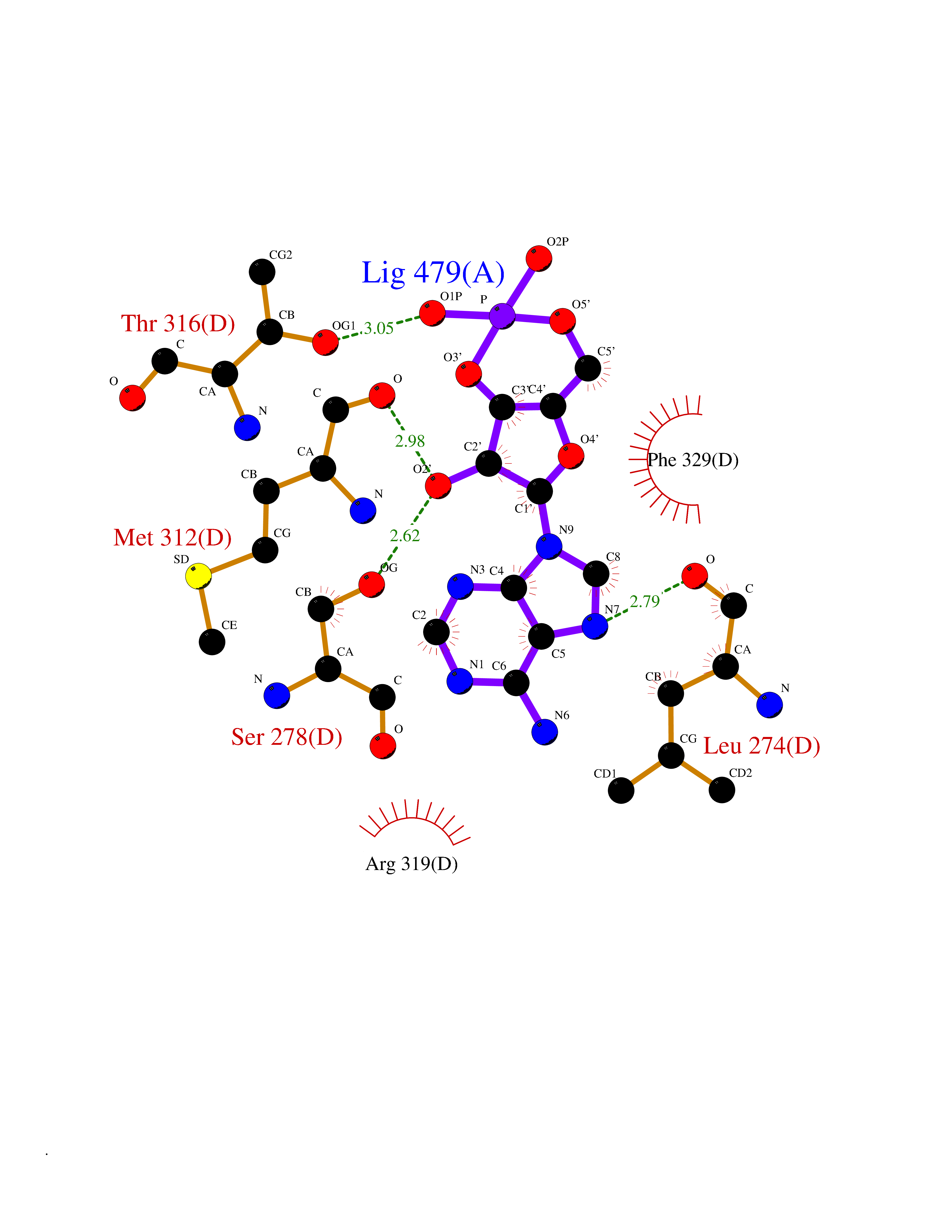 Ligplot Map 3D Binding mode Sequence QLTAAQELMIQQLVAAQLQCNKRSFSDQPKVTPWPSASQQRFAHFTELAIISVQEIVDFAKQVPGFLQLGREDQIALLKASTIEIMLLETARRYNHETECITFLKDFTYSKDDFHRAGLQVEFINPIFEFSRAMRRLGLDDAEYALLIAINIFSADRPNVQEPGRVEALQQPYVEALLSYTRIKRPQDQLRFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDVHEGSGSGSHKILHRLLQD Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Protein arginine methyltransferase 5 (PRMT5) | 7MXC | 6.64 | |
Target general information Gen name PRMT5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Shk1 kinase-binding protein 1 homolog; SKB1Hs; SKB1 homolog; SKB1; Protein arginine N-methyltransferase 5; Jak-binding protein 1; JBP1; IBP72; Histone-arginine N-methyltransferase PRMT5; HRMT1L5; 72 k Protein family Class I-like SAM-binding methyltransferase superfamily, Protein arginine N-methyltransferase family Biochemical class Methyltransferase Function Specifically mediates the symmetrical dimethylation of arginine residues in the small nuclear ribonucleoproteins Sm D1 (SNRPD1) and Sm D3 (SNRPD3); such methylation being required for the assembly and biogenesis of snRNP core particles. Methylates SUPT5H and may regulate its transcriptional elongation properties. Mono- and dimethylates arginine residues of myelin basic protein (MBP) in vitro. May play a role in cytokine-activated transduction pathways. Negatively regulates cyclin E1 promoter activity and cellular proliferation. Methylates histone H2A and H4 'Arg-3' during germ cell development. Methylates histone H3 'Arg-8', which may repress transcription. Methylates the Piwi proteins (PIWIL1, PIWIL2 and PIWIL4), methylation of Piwi proteins being required for the interaction with Tudor domain-containing proteins and subsequent localization to the meiotic nuage. Methylates RPS10. Attenuates EGF signaling through the MAPK1/MAPK3 pathway acting at 2 levels. First, monomethylates EGFR; this enhances EGFR 'Tyr-1197' phosphorylation and PTPN6 recruitment, eventually leading to reduced SOS1 phosphorylation. Second, methylates RAF1 and probably BRAF, hence destabilizing these 2 signaling proteins and reducing their catalytic activity. Required for induction of E-selectin and VCAM-1, on the endothelial cells surface at sites of inflammation. Methylates HOXA9. Methylates and regulates SRGAP2 which is involved in cell migration and differentiation. Acts as a transcriptional corepressor in CRY1-mediated repression of the core circadian component PER1 by regulating the H4R3 dimethylation at the PER1 promoter. Methylates GM130/GOLGA2, regulating Golgi ribbon formation. Methylates H4R3 in genes involved in glioblastomagenesis in a CHTOP- and/or TET1-dependent manner. Symmetrically methylates POLR2A, a modification that allows the recruitment to POLR2A of proteins including SMN1/SMN2 and SETX. This is required for resolving RNA-DNA hybrids created by RNA polymerase II, that form R-loop in transcription terminal regions, an important step in proper transcription termination. Along with LYAR, binds the promoter of gamma-globin HBG1/HBG2 and represses its expression. Symmetrically methylates NCL. Methylates TP53; methylation might possibly affect TP53 target gene specificity. Arginine methyltransferase that can both catalyze the formation of omega-N monomethylarginine (MMA) and symmetrical dimethylarginine (sDMA), with a preference for the formation of MMA. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P01019; Q9NX04; Q8WUW1; Q08289; P78371; Q16543; Q8N8U2; P54105; P21964-2; Q9NQ92; Q16526; Q9Y6K1; Q01094; Q08426; P38919; Q14241; O15197-2; Q6ZV65; P01100; O95995; P62993; Q8TE85; Q9BX10; P62805; P31269; Q00613; Q63ZY3; P03952; Q8TBB1; P06858; Q86UQ8-1; Q96HA8; Q8WVJ2; P24928; O14744; Q86U06; Q9BRS2; O75044; Q96RU7; P31930; P40337-2; Q9BQA1; P63104; Q96E35; Q91XC0; P03418; Q6ZV65-2 EC number EC 2.1.1.320 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Direct protein sequencing; Golgi apparatus; Methyltransferase; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 71188.3 Length 621 Aromaticity 0.11 Instability index 44.6 Isoelectric point 5.95 Charge (pH=7) -9.68 2D Binding mode Binding energy (Kcal/mol) -8.02 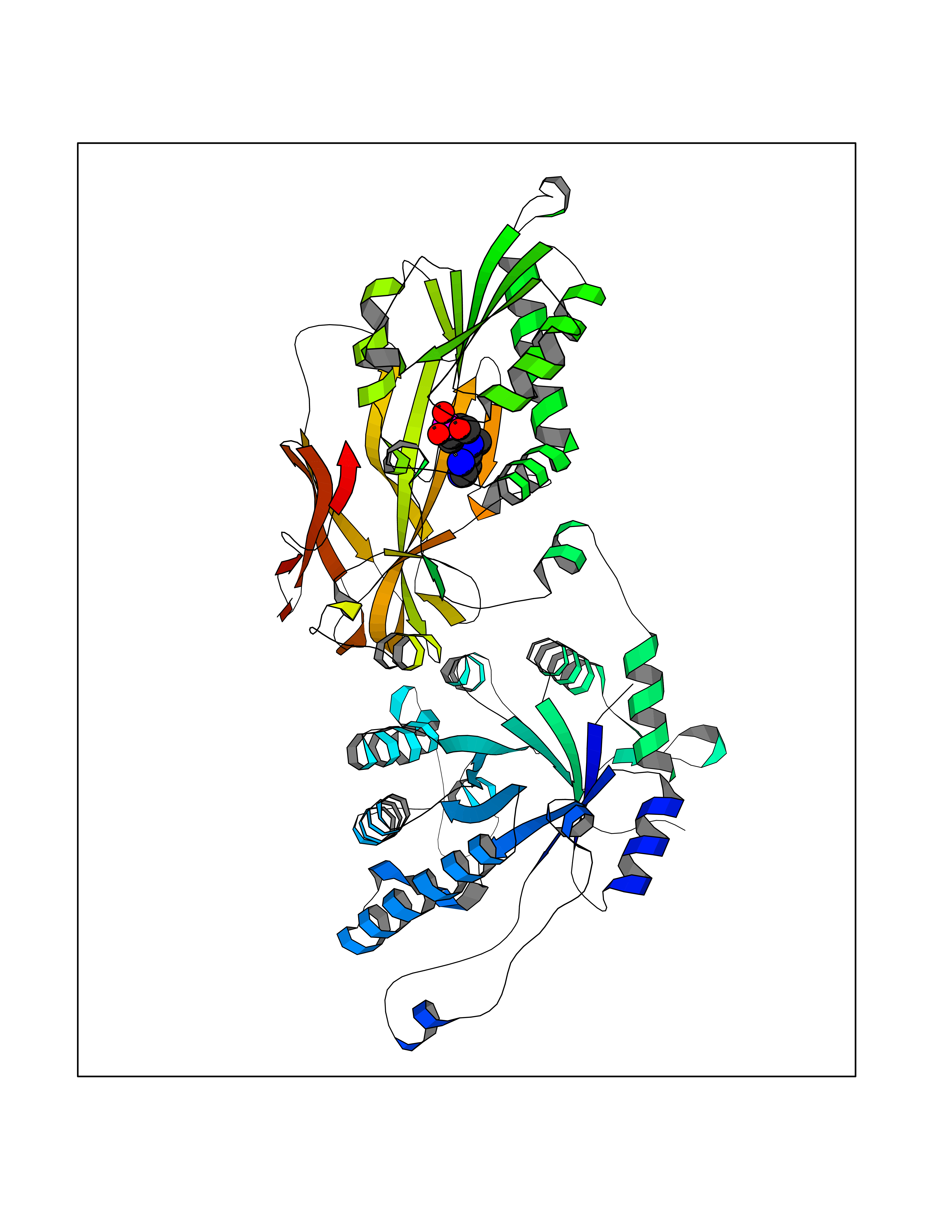 Molscript Map 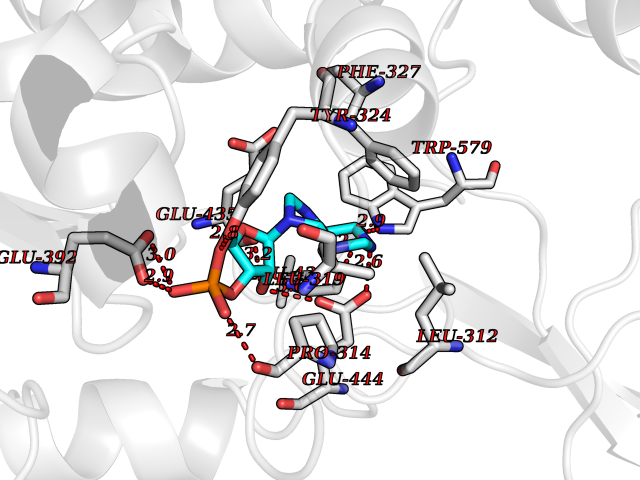 Pymol Map 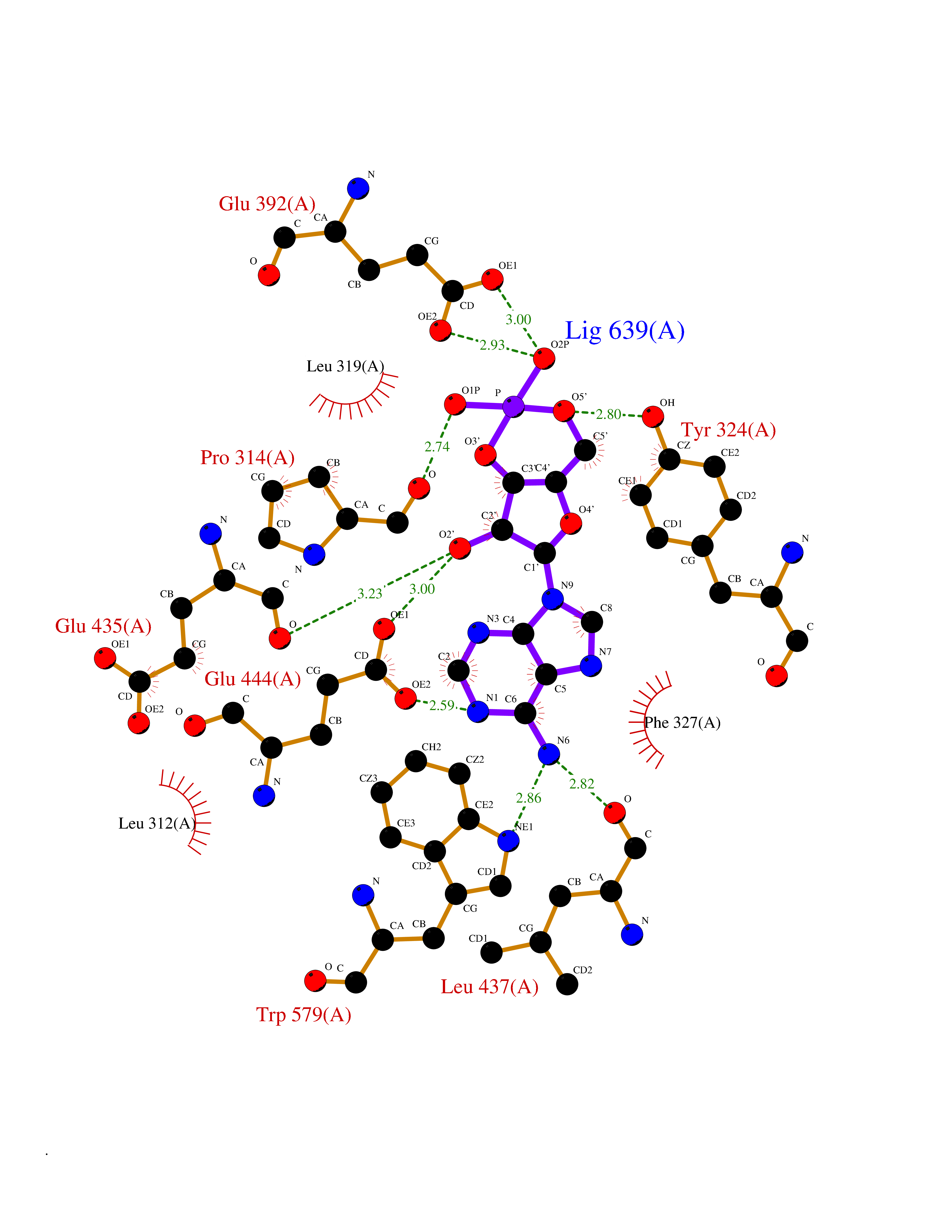 Ligplot Map 3D Binding mode Sequence RVSSGRDLNCVPEIADTLGAVAKQGFDFLCMPVFHPRFKREFIQEPAKNRPGPQTRSDLLLSGRDWNTLIVGKLSPWIRPDSKVEKIRRNSEAAMLQELNFGAYLGLPAFLLPLNQEDNTNLARVLTNHIHTGHHSSMFWMRVPLVAPEDLRDDIIENAPTTHTEEYSGEEKTWMWWHNFRTLCDYSKRIAVALEIGADLPSNHVIDRWLGEPIKAAILPTSIFLTNKKGFPVLSKMHQRLIFRLLKLEVQFIITGTNHHSCSYLQYLEYLSQNRPPPNAYELFAKGYEDYLQSPLQPLMDNLESQTYEVFEKDPIKYSQYQQAIYKCLLDRVPEEEKDTNVQVLMVLGAGRGPLVNASLRAAKQADRRIKLYAVEKNPNAVVTLENWQFEEWGSQVTVVSSDMREWVAPEKADIIVSELLGSFADNELSPECLDGAQHFLKDDGVSIPGEYTSFLAPISSSKLYNEVRACREKDRDPEAQFEMPYVVRLHNFHQLSAPQPCFTFSHPNRDPMIDNNRYCTLEFPVEVNTVLHGFAGYFETVLYQDITLSIRPETHSPGMFSWFPILFPIKQPITVREGQTICVRFWRCSNSKKVWYEWAVTAPVCSAIHNPTGRSYTIGL Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Voltage-gated sodium channel alpha Nav1.5 (SCN5A) | 6LQA | 6.63 | |
Target general information Gen name SCN5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-gated sodium channel subunit alpha Nav1.5; Sodium channel protein type V subunit alpha; Sodium channel protein type 5 subunit alpha; Sodium channel protein cardiac muscle subunit alpha; HH1 Protein family Sodium channel (TC 1.A.1.10) family, Nav1.5/SCN5A subfamily Biochemical class Voltage-gated ion channel Function Assuming opened or closed conformations in response to the voltage difference across the membrane, the protein forms a sodium-selective channel through which Na(+) ions may pass in accordance with their electrochemical gradient. It is a tetrodotoxin-resistant Na(+) channel isoform. This channel is responsible for the initial upstroke of the action potential. Channel inactivation is regulated by intracellular calcium levels. This protein mediates the voltage-dependent sodium ion permeability of excitable membranes. Related diseases Progressive familial heart block 1A (PFHB1A) [MIM:113900]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:11234013, ECO:0000269|PubMed:11804990, ECO:0000269|PubMed:12569159, ECO:0000269|PubMed:12574143, ECO:0000269|PubMed:19251209, ECO:0000269|PubMed:23420830}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Long QT syndrome 3 (LQT3) [MIM:603830]: A heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. {ECO:0000269|PubMed:10377081, ECO:0000269|PubMed:10508990, ECO:0000269|PubMed:10590249, ECO:0000269|PubMed:10627139, ECO:0000269|PubMed:10911008, ECO:0000269|PubMed:10973849, ECO:0000269|PubMed:11304498, ECO:0000269|PubMed:11410597, ECO:0000269|PubMed:11710892, ECO:0000269|PubMed:11889015, ECO:0000269|PubMed:11997281, ECO:0000269|PubMed:12209021, ECO:0000269|PubMed:12454206, ECO:0000269|PubMed:12673799, ECO:0000269|PubMed:15840476, ECO:0000269|PubMed:16414944, ECO:0000269|PubMed:16922724, ECO:0000269|PubMed:18060054, ECO:0000269|PubMed:18378609, ECO:0000269|PubMed:18451998, ECO:0000269|PubMed:18708744, ECO:0000269|PubMed:18848812, ECO:0000269|PubMed:18929331, ECO:0000269|PubMed:19716085, ECO:0000269|PubMed:26392562, ECO:0000269|PubMed:7651517, ECO:0000269|PubMed:7889574, ECO:0000269|PubMed:8541846, ECO:0000269|PubMed:9506831, ECO:0000269|PubMed:9686753, ECO:0000269|Ref.36}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 1 (BRGDA1) [MIM:601144]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:10532948, ECO:0000269|PubMed:10618304, ECO:0000269|PubMed:10690282, ECO:0000269|PubMed:11410597, ECO:0000269|PubMed:11748104, ECO:0000269|PubMed:11823453, ECO:0000269|PubMed:11901046, ECO:0000269|PubMed:12051963, ECO:0000269|PubMed:12106943, ECO:0000269|PubMed:15023552, ECO:0000269|PubMed:15338453, ECO:0000269|PubMed:15579534, ECO:0000269|PubMed:15851320, ECO:0000269|PubMed:16266370, ECO:0000269|PubMed:16325048, ECO:0000269|PubMed:16616735, ECO:0000269|PubMed:17075016, ECO:0000269|PubMed:17081365, ECO:0000269|PubMed:17198989, ECO:0000269|PubMed:18252757, ECO:0000269|PubMed:18341814, ECO:0000269|PubMed:18451998, ECO:0000269|PubMed:18456723, ECO:0000269|PubMed:18616619, ECO:0000269|PubMed:19251209, ECO:0000269|PubMed:19272188, ECO:0000269|PubMed:20129283, ECO:0000269|PubMed:23085483, ECO:0000269|PubMed:23420830, ECO:0000269|PubMed:24167619, ECO:0000269|PubMed:26279430, ECO:0000269|PubMed:26776555, ECO:0000269|PubMed:32850980, ECO:0000269|PubMed:9521325}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Sick sinus syndrome 1 (SSS1) [MIM:608567]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS1 onset is in utero, infancy, or early childhood. {ECO:0000269|PubMed:11748104, ECO:0000269|PubMed:14523039, ECO:0000269|PubMed:22795782}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Familial paroxysmal ventricular fibrillation 1 (VF1) [MIM:603829]: A cardiac arrhythmia marked by fibrillary contractions of the ventricular muscle due to rapid repetitive excitation of myocardial fibers without coordinated contraction of the ventricle and by absence of atrial activity. {ECO:0000269|PubMed:10940383}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Sudden infant death syndrome (SIDS) [MIM:272120]: SIDS is the sudden death of an infant younger than 1 year that remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of clinical history. Pathophysiologic mechanisms for SIDS may include respiratory dysfunction, cardiac dysrhythmias, cardiorespiratory instability, and inborn errors of metabolism, but definitive pathogenic mechanisms precipitating an infant sudden death remain elusive. {ECO:0000269|PubMed:18596570, ECO:0000269|PubMed:19302788}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Atrial standstill 1 (ATRST1) [MIM:108770]: A rare arrhythmia characterized by the absence of electrical and mechanical activity in the atria. Electrocardiographically, it is characterized by bradycardia, the absence of P waves, and a junctional narrow complex escape rhythm. {ECO:0000269|PubMed:12522116, ECO:0000269|PubMed:23420830}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry. A mutation in SCN5A has been detected in combination with a rare GJA5 genotype in a large family with atrial standstill.; DISEASE: Cardiomyopathy, dilated, 1E (CMD1E) [MIM:601154]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:15466643, ECO:0000269|PubMed:23420830}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Atrial fibrillation, familial, 10 (ATFB10) [MIM:614022]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:18088563, ECO:0000269|PubMed:18378609}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01426; DB09088; DB01429; DB00868; DB13746; DB05541; DB00564; DB01161; DB00527; DB00907; DB13269; DB00280; DB04855; DB01228; DB00754; DB13961; DB01195; DB01320; DB00473; DB00192; DB11633; DB00555; DB00281; DB00532; DB00379; DB00680; DB00776; DB11186; DB00252; DB09345; DB00750; DB01035; DB01069; DB01182; DB09342; DB00908; DB01346; DB00243; DB00740; DB09085; DB01056; DB00273; DB00313; DB06217; DB00909 Interacts with P0DP25; Q13557; P61328-2; P26045; Q49AR9; Q8N9N5-2; Q9Y3B6; Q8WW24; Q96E35 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Atrial fibrillation; Brugada syndrome; Calmodulin-binding; Cardiomyopathy; Cell junction; Cell membrane; Cytoplasm; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Long QT syndrome; Membrane; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Ubl conjugation; Voltage-gated channel Protein physicochemical properties Chain ID B Molecular weight (Da) 131190 Length 1151 Aromaticity 0.14 Instability index 32.8 Isoelectric point 7.56 Charge (pH=7) 1.76 2D Binding mode Binding energy (Kcal/mol) -7.1 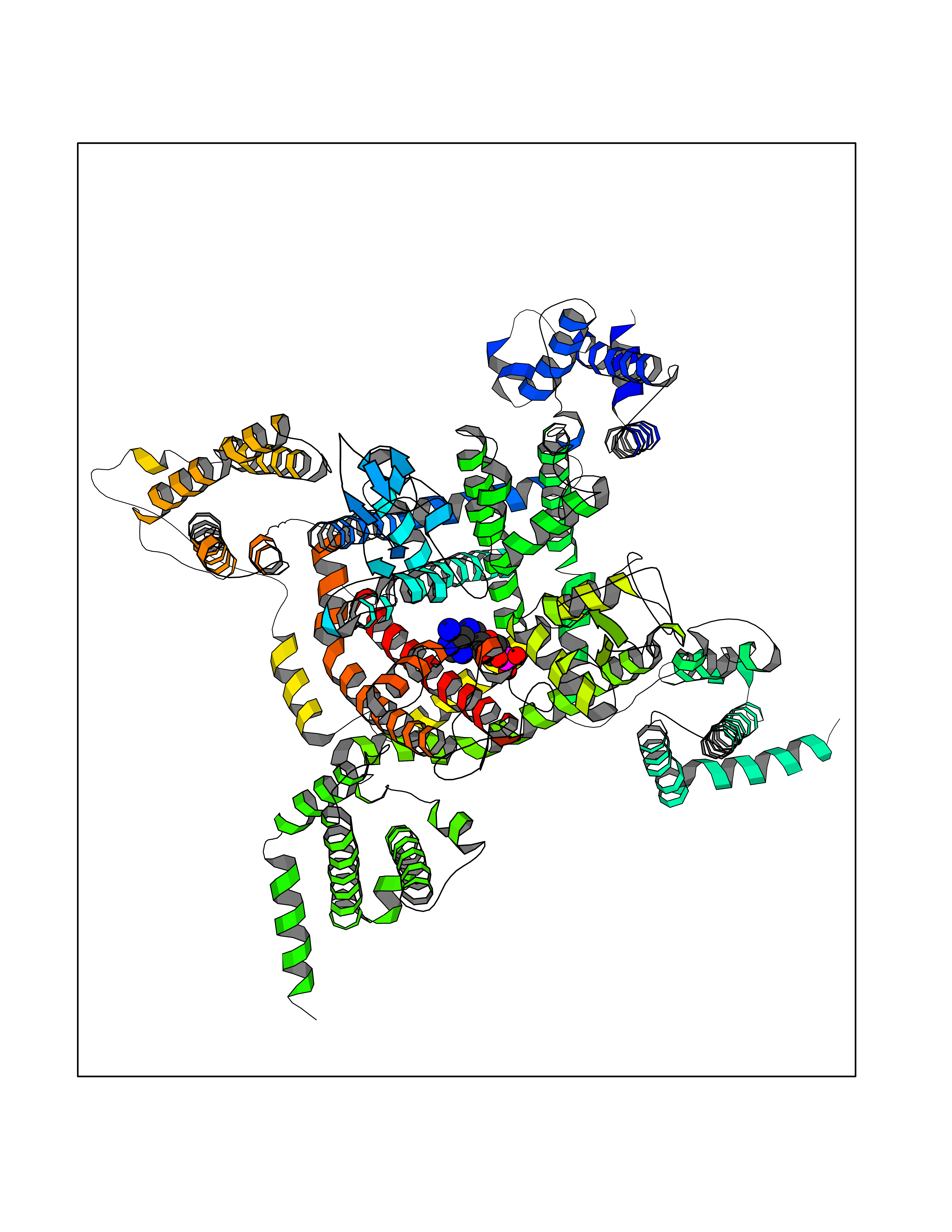 Molscript Map 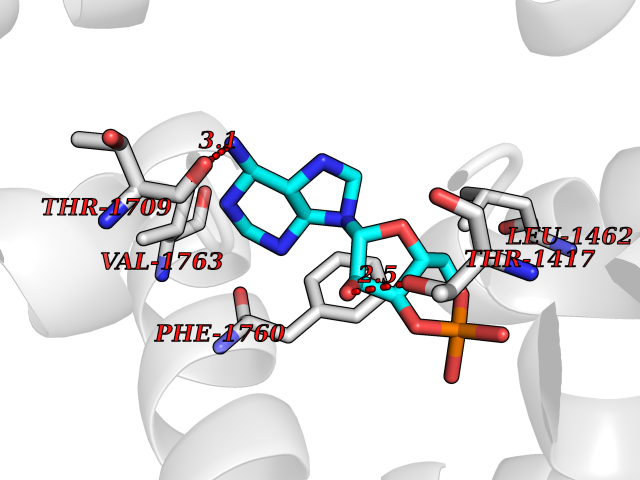 Pymol Map 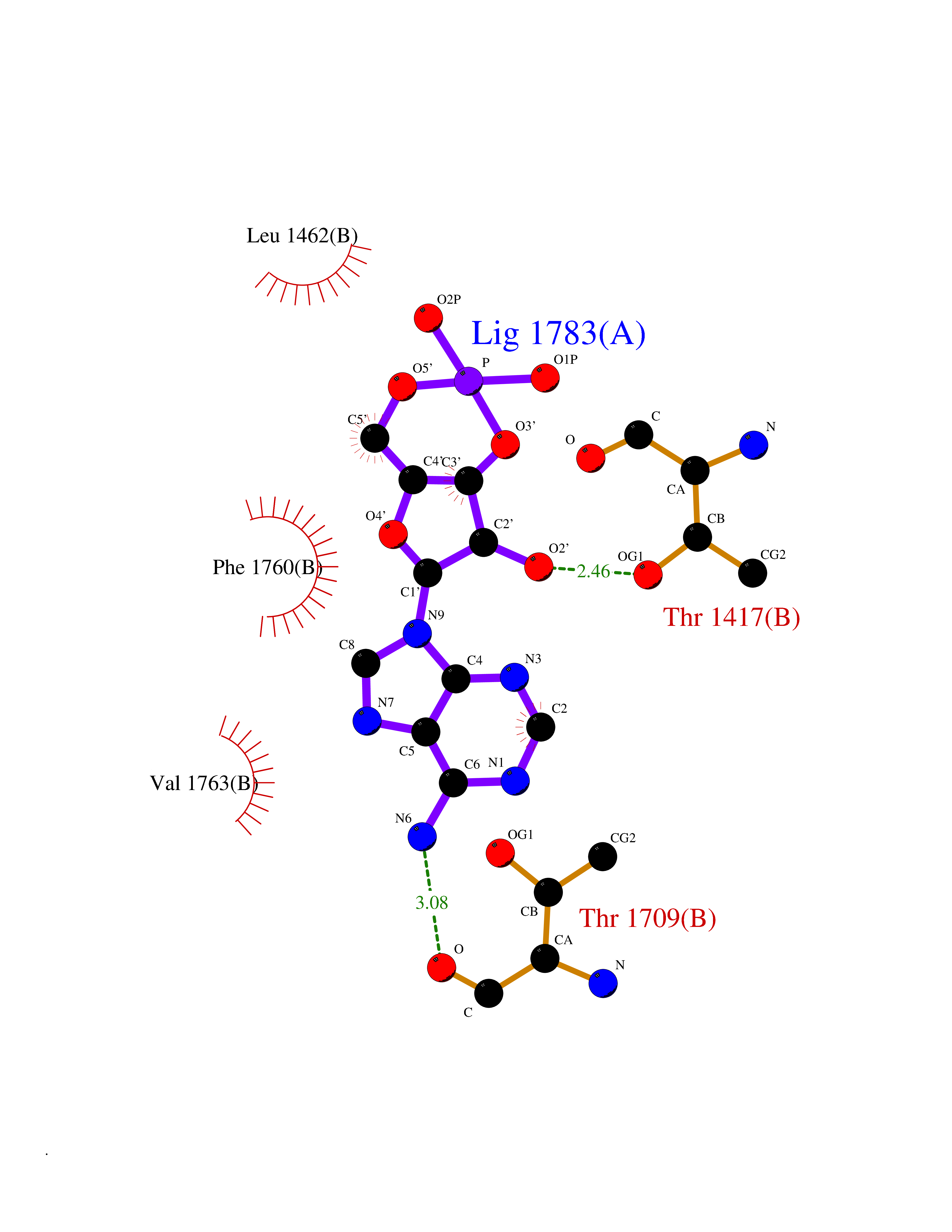 Ligplot Map 3D Binding mode Sequence PIRRAAVKILVHSLFNMLIMCTILTNCVFMAQHDPPPWTKYVEYTFTAIYTFESLVKILARGFCLHAFTFLRDPWNWLDFSVIIMAYTTEFVDLGNVSALRTFRVLRALKTISVISGLKTIVGALIQSVKKLADVMVLTVFCLSVFALIGLQLFMGNLRHKCVRNFTALNGTNGSVEADGLVWESLDLYLSDPENYLLKNGTSDVLLCGNSSDAGTCPEGYRCLKAGENPDHGYTSFDSFAWAFLALFRLMTQDCWERLYQQTLRSAGKIYMIFFMLVIFLGSFYLVNLILAVVAMAYEEQNQATIAETEECCPLWMSIKQGVKLVVMDPFTDLTITMCIVLNTLFMALEHYNMTSEFEEMLQVGNLVFTGIFTAEMTFKIIALDPYYYFQQGWNIFDSIIVILSLMELGLSRMSNLSVLRSFRLLRVFKLAKSWPTLNTLIKIIGNSVGALGNLTLVLAIIVFIFAVVGMQLFGKNYSELRDSDSGLLPRWHMMDFFHAFLIIFRILCGEWIETMWDCMEVSGQSLCLLVFLLVMVIGNLVVLNLFLALLLSSFSAGKVWWRLRKTCYHIVEHSWFETFIIFMILLSSGALAFEDIYLEERKTIKVLLEYADKMFTYVFVLEMLLKWVAYGFKKYFTNAWCWLDFLIVDVSLVSLVANTLGFAEMGPIKSLRTLRALRPLRALSRFEGMRVVVNALVGAIPSIMNVLLVCLIFWLIFSIMGVNLFAGKFGRCINQTEGDLPLNYTIVNNKSQCESLNLTGELYWTKVKVNFDNVGAGYLALLQVATFKGWMDIMYAAVDSRGYEEQPQWEYNLYMYIYFVIFIIFGSFFTLNLFIGVIIDNFNQQKKKLGGQDIFMTEEQKKYYNAMKKLGSKKPQKPIPRPLNKYQGFIFDIVTKQAFDVTIMFLICLNMVTMMVETDDQSPEKINILAKINLLFVAIFTGECIVKLAALRHYYFTNSWNIFDFVVVILSIVGTVLSDIIQKYFFSPTLFRVIRLARIGRILRLIRGAKGIRTLLFALMMSLPALFNIGLLLFLVMFIYSIFGMANFAYVKWEAGIDDMFNFQTFANSMLCLFQITTSAGWDGLLSPILNTGPPYCDPTLPNSNGSRGDCGSPAVGILFFTTYIIISFLIVVNMYIAIILENFSVATEE Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Fatty acid synthase (FASN) | 3TJM | 6.61 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -7.17 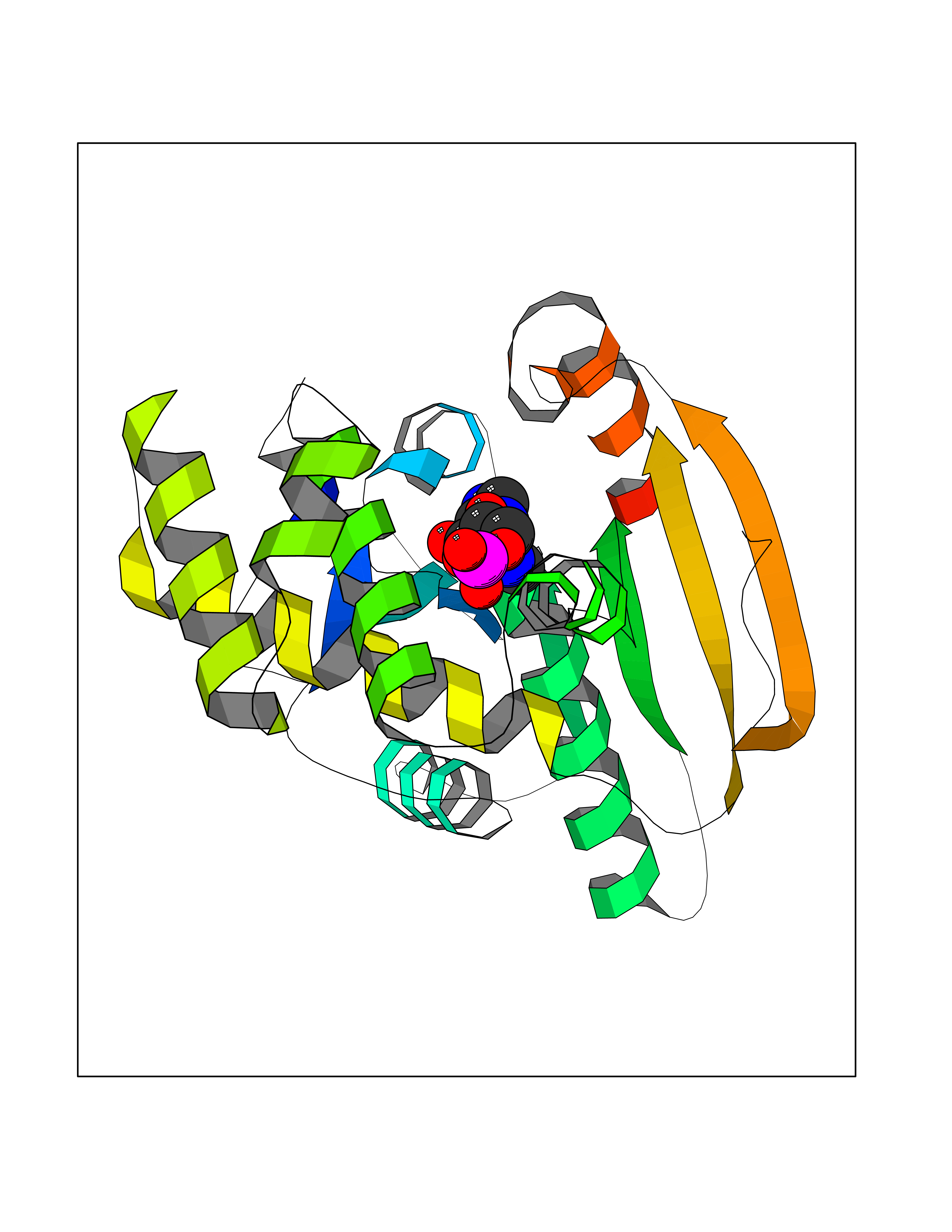 Molscript Map 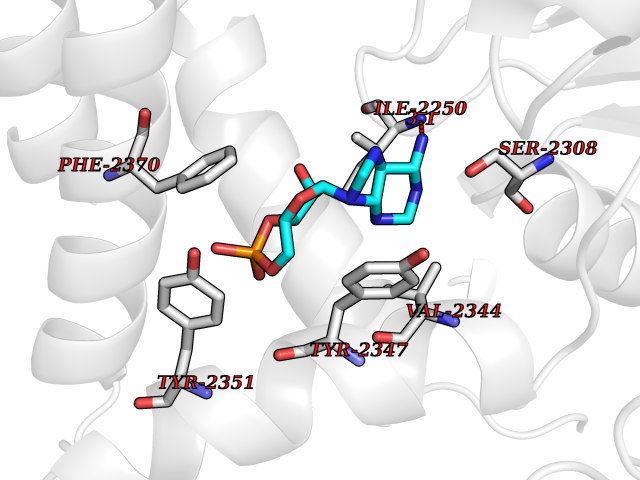 Pymol Map 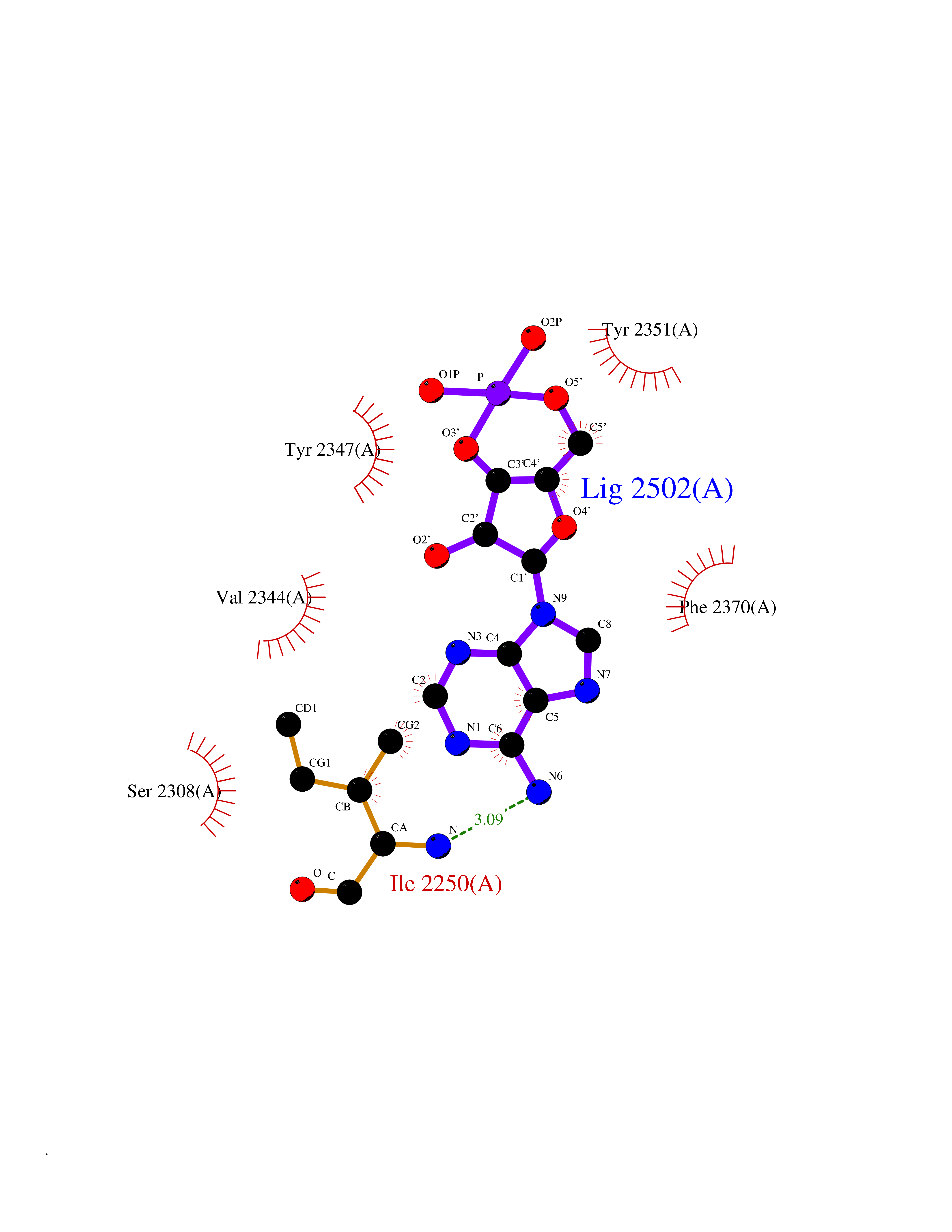 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Ubiquitin-activating enzyme E1 (UBAE1) | 6DC6 | 6.61 | |
Target general information Gen name UBA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-like modifier-activating enzyme 1; UBE1; Protein A1S9; A1S9T Protein family Ubiquitin-activating E1 family Biochemical class NA Function Catalyzes the first step in ubiquitin conjugation to mark cellular proteins for degradation through the ubiquitin-proteasome system. Activates ubiquitin by first adenylating its C-terminal glycine residue with ATP, and thereafter linking this residue to the side chain of a cysteine residue in E1, yielding a ubiquitin-E1 thioester and free AMP. Essential for the formation of radiation-induced foci, timely DNA repair and for response to replication stress. Promotes the recruitment of TP53BP1 and BRCA1 at DNA damage sites. Related diseases Spinal muscular atrophy X-linked 2 (SMAX2) [MIM:301830]: A lethal infantile form of spinal muscular atrophy, a neuromuscular disorder characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. Clinical features include hypotonia, areflexia, and multiple congenital contractures. {ECO:0000269|PubMed:18179898, ECO:0000269|PubMed:23518311}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: VEXAS syndrome (VEXAS) [MIM:301054]: A sporadic, often fatal, treatment-refractory inflammatory syndrome that develops in late adulthood. Clinical features include fevers, cytopenias, characteristic vacuoles in myeloid and erythroid precursor cells, dysplastic bone marrow, neutrophilic cutaneous and pulmonary inflammation, chondritis, and vasculitis. The disease affects only males and is associated with de novo somatic mutations. {ECO:0000269|PubMed:33108101}. The disease is caused by variants affecting the gene represented in this entry. Somatic variants affecting the initiator methionine of isoform 2 are recurrently found in VEXAS patients. These variants cause loss of isoform 2 and production of a shorter isoform with strongly reduced enzymatic activity from a downstream methionine (Met-67). {ECO:0000269|PubMed:33108101}. Drugs (DrugBank ID) DB04119; DB04216 Interacts with Q9H2C0; P42858; Q96FW1; Q9BUZ4; P63279; O00308; Q76353 EC number EC 6.2.1.45 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Ligase; Mitochondrion; Neurodegeneration; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 119604 Length 1068 Aromaticity 0.09 Instability index 36.15 Isoelectric point 5.4 Charge (pH=7) -26.22 2D Binding mode Binding energy (Kcal/mol) -6.84 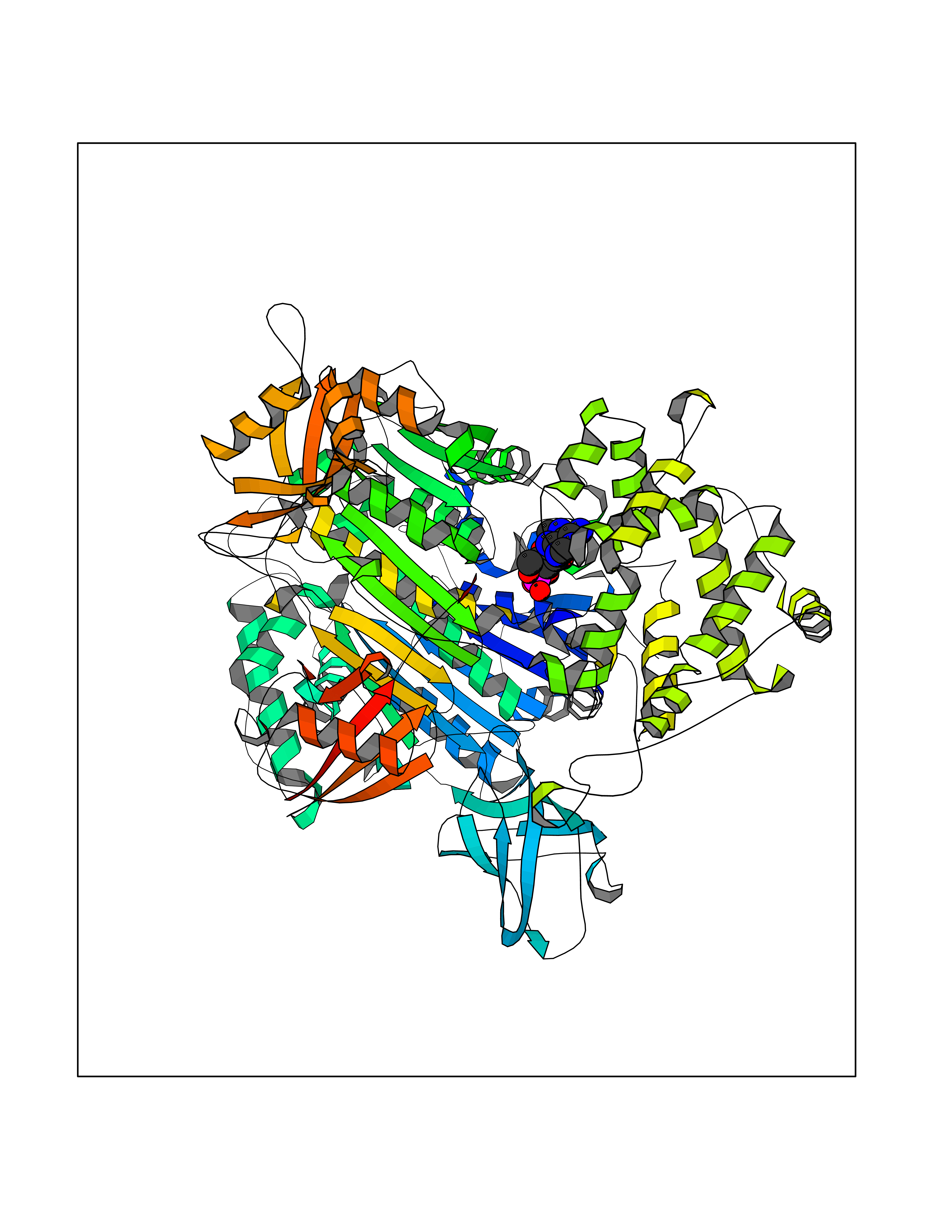 Molscript Map 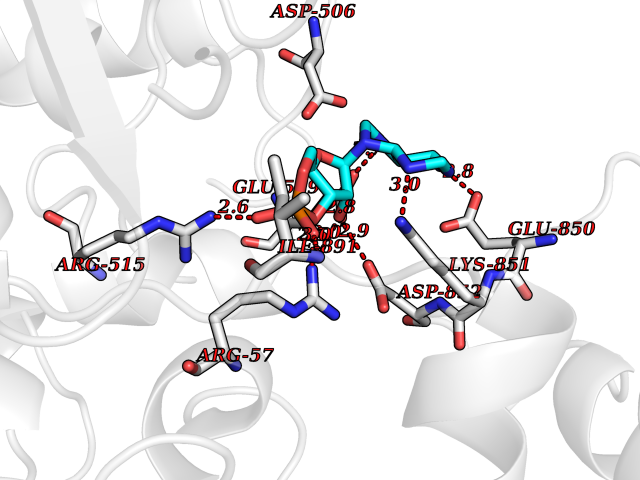 Pymol Map  Ligplot Map 3D Binding mode Sequence DIDEGLYSRQLYVLGHEAMKRLQTSSVLVSGLRGLGVEIAKNIILGGVKAVTLHDQGTAQWADLSSQFYLREEDIGKNRAEVSQPRLAELNSYVPVTAYTGPLVEDFLSGFQVVVLTNTPLEDQLRVGEFCHNRGIKLVVADTRGLFGQLFCDFGEEMILTDSNGEQPLSAMVSMVTKDNPGVVTCLDEARHGFESGDFVSFSEVQGMVELNGNQPMEIKVLGPYTFSICDTSNFSDYIRGGIVSQVKVPKKISFKSLVASLAEPDFVVTDFAKFSRPAQLHIGFQALHQFCAQHGRPPRPRNEEDAAELVALAQAVNARALPAVQQNNLDEDLIRKLAYVAAGDLAPINAFIGGLAAQEVMKACSGKFMPIMQWLYFDALECLPEDKEVLTEDKCLQRQNRYDGQVAVFGSDLQEKLGKQKYFLVGAGAIGCELLKNFAMIGLGCGEGGEIIVTDMDTIEKSNLNRQFLFRPWDVTKLKSDTAAAAVRQMNPHIRVTSHQNRVGPDTERIYDDDFFQNLDGVANALDNVDARMYMDRRCVYYRKPLLESGTLGTKGNVQVVIPFLTESYSSSQDPPEKSIPIATLKNFPNAIEHTLQWARDEFEGLFKQPAENVNQYLTDPKFVERTLRLAGTQPLEVLEAVQRSLVLQRPQTWADCVTWACHHWHTQYSNNIRQLLHNFPPDQLTSSGAPFWSGPKRCPHPLTFDVNNPLHLDYVMAAANLFAQTYGLTGSQDRAAVATFLQSVQVPEFTPKSVDDSRLEELKATLPSPDKLPGFKMYPIDFEKDDDSNFHMDFIVAASNLRAENYDIPSADRHKSKLIAGKIIPAIATTTAAVVGLVCLELYKVVQGHRQLDSYKNGFLNLALPFFGFSEPLAAPRHQYYNQEWTLWDRFEVQGLQPNGEEMTLKQFLDYFKTEHKLEITMLSQGVSMLYSFFMPAAKLKERLDQPMTEIVSRVSKRKLGRHVRALVLELCCNDESGEDVEVPYVRYTIMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG Hydrogen bonds contact Hydrophobic contact | ||||