Job Results:
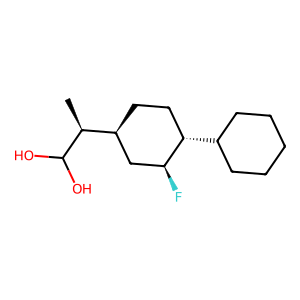
Ligand
Structure
Job ID
6a37619723fa756e073677fca12f4afa
Job name
NA
Time
2024-07-14 16:31:23
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Protein cereblon (CRBN) | 5FQD | 7.63 | |
Target general information Gen name CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein cereblon Protein family CRBN family Biochemical class NA Function Substrate recognition component of a DCX (DDB1-CUL4-X-box) E3 protein ligase complex that mediates the ubiquitination and subsequent proteasomal degradation of target proteins, such as MEIS2. Normal degradation of key regulatory proteins is required for normal limb outgrowth and expression of the fibroblast growth factor FGF8. May play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium channels in brain regions involved in memory and learning via its interaction with KCNT1. Binding of pomalidomide and other thalidomide-related drugs changes the substrate specificity of the human protein, leading to decreased degradation of MEIS2 and other target proteins and increased degradation of MYC, IRF4, IKZF1 and IKZF3. Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00480; DB08910; DB01041 Interacts with Q96A83-2; P48729; Q16531; O14901; Q8IVT2; Q9P286; A0A6Q8PF08; Q93062; Q16531; Q13422-7 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; Intellectual disability; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID B,E Molecular weight (Da) 38245.7 Length 337 Aromaticity 0.08 Instability index 40.62 Isoelectric point 5.7 Charge (pH=7) -6.53 2D Binding mode Binding energy (Kcal/mol) -9.32 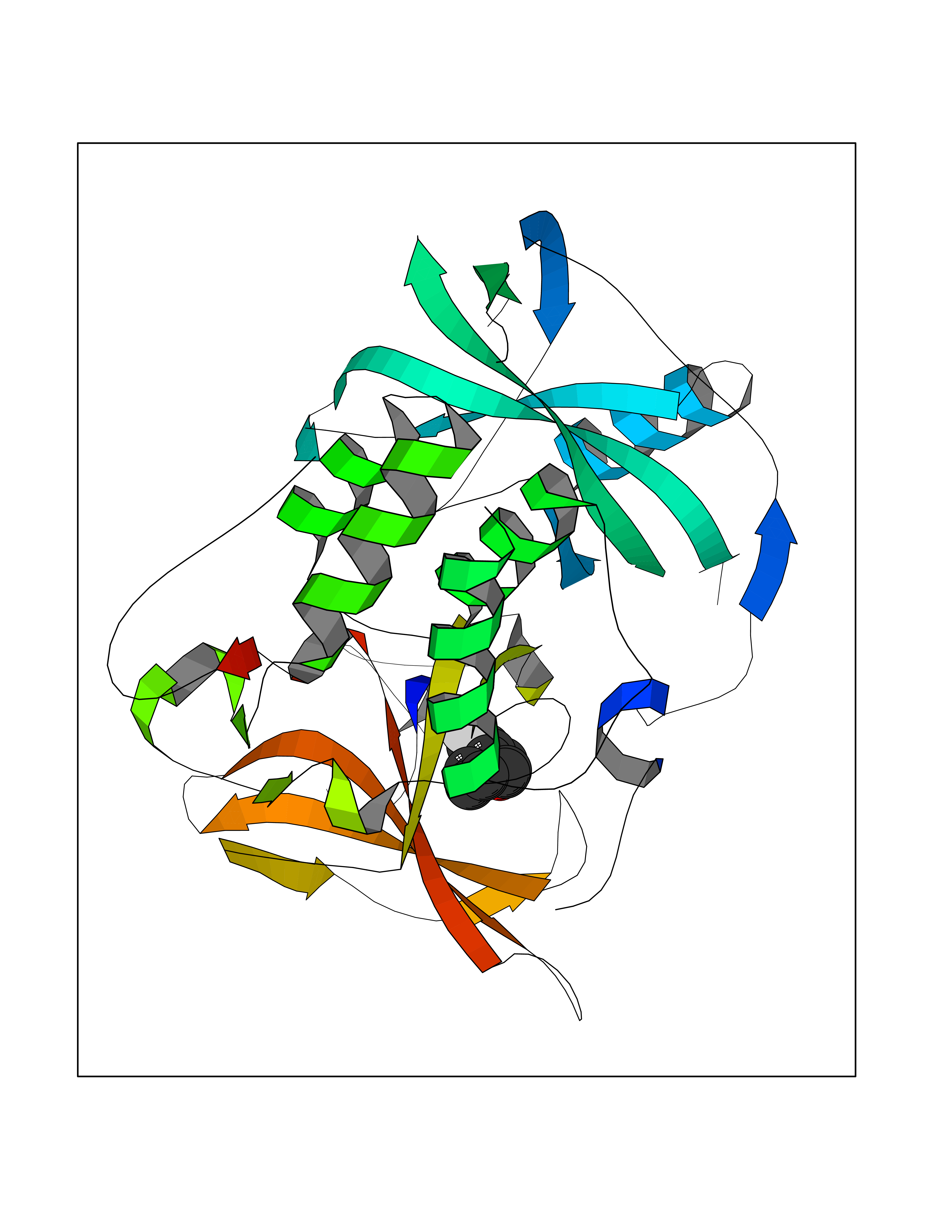 Molscript Map 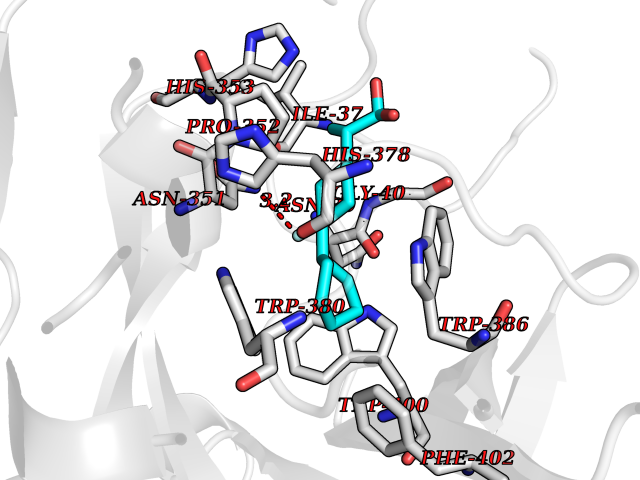 Pymol Map 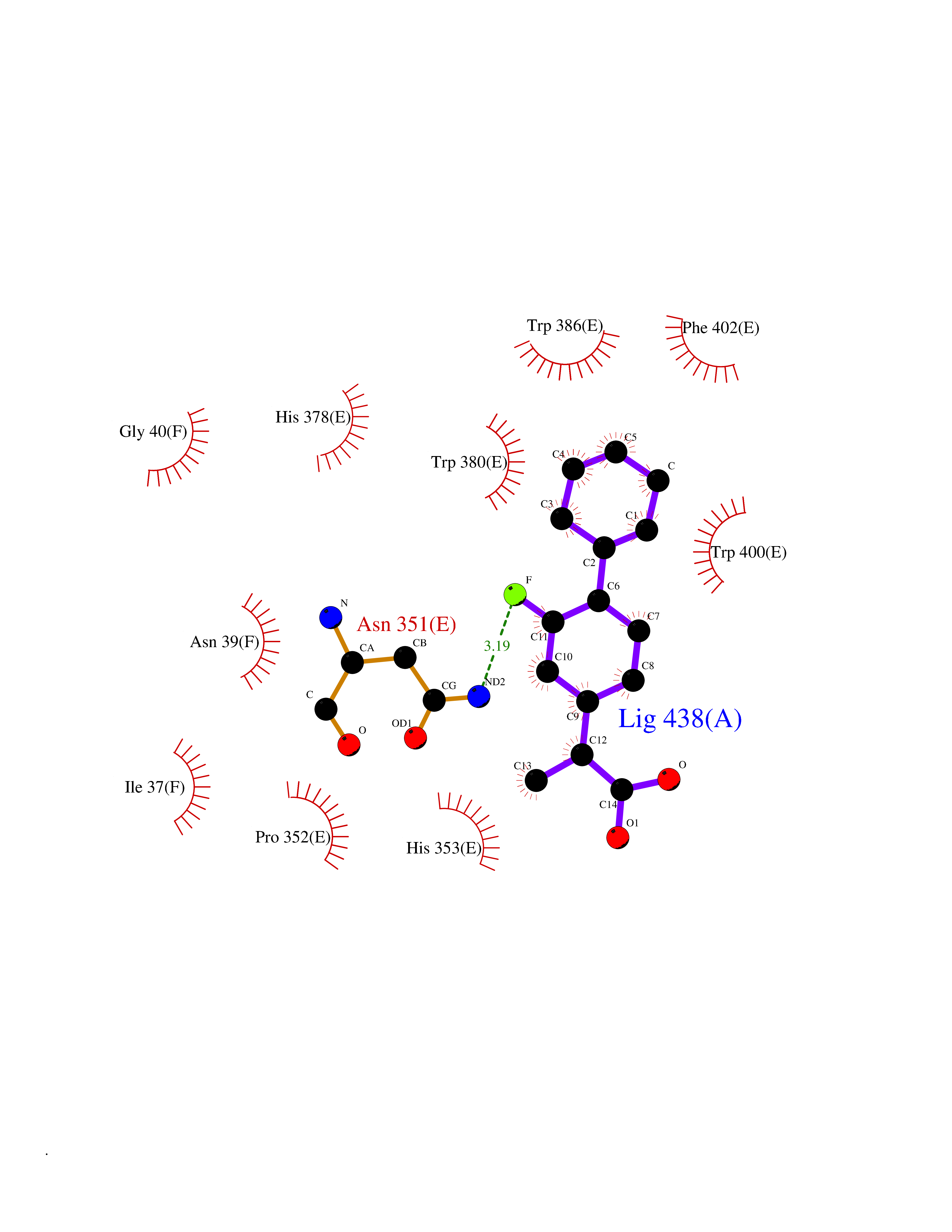 Ligplot Map 3D Binding mode Sequence EFIVGGKYKLNITNGEEVAVINFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEIVKVKAIGRQRFKVLEQQAKVQILPECVLAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPD Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Lanosterol 14-alpha demethylase (CYP51A1) | 4UHI | 7.63 | |
Target general information Gen name CYP51A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450LI; Cytochrome P45014DM; Cytochrome P450-14DM; Cytochrome P450 51A1 Protein family Cytochrome P450 family Biochemical class Cytochrome P450 family Function Catalyzes C14-demethylation of lanosterol; it transforms lanosterol into 4,4'-dimethyl cholesta-8,14,24-triene-3-beta-ol. Related diseases Spondyloepimetaphyseal dysplasia, short limb-hand type (SEMD-SL) [MIM:271665]: A bone disease characterized by short-limbed dwarfism, a narrow chest with pectus excavatum, brachydactyly in the hands and feet, a characteristic craniofacial appearance and premature calcifications. The radiological findings are distinctive and comprise short long bones throughout the skeleton with striking epiphyses that are stippled, flattened and fragmented and flared, irregular metaphyses. Platyspondyly in the spine with wide intervertebral spaces is observed and some vertebral bodies are pear-shaped with central humps, anterior protrusions and posterior scalloping. {ECO:0000269|PubMed:19110212, ECO:0000269|PubMed:20223752, ECO:0000269|PubMed:26463668}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Warburg-Cinotti syndrome (WRCN) [MIM:618175]: An autosomal dominant disease characterized by progressive corneal neovascularization, keloid formation, chronic skin ulcers, wasting of subcutaneous tissue, flexion contractures of the fingers, and acro-osteolysis. {ECO:0000269|PubMed:30449416}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07705; DB05667; DB01110; DB01007 Interacts with NA EC number EC 1.14.14.154 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid biosynthesis; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 53013.3 Length 462 Aromaticity 0.11 Instability index 47.66 Isoelectric point 8.8 Charge (pH=7) 7 2D Binding mode Binding energy (Kcal/mol) -7.98  Molscript Map 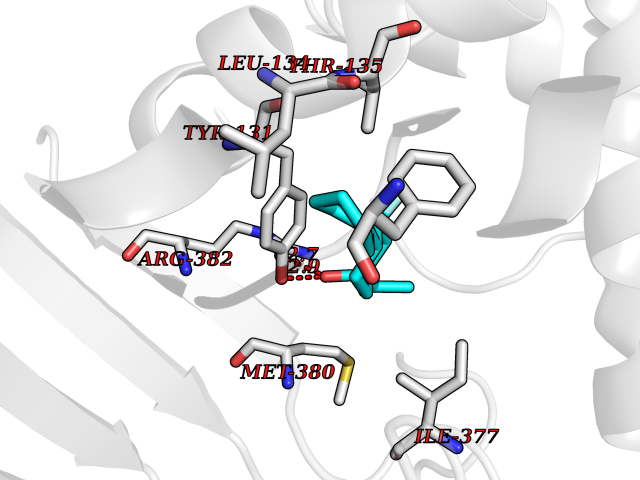 Pymol Map 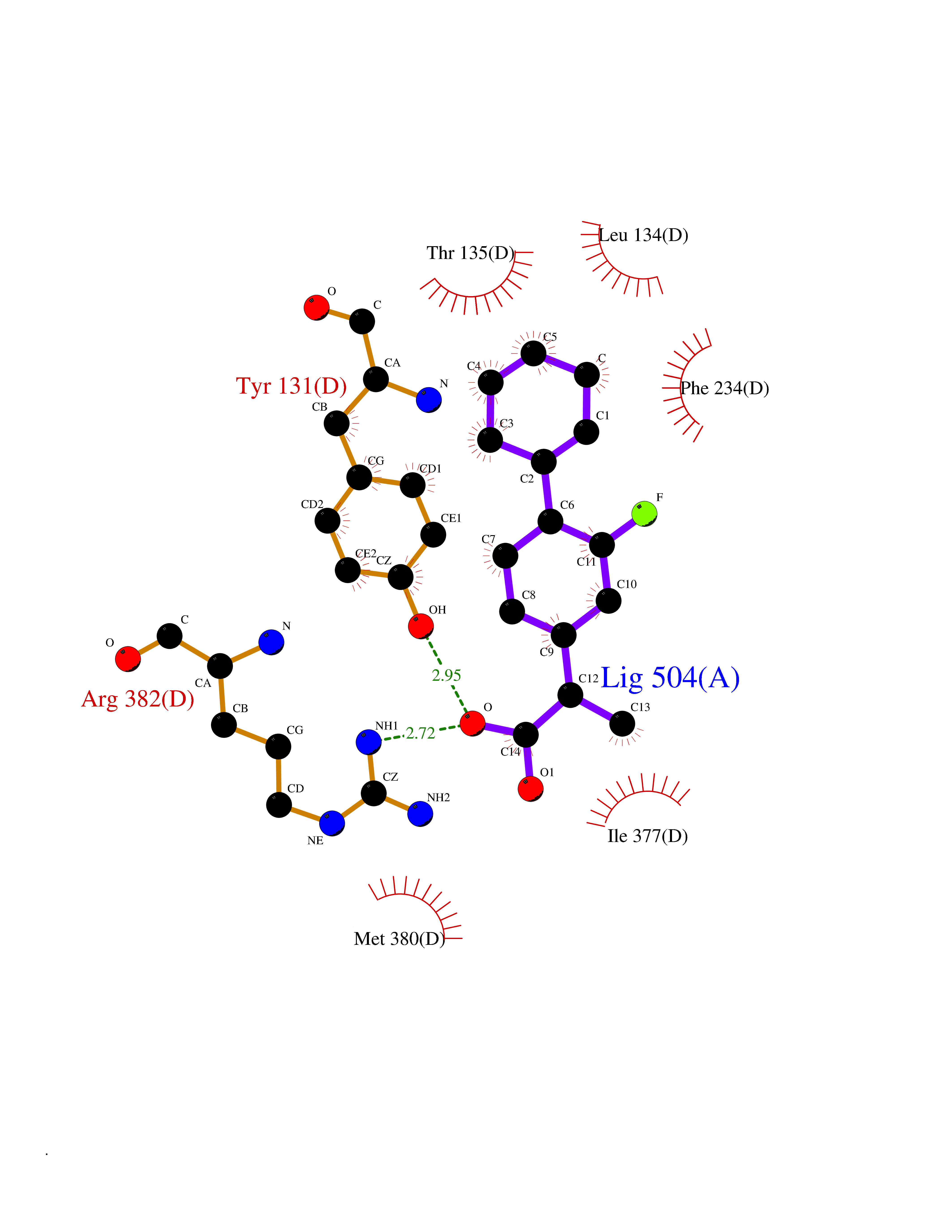 Ligplot Map 3D Binding mode Sequence PPYIFSPIPFLGHAIAFGKSPIEFLENAYEKYGPVFSFTMVGKTFTYLLGSDAAALLFNSKNEDLNAEDVYSRLTTPVFGKGVAYDVPNPVFLEQKKMLKSGLNIAHFKQHVSIIEKETKEYFESWGESGEKNVFEALSELIILTASHCLHGKEIRSQLNEKVAQLYADLDGGFSHAAWLLPGWLPLPSFRRRDRAHREIKDIFYKAIQKRRQSQEKIDDILQTLLDATYKDGRPLTDDEVAGMLIGLLLAGQHTSSTTSAWMGFFLARDKTLQKKCYLEQKTVCGENLPPLTYDQLKDLNLLDRCIKETLRLRPPIMIMMRMARTPQTVAGYTIPPGHQVCVSPTVNQRLKDSWVERLDFNPDRYLQDNPASGEKFAYVPFGAGRHRCIGENFAYVQIKTIWSTMLRLYEFDLIDGYFPTVNYTTMIHTPENPVIRYKRRSLPGWLPLPSFRRRDRAHREI Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Vitamin D3 receptor (VDR) | 3B0T | 7.62 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -8.92 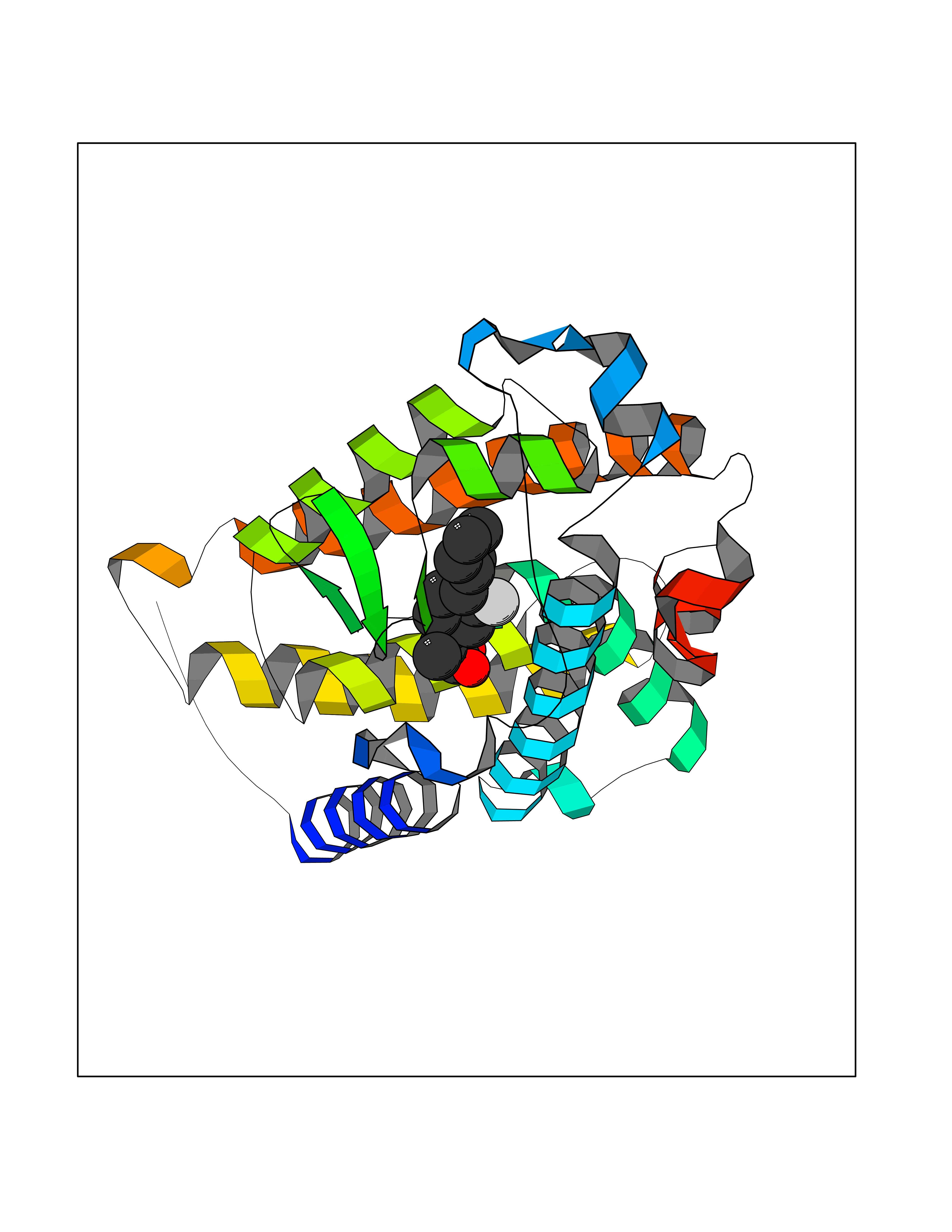 Molscript Map 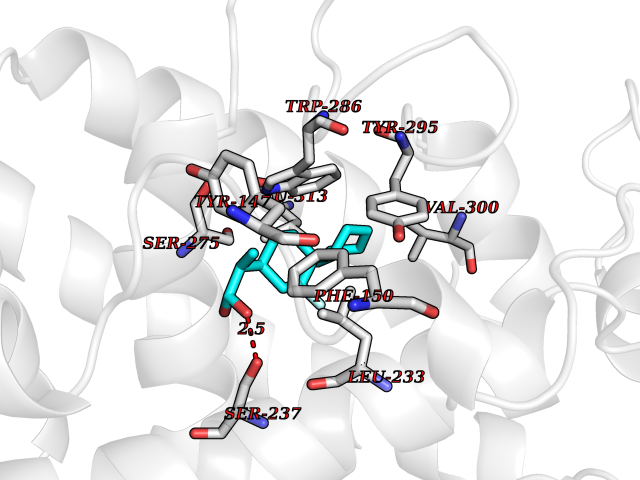 Pymol Map 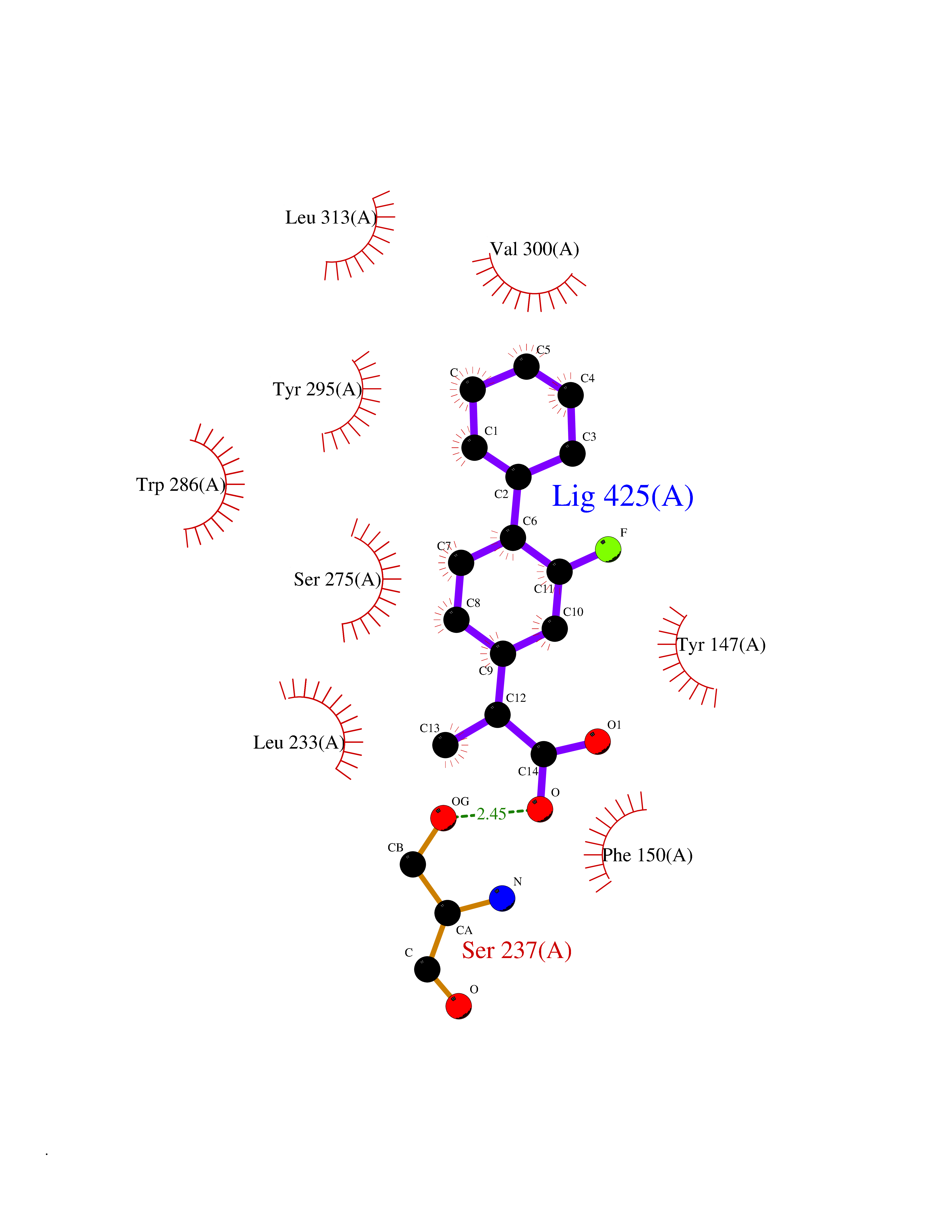 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Purine nucleoside phosphorylase (PNP) | 4EAR | 7.60 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -8.82  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | FkbI | 1R2J | 7.59 | |
Target general information Gen name fkbI Organism Streptomyces hygroscopicus subsp. ascomyceticus Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on the CH-CH group of donors. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 36670.3 Length 353 Aromaticity 0.04 Instability index 22.05 Isoelectric point 6.12 Charge (pH=7) -5.04 2D Binding mode Binding energy (Kcal/mol) -7.91  Molscript Map 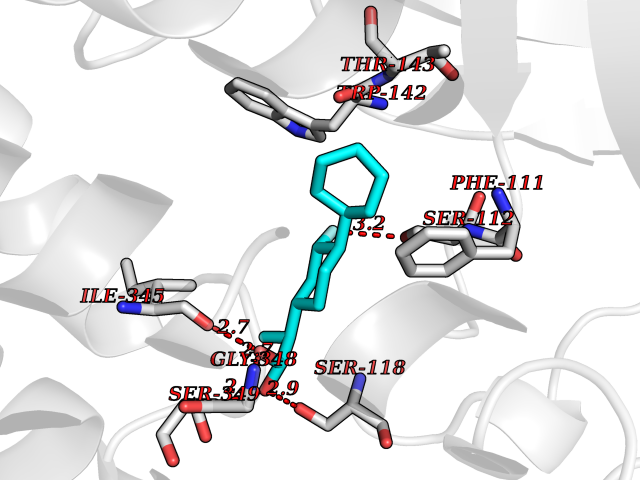 Pymol Map 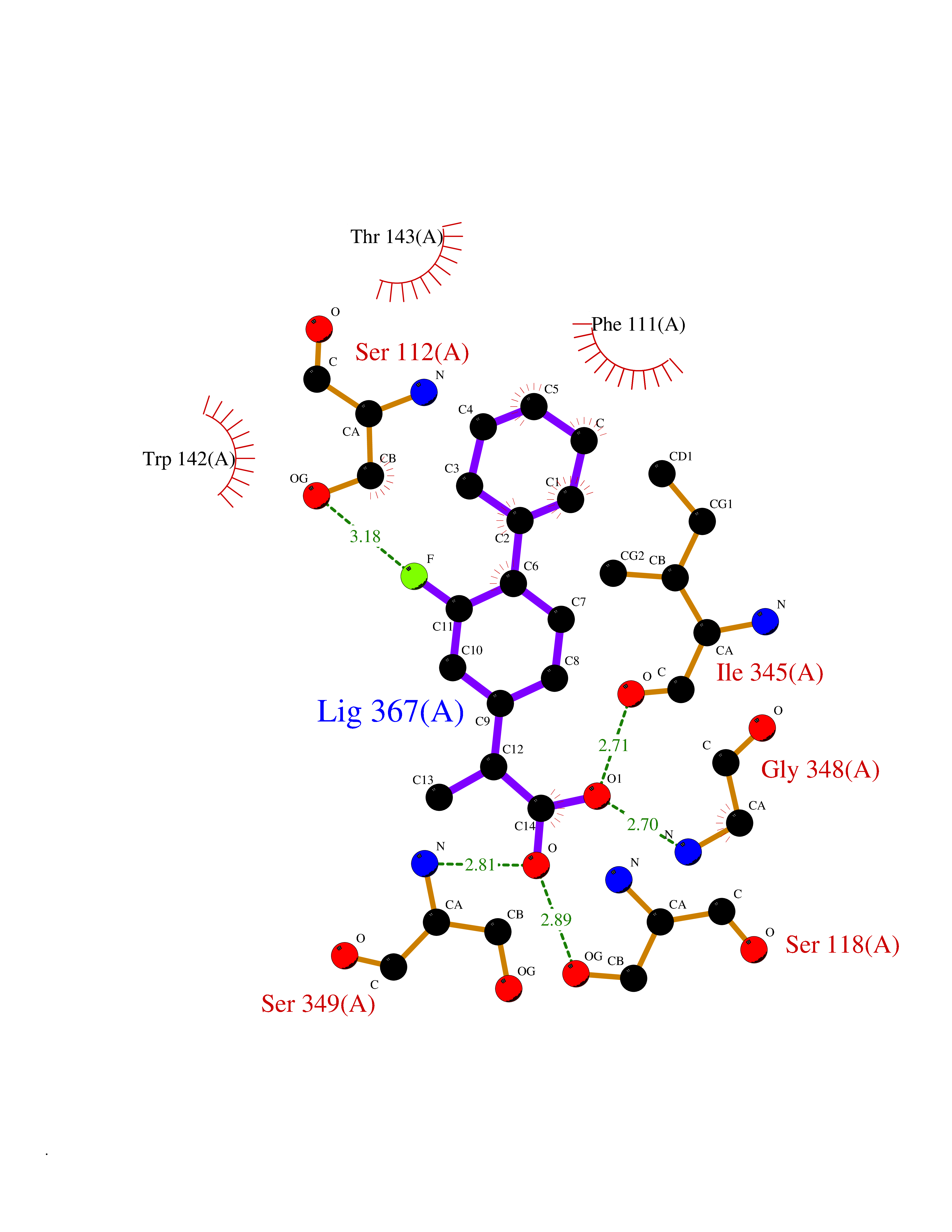 Ligplot Map 3D Binding mode Sequence ERDALLTDLVGDRAAEWDTSGELPRDLLVRLGADGLLCAEVAAEHGGLGLGSRENGEFTAHVGSLCSSLRSVMTSQGMAAWTVQRLGDAGQRATFLKELTSGLAAVGFSERQAGSDLSAMRTRVRLDGDTAVVDGHKVWTTAAAYADHLVVFGLQEDGSGAVVVVPADTPGVRVERVPKPSGCRAAGHADLHLDQVRVPAGAVLAGSGASLPMLVAASLAYGRKSVAWGCVGILRACRTAAVAHARTREQFGRPLGDHQLVAGHIADLWTAEQIAARVCEYASDHMVPATILAKHVAAERAAAGAATAAQVLASAGAGHVVERAYRDAKLMEIIEGSSEMCRVMLAQHALALP Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Serotonin transporter (SERT) | 5I6X | 7.58 | |
Target general information Gen name SLC6A4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 6 member 4; HTT; 5HTT; 5HT transporter Protein family Sodium:neurotransmitter symporter (SNF) (TC 2.A.22) family, SLC6A4 subfamily Biochemical class Neurotransmitter:sodium symporter Function Plays a key role in mediating regulation of the availability of serotonin to other receptors of serotonergic systems. Terminates the action of serotonin and recycles it in a sodium-dependent manner. Serotonin transporter whose primary function in the central nervous system involves the regulation of serotonergic signaling via transport of serotonin molecules from the synaptic cleft back into the pre-synaptic terminal for re-utilization. Related diseases Pancreatitis, hereditary (PCTT) [MIM:167800]: A disease characterized by pancreas inflammation, permanent destruction of the pancreatic parenchyma, maldigestion, and severe abdominal pain attacks. {ECO:0000269|PubMed:10204851, ECO:0000269|PubMed:10381903, ECO:0000269|PubMed:10930381, ECO:0000269|PubMed:11073545, ECO:0000269|PubMed:11788572, ECO:0000269|PubMed:11866271, ECO:0000269|PubMed:14695529, ECO:0000269|PubMed:15776435, ECO:0000269|PubMed:8841182, ECO:0000269|PubMed:9322498, ECO:0000269|PubMed:9633818}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB04836; DB05964; DB00321; DB00543; DB00182; DB01238; DB00289; DB00245; DB04889; DB09016; DB01114; DB00215; DB01242; DB00907; DB05688; DB00924; DB12305; DB01151; DB06700; DB01191; DB06701; DB00514; DB00988; DB09167; DB01142; DB00476; DB01363; DB01175; DB09194; DB00574; DB00472; DB00176; DB00458; DB08918; DB09195; DB00408; DB06077; DB00579; DB00454; DB01577; DB06148; DB01454; DB04896; DB00805; DB00370; DB01442; DB01149; DB09186; DB04821; DB00540; DB05422; DB00715; DB00191; DB00721; DB00344; DB00852; DB08839; DB01104; DB01105; DB06204; DB01079; DB06156; DB00193; DB00656; DB00726; DB00285; DB00661; DB06684; DB09068; DB04832; DB09225 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Antiport; Cell junction; Cell membrane; Cell projection; Disulfide bond; Endosome; Glycoprotein; Membrane; Metal-binding; Neurotransmitter transport; Phosphoprotein; Proteomics identification; Reference proteome; Sodium; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 61081.8 Length 544 Aromaticity 0.17 Instability index 32.58 Isoelectric point 8.14 Charge (pH=7) 3.09 2D Binding mode Binding energy (Kcal/mol) -8.68 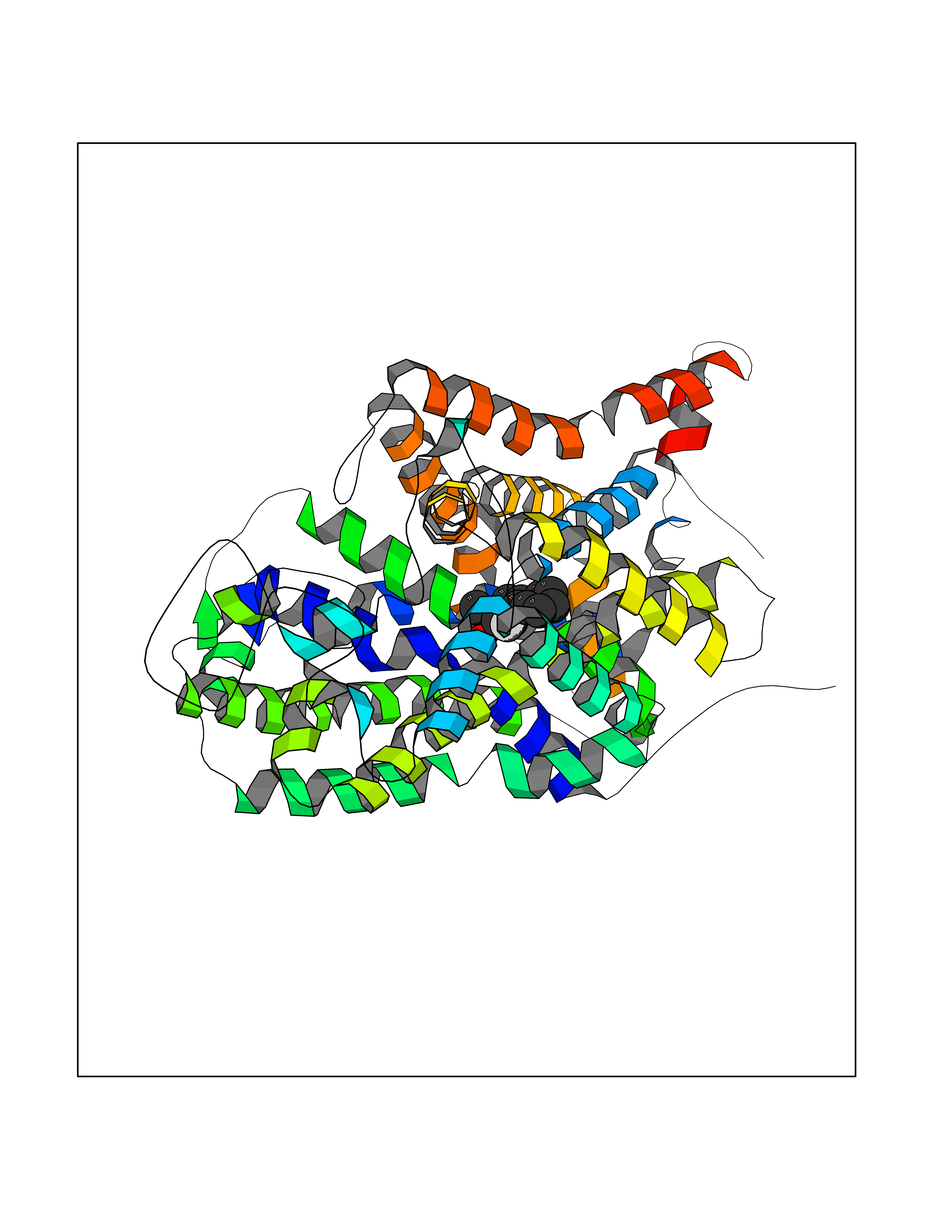 Molscript Map 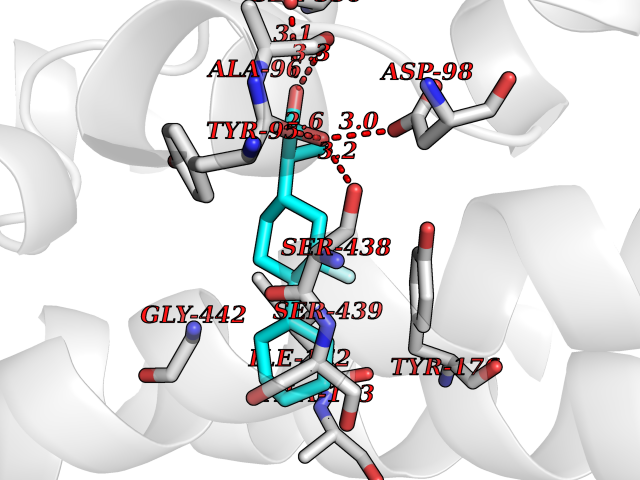 Pymol Map 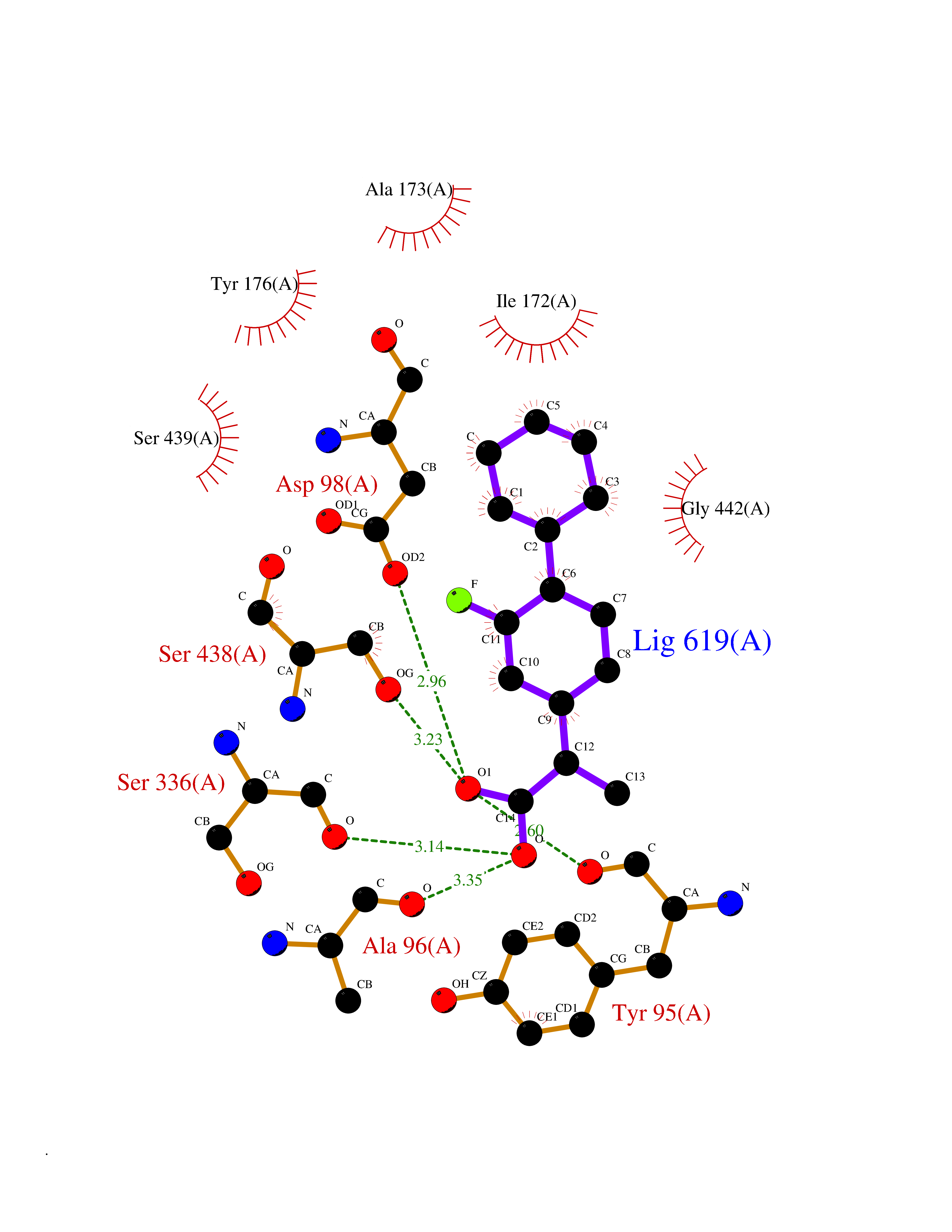 Ligplot Map 3D Binding mode Sequence GSQGERETWGKKVDFLLSVIGYAVDLGNVWRFPYICAQNGGGAFLLPYTIMAIFGGIPLFYMELALGQYHRNGCISIWRKICPIFKGIGYAICIIAFYIASYYNTIMAWALYYLISSFTDQLPWTSCKNSWNTGNCTNYFSEDNITWTLHSTSPAEEFYTRHVLQIHRSKGLQDLGGISWQLALCIMLIFTVIYFSIWKGVKTSGKVVWVTATFPYIALSVLLVRGATLPGAWRGVLFYLKPNWQKLLETGVWIDAAAQIFFSLGPGFGVLLAFASYNKFNNNCYQDALVTSVVNCMTSFVSGFVIFTVLGYMAEMRNEDVSEVAKDAGPSLLFITYAEAIANMPASTFFAIIFFLMLITLGLDSSFAGLEGVITAVLDEFPHVWAKRRERFVLAVVITCFFGSLVTLTFGGAYVVKLLEEYATGPAVLTVALIEAVAVSWFYGITQFCRDVKEMLGFSPGWFWRICWVAISPLFLLFIIASFLMSPPQLRLFQYNYPYWSIILGYAIGTSSFICIPTYIAYRLIITPGTFKERIIKSITPETP Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Dopamine beta-hydroxylase | 4ZEL | 7.57 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -8.85 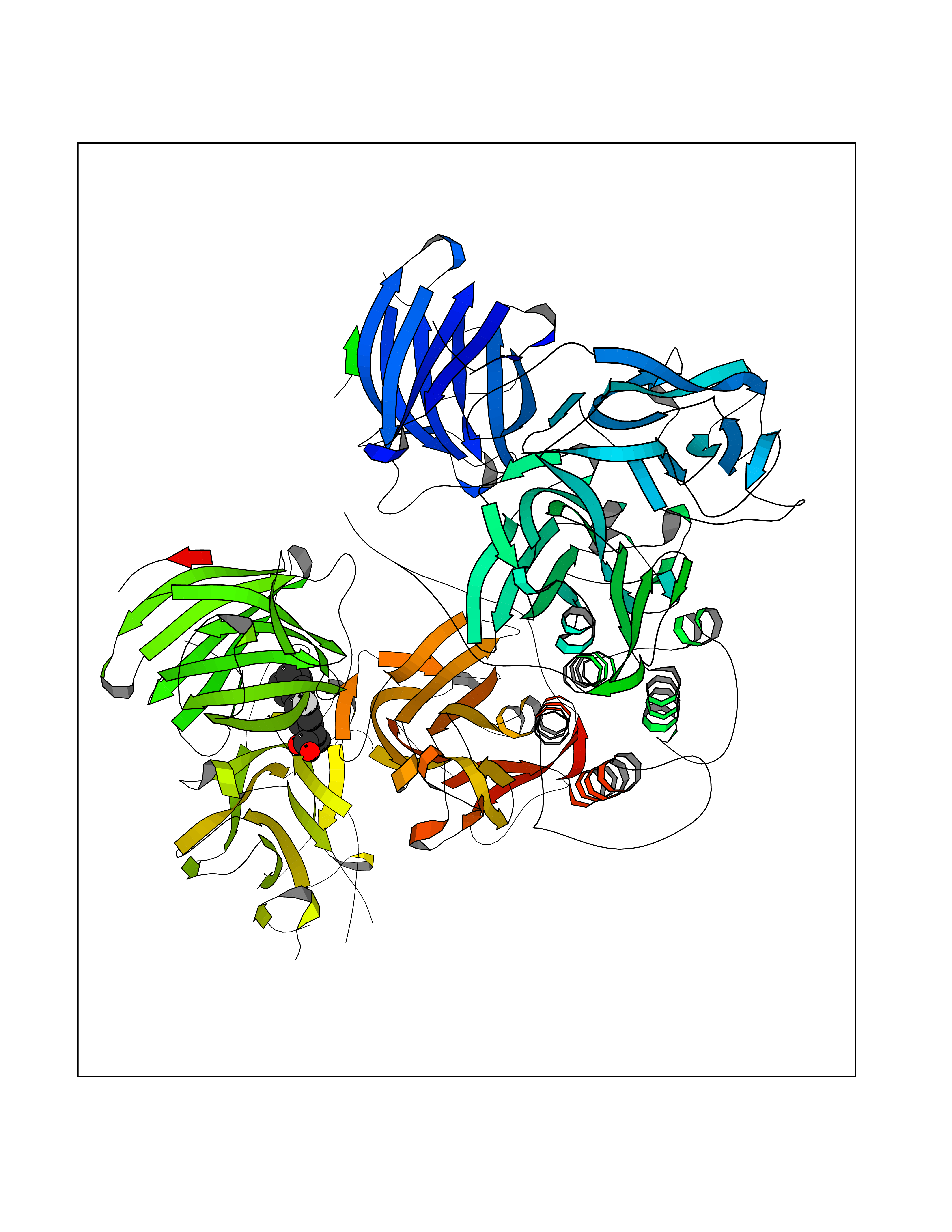 Molscript Map 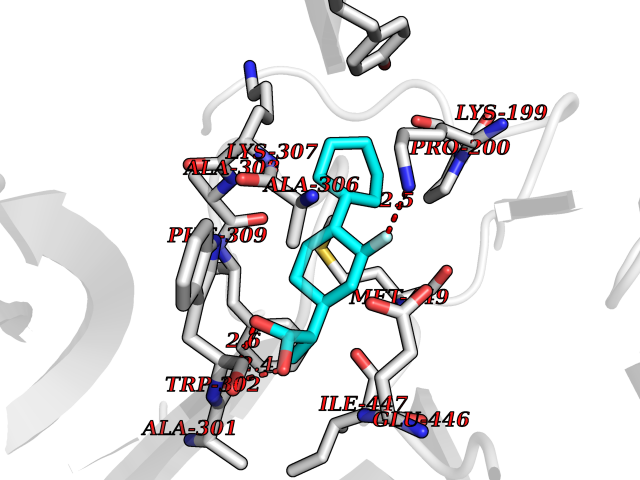 Pymol Map 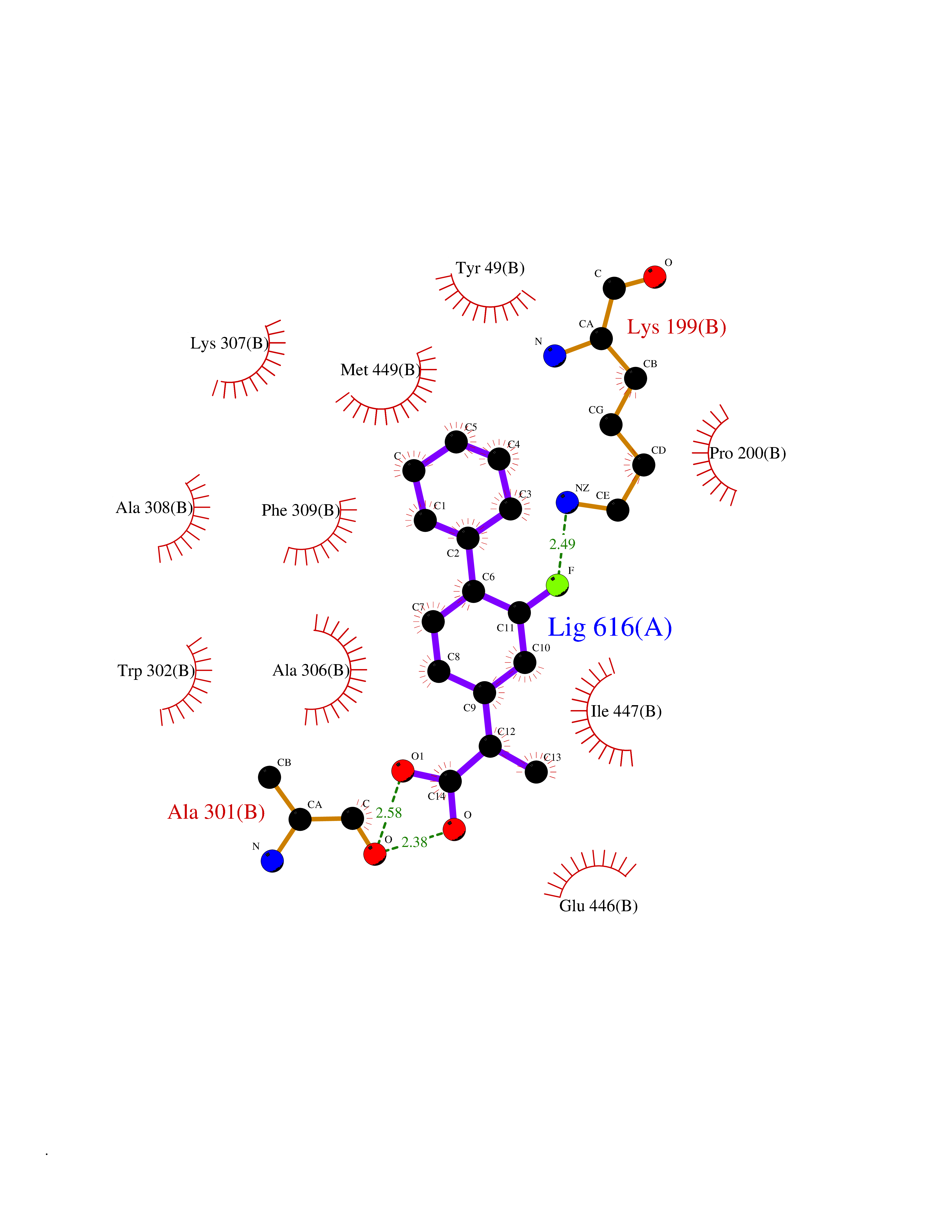 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Opioid receptor delta (OPRD1) | 4N6H | 7.56 | |
Target general information Gen name OPRD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OPRD; Delta-type opioid receptor; Delta opioid receptor; DOR-1; D-OR-1 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling leads to the inhibition of adenylate cyclase activity. Inhibits neurotransmitter release by reducing calcium ion currents and increasing potassium ion conductance. Plays a role in the perception of pain and in opiate-mediated analgesia. Plays a role in developing analgesic tolerance to morphine. G-protein coupled receptor that functions as receptor for endogenous enkephalins and for a subset of other opioids. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB01571; DB01439; DB05050; DB06274; DB06288; DB00321; DB01238; DB00921; DB00611; DB09173; DB09061; DB01535; DB00318; DB00514; DB00647; DB01452; DB01565; DB01444; DB01081; DB01548; DB09272; DB01497; DB00813; DB00956; DB00327; DB01221; DB06738; DB00854; DB00836; DB14146; DB14009; DB12668; DB00333; DB00295; DB06409; DB14011; DB00844; DB11691; DB06230; DB01183; DB00704; DB11130; DB00497; DB01192; DB09209; DB00899; DB12543; DB00708; DB06204; DB00193 Interacts with P16615; P27824; Q4LDR2; Q5JY77; Q9NS64; Q9Y666-2; Q9UKG4; Q0VAQ4; Q96Q45-2; P11607 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32859.3 Length 294 Aromaticity 0.11 Instability index 33.86 Isoelectric point 9.38 Charge (pH=7) 13.6 2D Binding mode Binding energy (Kcal/mol) -8.47 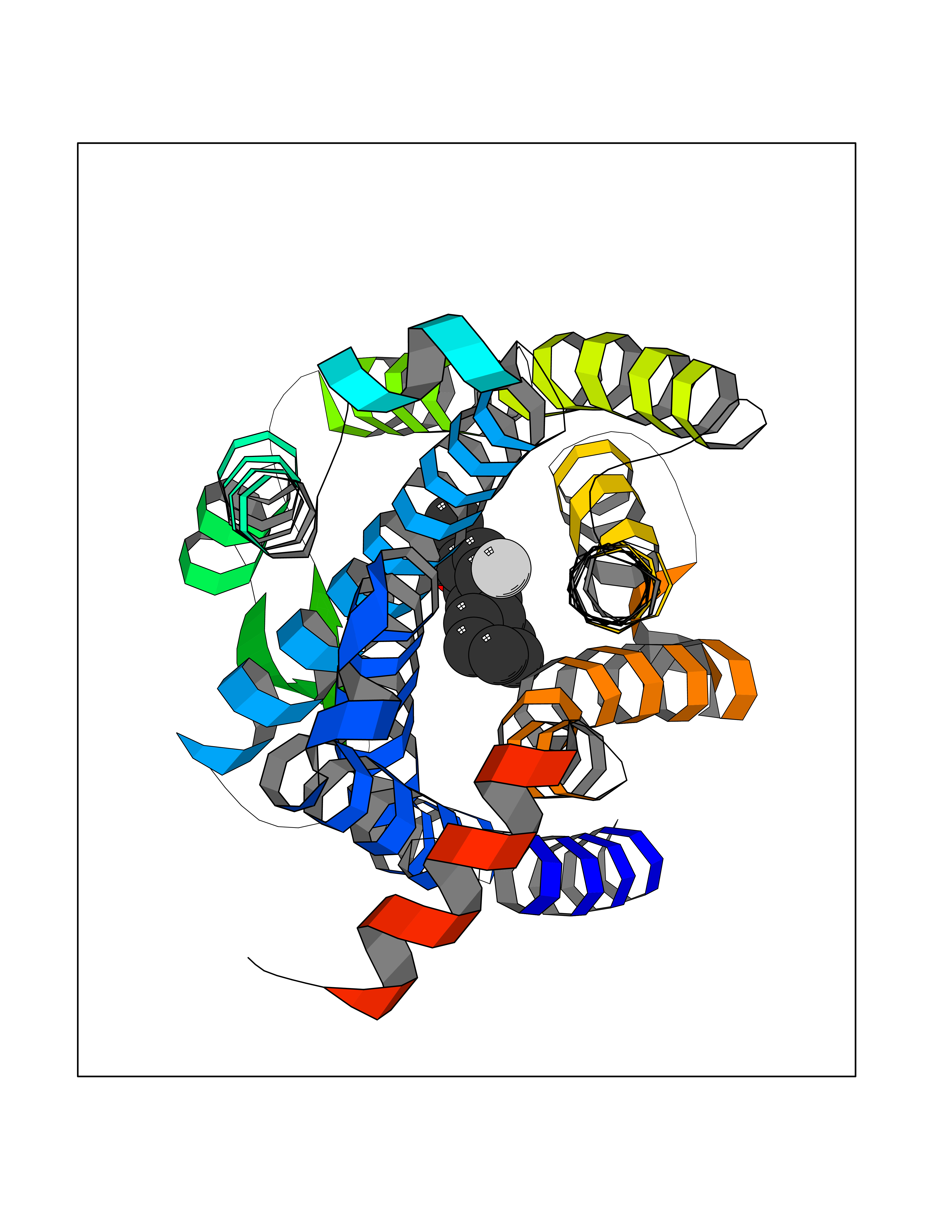 Molscript Map 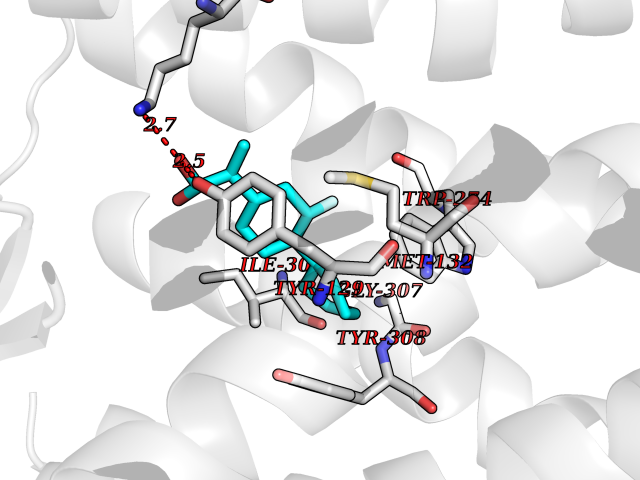 Pymol Map 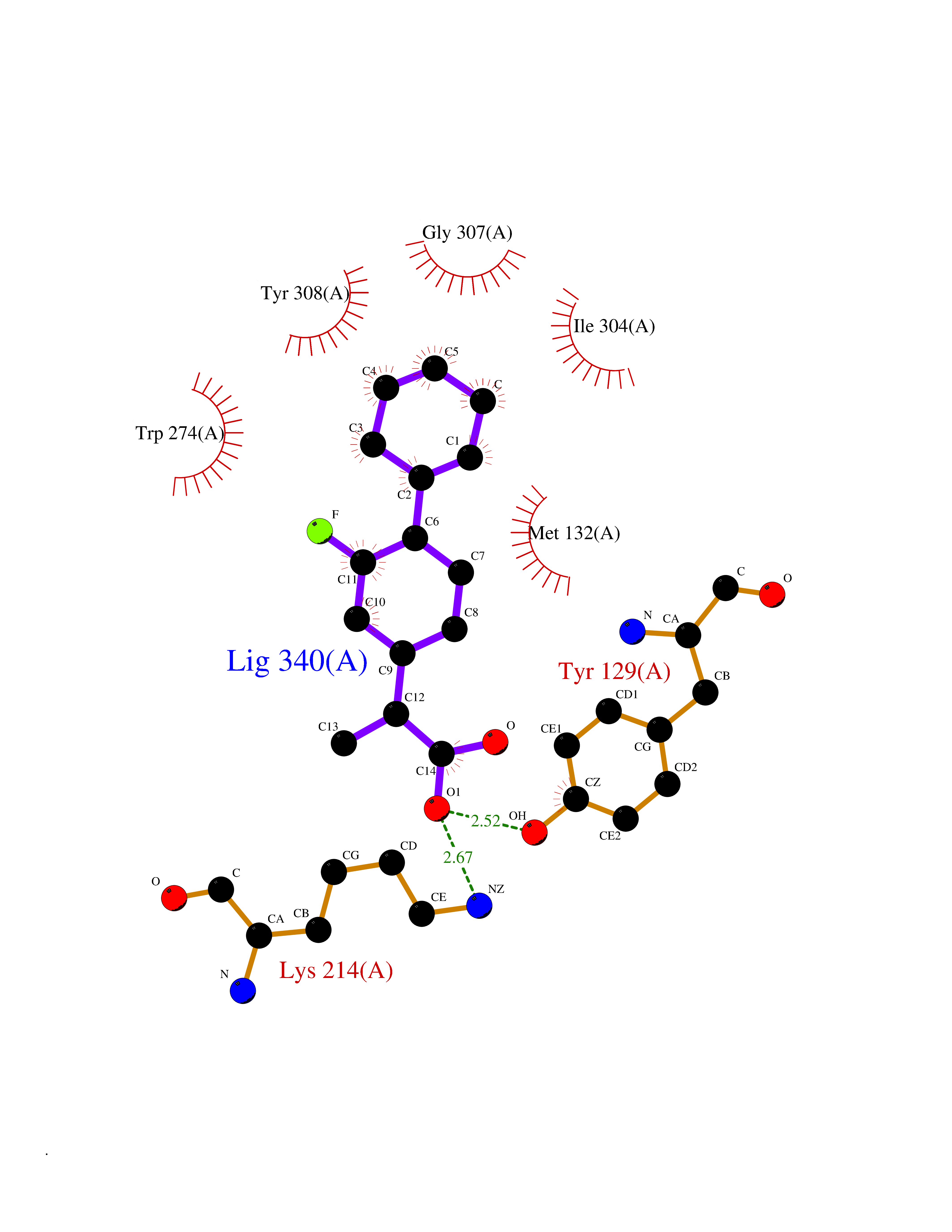 Ligplot Map 3D Binding mode Sequence SLALAIAITALYSAVCAVGLLGNVLVMFGIVRYTKMKTATNIYIFNLALADALATSTLPFQSAKYLMETWPFGELLCKAVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPAKAKLINICIWVLASGVGVPIMVMAVTRPRDGAVVCMLQFPSPSWYWDTVTKICVFLFAFVVPILIITVCYGLMLLRLRSVRLLSGSKEKDRSLRRITRMVLVVVGAFVVCWAPIHIFVIVWTLVDIDRRDPLVVAALHLCIALGYANSSLNPVLYAFLDENFKRCFRQLCRKPCG Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Solute carrier family 19 member 1 (SLC19A1) | 8GOF | 7.56 | |
Target general information Gen name SLC19A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Reduced folate carrier protein; RFC1; RFC; Placental folate transporter; Intestinal folate carrier 1; IFC-1; Folate transporter 1; FOLT; FLOT1 Protein family Reduced folate carrier (RFC) transporter (TC 2.A.48) family Biochemical class NA Function Transporter for the intake of folate. Uptake of folate in human placental choriocarcinoma cells occurs by a novel mechanism called potocytosis which functionally couples three components, namely the folate receptor, the folate transporter, and a V-type H(+)-pump. Related diseases Megaloblastic anemia, folate-responsive (MEGAF) [MIM:601775]: An autosomal recessive metabolic disorder characterized by megaloblastic anemia resulting from decreased folate transport into erythrocytes. Disease manifestations include hemolytic anemia, hyperhomocysteinemia, and low vitamin B12. Serum folate levels are normal, but erythrocyte folate levels are decreased. Treatment with oral folate corrects the anemia and normalizes homocysteine. {ECO:0000269|PubMed:32276275}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 114, folate-responsive (IMD114) [MIM:620603]: An autosomal recessive immunologic disorder manifesting in early infancy and characterized by recurrent skin and respiratory infections, mucosal bleeding, oral ulcers, chronic diarrhea, and poor overall growth. Affected individuals have lymphopenia, low serum immunoglobulins, and impaired T cell proliferation. Some patients have global developmental delay. {ECO:0000269|PubMed:36517554, ECO:0000269|PubMed:36745868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11256; DB00563; DB00642; DB06813; DB01157 Interacts with Q7Z3Y9 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Antiport; Cell membrane; Disease variant; Folate-binding; Glycoprotein; Hereditary hemolytic anemia; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46087.7 Length 407 Aromaticity 0.15 Instability index 34.62 Isoelectric point 9.82 Charge (pH=7) 17.33 2D Binding mode Binding energy (Kcal/mol) -8.49 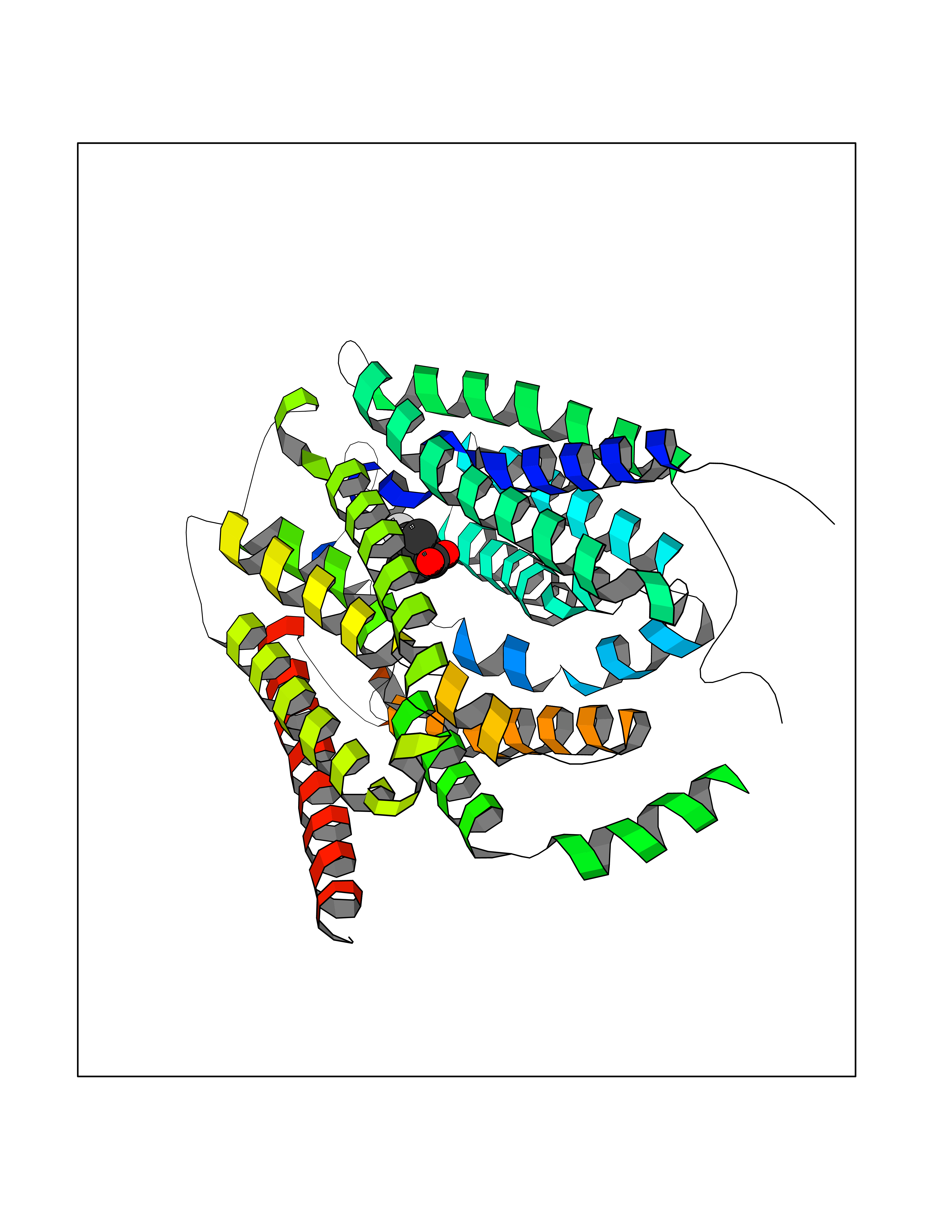 Molscript Map 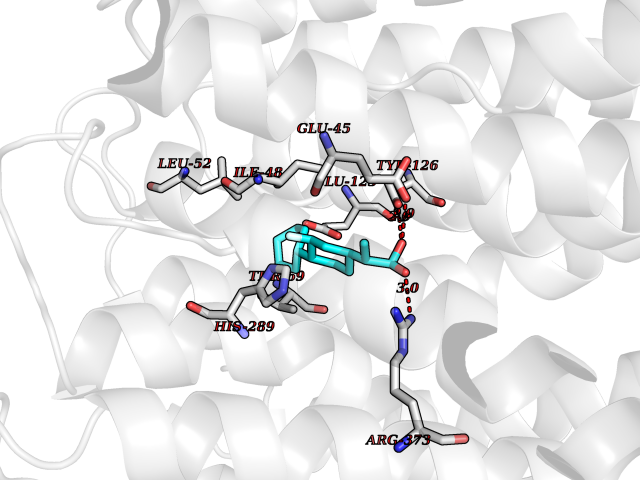 Pymol Map 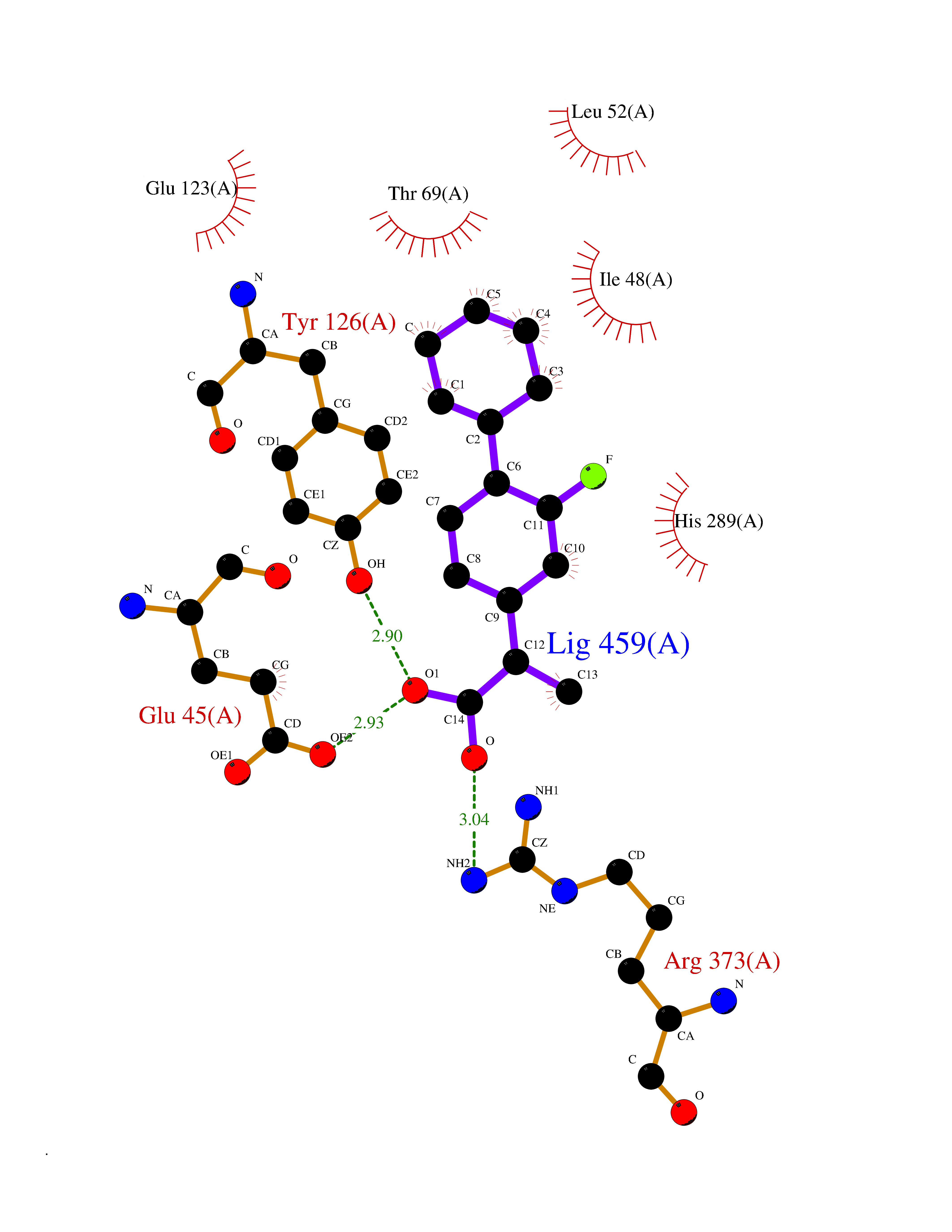 Ligplot Map 3D Binding mode Sequence DPELRSWRHLVCYLCFYGFMAQIRPGESFITPYLLGPDKNFTREQVTNEITPVLSYSYLAVLVPVFLLTDYLRYTPVLLLQGLSFVSVWLLLLLGHSVAHMQLMELFYSVTMAARIAYSSYIFSLVRPARYQRVAGYSRAAVLLGVFTSSVLGQLLVTVGRVSFSTLNYISLAFLTFSVVLALFLKRPKRSLFFNRDDSVLARMLRELGDSLRRPQLRLWSLWWVFNSAGYYLVVYYVHILWNEVDPTTNSARVYNGAADAASTLLGAITSFAAGFVKIRWARWSKLLIAGVTATQAGLVFLLAHTRHPSSIWLCYAAFVLFRGSYQFLVPIATFQIASSLSKELCALVFGVNTFFATIVKTIITFIVSDVRGLGLPVRKQFQLYSVYFLILSIIYFLGAMLDGLRH Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Ferredoxin reductase | 2GQW | 7.56 | |
Target general information Gen name bphA4 Organism Pseudomonas sp. (strain KKS102) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Oxidoreductase activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03147 Interacts with NA EC number NA Uniprot keywords 3D-structure; FAD; Flavoprotein; Nucleotide-binding Protein physicochemical properties Chain ID A Molecular weight (Da) 42484 Length 401 Aromaticity 0.05 Instability index 32.45 Isoelectric point 6.13 Charge (pH=7) -2.69 2D Binding mode Binding energy (Kcal/mol) -8.42  Molscript Map 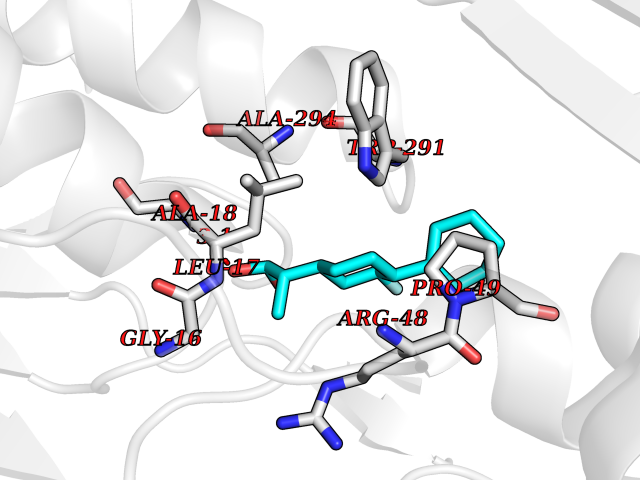 Pymol Map 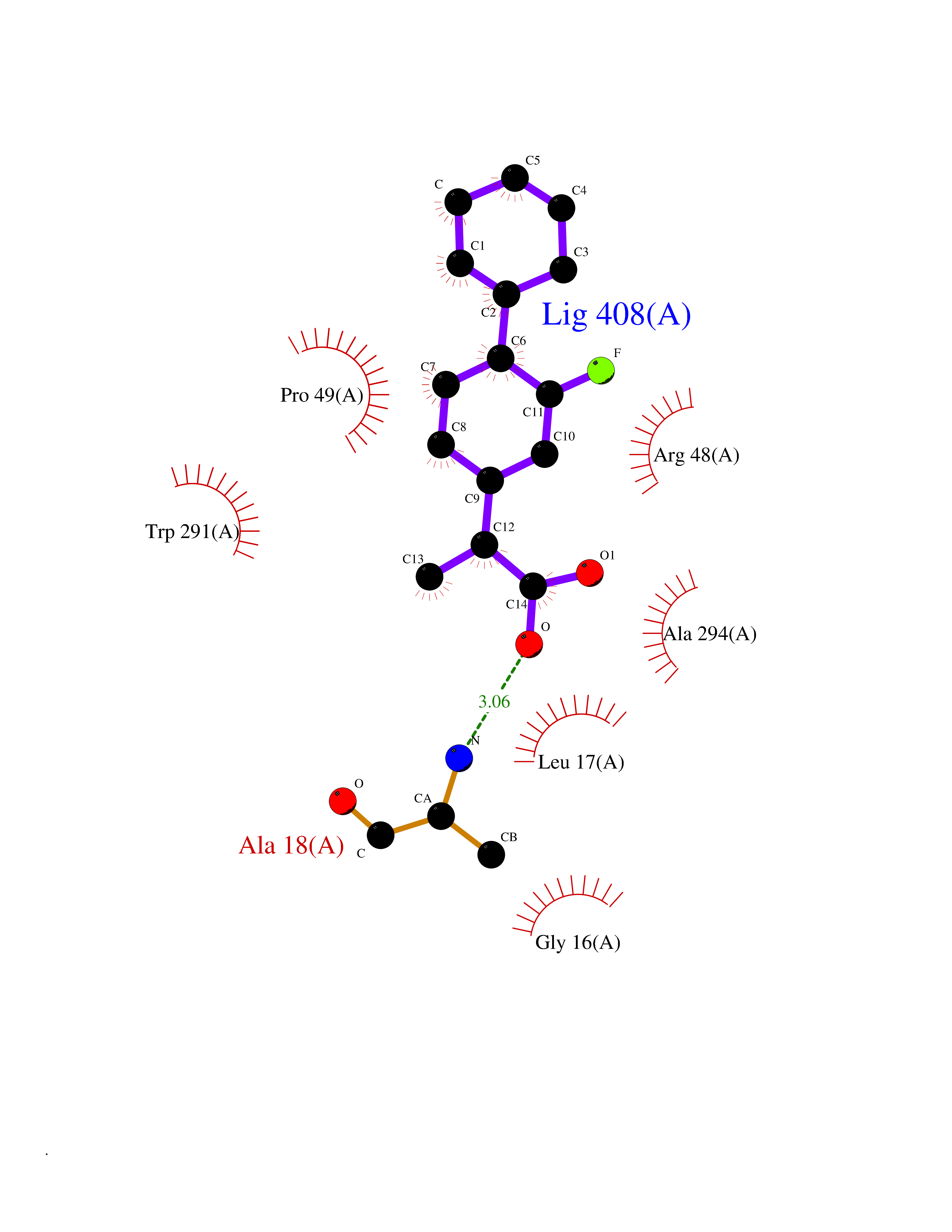 Ligplot Map 3D Binding mode Sequence LKAPVVVLGAGLASVSFVAELRQAGYQGLITVVGDEAERPYDRPPLSKDFMAHGDAEKIRLDCKRAPEVEWLLGVTAQSFDPQAHTVALSDGRTLPYGTLVLATGAAPRALPTLQGATMPVHTLRTLEDARRIQAGLRPQSRLLIVGGGVIGLELAATARTAGVHVSLVETQPRLMSRAAPATLADFVARYHAAQGVDLRFERSVTGSVDGVVLLDDGTRIAADMVVVGIGVLANDALARAAGLACDDGIFVDAYGRTTCPDVYALGDVTRQRNPLSGRFERIETWSNAQNQGIAVARHLVDPTAPGYAELPWYWSDQGALRIQVAGLASGDEEIVRGEVSLDAPKFTLIELQKGRIVGATCVNNARDFAPLRRLLAVGAKPDRAALADPATDLRKLAAAV Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 7.54 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -8.92  Molscript Map 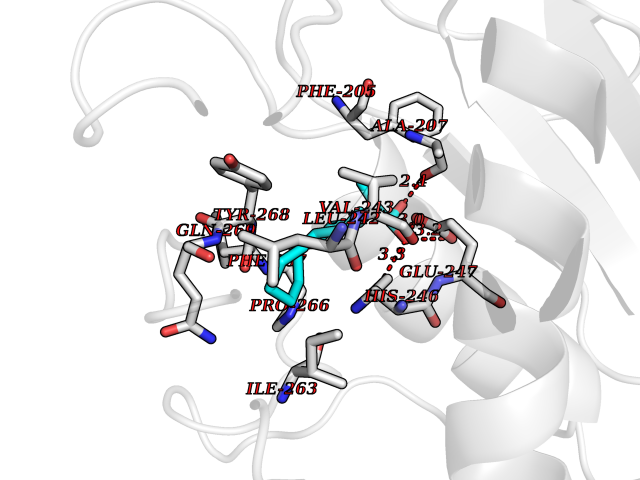 Pymol Map 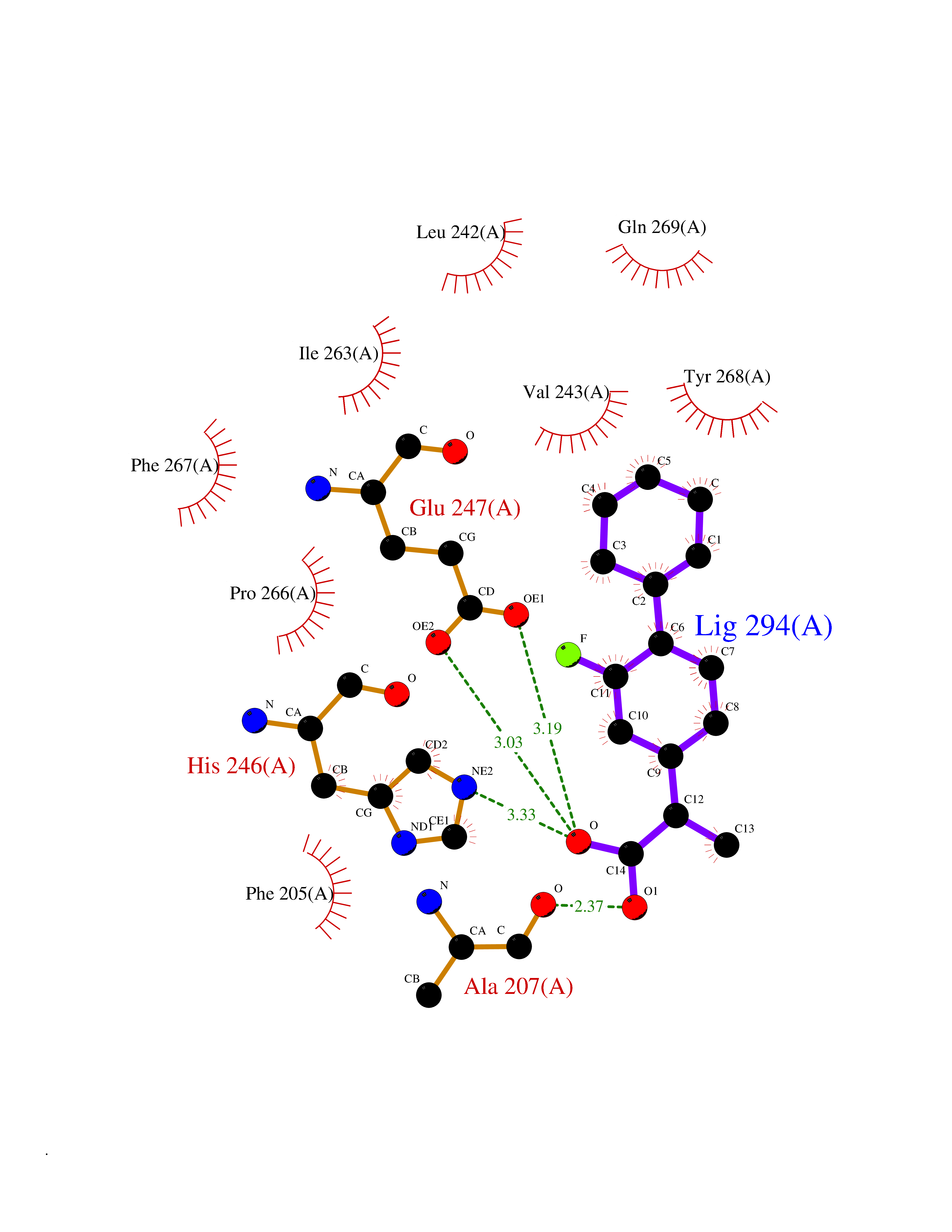 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Plasmodium DOXP reductoisomerase (Malaria DXR) | 3AU9 | 7.53 | |
Target general information Gen name Malaria DXR Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID Synonyms IspC; DXR; DXP reductoisomerase; DOXP reductoisomerase; 2-C-Methyl-d-erythritol 4-phosphate synthase; 1-deoxyxylulose-5-phosphate reductoisomerase Protein family DXR family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADP-dependent rearrangement and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol 4-phosphate (MEP). Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.1.267 Uniprot keywords 3D-structure; Apicoplast; Isoprene biosynthesis; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Plastid; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46644.4 Length 410 Aromaticity 0.09 Instability index 36.77 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -8.58  Molscript Map 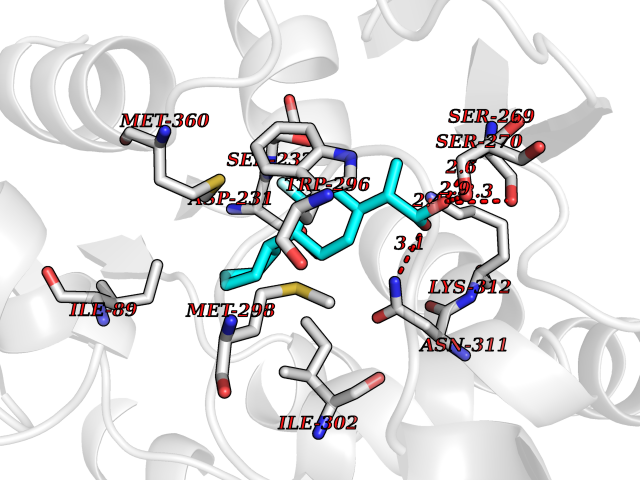 Pymol Map 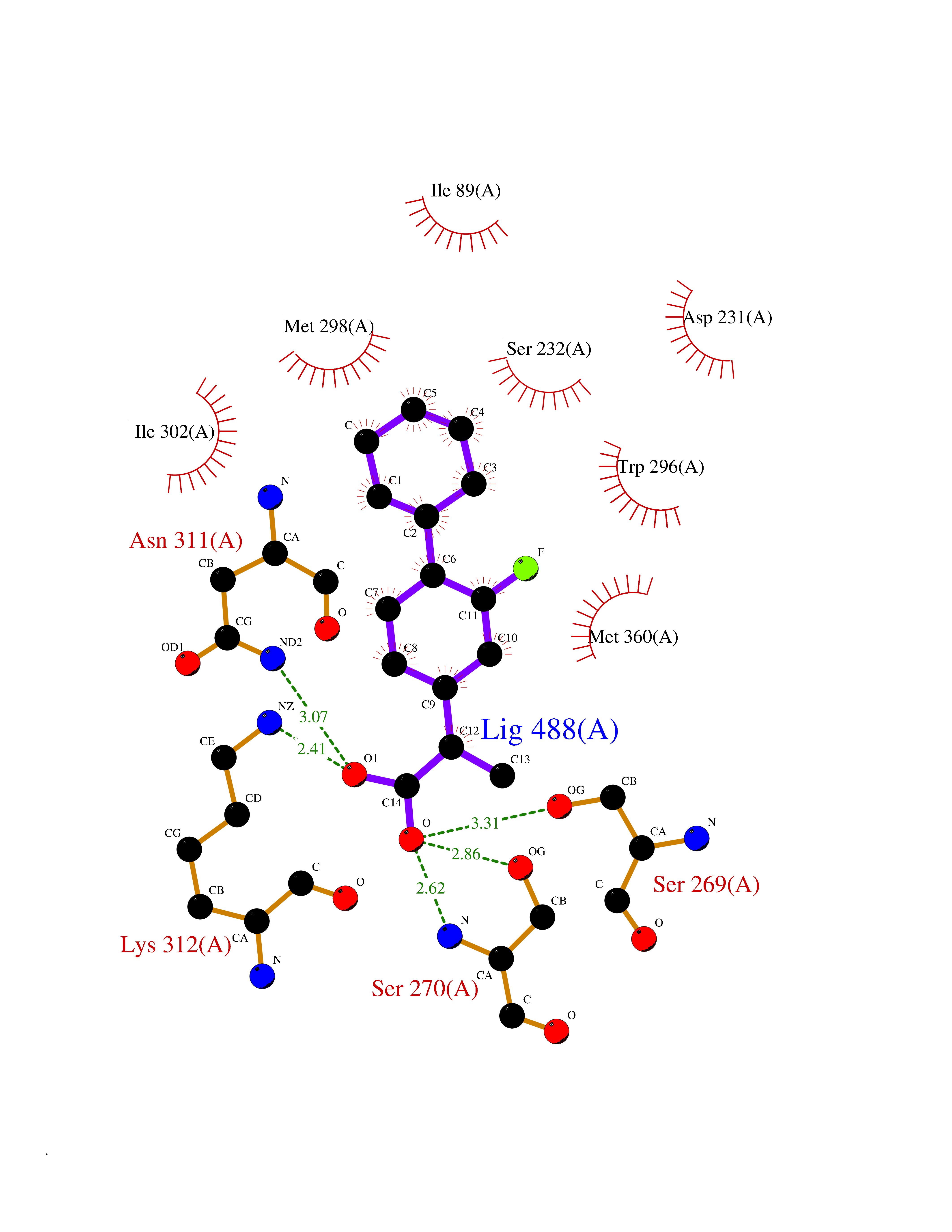 Ligplot Map 3D Binding mode Sequence PINVAIFGSTGSIGTNALNIIRECNKIENVFNVKALYVNKSVNELYEQAREFLPEYLCIHDKSVYEELKELVKNIKDYKPIILCGDEGMKEICSSNSIDKIVIGIDSFQGLYSTMYAIMNNKIVALANKESIVSAGFFLKKLLNIHKNAKIIPVDSEHSAIFQCLDNNKVLKTKCLQDNFSKINNINKIFLCSSGGPFQNLTMDELKNVTSENALKHPKWKMGKKITIDSATMMNKGLEVIETHFLFDVDYNDIEVIVHKECIIHSCVEFIDKSVISQMYYPDMQIPILYSLTWPDRIKTNLKPLDLAQVSTLTFHKPSLEHFPCIKLAYQAGIKGNFYPTVLNASNEIANNLFLNNKIKYFDISSIISQVLESFNSQKVSENSEDLMKQILQIHSWAKDKATDIYNKHN Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 7.51 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -8.15 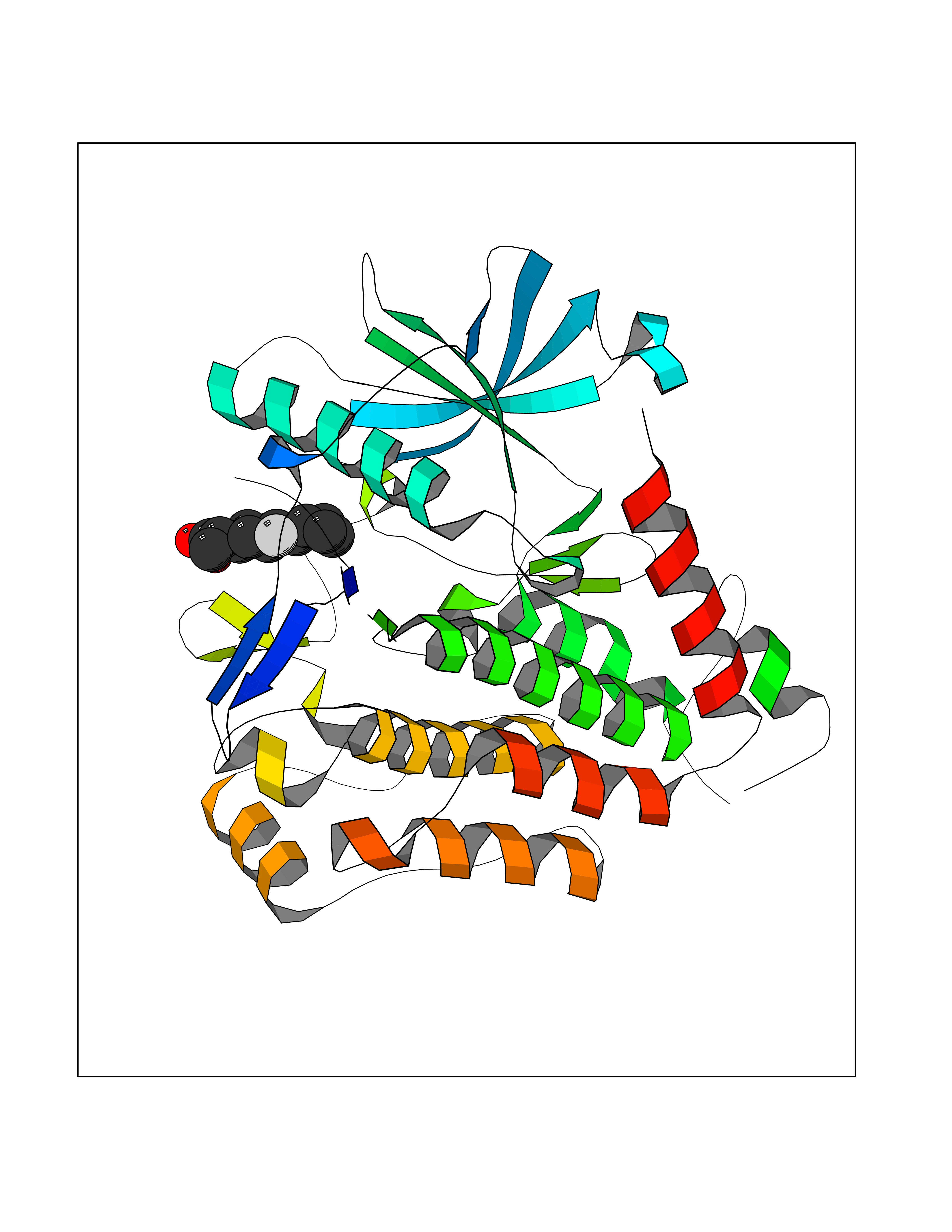 Molscript Map 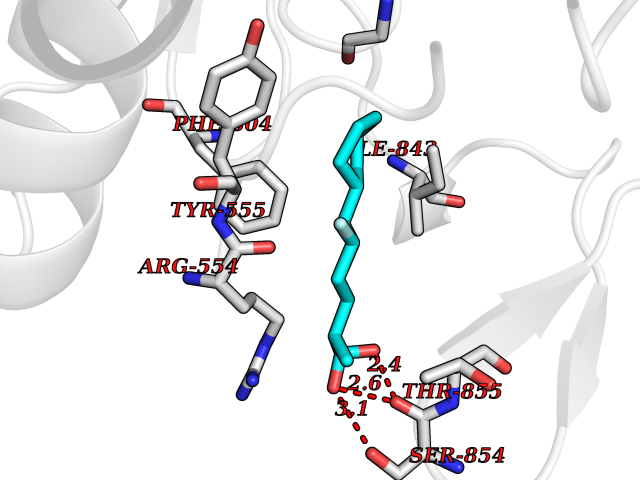 Pymol Map 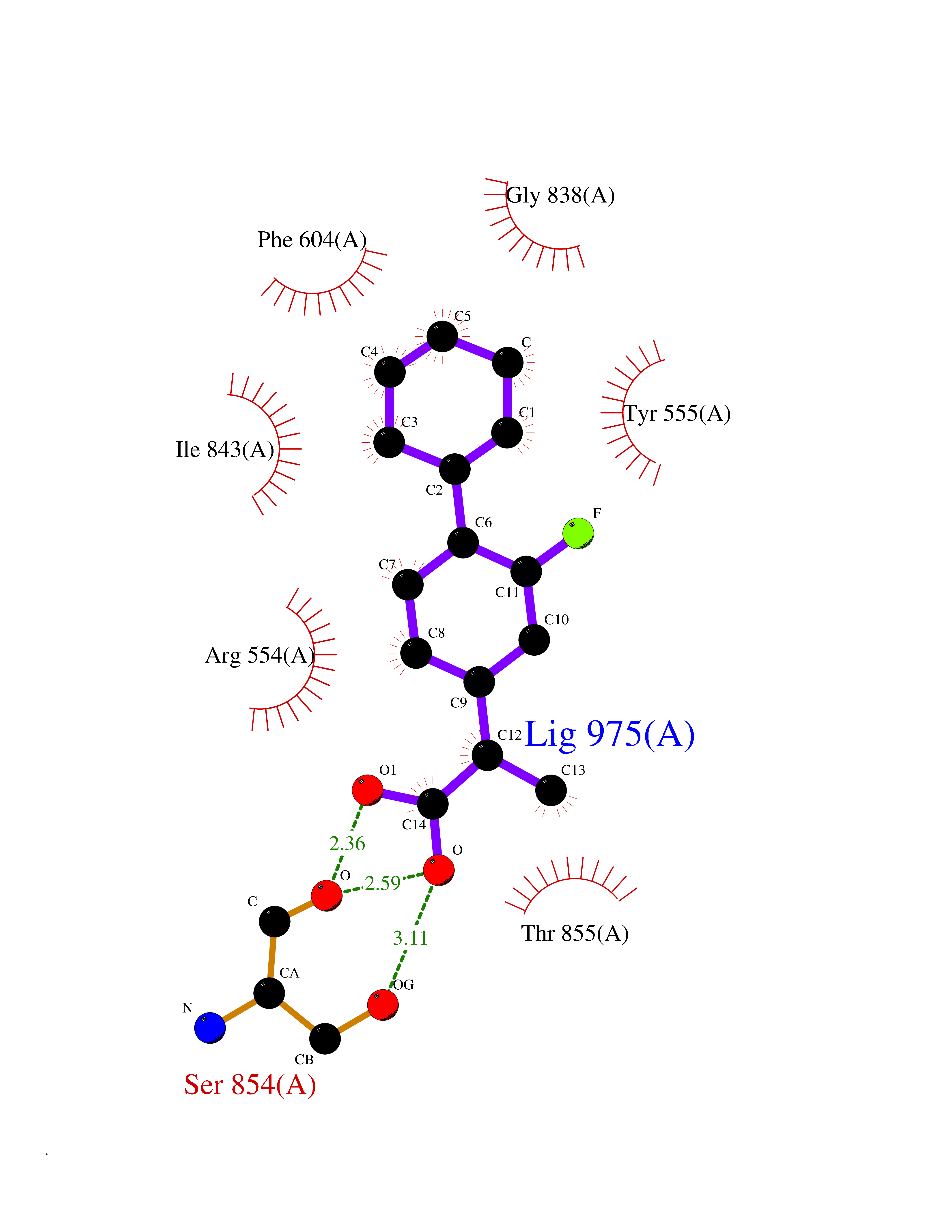 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Fatty acid synthase (FASN) | 3TJM | 7.50 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -9.13  Molscript Map 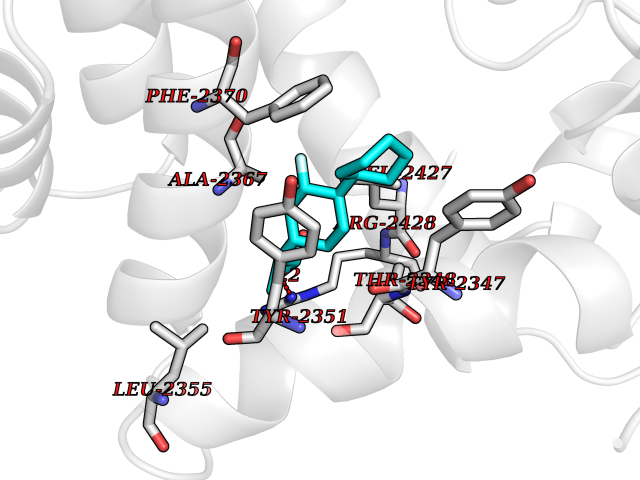 Pymol Map 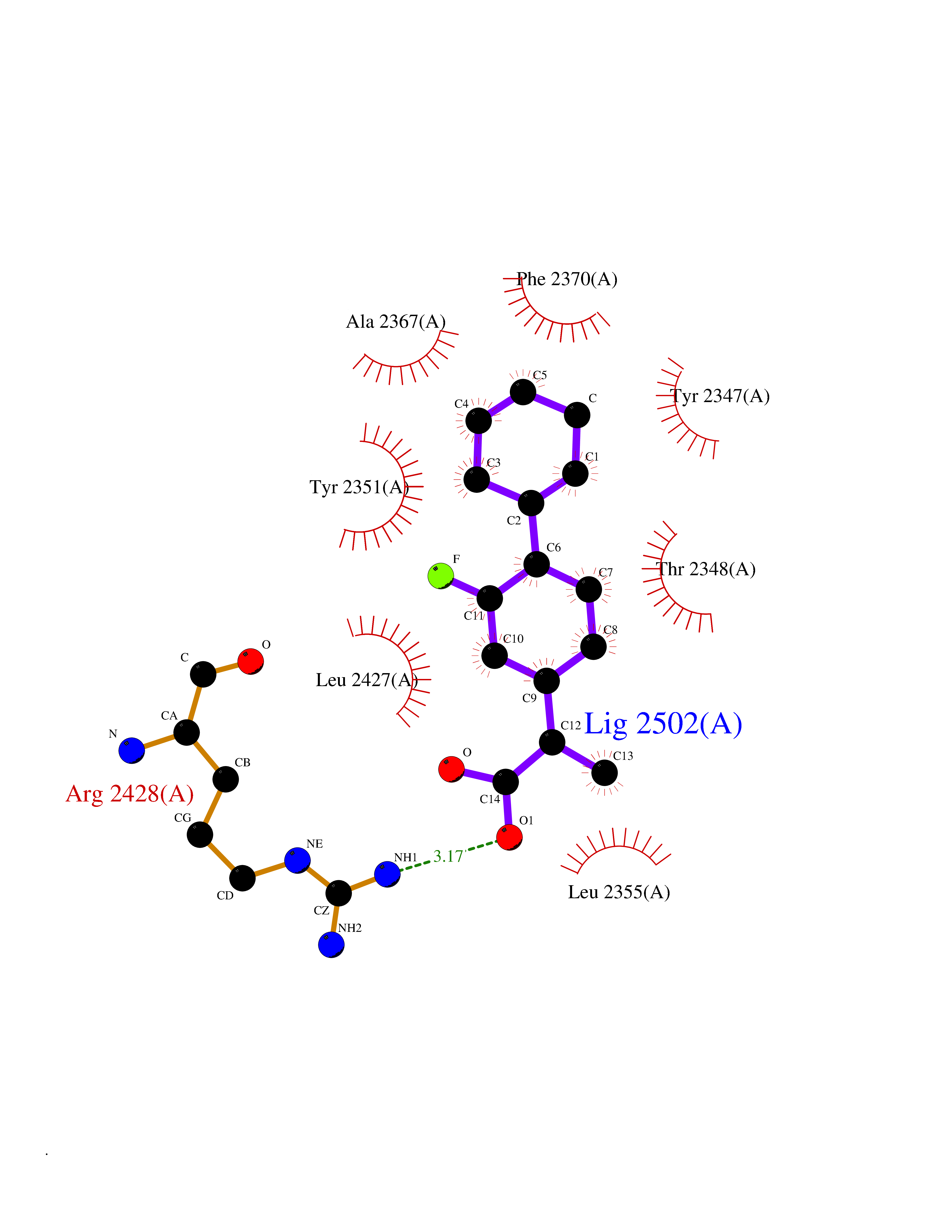 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Human immunodeficiency virus Protease (HIV PR) | 3TL9 | 7.49 | |
Target general information Gen name HIV PR Organism Human immunodeficiency virus type 1 group M subtype B (isolate BH10) (HIV-1) Uniprot ID TTD ID Synonyms HIV Retropepsin; HIV PR Protein family NA Biochemical class Peptidase Function Gag-Pol polyprotein: Mediates, with Gag polyprotein, the essential events in virion assembly, including binding the plasma membrane, making the protein-protein interactions necessary to create spherical particles, recruiting the viral Env proteins, and packaging the genomic RNA via direct interactions with the RNA packaging sequence (Psi). Gag-Pol polyprotein may regulate its own translation, by the binding genomic RNA in the 5'-UTR. At low concentration, the polyprotein would promote translation, whereas at high concentration, the polyprotein would encapsidate genomic RNA and then shut off translation. Related diseases Sitosterolemia 2 (STSL2) [MIM:618666]: A form of sitosterolemia, an autosomal recessive metabolic disorder characterized by unregulated intestinal absorption of cholesterol, phytosterols and shellfish sterols, and decreased biliary excretion of dietary sterols into bile. Patients have hypercholesterolemia, very high levels of plant sterols in the plasma, and frequently develop tendon and tuberous xanthomas, accelerated atherosclerosis and premature coronary artery disease. {ECO:0000269|PubMed:11138003, ECO:0000269|PubMed:11452359, ECO:0000269|PubMed:11668628, ECO:0000269|PubMed:15054092, ECO:0000269|PubMed:35557526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07035; DB02704; DB07806; DB02785; DB01824; DB01732; DB06874; DB08034; DB07961; DB07451; DB08212; DB08372; DB02972; DB04190; DB04042; DB08428; DB03076; DB03141; DB08457; DB07343; DB07337; DB07018; DB07332; DB05398; DB07578; DB08639; DB06414; DB04255; DB04547; DB02683; DB02009; DB03908; DB02629; DB01887; DB03803; DB02033; DB08281; DB08282; DB08284; DB08414; DB08598; DB07327; DB07885; DB02768; DB08600; DB01891; DB05871 Interacts with NA EC number EC 3.4.23.16 Uniprot keywords 3D-structure; Activation of host caspases by virus; AIDS; Aspartyl protease; Capsid protein; Direct protein sequencing; DNA integration; DNA recombination; DNA-binding; DNA-directed DNA polymerase; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host translation shutoff by virus; Host cell membrane; Host cytoplasm; Host endosome; Host gene expression shutoff by virus; Host membrane; Host nucleus; Host-virus interaction; Hydrolase; Lipid-binding; Lipoprotein; Magnesium; Membrane; Metal-binding; Methylation; Modulation of host cell apoptosis by virus; Multifunctional enzyme; Myristate; Nuclease; Nucleotidyltransferase; Phosphoprotein; Protease; Repeat; Ribosomal frameshifting; RNA-binding; RNA-directed DNA polymerase; Transferase; Viral genome integration; Viral nucleoprotein; Viral penetration into host nucleus; Viral release from host cell; Virion; Virion maturation; Virus entry into host cell; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 21934.7 Length 202 Aromaticity 0.06 Instability index 48.65 Isoelectric point 9.66 Charge (pH=7) 6.15 2D Binding mode Binding energy (Kcal/mol) -7.28 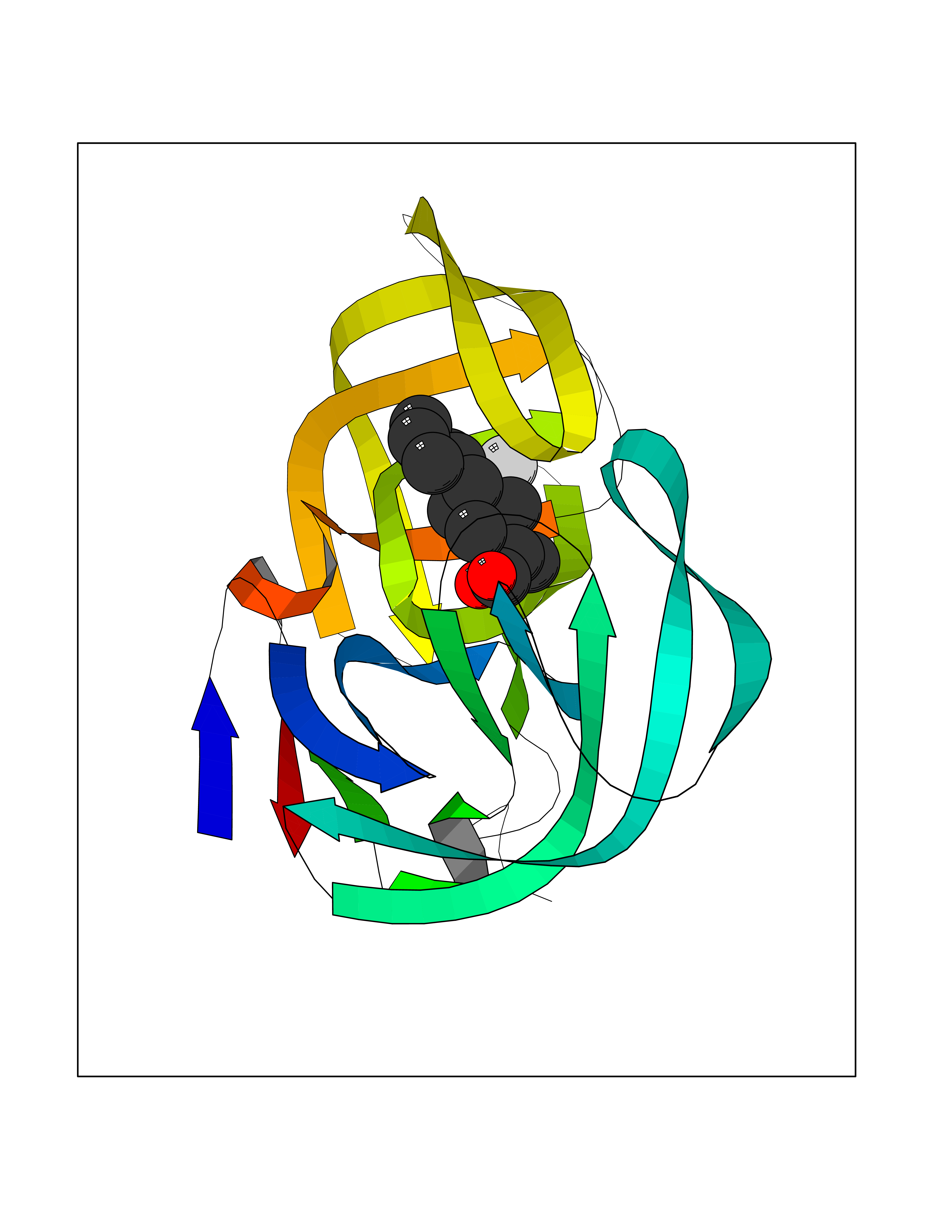 Molscript Map 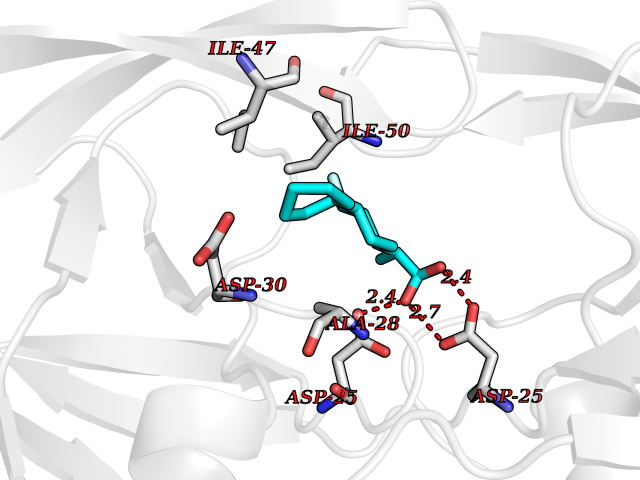 Pymol Map 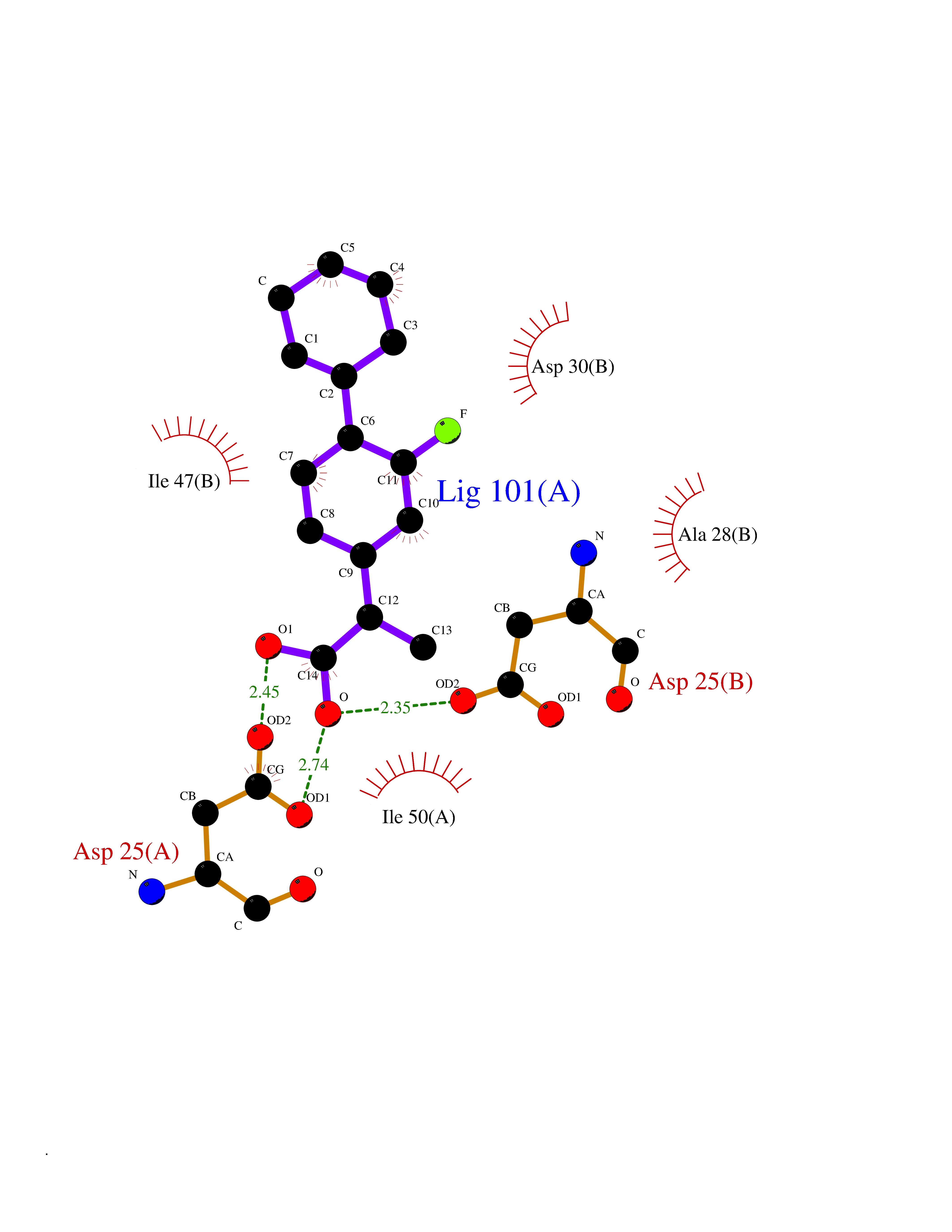 Ligplot Map 3D Binding mode Sequence PQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNFSFNFPQITLWKRPLVTIKIGGQLKEALLDTGADDTVIEEMSLPGRWKPKMIGGIGGFIKVRQYDQIIIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNF Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 7.49 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -7.76 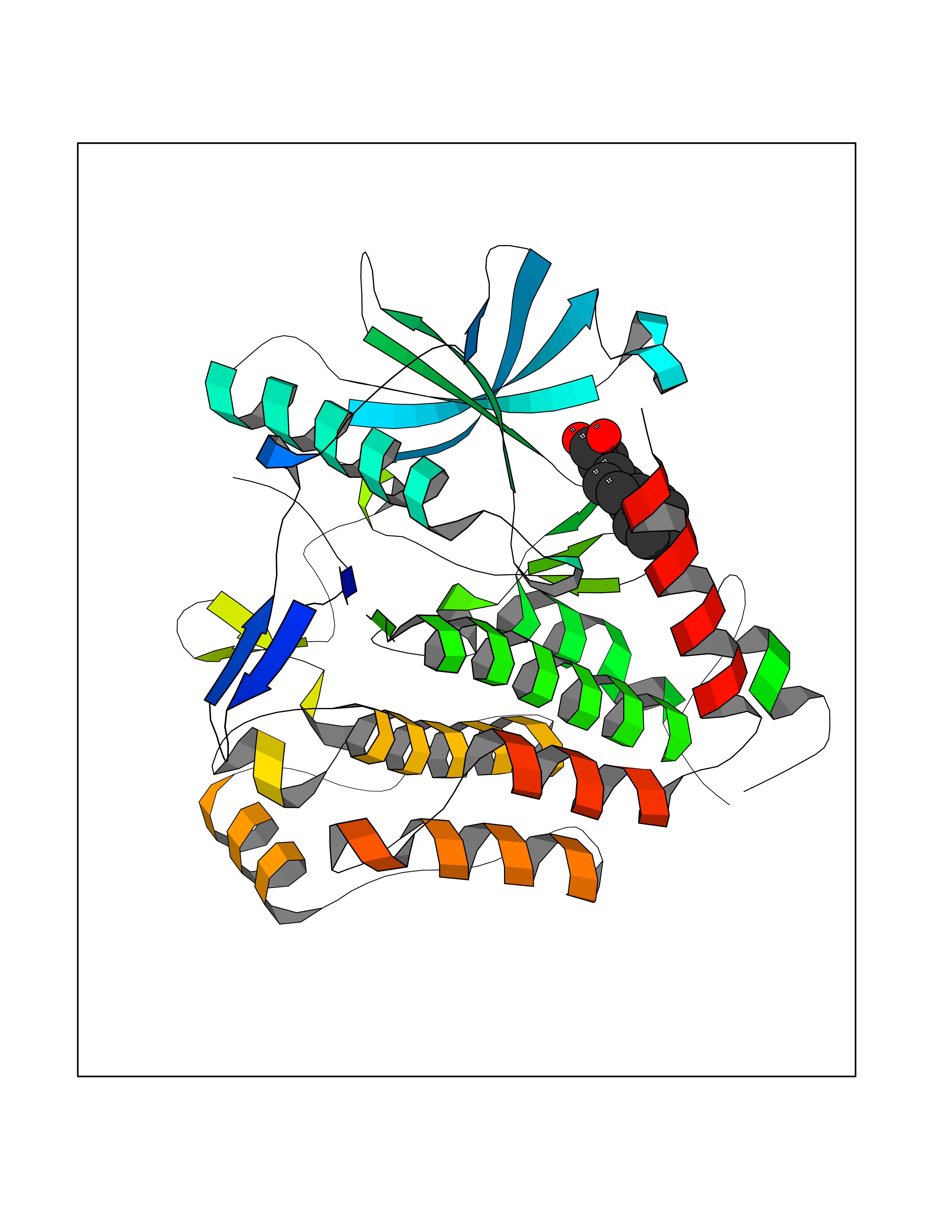 Molscript Map 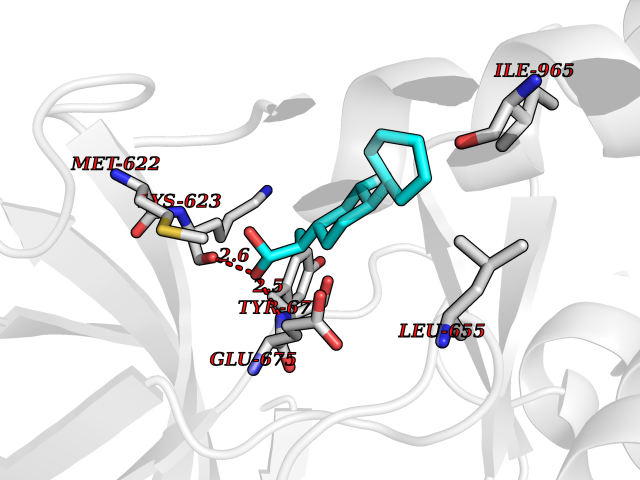 Pymol Map 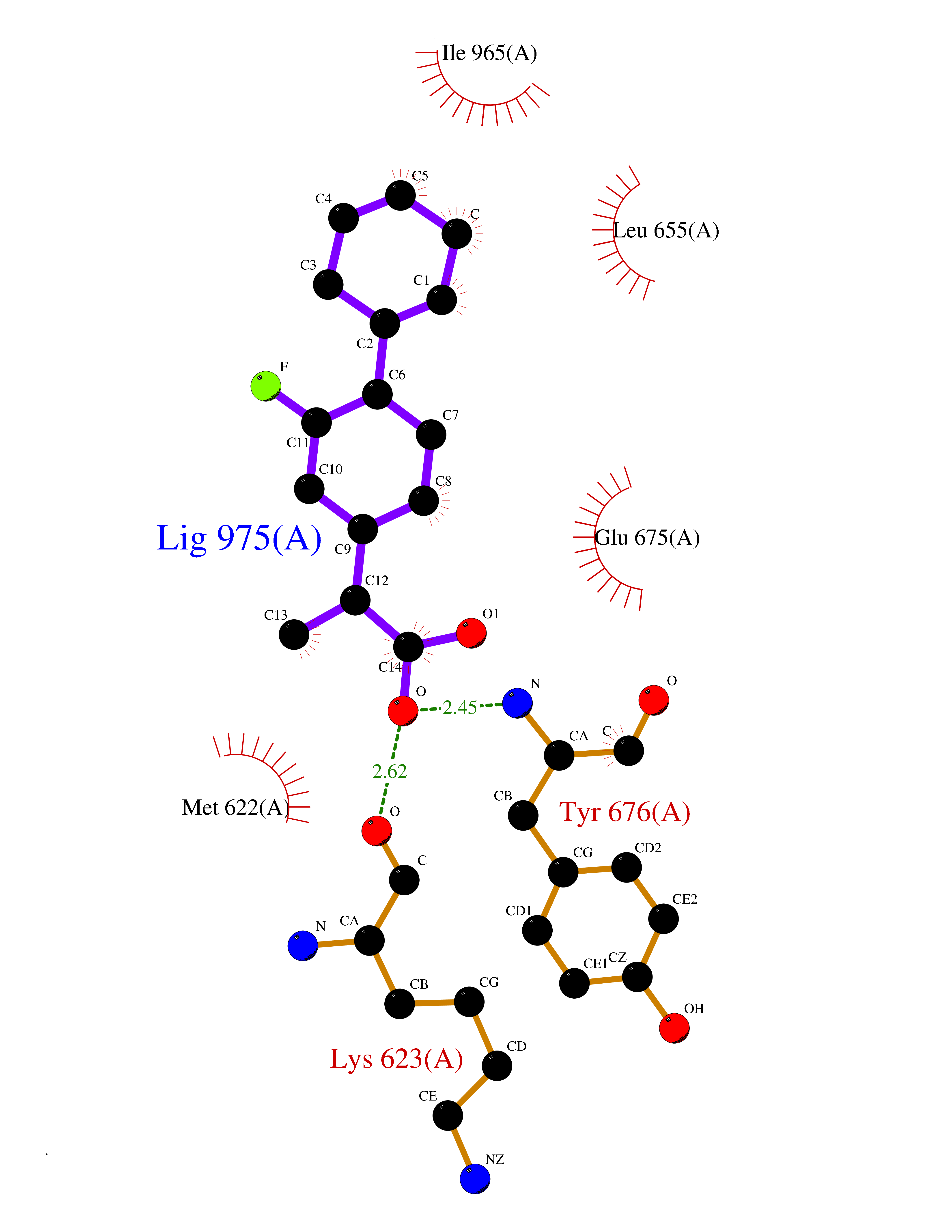 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Extracellular calcium-sensing receptor (CASR) | 5FBK | 7.47 | |
Target general information Gen name CASR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCasR; Parathyroid cell calciumreceptor; Parathyroid cell calcium-sensing receptor 1; Parathyroid calcium receptor; Parathyroid Cell calcium-sensing receptor; PCaR1; GPRC2A; CaSR Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Senses fluctuations in the circulating calcium concentration and modulates the production of parathyroid hormone (PTH) in parathyroid glands. The activity of this receptor is mediated by a G-protein that activates a phosphatidylinositol-calcium second messenger system. The G-protein-coupled receptor activity is activated by a co-agonist mechanism: aromatic amino acids, such as Trp or Phe, act concertedly with divalent cations, such as calcium or magnesium, to achieve full receptor activation. G-protein-coupled receptor that senses changes in the extracellular concentration of calcium ions and plays a key role in maintaining calcium homeostasis. Related diseases Hypocalciuric hypercalcemia, familial 1 (HHC1) [MIM:145980]: A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults. {ECO:0000269|PubMed:11762699, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:15579740, ECO:0000269|PubMed:15879434, ECO:0000269|PubMed:16598859, ECO:0000269|PubMed:16740594, ECO:0000269|PubMed:17473068, ECO:0000269|PubMed:17698911, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:19789209, ECO:0000269|PubMed:21566075, ECO:0000269|PubMed:21643651, ECO:0000269|PubMed:22114145, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25104082, ECO:0000269|PubMed:25292184, ECO:0000269|PubMed:26386835, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:7673400, ECO:0000269|PubMed:7726161, ECO:0000269|PubMed:7916660, ECO:0000269|PubMed:8636323, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9298824}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperparathyroidism, neonatal severe (NSHPT) [MIM:239200]: A disorder characterized by severe hypercalcemia, bone demineralization, and failure to thrive usually manifesting in the first 6 months of life. If untreated, NSHPT can be a devastating neurodevelopmental disorder, which in some cases is lethal without parathyroidectomy. {ECO:0000269|PubMed:14985373, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:17555508, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:8675635, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypocalcemia, autosomal dominant 1 (HYPOC1) [MIM:601198]: A disorder of mineral homeostasis characterized by blood calcium levels below normal, and low or normal serum parathyroid hormone concentrations. Disease manifestations include mild or asymptomatic hypocalcemia, paresthesias, carpopedal spasm, seizures, hypercalciuria with nephrocalcinosis or kidney stones, and ectopic and basal ganglia calcifications. Few patients manifest hypocalcemia and features of Bartter syndrome, including hypomagnesemia, hypokalemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia. {ECO:0000269|PubMed:10487661, ECO:0000269|PubMed:12050233, ECO:0000269|PubMed:12107202, ECO:0000269|PubMed:12241879, ECO:0000269|PubMed:12574188, ECO:0000269|PubMed:12915654, ECO:0000269|PubMed:15551332, ECO:0000269|PubMed:16608894, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:22789683, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25766501, ECO:0000269|PubMed:7874174, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8733126, ECO:0000269|PubMed:8813042, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253358, ECO:0000269|PubMed:9661634, ECO:0000269|PubMed:9920108}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 8 (EIG8) [MIM:612899]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Seizure types are variable, but include myoclonic seizures, absence seizures, febrile seizures, complex partial seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:18756473}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11093; DB11348; DB14481; DB01012; DB12865; DB00994; DB05695; DB05255; DB00127 Interacts with Q15363; P41180-1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 52438.9 Length 467 Aromaticity 0.12 Instability index 39.62 Isoelectric point 5.63 Charge (pH=7) -10.18 2D Binding mode Binding energy (Kcal/mol) -8.45 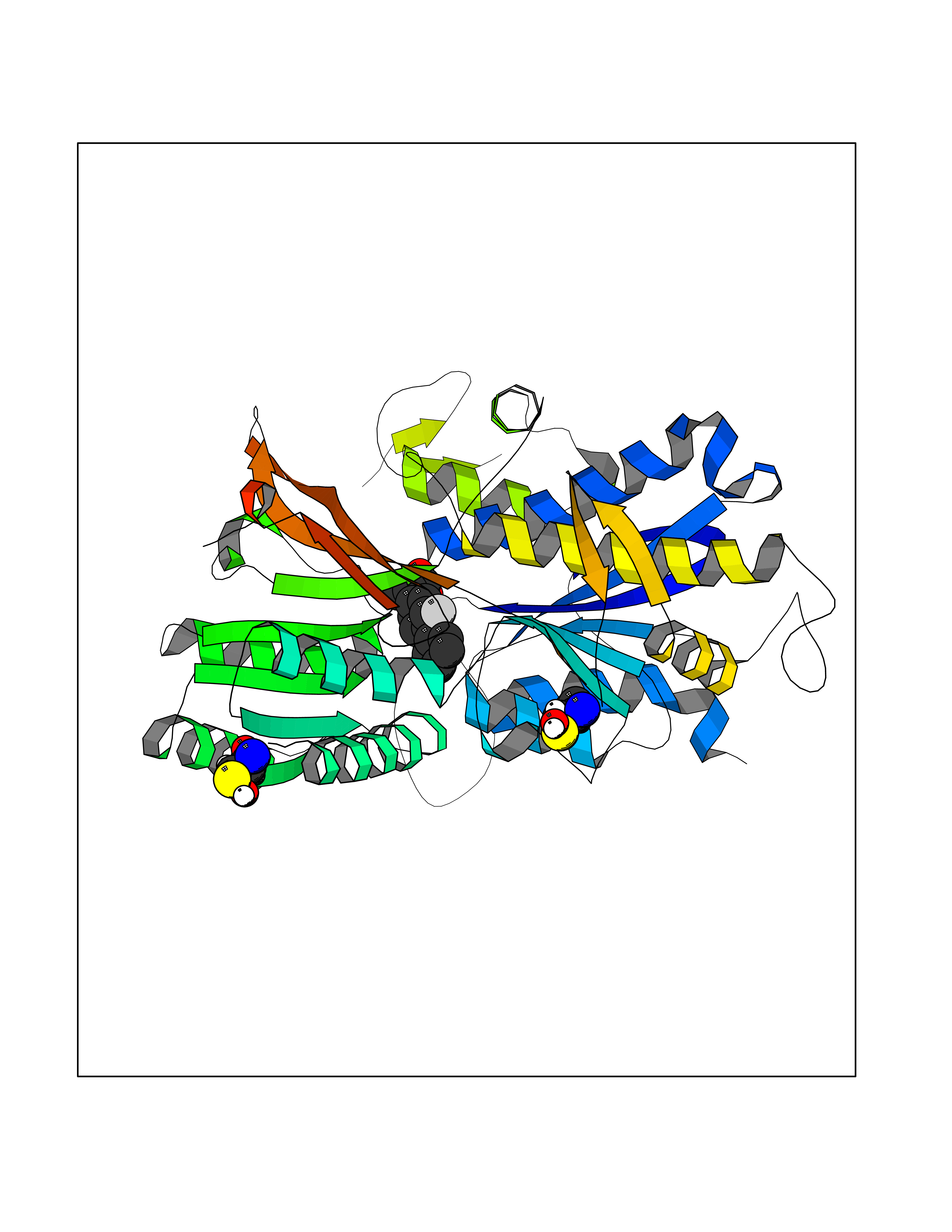 Molscript Map 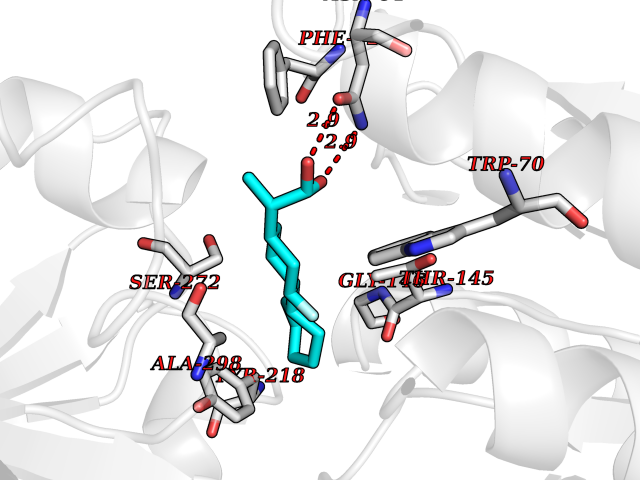 Pymol Map 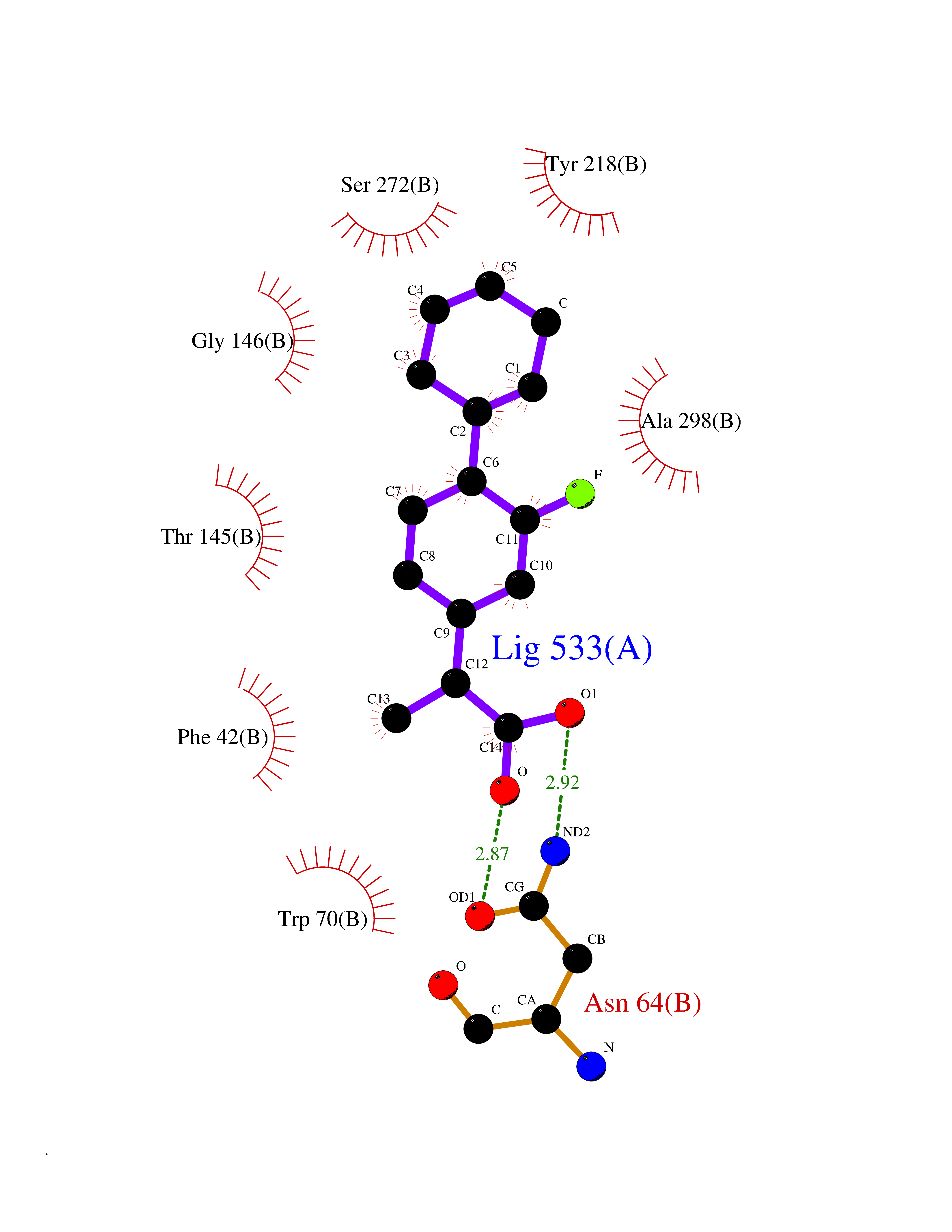 Ligplot Map 3D Binding mode Sequence GPDQRAQKKGDIILGGLFPIHFGVAAKDQDLKSRPESVECIRYNFRGFRWLQAMIFAIEEINSSPALLPNLTLGYRIFDTCNTVSKALEATLSFVAQNKIDSTIAVVGATGSGVSTAVANLLGLFYIPQVSYASSSRLLSNKNQFKSFLRTIPNDEHQATAMADIIEYFRWNWVGTIAADDDYGRPGIEKFREEAEERDIXIDFSELISQYSDEEEIQHVVEVIQNSTAKVIVVFSSGPDLEPLIKEIVRRNITGKIWLASEAWASSSLIAMPQYFHVVGGTIGFALKAGQIPGFREFLKKVHPRKSVHNGFAKEFWEETFNCHLQFRPLCTGDENISSVETPYIDYTHLRISYNVYLAVYSIAHALQDIYTCLPGRGLFTNGSCADIKKVEAWQVLKHLRHLNFTNNMGEQVTFDEXGDLVGNYSIINWHLSPEDGSIVFKEVGYYNVYAKKGERLFINEEKILWS Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Vasopressin V1a receptor | 1YTV | 7.44 | |
Target general information Gen name AVPR1A Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AVPR1 Protein family G-protein coupled receptor 1 family, Vasopressin/oxytocin receptor subfamily Biochemical class Sugar binding protein Function Peptide binding.Peptide hormone binding.Protein kinase C binding.V1A vasopressin receptor binding.Vasopressin receptor activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB09059; DB00872; DB00035; DB00093; DB14642; DB16279; DB05452; DB13929; DB02638; DB06212; DB00067 Interacts with P25106 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID M,N Molecular weight (Da) 40697.6 Length 371 Aromaticity 0.1 Instability index 20.94 Isoelectric point 5.07 Charge (pH=7) -9.93 2D Binding mode Binding energy (Kcal/mol) -8.82  Molscript Map 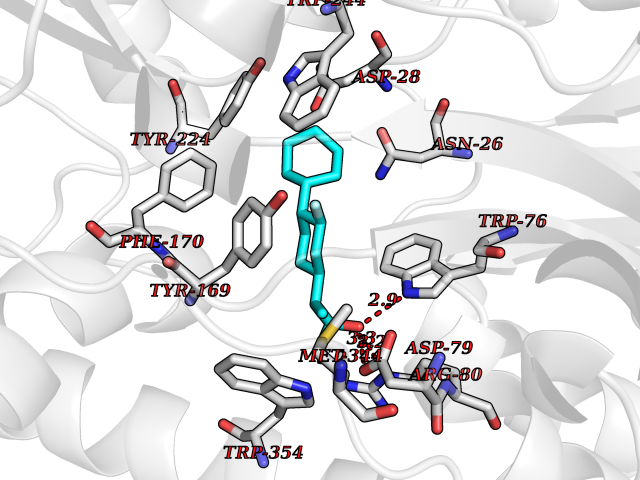 Pymol Map 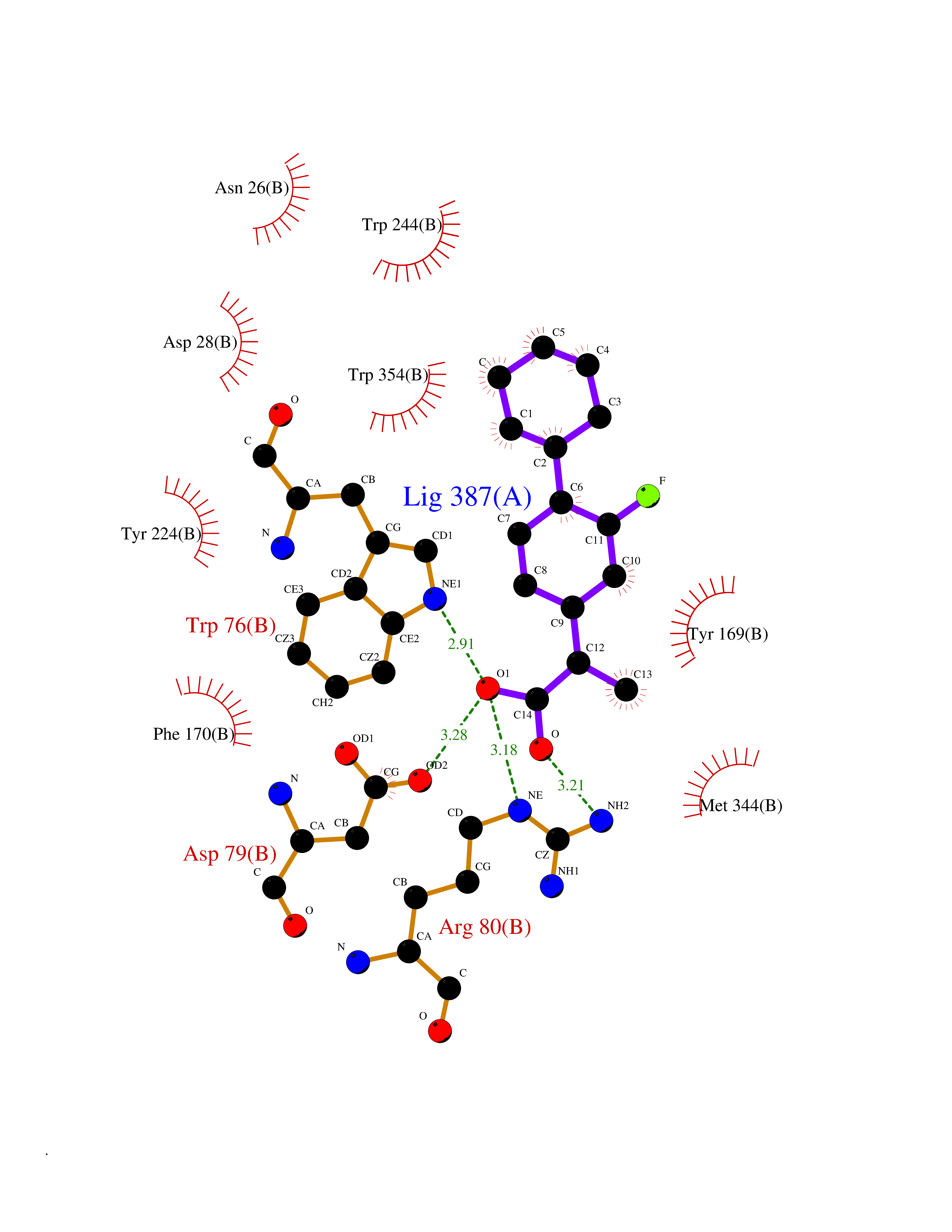 Ligplot Map 3D Binding mode Sequence NSSSNKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQT Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Angiotensin-converting enzyme | 5AMC | 7.43 | |
Target general information Gen name ACE Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms DCP1;DCP Protein family Peptidase M2 family Biochemical class Hydrolase Function Actin binding.Bradykinin receptor binding.Carboxypeptidase activity.Chloride ion binding.Drug binding.Endopeptidase activity.Exopeptidase activity.Metallodipeptidase activity.Metallopeptidase activity.Mitogen-activated protein kinase binding.Mitogen-activated protein kinase kinase binding.Peptidyl-dipeptidase activity.Tripeptidyl-peptidase activity.Zinc ion binding. Related diseases Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Renal tubular dysgenesis (RTD) [MIM:267430]: Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). {ECO:0000269|PubMed:16116425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Microvascular complications of diabetes 3 (MVCD3) [MIM:612624]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. {ECO:0000269|PubMed:10099885}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Intracerebral hemorrhage (ICH) [MIM:614519]: A pathological condition characterized by bleeding into one or both cerebral hemispheres including the basal ganglia and the cerebral cortex. It is often associated with hypertension and craniocerebral trauma. Intracerebral bleeding is a common cause of stroke. {ECO:0000269|PubMed:15277638}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00542; DB00616; DB01197; DB01340; DB15565; DB00584; DB09477; DB02032; DB00492; DB00722; DB00691; DB03740; DB00886; DB00790; DB00881; DB00178; DB01180; DB01348; DB08836; DB00519; DB13166 Interacts with P05556 EC number 3.4.15.1 Uniprot keywords 3D-structure; Alternative promoter usage; Alternative splicing; Calmodulin-binding; Carboxypeptidase; Cell membrane; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 70008.2 Length 607 Aromaticity 0.13 Instability index 46.01 Isoelectric point 5.71 Charge (pH=7) -13.56 2D Binding mode Binding energy (Kcal/mol) -8.14  Molscript Map 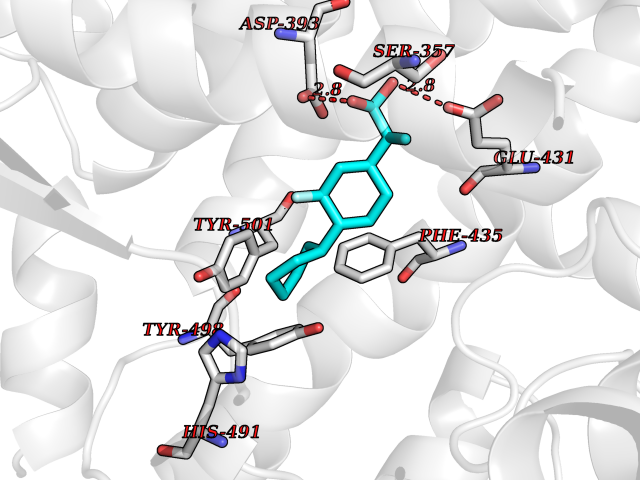 Pymol Map 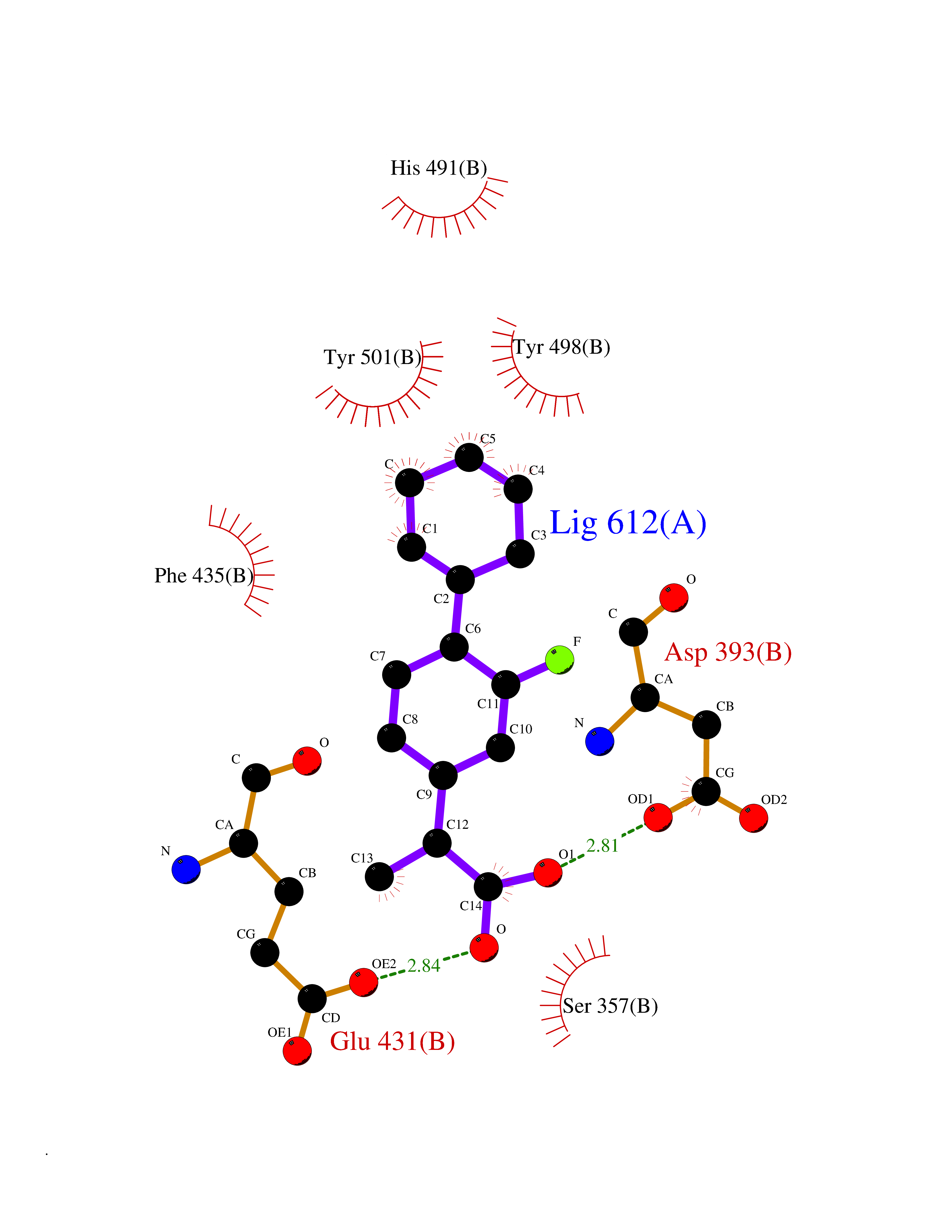 Ligplot Map 3D Binding mode Sequence LDPGLQPGQFSADEAGAQLFAQSYQSSAEQVLFQSVAASWAHDTNITAENARRQEEAALLSQEFAEAWGQKAKELYEPIWQQFTDPQLRRIIGAVRTLGSANLPLAKRQQYNALLSQMSRIYSTAKVCLTATCWSLDPDLTNILASSRSYAMLLFAWEGWHNAAGIPLKPLYEDFTALSNEAYKQDGFTDTGAYWRSWYNSPTFEDDLEHLYQQLEPLYLNLHAFVRRALHRRYGDRYINLRGPIPAHLLGDMWAQSWENIYDMVVPFPDKPNLDVTSTMLQQGWQATHMFRVAEEFFTSLELSPMPPEFWEGSMLEKPADGREVVCHASAWDFYNRKDFRIKQCTRVTMDQLSTVHHEMGHIQYYLQYKDLPVSLRRGANPGFHEAIGDVLALSVSTPEHLHKIGLLDRVTNDTESDINYLLKMALEKIAFLPFGYLVDQWRWGVFSGRTPPSRYNFDWWYLRTKYQGICPPVTRNETHFDAGAKFHVPNVTPYIRYFVSFVLQFQFHEALCKEAGYEGPLHQCDIYRSTKAGAKLRKVLRAGSSRPWQEVLKDMVGLDALDAQPLLKYFQLVTQWLQEQNQQNGEVLGWPEYQWHPPLPDNYPEG Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Interleukin 21 receptor (IL21R) | 6PLH | 7.43 | |
Target general information Gen name IL21R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ3121/PRO10273; Novel interleukin receptor; NILR; Interleukin-21 receptor; IL21 receptor; IL-21R; IL-21 receptor; CD360 Protein family Type I cytokine receptor family, Type 4 subfamily Biochemical class Cytokine receptor Function This is a receptor for interleukin-21. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) NA Interacts with P29972 EC number NA Uniprot keywords 3D-structure; Chromosomal rearrangement; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C,B Molecular weight (Da) 48376.5 Length 446 Aromaticity 0.1 Instability index 43.94 Isoelectric point 8.24 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -7.36  Molscript Map  Pymol Map 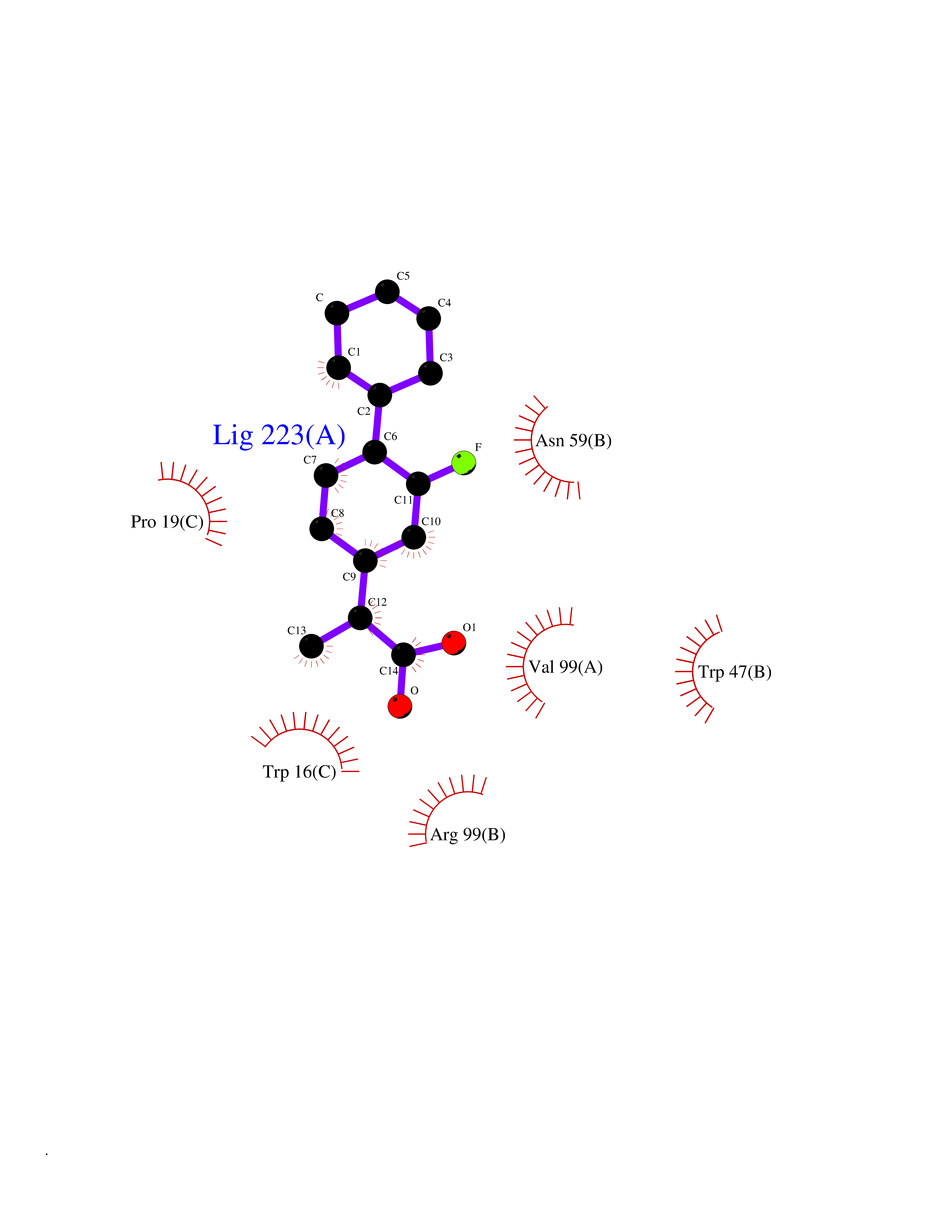 Ligplot Map 3D Binding mode Sequence DVVMTHTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGADFTLKISRVEAEDLGVYFCSQSTHVPRTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNECXVHLQQPGADLVKPGASVKMSCKASGYTFTSYWITWVKLRPGQGLEWIGDIYPGSGSTNFIEKFKSKATLTVDTSSSTAYMQLRSLTSEDSAVYYCARRGHGNYEDYWGQGTTLIVSSAKTTAPSVYPLAPVCGTGSSVTLGCLVKGYFPEPVTLTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVTSSTWPSQSITCNVAHPASSTKVDKKIEPRGPTTWSEWSDP Hydrogen bonds contact Hydrophobic contact | ||||