Job Results:
Ligand
Structure
Job ID
ea4aeeea43c0082ff76353d76181fa87
Job name
NA
Time
2024-06-15 15:52:32
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Peptidyl-prolyl cis-trans isomerase C | 2ESL | 6.40 | |
Target general information Gen name PPIC Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYPC Protein family Cyclophilin-type PPIase family Biochemical class Isomerase / immunosuppressant Function Cyclosporin A binding.Peptidyl-prolyl cis-trans isomerase activity. Related diseases Canavan disease (CAND) [MIM:271900]: A rare neurodegenerative condition of infancy or childhood characterized by white matter vacuolization and demyelination that gives rise to a spongy appearance. The clinical features are onset in early infancy, atonia of neck muscles, hypotonia, hyperextension of legs and flexion of arms, blindness, severe mental defect, megalocephaly, and death by 18 months on the average. {ECO:0000269|PubMed:10407784, ECO:0000269|PubMed:10564886, ECO:0000269|PubMed:10909858, ECO:0000269|PubMed:12205125, ECO:0000269|PubMed:12638939, ECO:0000269|PubMed:12706335, ECO:0000269|PubMed:24036223, ECO:0000269|PubMed:28101991, ECO:0000269|PubMed:7599639, ECO:0000269|PubMed:7668285, ECO:0000269|PubMed:8023850, ECO:0000269|PubMed:8252036, ECO:0000269|PubMed:8659549, ECO:0000269|PubMed:9452117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00172 Interacts with Q8N9N5; Q8N9N5-2; Q96L46; B3EWG5; O43765; Q96EQ0; Q9NZ09; Q9UMX0; Q9UMX0-2; Q9UHD9 EC number 5.2.1.8 Uniprot keywords 3D-structure; Cytoplasm; Isomerase; Proteomics identification; Reference proteome; Rotamase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 19625.2 Length 181 Aromaticity 0.1 Instability index 4.3 Isoelectric point 7.14 Charge (pH=7) 0.2 2D Binding mode Binding energy (Kcal/mol) -6.48 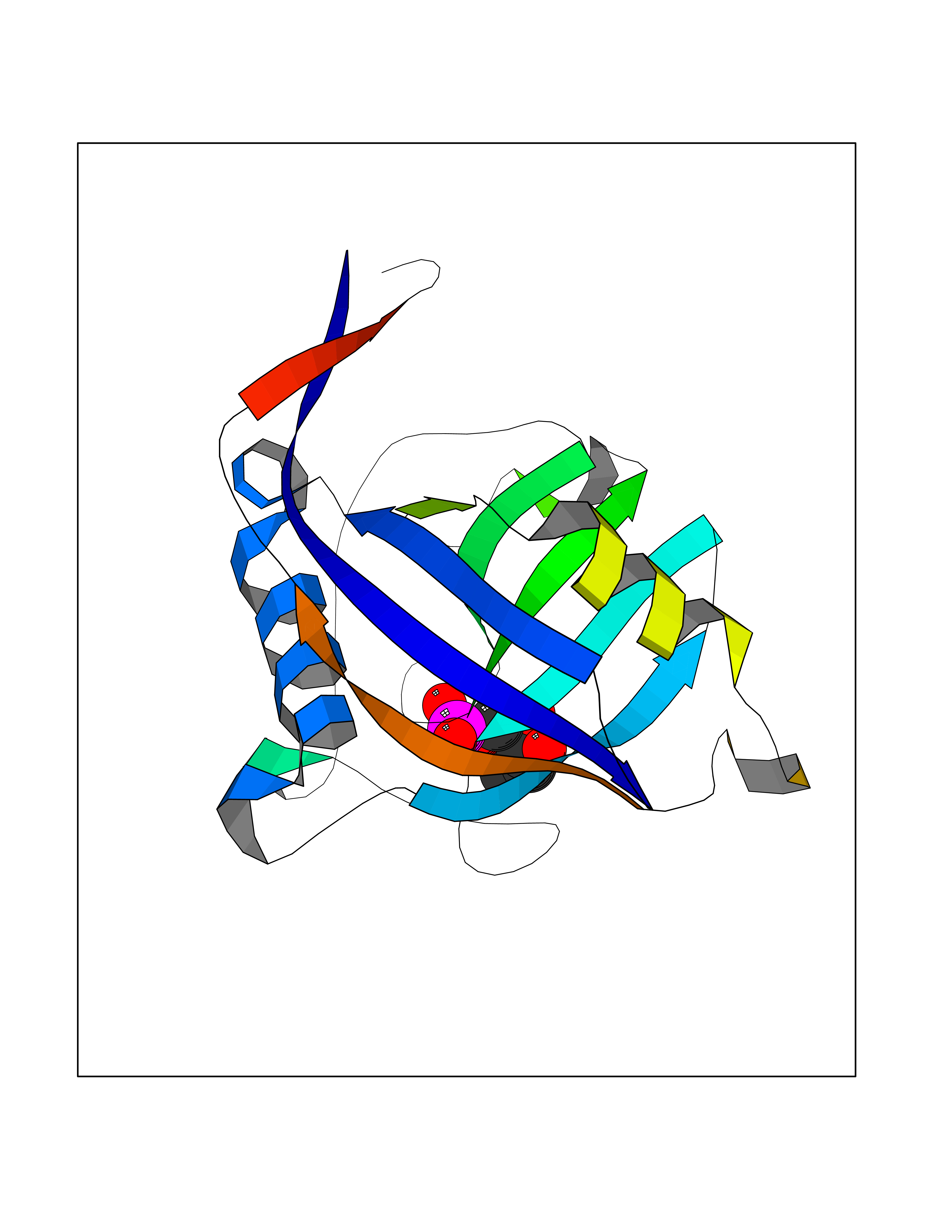 Molscript Map 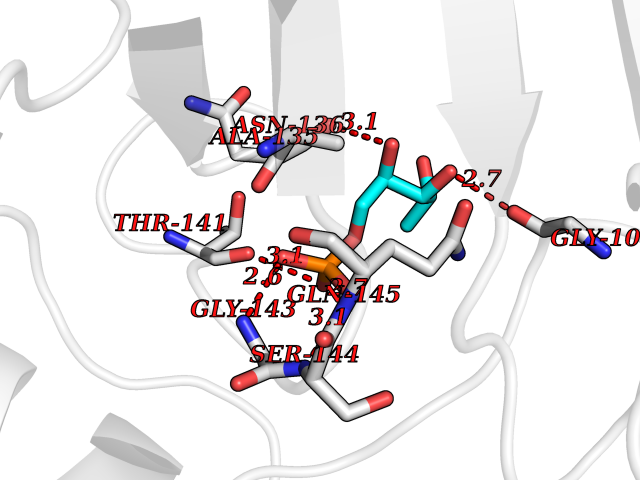 Pymol Map 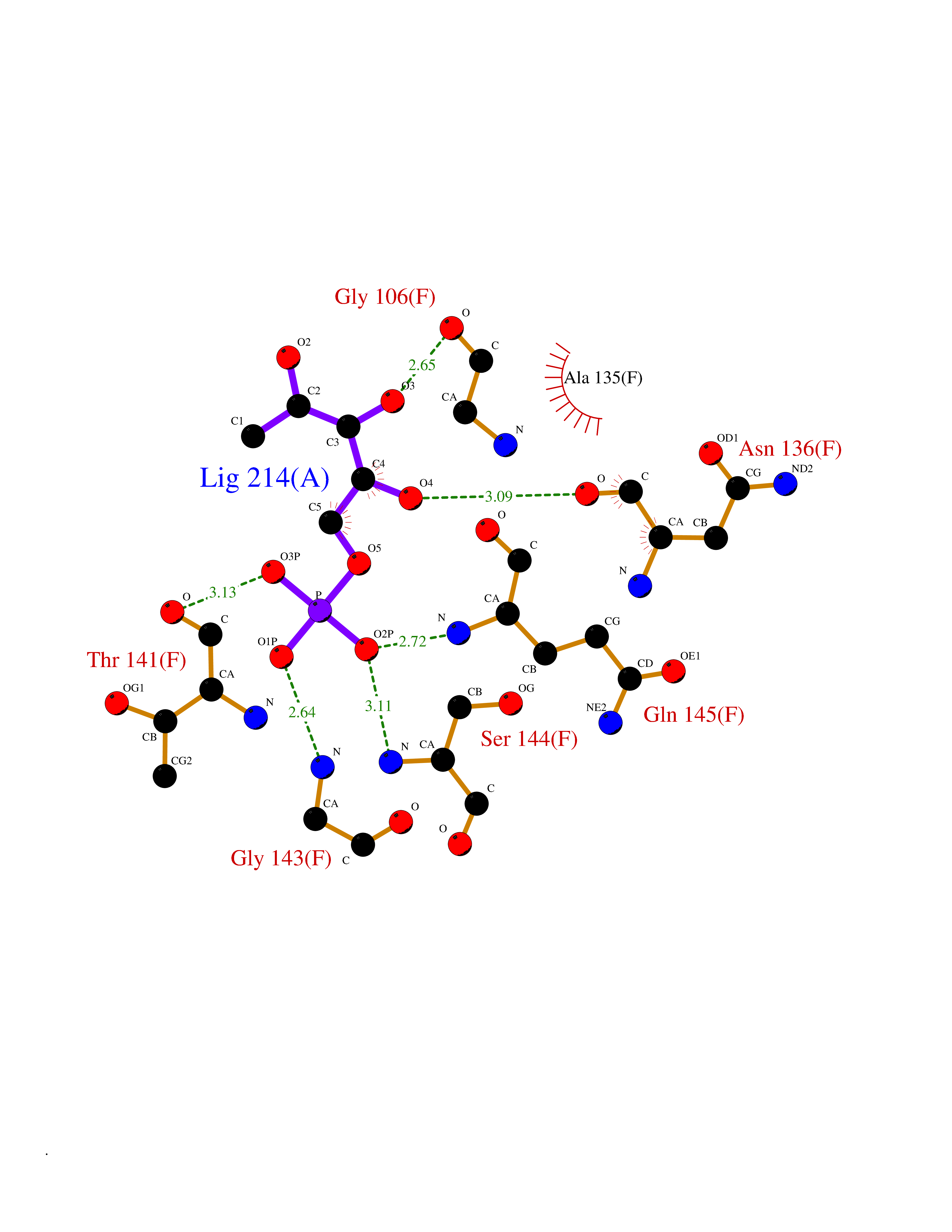 Ligplot Map 3D Binding mode Sequence RGPSVTAKVFFDVRIGDKDVGRIVIGLFGKVVPKTVENFVALATGEKGYGYKGSKFHRVIKDFMIQGGDITTGDGTGGVSIYGETFPDENFKLKHYGIGWVSMANAGPDTNGSQFFITLTKPTWLDGKHVVFGKVIDGMTVVHSIELQATDGHDRPLTNCSIINSGKIDVKTPFVVEIADW Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Myeloperoxidase (MPO) | 4DL1 | 6.40 | |
Target general information Gen name MPO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MPO Protein family Peroxidase family, XPO subfamily Biochemical class Peroxidases Function Part of the host defense system of polymorphonuclear leukocytes. It is responsible for microbicidal activity against a wide range of organisms. In the stimulated PMN, MPO catalyzes the production of hypohalous acids, primarily hypochlorous acidin physiologic situations, and other toxic intermediates that greatly enhance PMN microbicidal activity. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06111; DB00233; DB00006; DB02300; DB06774; DB00958; DB06468; DB00833; DB00535; DB00515; DB00847; DB00250; DB05161; DB01225; DB00583; DB01065; DB00461; DB04821; DB00104; DB00526; DB00550; DB00208; DB06823; DB00500; DB04827 Interacts with P27918; Q9UNE7 EC number EC 1.11.2.2 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Heme; Hydrogen peroxide; Iron; Lysosome; Metal-binding; Oxidation; Oxidoreductase; Peroxidase; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A,B,E,F,I,J,M,N Molecular weight (Da) 53052.6 Length 466 Aromaticity 0.08 Instability index 40.64 Isoelectric point 9.48 Charge (pH=7) 15.12 2D Binding mode Binding energy (Kcal/mol) -6.13  Molscript Map 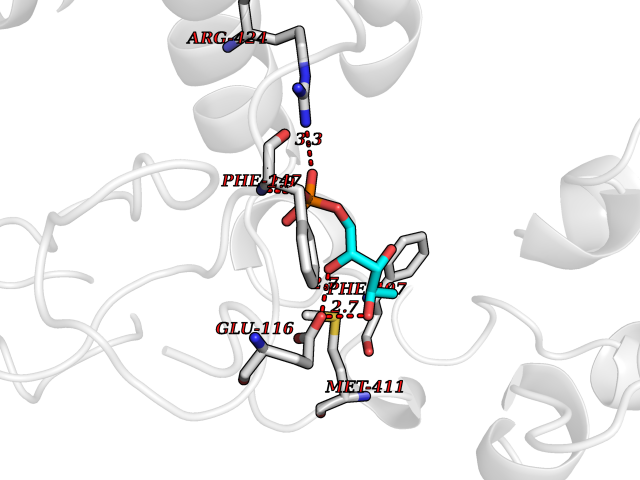 Pymol Map 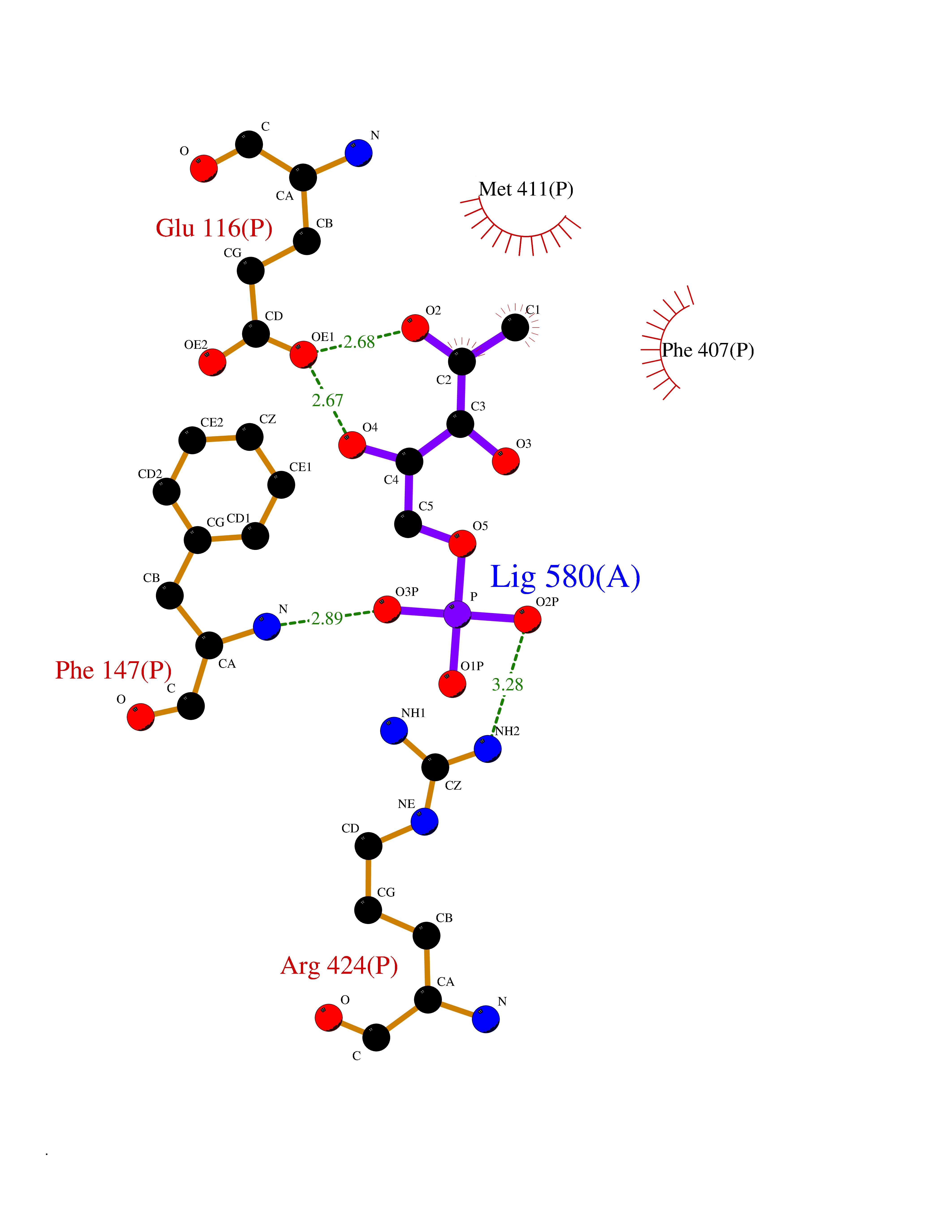 Ligplot Map 3D Binding mode Sequence VNCETSCVQQPPCFPLKIPPNDPRIKNQADCIPFFRSXPACPGSNITIRNQINALTSFVDASMVYGSEEPLARNLRNMSNQLGLLAVNQRFQDNGRALLPFDNLHDDPCLLTNRSARIPCFLAGDTRSSEMPELTSMHTLLLREHNRLATELKSLNPRWDGERLYQEARKIVGAMVQIITYRDYLPLVLGPTAMRKYLPTYRSYNDSVDPRIANVFTNAFRYGHTLIQPFMFRLDNRYQPMEPNPRVPLSRVFFASWRVVLEGGIDPILRGLMATPAKLNRQNQIAVDEIRERLFEQVMRIGLDLPALNMQRSRDHGLPGYNAWRRFCGLPQPETVGQLGTVLRNLKLARKLMEQYGTPNNIDIWMGGVSEPLKRKGRVGPLLACIIGTQFRKLRDGDRFWWENEGVFSMQQRQALAQISLPRIICDNTGITTVSKNNIFMSNSYPRDFVNCSTLPALNLASWREA Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Carbonic anhydrase IX (CA-IX) | 5FL4 | 6.39 | |
Target general information Gen name CA9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Renal cell carcinoma-associated antigen G250; RCC-associated antigen G250; PMW1; P54/58N; Membrane antigen MN; MN; G250 antigen (MN/CA IX/G250); G250; Carbonic anhydrase 9; Carbonate dehydratase IX; C Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Participates in pH regulation. May be involved in the control of cell proliferation and transformation. Appears to be a novel specific biomarker for a cervical neoplasia. Reversible hydration of carbon dioxide. Related diseases Hydroxykynureninuria (HYXKY) [MIM:236800]: An inborn error of amino acid metabolism characterized by massive urinary excretion of large amounts of kynurenine, 3-hydroxykynurenine and xanthurenic acid. Affected individuals manifest renal tubular dysfunction, metabolic acidosis, psychomotor retardation, non-progressive encephalopathy, and muscular hypertonia. {ECO:0000269|PubMed:17334708, ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Vertebral, cardiac, renal, and limb defects syndrome 2 (VCRL2) [MIM:617661]: An autosomal recessive congenital malformation syndrome characterized by vertebral segmentation abnormalities, congenital cardiac defects, renal defects, and distal mild limb defects. {ECO:0000269|PubMed:28792876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00562; DB00606; DB12741; DB08846; DB05304; DB00774; DB09460; DB00909 Interacts with P21291; O76003 EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Cell membrane; Cell projection; Direct protein sequencing; Disulfide bond; Glycoprotein; Lyase; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27522.8 Length 251 Aromaticity 0.08 Instability index 48.97 Isoelectric point 5.48 Charge (pH=7) -7.5 2D Binding mode Binding energy (Kcal/mol) -6.33  Molscript Map 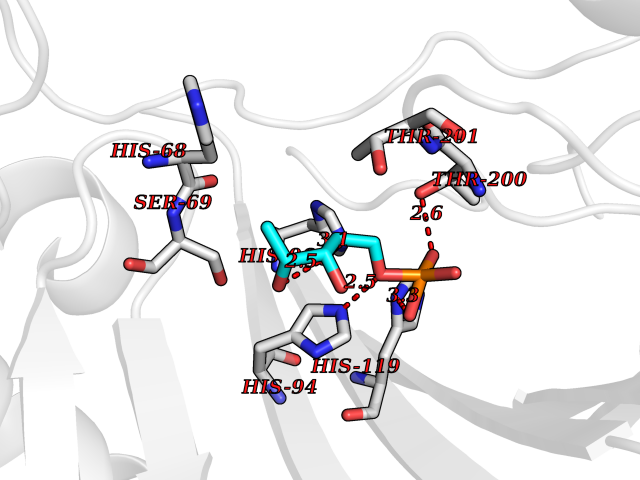 Pymol Map 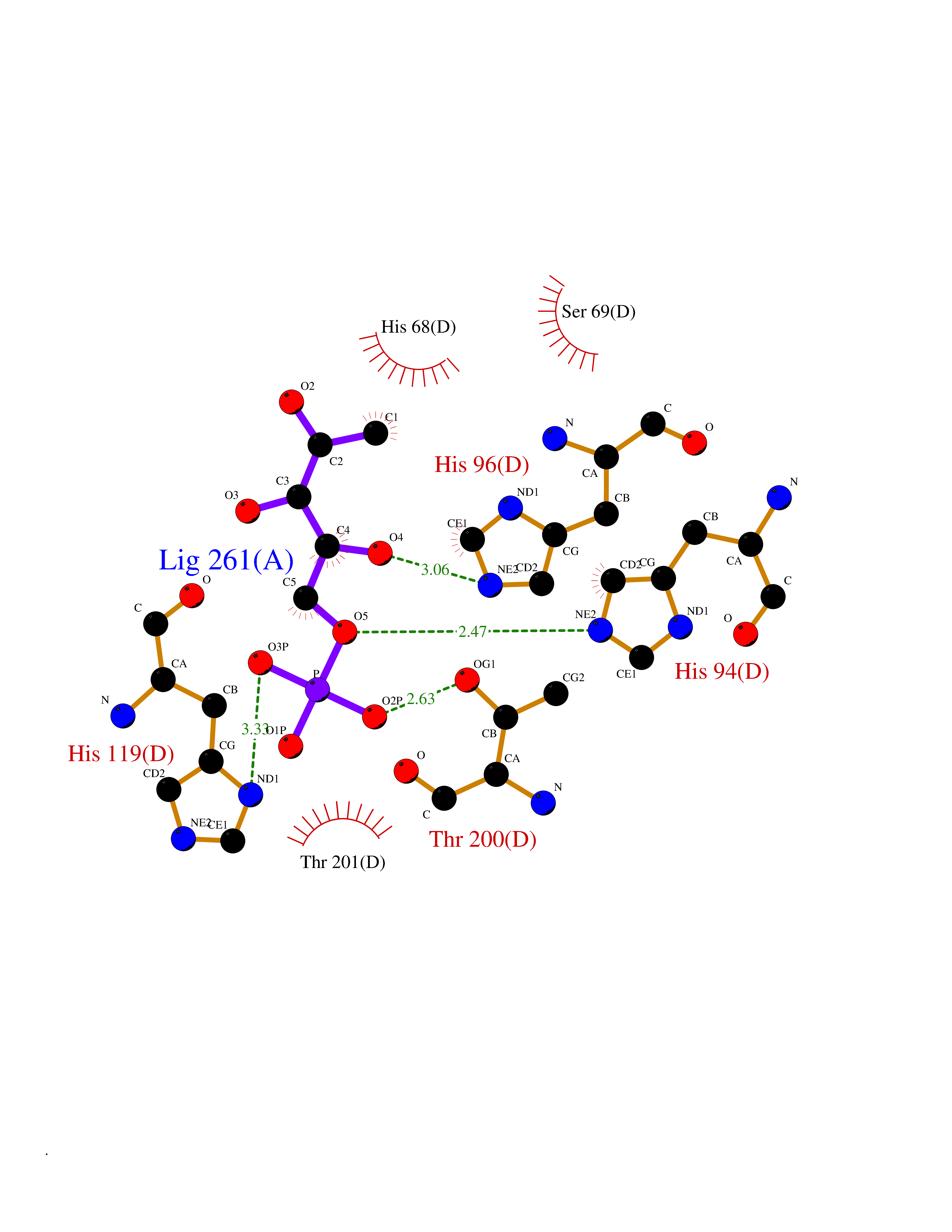 Ligplot Map 3D Binding mode Sequence WRYGGDPPWPRVSPACAGRFQSPVDIRPQLAAFSPALRPLELLGFQLPPLPELRLRNNGHSVQLTLPPGLEMALGPGREYRALQLHLHWGAAGRPGSEHTVEGHRFPAEIHVVHLSTAFARVDEALGRPGGLAVLAAFLEEGPEENSAYEQLLSRLEEIAEEGSETQVPGLDISALLPSDFSRYFQYEGSLTTPPCAQGVIWTVFNQTVMLSAKQLHTLSDTLWGPGDSRLQLNFRATQPLNGRVIEASFP Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Pyruvate kinase PKLR | 4IP7 | 6.38 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -5.77 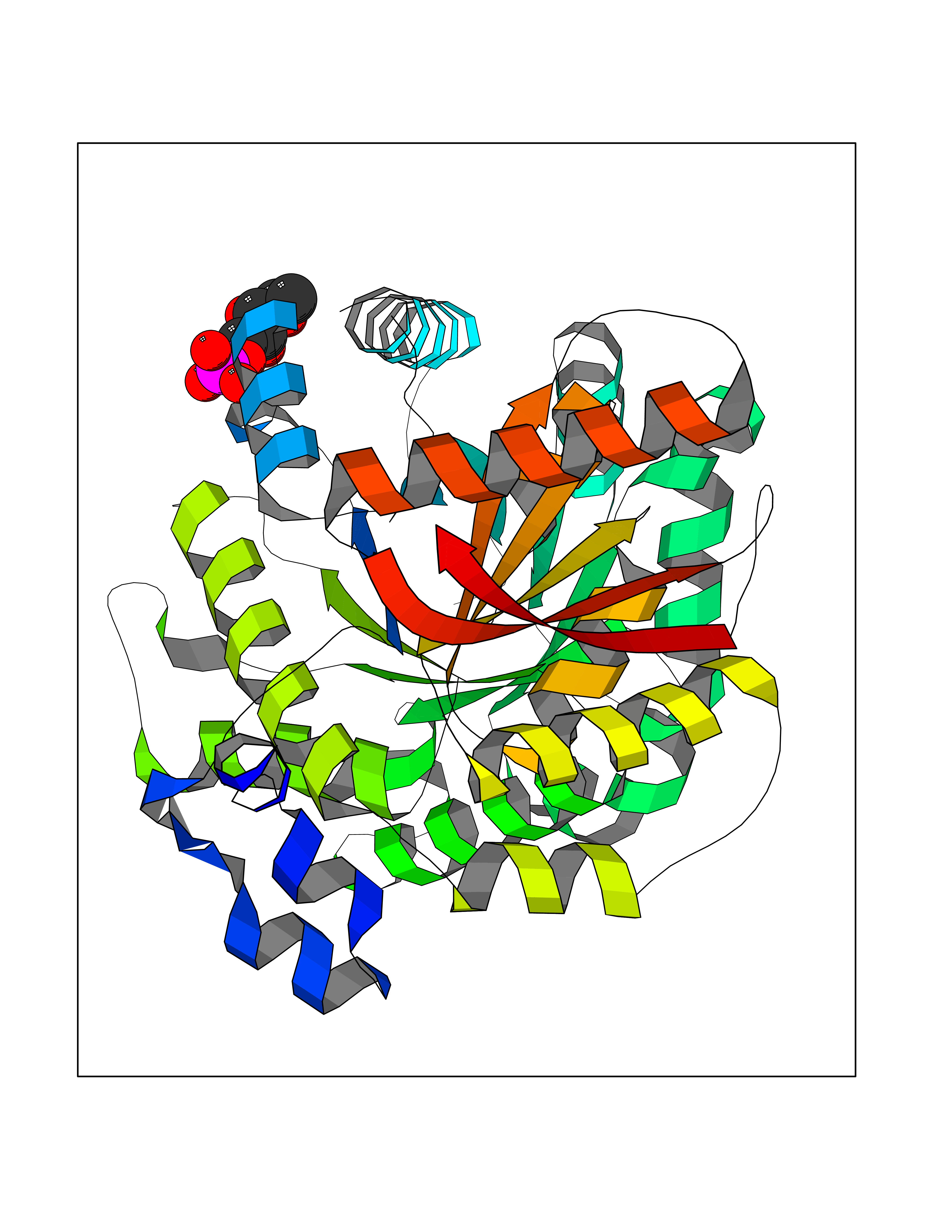 Molscript Map 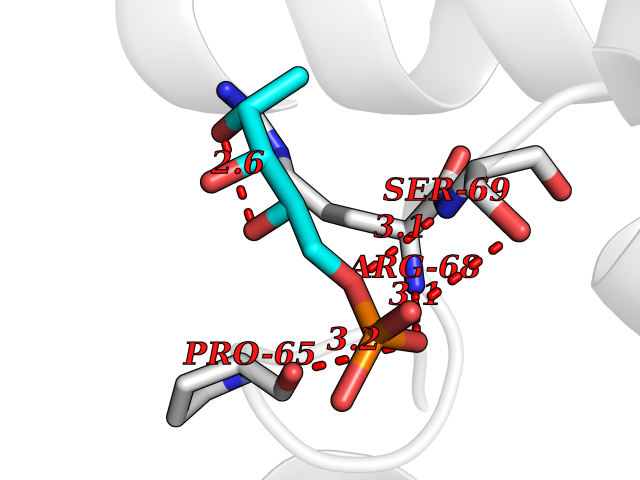 Pymol Map  Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | Extracellular calcium-sensing receptor (CASR) | 5FBK | 6.36 | |
Target general information Gen name CASR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCasR; Parathyroid cell calciumreceptor; Parathyroid cell calcium-sensing receptor 1; Parathyroid calcium receptor; Parathyroid Cell calcium-sensing receptor; PCaR1; GPRC2A; CaSR Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Senses fluctuations in the circulating calcium concentration and modulates the production of parathyroid hormone (PTH) in parathyroid glands. The activity of this receptor is mediated by a G-protein that activates a phosphatidylinositol-calcium second messenger system. The G-protein-coupled receptor activity is activated by a co-agonist mechanism: aromatic amino acids, such as Trp or Phe, act concertedly with divalent cations, such as calcium or magnesium, to achieve full receptor activation. G-protein-coupled receptor that senses changes in the extracellular concentration of calcium ions and plays a key role in maintaining calcium homeostasis. Related diseases Hypocalciuric hypercalcemia, familial 1 (HHC1) [MIM:145980]: A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults. {ECO:0000269|PubMed:11762699, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:15579740, ECO:0000269|PubMed:15879434, ECO:0000269|PubMed:16598859, ECO:0000269|PubMed:16740594, ECO:0000269|PubMed:17473068, ECO:0000269|PubMed:17698911, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:19789209, ECO:0000269|PubMed:21566075, ECO:0000269|PubMed:21643651, ECO:0000269|PubMed:22114145, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25104082, ECO:0000269|PubMed:25292184, ECO:0000269|PubMed:26386835, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:7673400, ECO:0000269|PubMed:7726161, ECO:0000269|PubMed:7916660, ECO:0000269|PubMed:8636323, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9298824}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperparathyroidism, neonatal severe (NSHPT) [MIM:239200]: A disorder characterized by severe hypercalcemia, bone demineralization, and failure to thrive usually manifesting in the first 6 months of life. If untreated, NSHPT can be a devastating neurodevelopmental disorder, which in some cases is lethal without parathyroidectomy. {ECO:0000269|PubMed:14985373, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:17555508, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:8675635, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypocalcemia, autosomal dominant 1 (HYPOC1) [MIM:601198]: A disorder of mineral homeostasis characterized by blood calcium levels below normal, and low or normal serum parathyroid hormone concentrations. Disease manifestations include mild or asymptomatic hypocalcemia, paresthesias, carpopedal spasm, seizures, hypercalciuria with nephrocalcinosis or kidney stones, and ectopic and basal ganglia calcifications. Few patients manifest hypocalcemia and features of Bartter syndrome, including hypomagnesemia, hypokalemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia. {ECO:0000269|PubMed:10487661, ECO:0000269|PubMed:12050233, ECO:0000269|PubMed:12107202, ECO:0000269|PubMed:12241879, ECO:0000269|PubMed:12574188, ECO:0000269|PubMed:12915654, ECO:0000269|PubMed:15551332, ECO:0000269|PubMed:16608894, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:22789683, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25766501, ECO:0000269|PubMed:7874174, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8733126, ECO:0000269|PubMed:8813042, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253358, ECO:0000269|PubMed:9661634, ECO:0000269|PubMed:9920108}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 8 (EIG8) [MIM:612899]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Seizure types are variable, but include myoclonic seizures, absence seizures, febrile seizures, complex partial seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:18756473}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11093; DB11348; DB14481; DB01012; DB12865; DB00994; DB05695; DB05255; DB00127 Interacts with Q15363; P41180-1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 52438.9 Length 467 Aromaticity 0.12 Instability index 39.62 Isoelectric point 5.63 Charge (pH=7) -10.18 2D Binding mode Binding energy (Kcal/mol) -6.72  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GPDQRAQKKGDIILGGLFPIHFGVAAKDQDLKSRPESVECIRYNFRGFRWLQAMIFAIEEINSSPALLPNLTLGYRIFDTCNTVSKALEATLSFVAQNKIDSTIAVVGATGSGVSTAVANLLGLFYIPQVSYASSSRLLSNKNQFKSFLRTIPNDEHQATAMADIIEYFRWNWVGTIAADDDYGRPGIEKFREEAEERDIXIDFSELISQYSDEEEIQHVVEVIQNSTAKVIVVFSSGPDLEPLIKEIVRRNITGKIWLASEAWASSSLIAMPQYFHVVGGTIGFALKAGQIPGFREFLKKVHPRKSVHNGFAKEFWEETFNCHLQFRPLCTGDENISSVETPYIDYTHLRISYNVYLAVYSIAHALQDIYTCLPGRGLFTNGSCADIKKVEAWQVLKHLRHLNFTNNMGEQVTFDEXGDLVGNYSIINWHLSPEDGSIVFKEVGYYNVYAKKGERLFINEEKILWS Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Carbonic anhydrase II (CA-II) | 3K34 | 6.35 | |
Target general information Gen name CA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase C; Carbonic anhydrase 2; Carbonate dehydratase II; CAC Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Can hydrate cyanamide to urea. Involved in the regulation of fluid secretion into the anterior chamber of the eye. Contributes to intracellular pH regulation in the duodenal upper villous epithelium during proton-coupled peptide absorption. Stimulates the chloride-bicarbonate exchange activity of SLC26A6. Essential for bone resorption and osteoclast differentiation. Related diseases Osteopetrosis, autosomal recessive 3 (OPTB3) [MIM:259730]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability. {ECO:0000269|PubMed:15300855, ECO:0000269|PubMed:1542674, ECO:0000269|PubMed:1928091, ECO:0000269|PubMed:8834238, ECO:0000269|PubMed:9143915}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07596; DB03333; DB08418; DB04081; DB08416; DB02479; DB07467; DB03950; DB03594; DB03294; DB04763; DB03270; DB08083; DB06954; DB08659; DB08046; DB02087; DB08156; DB04203; DB04394; DB08782; DB04549; DB02221; DB04180; DB03039; DB02861; DB02429; DB08202; DB04600; DB04601; DB01784; DB03385; DB03697; DB04002; DB07632; DB07050; DB06891; DB08645; DB08765; DB00819; DB03877; DB03262; DB03598; DB04089; DB01964; DB03526; DB04371; DB02220; DB03221; DB02602; DB02535; DB00436; DB00562; DB01194; DB00482; DB00880; DB02679; DB00606; DB02866; DB01144; DB00869; DB08846; DB01031; DB00311; DB08157; DB01942; DB00695; DB00774; DB08165; DB02292; DB03975; DB00703; DB00232; DB02610; DB07742; DB03844; DB02069; DB02986; DB08301; DB07048; DB03596; DB07476; DB01748; DB08155; DB01671; DB07710; DB01325; DB09460; DB09472; DB02894; DB00391; DB08329; DB07363; DB00273; DB01021; DB03904; DB00580; DB14533; DB14548; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Lyase; Membrane; Metal-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29027.4 Length 258 Aromaticity 0.1 Instability index 20.07 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -6.11 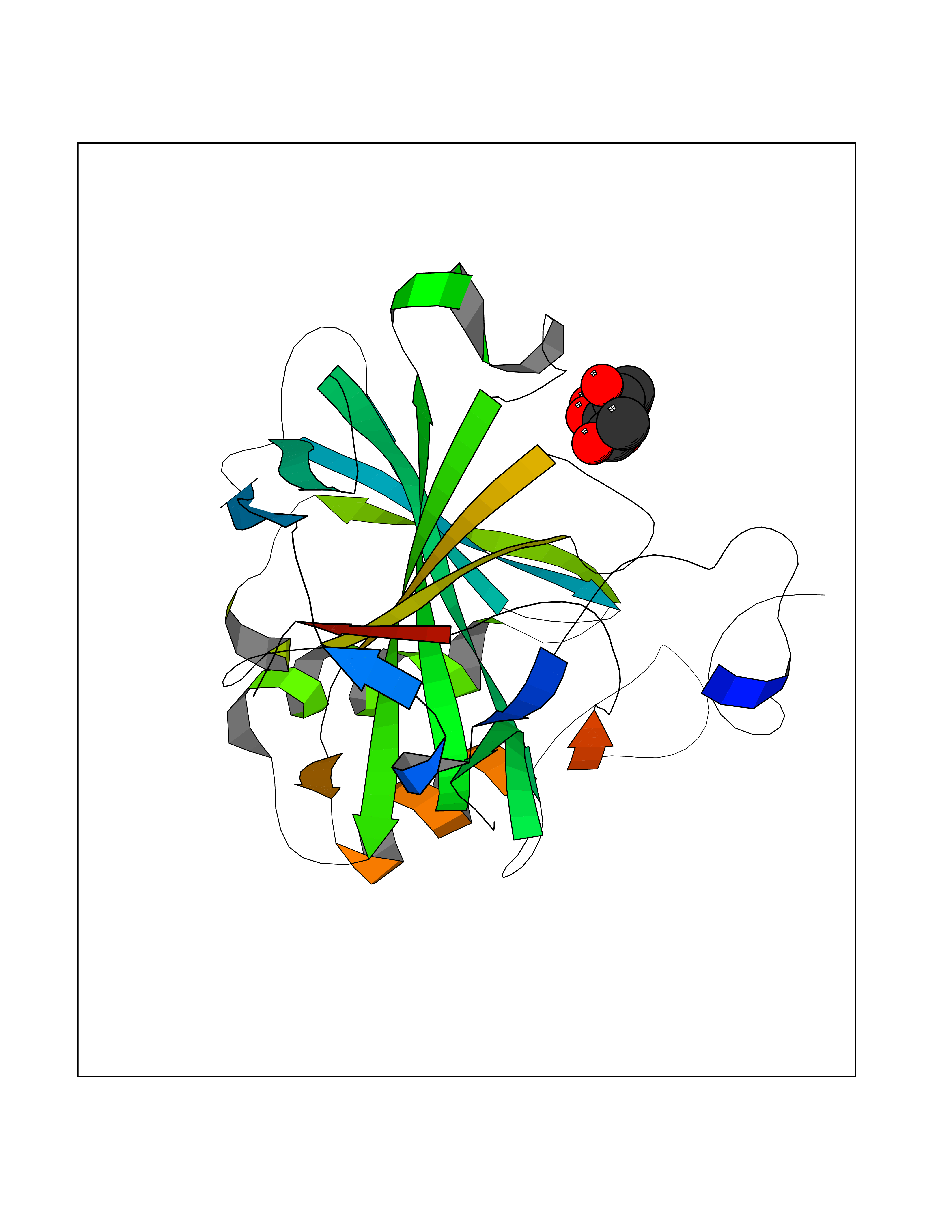 Molscript Map 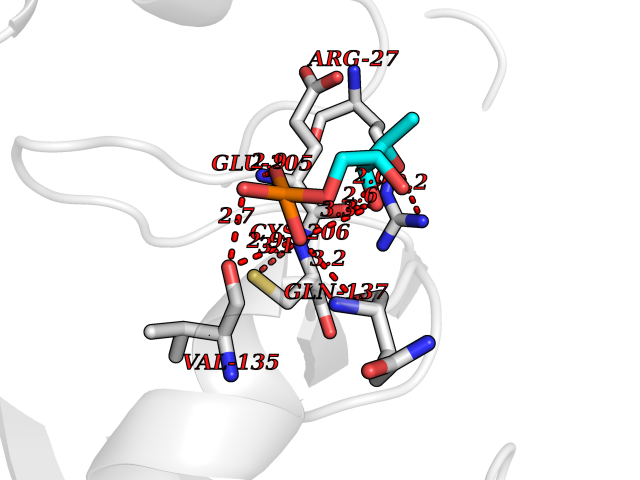 Pymol Map 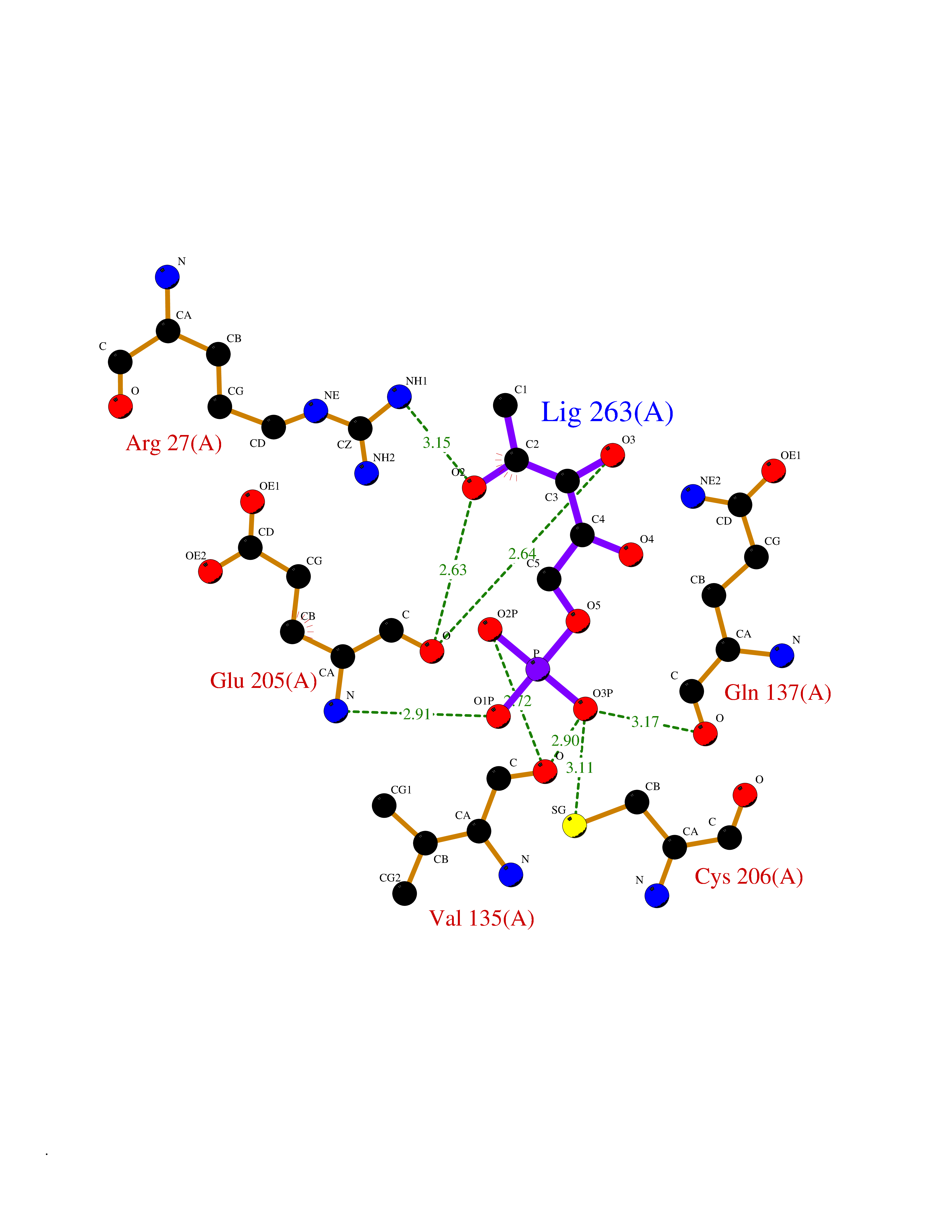 Ligplot Map 3D Binding mode Sequence HHWGYGKHNGPEHWHKDFPIAKGERQSPVDIDTHTAKYDPSLKPLSVSYDQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPPLLECVTWIVLKEPISVSSEQVLKFRKLNFNGEGEPEELMVDNWRPAQPLKNRQIKASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Serine/threonine PP1-alpha (PPP1CA) | 3E7B | 6.34 | |
Target general information Gen name PPP1CA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein phosphatase PP1-alpha catalytic subunit; Protein phosphatase 1alpha; PPP1A; PP-1A Protein family PPP phosphatase family, PP-1 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein phosphatase 1 (PP1) is essential for cell division, and participates in the regulation of glycogen metabolism, muscle contractility and protein synthesis. Involved in regulation of ionic conductances and long-term synaptic plasticity. May play an important role in dephosphorylating substrates such as the postsynaptic density-associated Ca(2+)/calmodulin dependent protein kinase II. Component of the PTW/PP1 phosphatase complex, which plays a role in the control of chromatin structure and cell cycle progression during the transition from mitosis into interphase. Regulates NEK2 function in terms of kinase activity and centrosome number and splitting, both in the presence and absence of radiation-induced DNA damage. Regulator of neural tube and optic fissure closure, and enteric neural crest cell (ENCCs) migration during development. In balance with CSNK1D and CSNK1E, determines the circadian period length, through the regulation of the speed and rhythmicity of PER1 and PER2 phosphorylation. May dephosphorylate CSNK1D and CSNK1E. Dephosphorylates the 'Ser-418' residue of FOXP3 in regulatory T-cells (Treg) from patients with rheumatoid arthritis, thereby inactivating FOXP3 and rendering Treg cells functionally defective. Dephosphorylates CENPA. Dephosphorylates the 'Ser-139' residue of ATG16L1 causing dissociation of ATG12-ATG5-ATG16L1 complex, thereby inhibiting autophagy. Protein phosphatase that associates with over 200 regulatory proteins to form highly specific holoenzymes which dephosphorylate hundreds of biological targets. Related diseases A chromosomal aberration involving FHIT has been found in a lymphoblastoid cell line established from a family with renal cell carcinoma and thyroid carcinoma. Translocation t(3;8)(p14.2;q24.1) with RNF139. Although the 3p14.2 breakpoint has been shown to interrupt FHIT in its 5-prime non-coding region, it is unlikely that FHIT is causally related to renal or other malignancies. {ECO:0000269|PubMed:15007172}.; DISEASE: Associated with digestive tract cancers. Numerous tumor types are found to have aberrant forms of FHIT protein due to deletions in a coding region of chromosome 3p14.2 including the fragile site locus FRA3B. {ECO:0000269|PubMed:15007172}. Drugs (DrugBank ID) DB02506 Interacts with Q6ZMQ8; P31749; O14727; P05067; O15169; P38398; O95400; Q99459; P12830; Q8TEP8; Q9NX63; Q6PJW8; Q96S65; Q9H175; Q92796; P05198; P55199; Q9BZS1; P42858; Q8NI77; Q8NG31; Q5S007; O00566; Q9UPR0; Q96QC0; Q96KQ4; Q8WUF5; O75807; Q5SWA1; Q96T49; Q6NYC8; P41236; Q5T8A7; Q86WC6; Q6NXS1; O14990; O75864; Q86XI6; Q9UQK1; O95685; Q15435; Q12972; Q12972-1; Q12972-2; Q96SB3; P60484; P06400; Q5UIP0; Q14684; P04271; Q7Z5V6; A8K8P3; Q562F6; Q9H788; Q8TEC5; P63208; Q7Z699; P43405-2; Q9HCH5; Q14C87; Q5JTV8; Q05BL1; Q13625; Q4KMQ1; Q4KMQ1-2; Q8TEL6; P49815; P55072; Q9Y2W2; Q9H4A3; P16989; P49750; Q9HBF4; Q7Z3T8; O95405; O08785; P36313; K9N4V7; Q76TK5; O35867; O35274 EC number EC 3.1.3.16 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Carbohydrate metabolism; Cell cycle; Cell division; Cytoplasm; Direct protein sequencing; Glycogen metabolism; Host-virus interaction; Hydrolase; Manganese; Metal-binding; Nucleus; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33626.3 Length 294 Aromaticity 0.11 Instability index 44.81 Isoelectric point 5.16 Charge (pH=7) -10.09 2D Binding mode Binding energy (Kcal/mol) -6.61 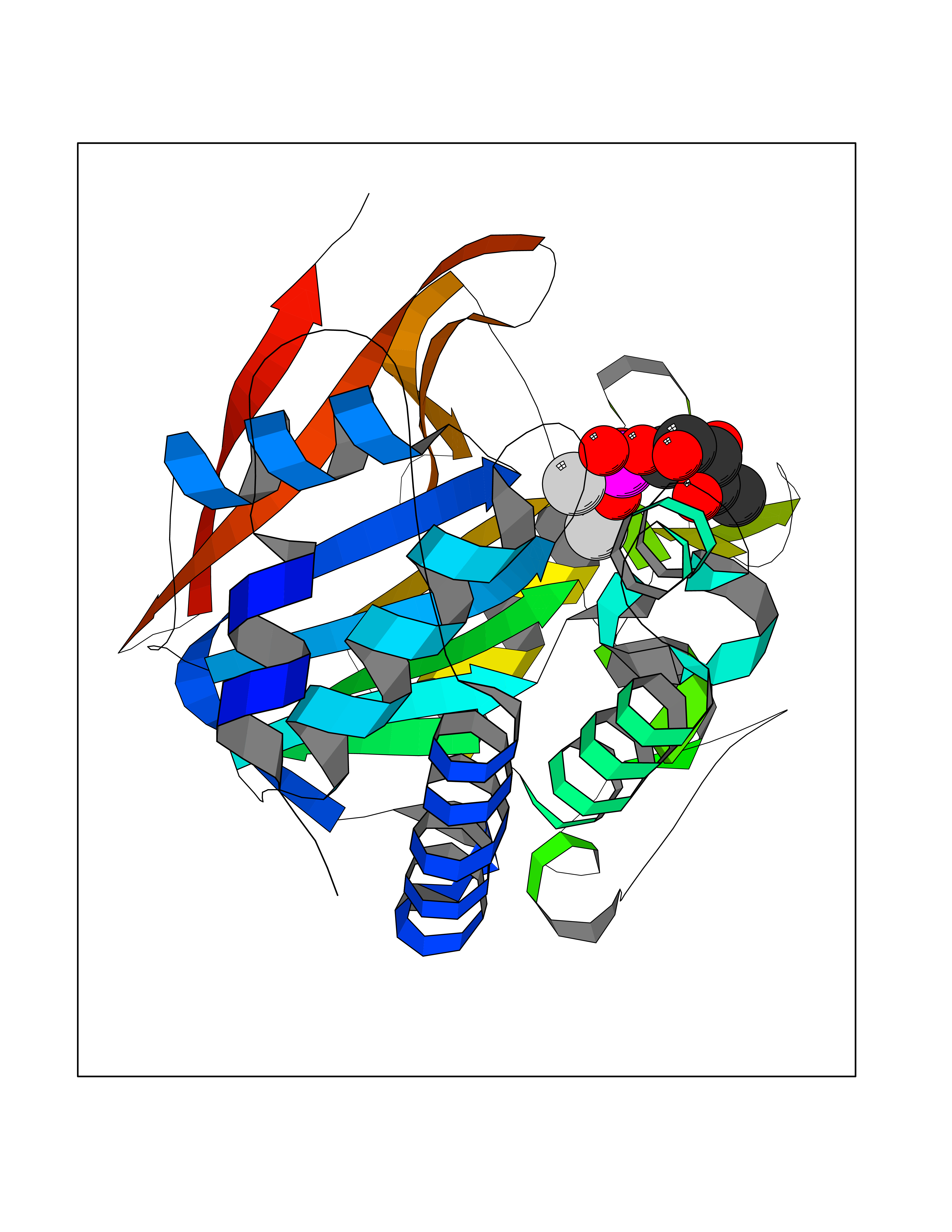 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SLNLDSIIGRLLEVQGSRPGKNVQLTENEIRGLCLKSREIFLSQPILLELEAPLKICGDIHGQYYDLLRLFEYGGFPPESNYLFLGDYVDRGKQSLETICLLLAYKIKYPENFFLLRGNHECASINRIYGFYDECKRRYNIKLWKTFTDCFNCLPIAAIVDEKIFCCHGGLSPDLQSMEQIRRIMRPTDVPDQGLLCDLLWSDPDKDVQGWGENDRGVSFTFGAEVVAKFLHKHDLDLICRAHQVVEDGYEFFAKRQLVTLFSAPNYCGEFDNAGAMMSVDETLMCSFQILKPA Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Tripartite motif-containing 24 (TRIM24) | 4YAD | 6.32 | |
Target general information Gen name TRIM24 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tripartite motif-containing protein 24; Transcription intermediary factor 1-alpha; TIF1A; TIF1-alpha; TIF1; RNF82; RING-type E3 ubiquitin transferase TIF1-alpha; RING finger protein 82; E3 ubiquitin-p Protein family NA Biochemical class Acyltransferase Function Interacts with chromatin depending on histone H3 modifications, having the highest affinity for histone H3 that is both unmodified at 'Lys-4' (H3K4me0) and acetylated at 'Lys-23' (H3K23ac). Has E3 protein-ubiquitin ligase activity. Promotes ubiquitination and proteasomal degradation of p53/TP53. Plays a role in the regulation of cell proliferation and apoptosis, at least in part via its effects on p53/TP53 levels. Up-regulates ligand-dependent transcription activation by AR, GCR/NR3C1, thyroid hormone receptor (TR) and ESR1. Modulates transcription activation by retinoic acid (RA) receptors, including RARA. Plays a role in regulating retinoic acid-dependent proliferation of hepatocytes. Transcriptional coactivator that interacts with numerous nuclear receptors and coactivators and modulates the transcription of target genes. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) NA Interacts with P03372; P04637; Q9UPN9 EC number EC 2.3.2.27 Uniprot keywords 3D-structure; Alternative splicing; Bromodomain; Chromosomal rearrangement; Coiled coil; Cytoplasm; Direct protein sequencing; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Mitochondrion; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Repressor; Transcription; Transcription regulation; Transferase; Tumor suppressor; Ubl conjugation; Ubl conjugation pathway; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 20651.6 Length 178 Aromaticity 0.11 Instability index 40.58 Isoelectric point 4.97 Charge (pH=7) -7.86 2D Binding mode Binding energy (Kcal/mol) -6.69  Molscript Map  Pymol Map 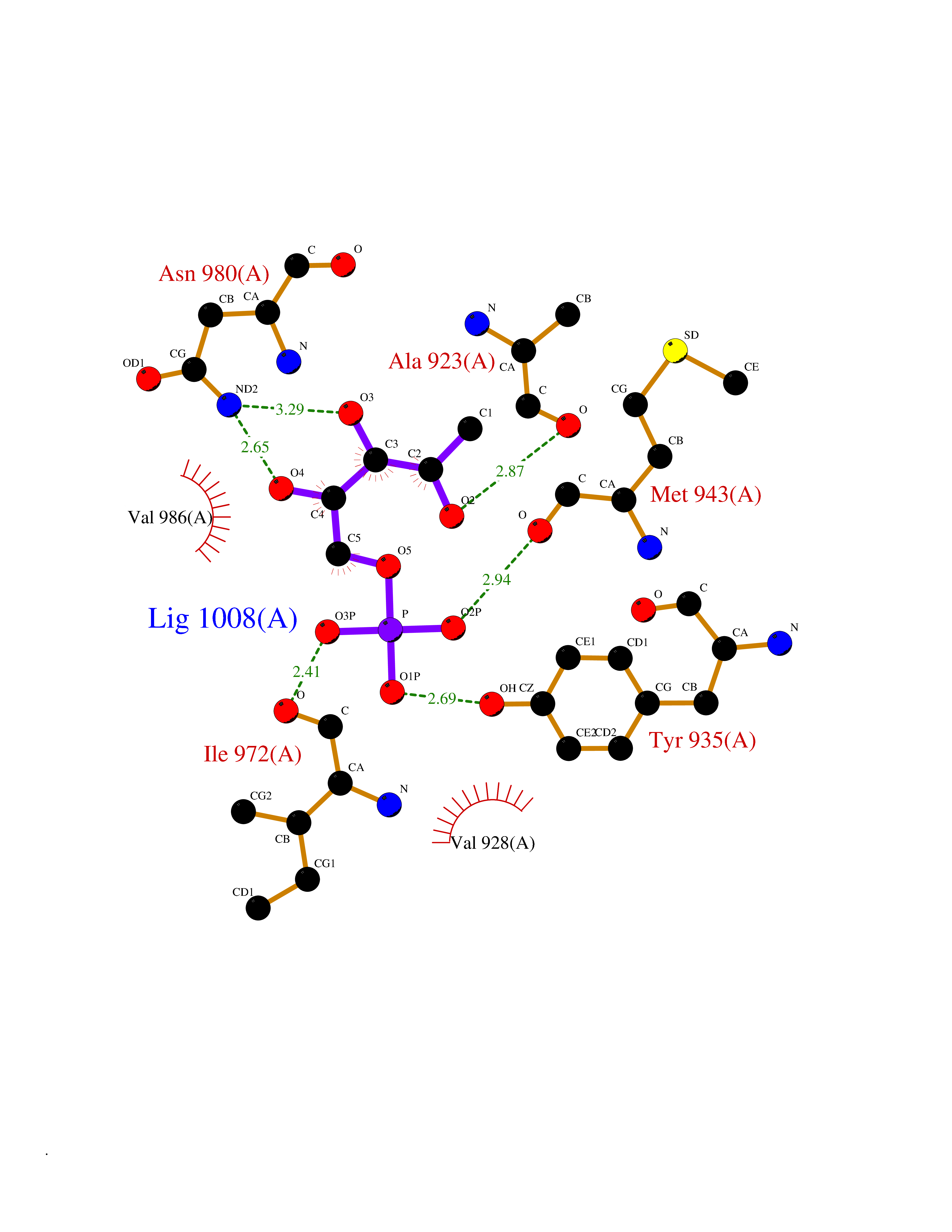 Ligplot Map 3D Binding mode Sequence PNEDWCAVCQNGGELLCCEKCPKVFHLSCHVPTLTNFPSGEWICTFCRDLSKPEVEYDCDAPKKKTEGLVKLTPIDKRKCERLLLFLYCHEMSLAFQDPVPLTVPDYYKIIKNPMDLSTIKKRLQEDYSMYSKPEDFVADFRLIFQNCAEFNEPDSEVANAGIKLENYFEELLKNLYP Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Plasmodium DOXP reductoisomerase (Malaria DXR) | 3AU9 | 6.32 | |
Target general information Gen name Malaria DXR Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID Synonyms IspC; DXR; DXP reductoisomerase; DOXP reductoisomerase; 2-C-Methyl-d-erythritol 4-phosphate synthase; 1-deoxyxylulose-5-phosphate reductoisomerase Protein family DXR family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADP-dependent rearrangement and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol 4-phosphate (MEP). Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.1.267 Uniprot keywords 3D-structure; Apicoplast; Isoprene biosynthesis; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Plastid; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46644.4 Length 410 Aromaticity 0.09 Instability index 36.77 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -7.14 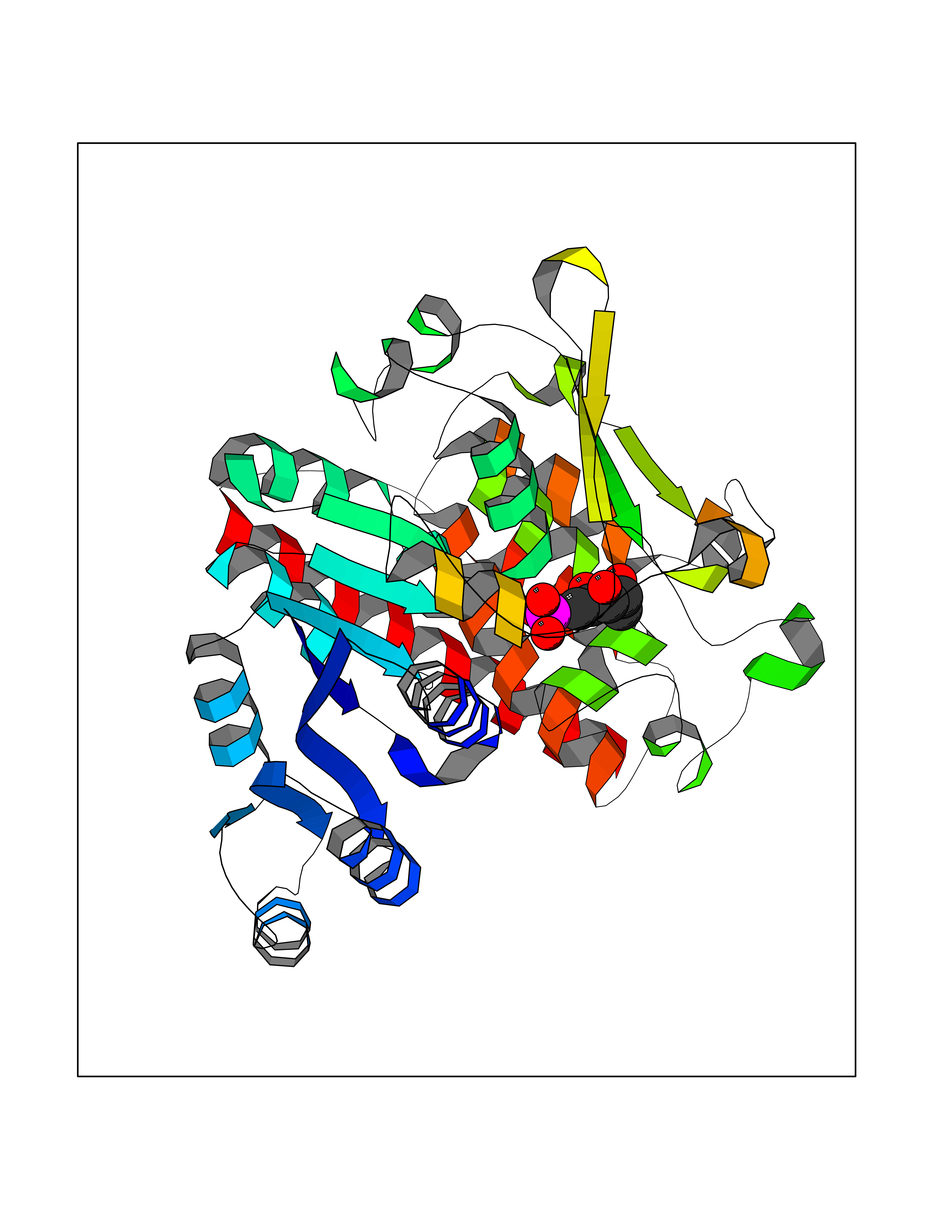 Molscript Map  Pymol Map 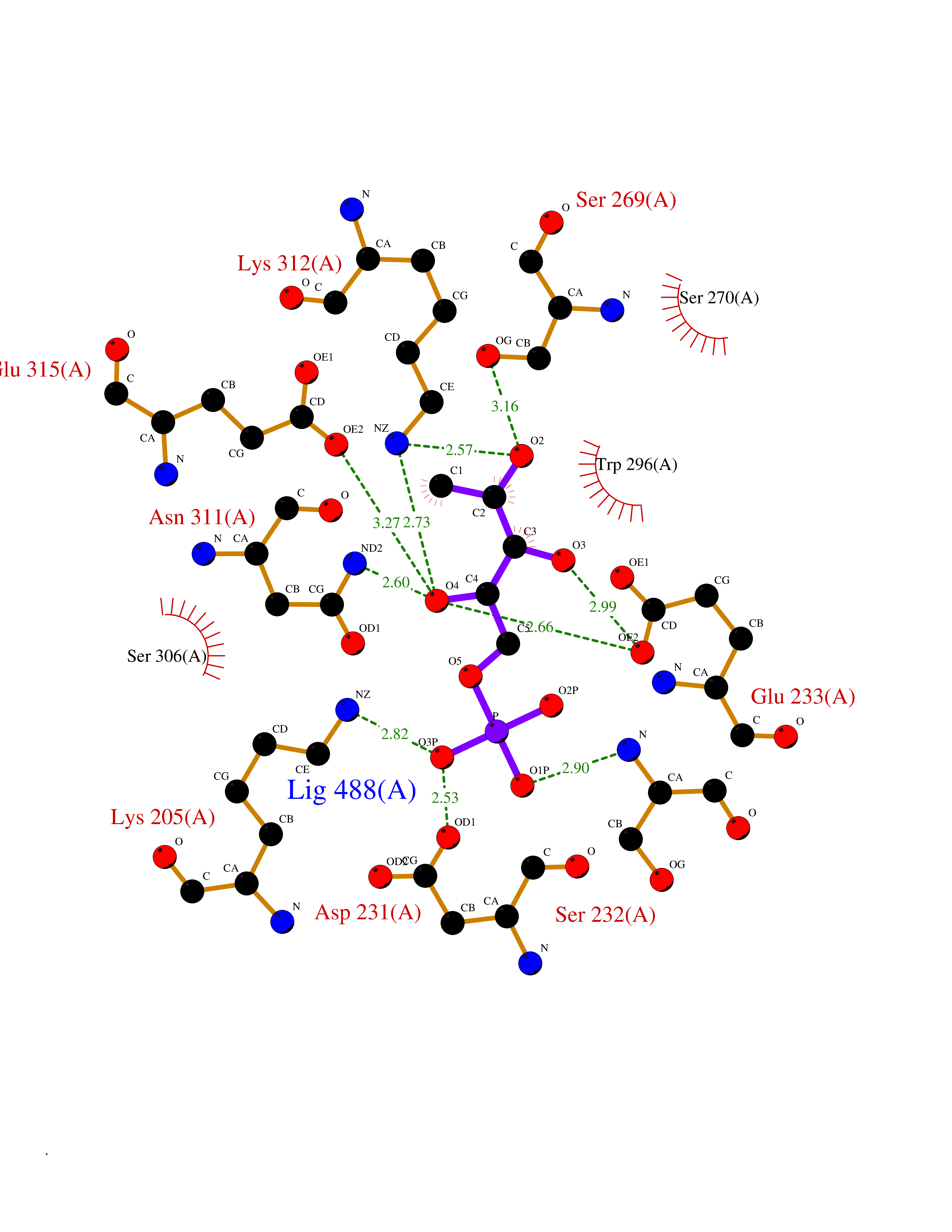 Ligplot Map 3D Binding mode Sequence PINVAIFGSTGSIGTNALNIIRECNKIENVFNVKALYVNKSVNELYEQAREFLPEYLCIHDKSVYEELKELVKNIKDYKPIILCGDEGMKEICSSNSIDKIVIGIDSFQGLYSTMYAIMNNKIVALANKESIVSAGFFLKKLLNIHKNAKIIPVDSEHSAIFQCLDNNKVLKTKCLQDNFSKINNINKIFLCSSGGPFQNLTMDELKNVTSENALKHPKWKMGKKITIDSATMMNKGLEVIETHFLFDVDYNDIEVIVHKECIIHSCVEFIDKSVISQMYYPDMQIPILYSLTWPDRIKTNLKPLDLAQVSTLTFHKPSLEHFPCIKLAYQAGIKGNFYPTVLNASNEIANNLFLNNKIKYFDISSIISQVLESFNSQKVSENSEDLMKQILQIHSWAKDKATDIYNKHN Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Dopamine beta-hydroxylase | 4ZEL | 6.31 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -6.53 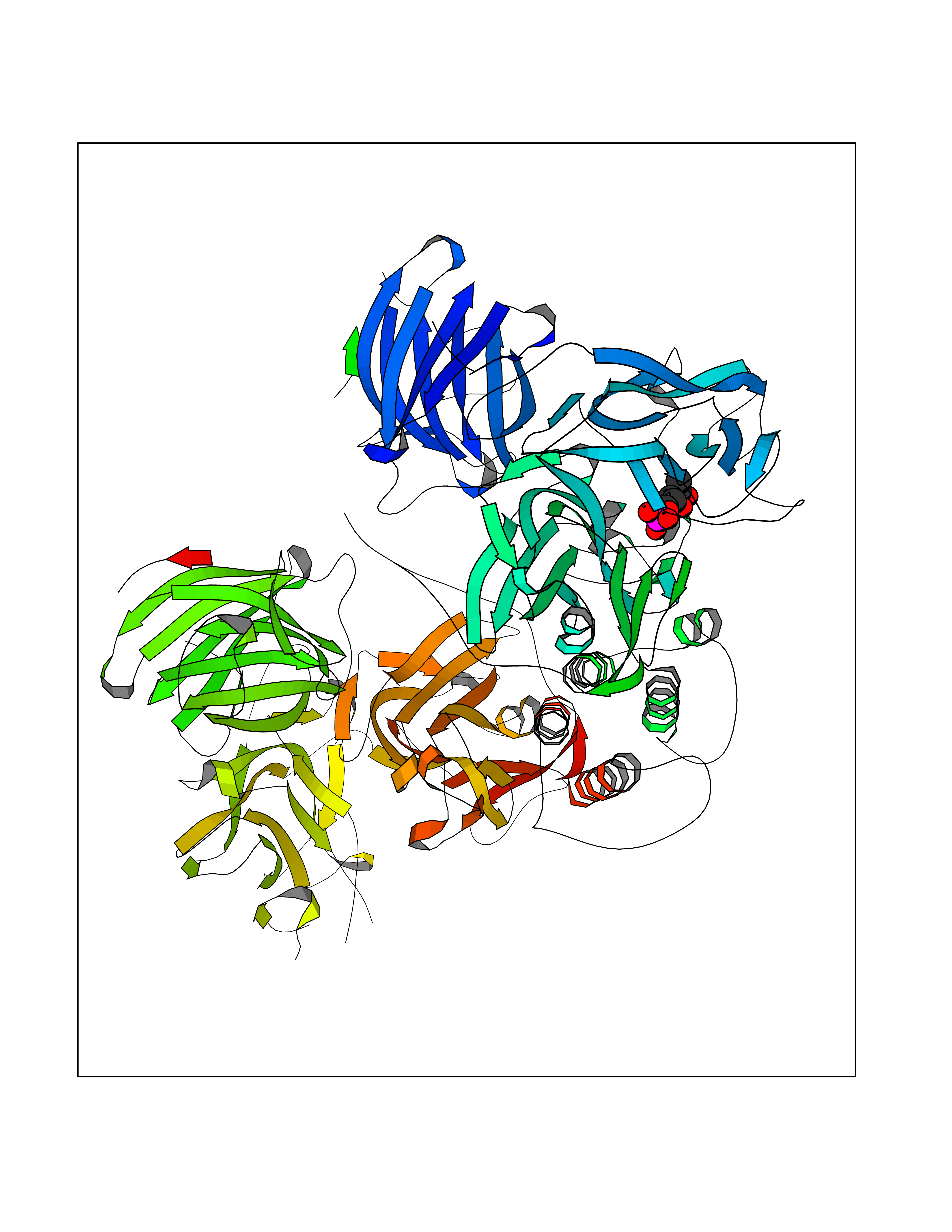 Molscript Map 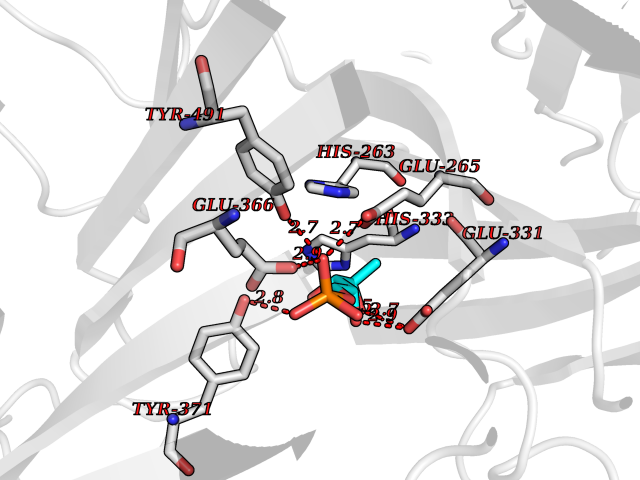 Pymol Map 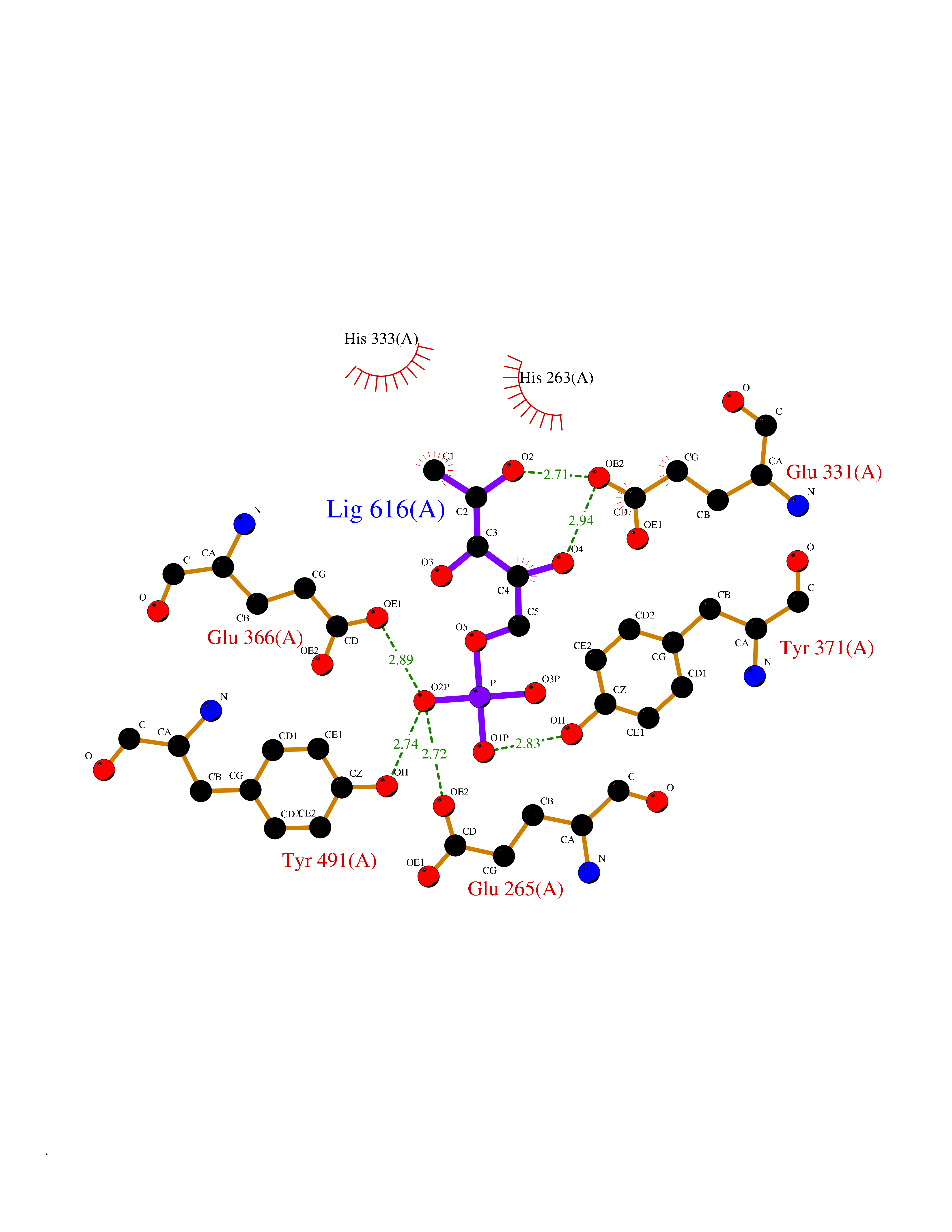 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | Carbonic anhydrase VII (CA-VII) | 3ML5 | 6.31 | |
Target general information Gen name CA7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase 7; Carbonate dehydratase VII Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Related diseases Neurodegeneration with brain iron accumulation 8 (NBIA8) [MIM:617917]: A neurodegenerative disorder associated with iron accumulation, primarily in the basal ganglia. Disease onset is in early childhood. Clinical features include speech delay, progressive cerebellar ataxia, unbalanced gait, and loss of ambulation. NBIA8 transmission pattern is consistent with autosomal recessive inheritance. {ECO:0000269|PubMed:29395073}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00819; DB00562; DB00606; DB01144; DB08846; DB00311; DB00774; DB00703; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lyase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29393.6 Length 262 Aromaticity 0.11 Instability index 40.14 Isoelectric point 7 Charge (pH=7) -0.01 2D Binding mode Binding energy (Kcal/mol) -6.68 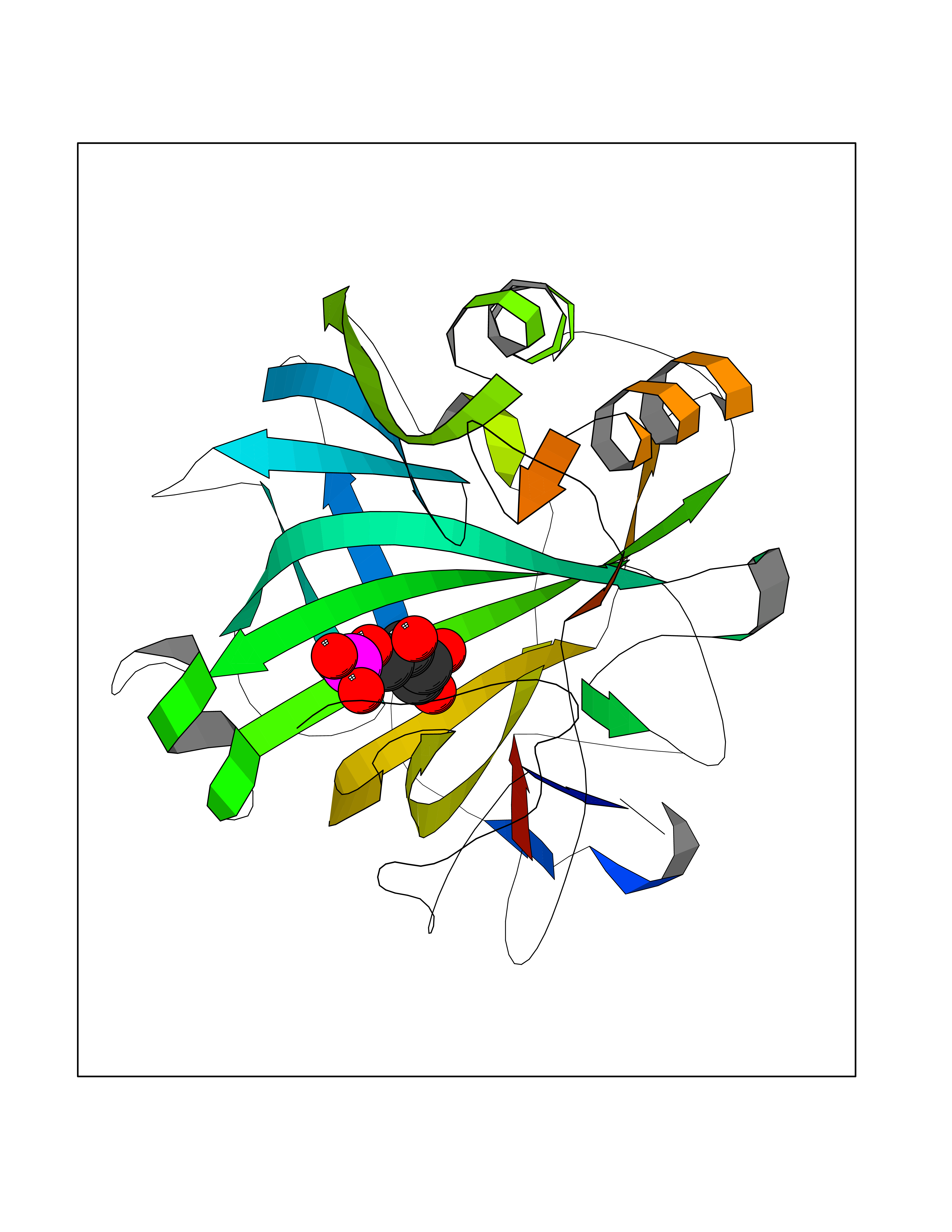 Molscript Map 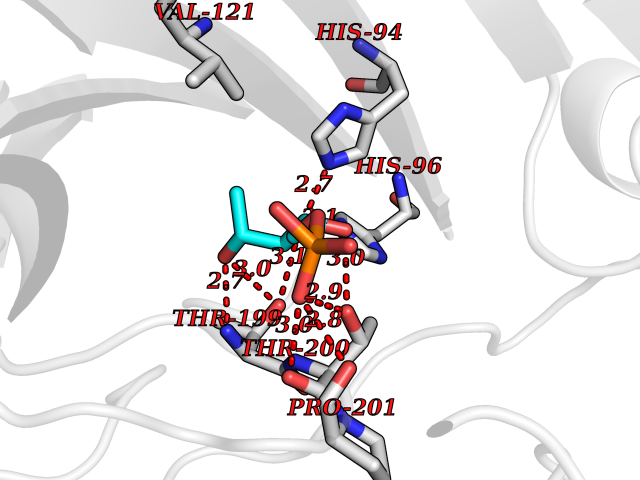 Pymol Map 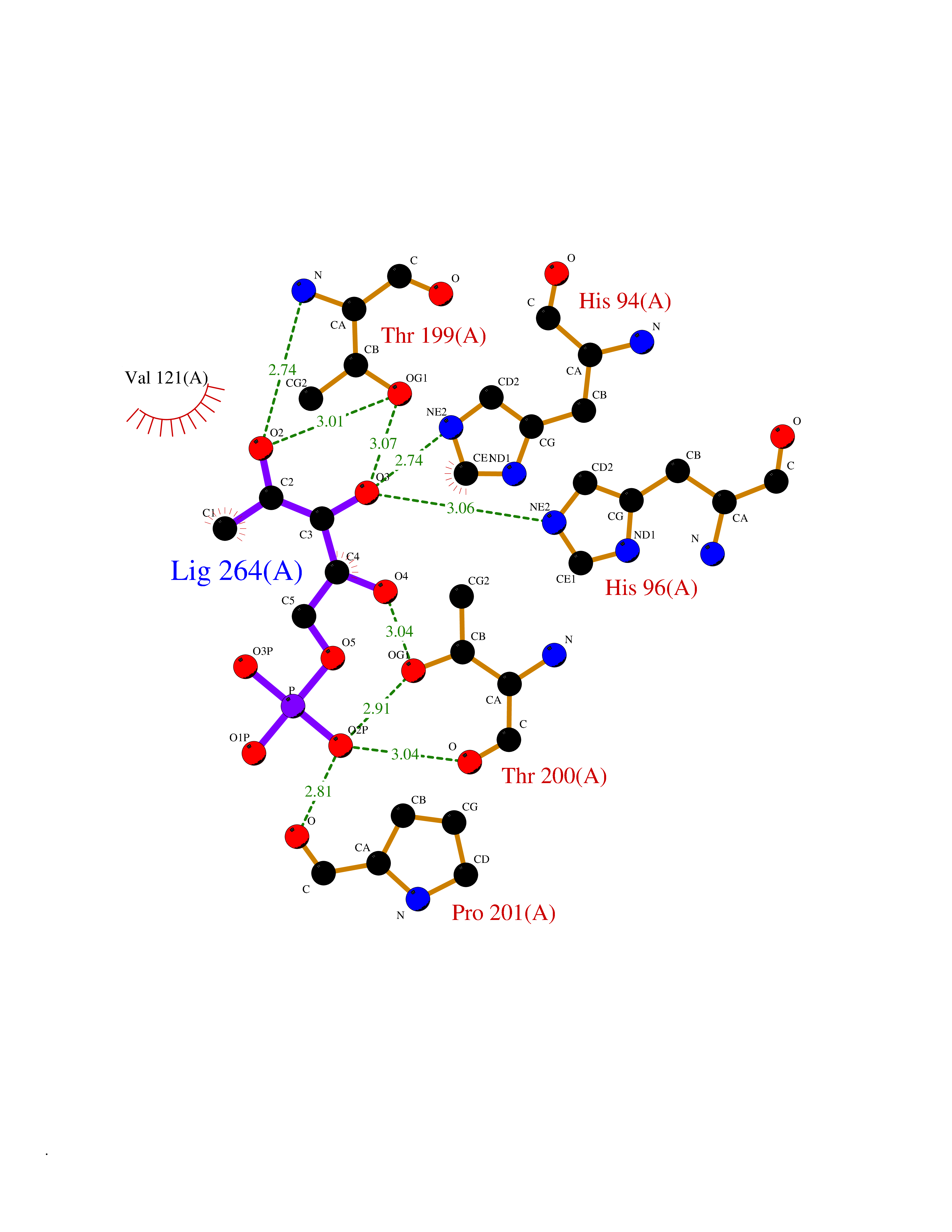 Ligplot Map 3D Binding mode Sequence GHHGWGYGQDDGPSHWHKLYPIAQGDRQSPINIISSQAVYSPSLQPLELSYEACMSLSITNNGHSVQVDFNDSDDRTVVTGGPLEGPYRLKQFHFHWGKKHDVGSEHTVDGKSFPSELHLVHWNAKKYSTFGEAASAPDGLAVVGVFLETGDEHPSMNRLTDALYMVRFKGTKAQFSCFNPKSLLPASRHYWTYPGSLTTPPLSESVTWIVLREPISISERQMGKFRSLLFTSEDDERIHMVNNFRPPQPLKGRVVKASFRA Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Cytochrome P450 1A2 | 2HI4 | 6.30 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -6.9 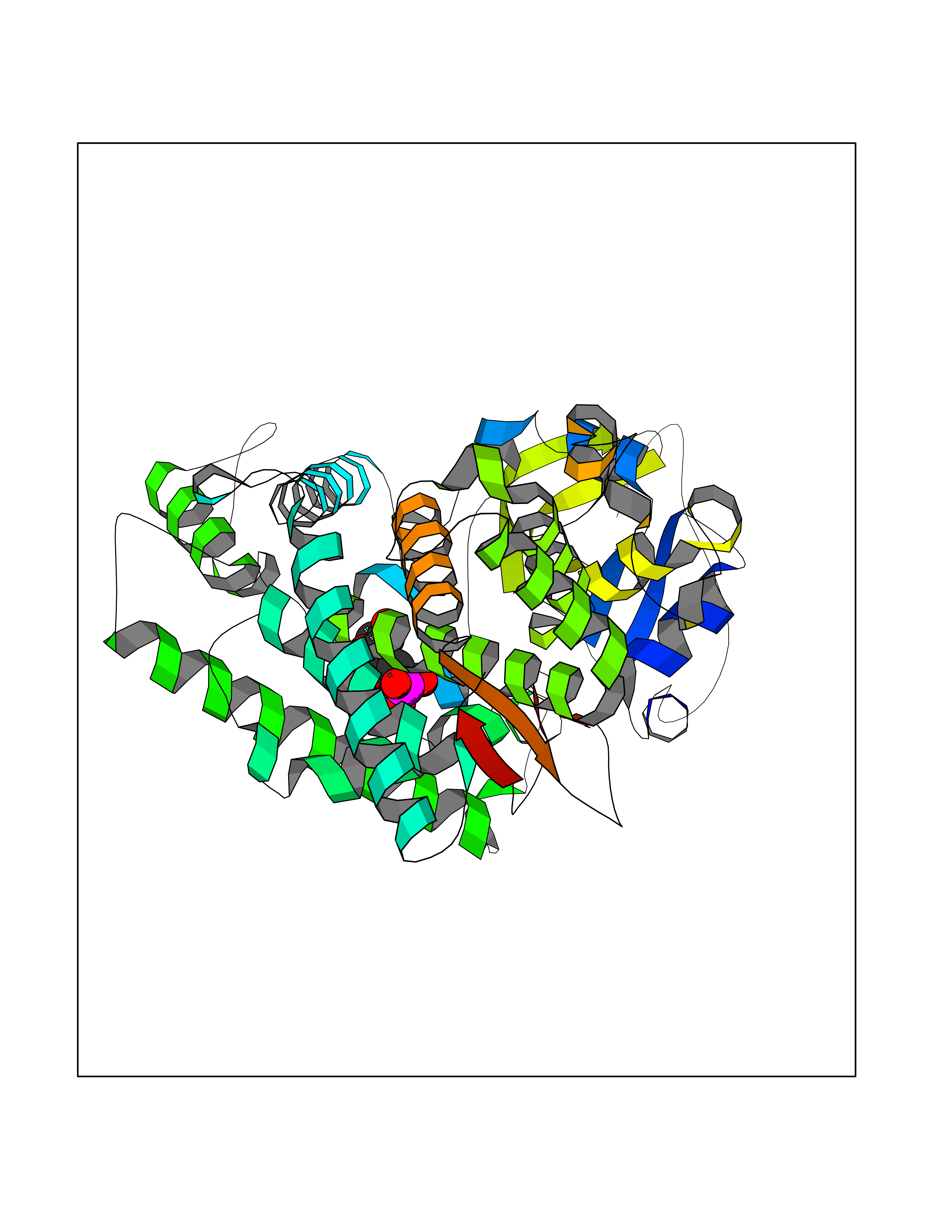 Molscript Map 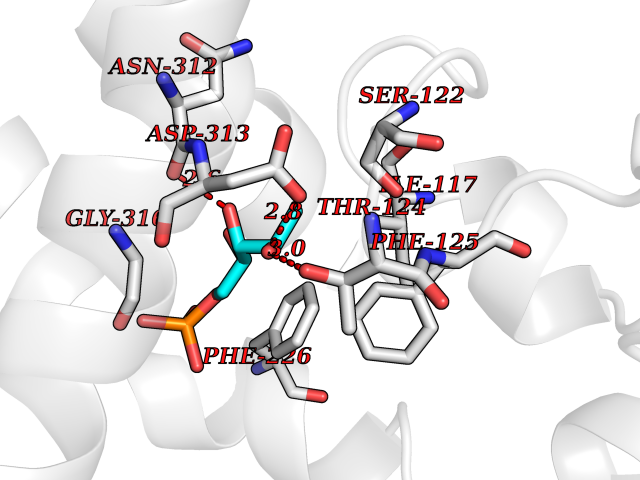 Pymol Map 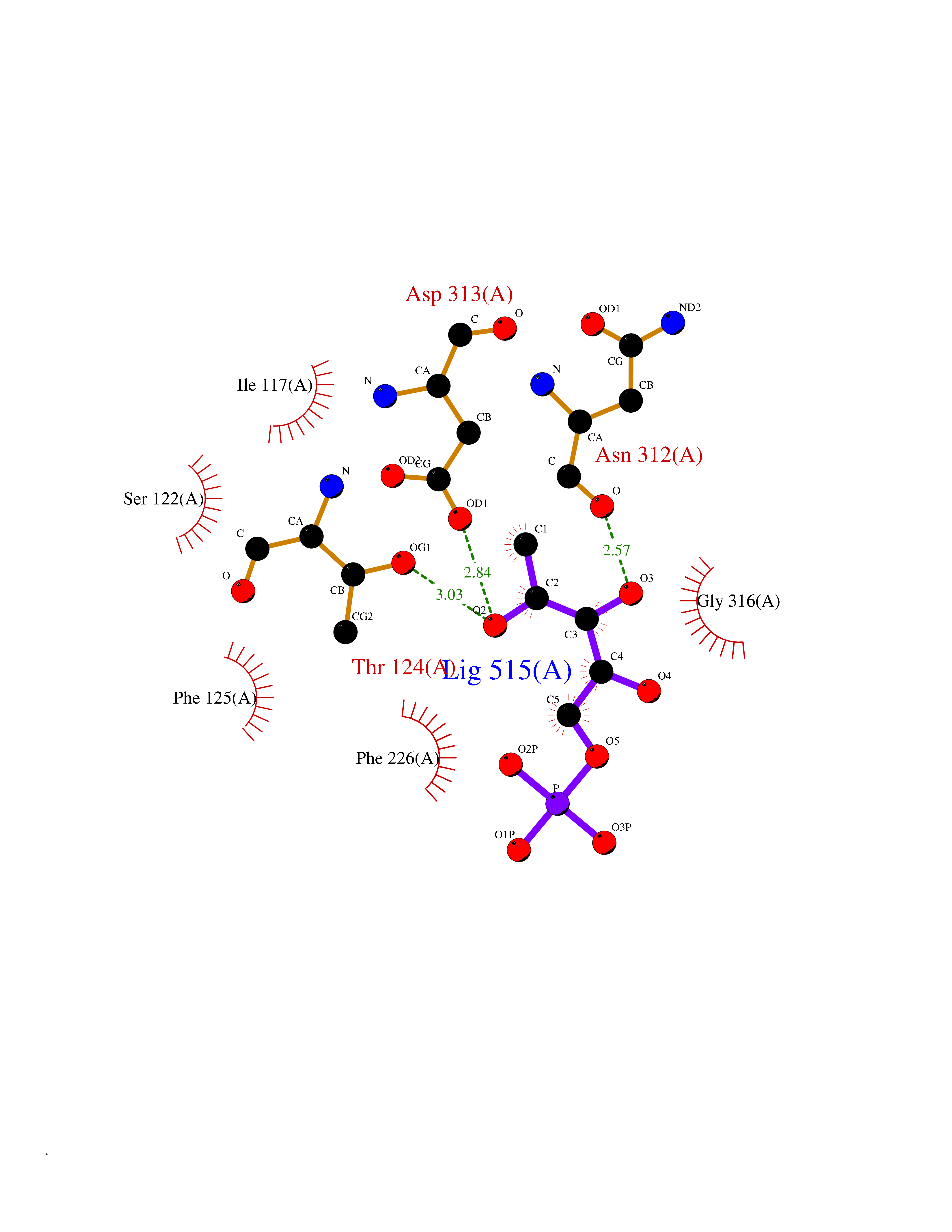 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | Purine nucleoside phosphorylase (PNP) | 4EAR | 6.30 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -6.97  Molscript Map 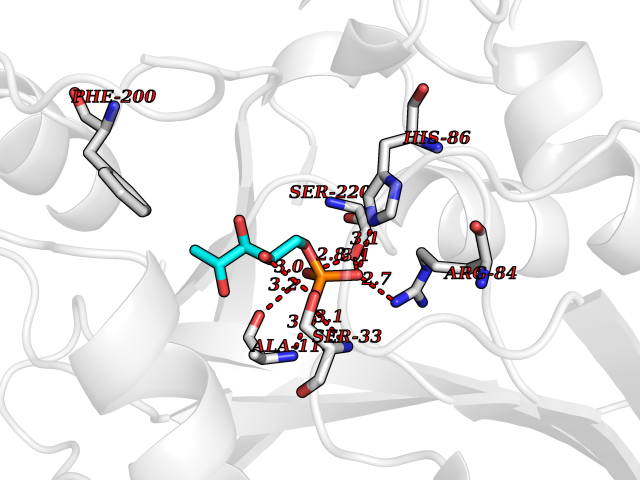 Pymol Map 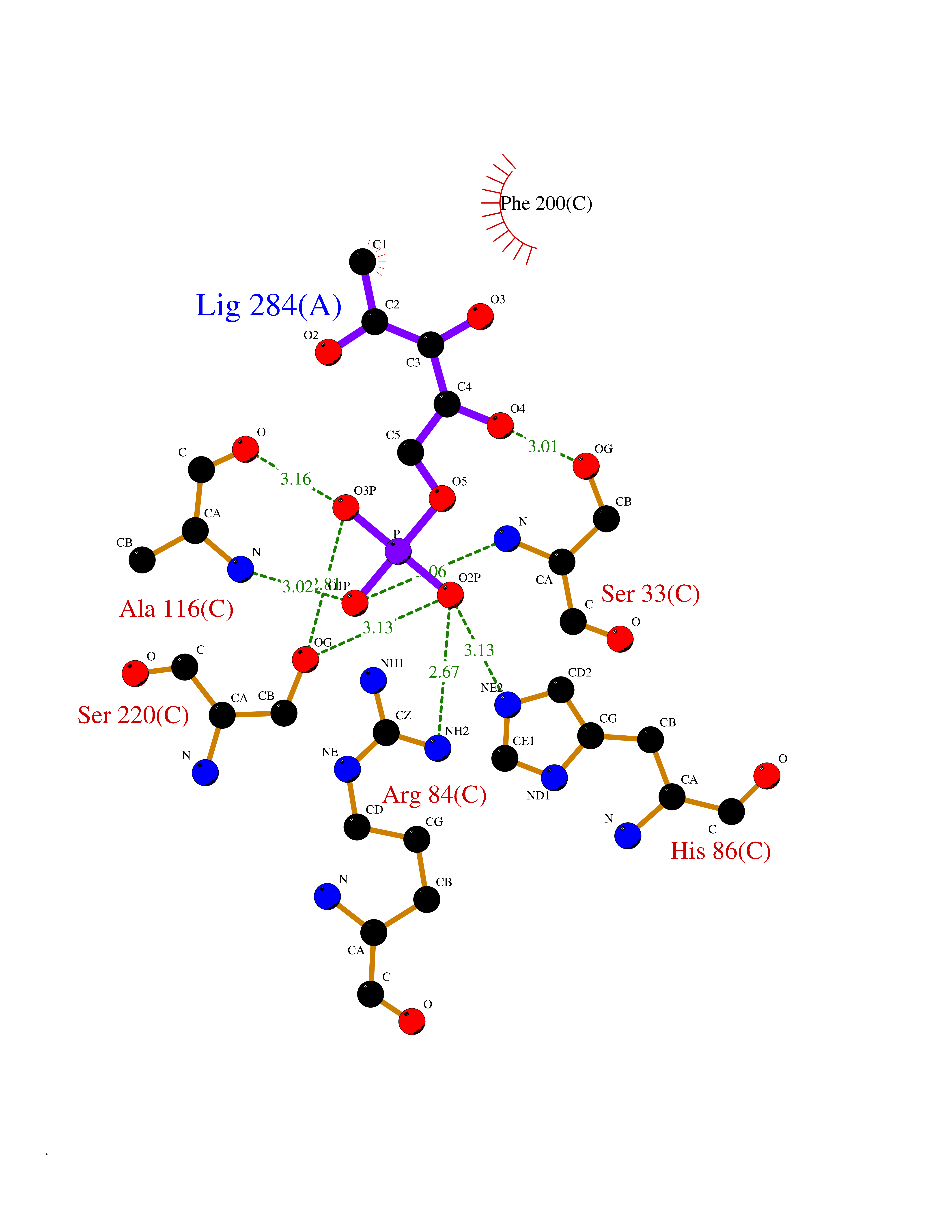 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | HMG-CoA reductase (HMGCR) | 2R4F | 6.30 | |
Target general information Gen name HMGCR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms 3-hydroxy-3-methylglutaryl-coenzyme A reductase Protein family HMG-CoA reductase family Biochemical class CH-OH donor oxidoreductase Function Transmembrane glycoprotein that is the rate-limiting enzyme in cholesterol biosynthesis as well as in the biosynthesis of nonsterol isoprenoids that are essential for normal cell function including ubiquinone and geranylgeranyl proteins. Related diseases Muscular dystrophy, limb-girdle, autosomal recessive 28 (LGMDR28) [MIM:620375]: An autosomal recessive form of limb girdle muscular dystrophy, a group of genetically heterogeneous muscular disorders that share proximal muscle weakness as the major attribute. Most limb girdle muscular dystrophies present with elevated creatinine kinase and myopathic electromyographic features. Disease is usually progressive to a variable degree, ranging from minor disability to complete inability to ambulate, and can involve the large proximal muscles, as well as axial and facial muscles. Different disease forms may exhibit skeletal muscle hypertrophy, kyphoscoliosis, and contractures or involve other muscle groups and manifest with distal weakness, cardiomyopathy, dysphagia, and respiratory difficulties. LGMDR28 is characterized by progressive muscle weakness affecting the proximal and axial muscles of the upper and lower limbs, and highly variable age at onset. Most patients have limited ambulation or become wheelchair-bound within a few decades, and respiratory insufficiency commonly occurs. {ECO:0000269|PubMed:36745799, ECO:0000269|PubMed:37167966}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03169; DB04447; DB01076; DB09061; DB00439; DB01992; DB01095; DB00227; DB14009; DB04377; DB06693; DB14011; DB00157; DB03461; DB08860; DB00175; DB01098; DB00641; DB05317; DB09270 Interacts with Q9Y5Z9; Q9Y5Z9-1 EC number EC 1.1.1.34 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Endoplasmic reticulum; Glycoprotein; Isopeptide bond; Limb-girdle muscular dystrophy; Lipid biosynthesis; Lipid metabolism; Membrane; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84796.9 Length 798 Aromaticity 0.05 Instability index 47.61 Isoelectric point 6.2 Charge (pH=7) -3.71 2D Binding mode Binding energy (Kcal/mol) -6.24  Molscript Map 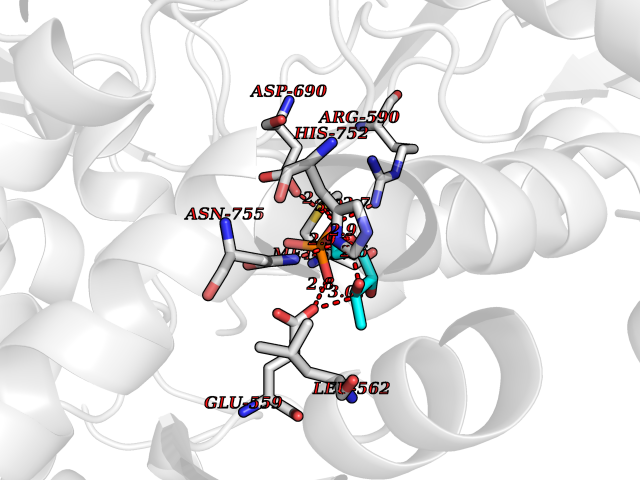 Pymol Map 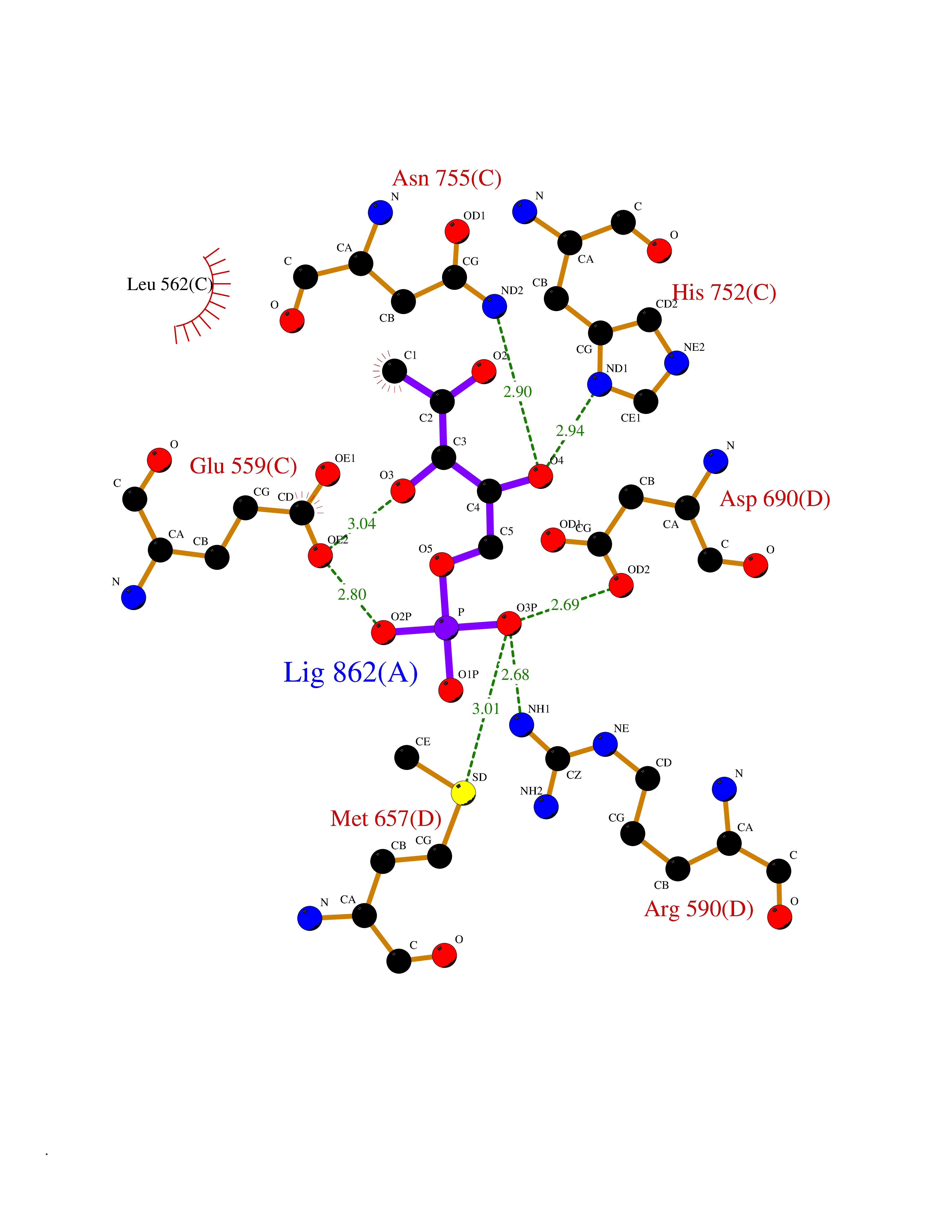 Ligplot Map 3D Binding mode Sequence GAKFLSDAEIIQLVNETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAGPNEECLQILGNGAKFLSDAEIIQLVETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAG Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Choline O-acetyltransferase | 2FY3 | 6.27 | |
Target general information Gen name CHAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Carnitine/choline acetyltransferase family Biochemical class Transferase Function Choline O-acetyltransferase activity. Related diseases Myasthenic syndrome, congenital, 6, presynaptic (CMS6) [MIM:254210]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive. {ECO:0000269|PubMed:11172068, ECO:0000269|PubMed:12756141}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006; DB00184 Interacts with Q6H8Q1-8; Q8N302-2; Q9NXL2-1; Q6XD76; Q9UII2; Q8TBE0; Q9UQB8-6; Q9ULD4-2; Q9NSI6-4; Q6P5X5; Q96LL4; P20807-4; O00257-3; Q6ZP82-1; O95674; Q9H3R5; Q8WUX9; Q9H2A9; Q3SX64; Q92782-2; Q14117; O14641; Q658K8; Q6UXG2-3; O00472; Q6NXG1; Q15910-2; Q8IZU1; P15407; P55318; Q06547-3; P23769-2; P23771; Q15486; Q8IV36; Q4VB01; Q53GQ0; P10809; P41134; Q9NZH6; Q8NA54; Q86U28; P17275; Q8N5Z5; Q6P597; P08727; Q14525; Q8IUC2; Q6IAA8; Q14847-2; P27338; Q9GZQ8; Q53S70; Q5JXC2; A0A0A0MR05; Q8NEH6; Q8TCY5; Q6IN84-2; Q96H12; P01106; P41271-2; P14598; Q9GZM8; Q5BJF6-2; Q9H8K7; Q9NR21-5; Q5VU43-8; Q13956; Q5SXH7-1; Q96T60; Q96I34; Q86UA1; Q15311; Q8TBY0; Q04206; P47804-3; Q9H0X6; P62899; Q66K80; Q9BY12-3; Q86SQ7-2; Q7Z6I5; Q496A3; Q7Z698; Q9C004; Q92783-2; Q8N4C7; O75528; Q15814; O15273; Q96A09; Q8WTV1; Q53NU3; Q71RG4-4; Q86WT6-2; Q9Y3Q8; Q99598; P49459; P11441; Q9H270; P19544-6; Q53FD0-2; Q3KNS6-3 EC number 2.3.1.6 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Neurotransmitter biosynthesis; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 66365.9 Length 595 Aromaticity 0.08 Instability index 53.36 Isoelectric point 8.16 Charge (pH=7) 4.64 2D Binding mode Binding energy (Kcal/mol) -6.64  Molscript Map 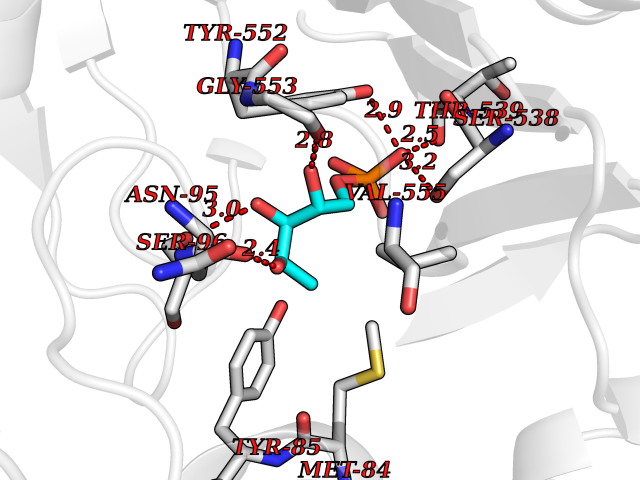 Pymol Map 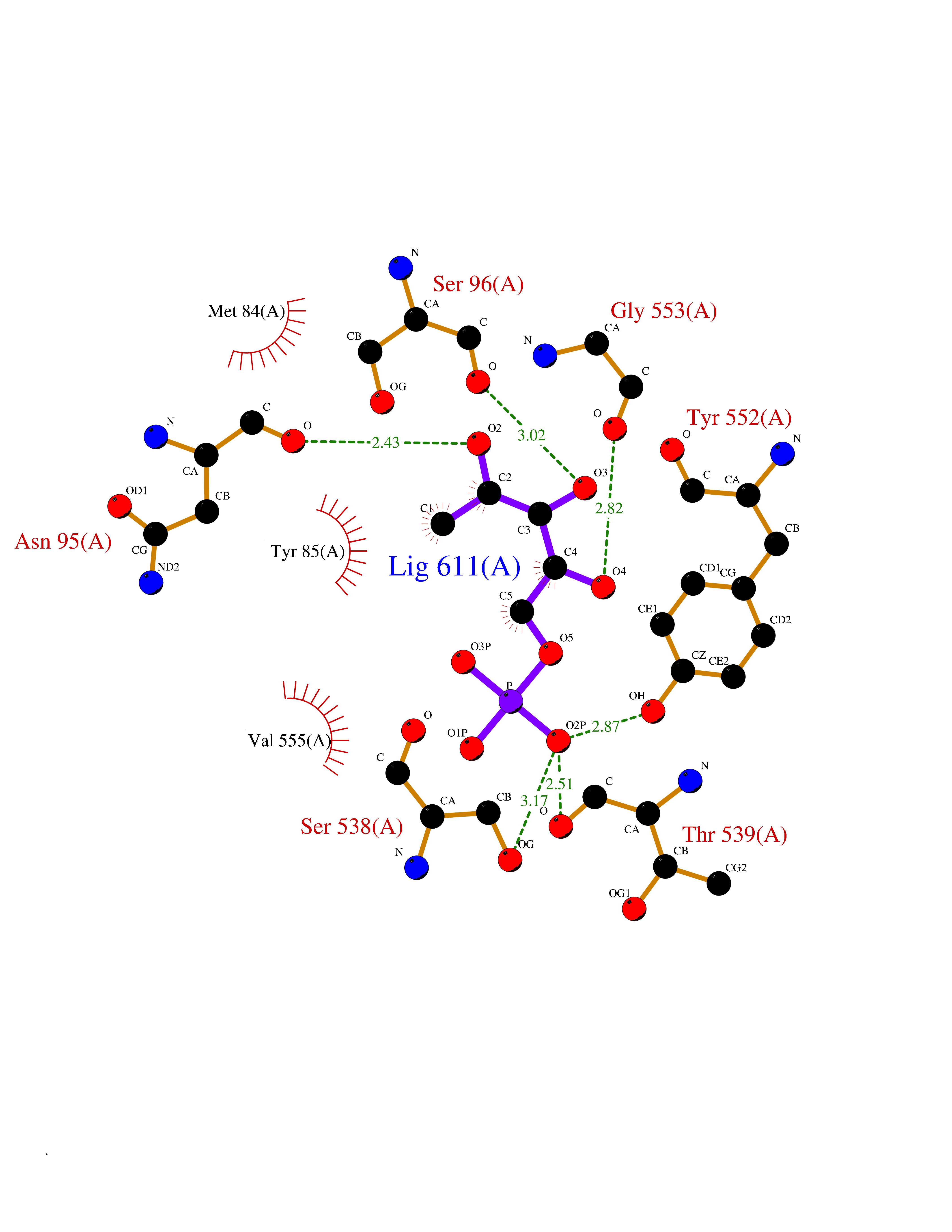 Ligplot Map 3D Binding mode Sequence SEESGLPKLPVPPLQQTLATYLQCMRHLVSEEQFRKSQAIVQQFGAPGGLGETLQQKLLERQEKTANWVSEYWLNDMYLNNRLALPVNSSPAVIFARQHFPGTDDQLRFAASLISGVLSYKALLDSHSIPTDCAKGQPLCMKQYYGLFSSYRLPGHTQDTLVAQNSSIMPEPEHVIVACCNQFFVLDVVINFRRLSEGDLFTQLRKIVKMASNAAARLPPIGLLTSDGRSEWAEARTVLVKDSTNRDSLDMIERCICLVCLDAPGGVELSDTHRALQLLHGGGYSKNGANRWYDKSLQFVVGRDGTCGVVCEHSPFDGIVLVQCTEHLLKHMTQPELVRSPMVPLPAPRRLRWKCSPEIQGHLASSAEKLQRIVKNLDFIVYKFDNYGKTFIKKQKCSPDAFIQVALQLAFYRLHRRLVPTYESASIRRFQEGRVDNIRSATPEALAFVRAVTDHKAAVPASEKLLLLKDAIRAQTAYTVMAITGMAIDNHLLALRELARAMCAALPEMFMDETYLMSNRFVLSTSQVPTTTEMFCCYGPVVPNGYGACYNPQPETILFCISSFHSCAATSSSKFAKAVEESLIDMRDLCSLLPP Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | ATP-dependent protease Lon (LONP1) | 7P09 | 6.27 | |
Target general information Gen name LONP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine protease 15; Mitochondrial ATP-dependent protease Lon; Lon protease-like protein; LONP1; LONP; LONHs Protein family Peptidase S16 family Biochemical class Peptidase Function ATP-dependent serine protease that mediates the selective degradation of misfolded, unassembled or oxidatively damaged polypeptides as well as certain short-lived regulatory proteins in the mitochondrial matrix. May also have a chaperone function in the assembly of inner membrane protein complexes. Participates in the regulation of mitochondrial gene expression and in the maintenance of the integrity of the mitochondrial genome. Binds to mitochondrial promoters and RNA in a single- stranded, site-specific, and strand-specific manner. May regulate mitochondrial DNA replication and/or gene expression using site- specific, single-stranded DNA binding to target the degradation of regulatory proteins binding to adjacent sites in mitochondrial promoters. Endogenous substrates include mitochondrial steroidogenic acute regulatory (StAR) protein. Related diseases CODAS syndrome (CODASS) [MIM:600373]: A rare syndrome characterized by the combination of cerebral, ocular, dental, auricular, and skeletal features. These include developmental delay, craniofacial anomalies, cataracts, ptosis, median nasal groove, delayed tooth eruption, hearing loss, short stature, delayed epiphyseal ossification, metaphyseal hip dysplasia, and vertebral coronal clefts. {ECO:0000269|PubMed:25574826, ECO:0000269|PubMed:25808063}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02666; P36776-1 EC number EC 3.4.21.53 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cataract; Deafness; Disease variant; DNA-binding; Dwarfism; Hydrolase; Mitochondrion; Nucleotide-binding; Protease; Proteomics identification; Reference proteome; Serine protease; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 118797 Length 1070 Aromaticity 0.07 Instability index 35.44 Isoelectric point 5.72 Charge (pH=7) -18.18 2D Binding mode Binding energy (Kcal/mol) -7.04  Molscript Map 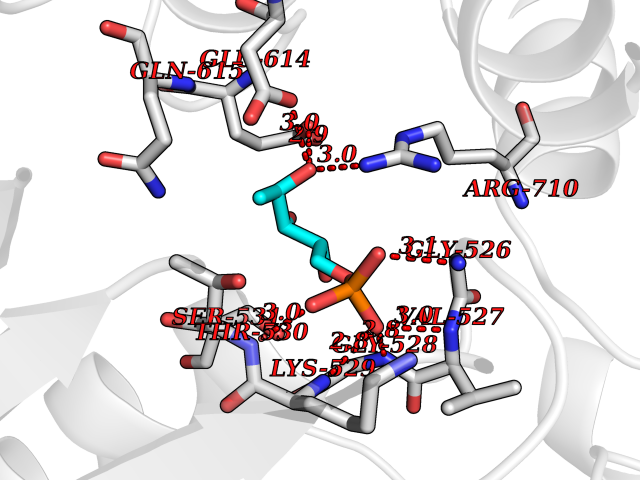 Pymol Map 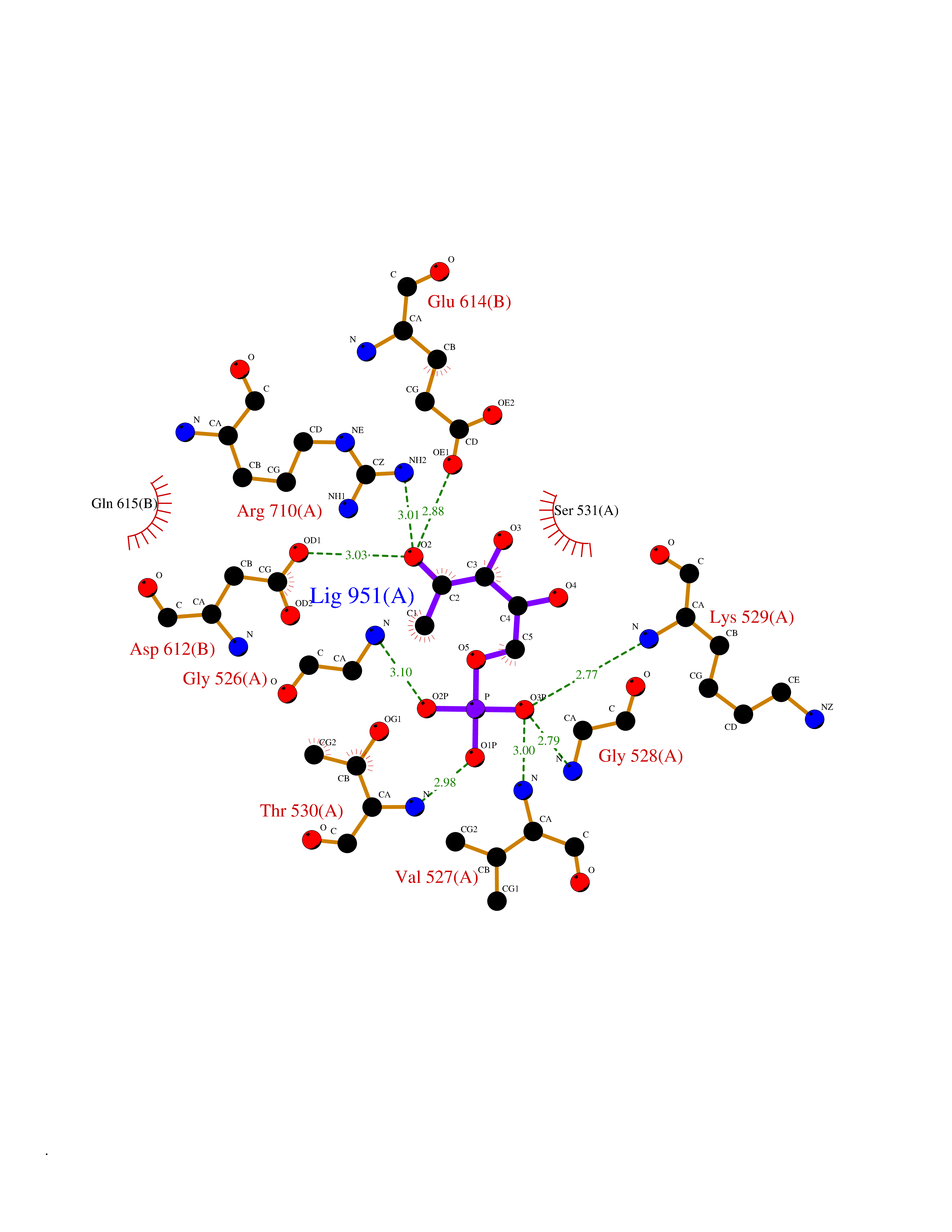 Ligplot Map 3D Binding mode Sequence EKDDKDAIEEKFRERLKELVVPKHVMDVVDEELSKLGLLDNHSSEFNVTRNYLDWLTSIPWGKYSNENLDLARAQAVLEEDHYGMEDVKKRILEFIAVSQLRGSTQGKILCFYGPPGVGKTSIARSIARALNREYFRFSVGGMTDVAEIKGHRRTYVGAMPGKIIQCLKKTKTENPLILIDEVDKIGRGYQGDPSSALLELLDPEQNANFLDHYLDVPVDLSKVLFICTANVTDTIPEPLRDRMEMINVSGYVAQEKLAIAERYLVPQARALCGLDESKAKLSSDVLTLLIKQYCRESGVRNLQKQVEKVLRKSAYKIVSGEAESVEVTPENLQDFVGKPVFTVERMYDVTPPGVVMGLAWTAMGGSTLFVETSLRRPGDKDGSLEVTGQLGEVMKESARIAYTFARAFLMQHAPANDYLVTSHIHLHVPEGATPKDGPSAGCTIVTALLSLAMGRPVRQNLAMTGEVSLTGKILPVGGIKEKTIAAKRAGVTCIVLPAENKKDFYDLAAFITEGLEVHFVEHYREIFDIAFPDEKDDKDAIEEKFRERLKELVVPKHVMDVVDEELSKLGLLDNHSSEFNVTRNYLDWLTSIPWGKYSNENLDLARAQAVLEEDHYGMEDVKKRILEFIAVSQLRGSTQGKILCFYGPPGVGKTSIARSIARALNREYFRFSVGGMTDVAEIKGHRRTYVGAMPGKIIQCLKKTKTENPLILIDEVDKIGRGYQGDPSSALLELLDPEQNANFLDHYLDVPVDLSKVLFICTANVTDTIPEPLRDRMEMINVSGYVAQEKLAIAERYLVPQARALCGLDESKAKLSSDVLTLLIKQYCRESGVRNLQKQVEKVLRKSAYKIVSGEAESVEVTPENLQDFVGKPVFTVERMYDVTPPGVVMGLAWTAMGGSTLFVETSLRRPQDKDKDGSLEVTGQLGEVMKESARIAYTFARAFLMQHAPANDYLVTSHIHLHVPEGATPKDGPSAGCTIVTALLSLAMGRPVRQNLAMTGEVSLTGKILPVGGIKEKTIAAKRAGVTCIVLPAENKKDFYDLAAFITEGLEVHFVEHYREIFDIAFPD Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | SET domain containing 8 (KMT5A) | 5TEG | 6.26 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -7.19  Molscript Map 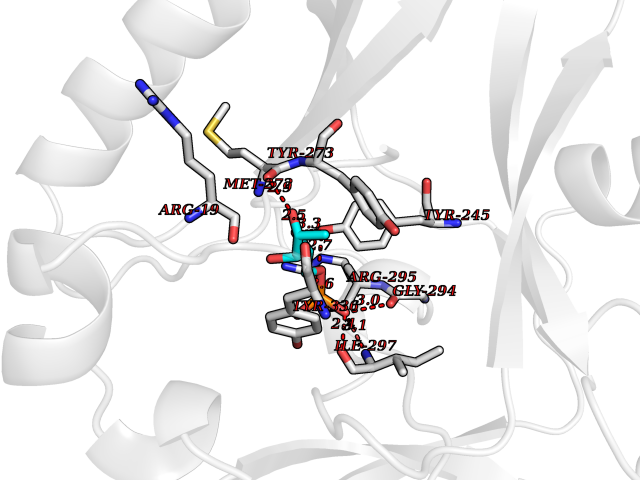 Pymol Map 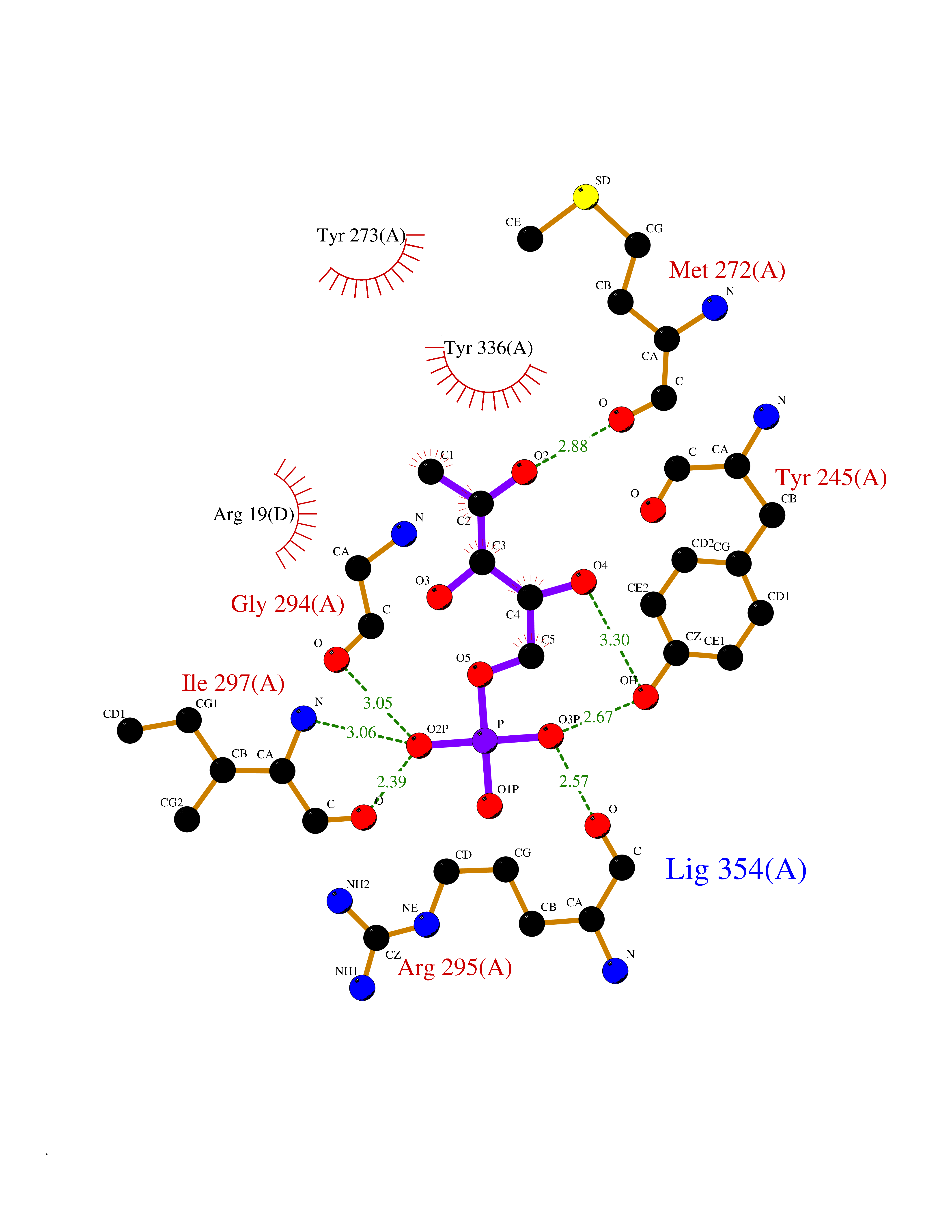 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Mucin-1 (MUC1) | 6KX1 | 6.26 | |
Target general information Gen name MUC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumour-associated antigen mucin 1; Tumor-associated mucin; Tumor-associated epithelial membraneantigen; Tumor-associated epithelial membrane antigen; Polymorphic epithelial mucin; Peanut-reactive urin Protein family NA Biochemical class NA Function Can act both as an adhesion and an anti-adhesion protein. May provide a protective layer on epithelial cells against bacterial and enzyme attack. The alpha subunit has cell adhesive properties. Related diseases MUC1/CA 15-3 is used as a serological clinical marker of breast cancer to monitor response to breast cancer treatment and disease recurrence (PubMed:20816948). Decreased levels over time may be indicative of a positive response to treatment. Conversely, increased levels may indicate disease progression. At an early stage disease, only 21% of patients exhibit high MUC1/CA 15-3 levels, that is why CA 15-3 is not a useful screening test. Most antibodies target the highly immunodominant core peptide domain of 20 amino acid (APDTRPAPGSTAPPAHGVTS) tandem repeats. Some antibodies recognize glycosylated epitopes. {ECO:0000269|PubMed:20816948}.; DISEASE: Tubulointerstitial kidney disease, autosomal dominant, 2 (ADTKD2) [MIM:174000]: A form of autosomal dominant tubulointerstitial kidney disease, a genetically heterogeneous disorder characterized by slowly progressive loss of kidney function, bland urinary sediment, hyperuricemia, absent or mildly increased albuminuria, lack of severe hypertension during the early stages, and normal or small kidneys on ultrasound. Renal histology shows variable abnormalities including interstitial fibrosis with tubular atrophy, microcystic dilatation of the tubules, thickening of tubular basement membranes, medullary cysts, and secondary glomerulosclerotic or glomerulocystic changes with abnormal glomerular tufting. There is significant variability, as well as incomplete penetrance. {ECO:0000269|PubMed:23396133}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11090; DB06584 Interacts with P00519; P00533; P08581; P15941-7; Q08AM2; O60242; Q15848; Q86W74-2; P02652; P05067-2; P29972; P41181; Q92482; Q9H2C2; Q92843; Q6PL45-2; Q8WVV5; P06681; O14523; Q06432; Q9P0B6; Q08722-3; P19397; P34810; Q8N6F1-2; P56747; Q8NHS1; Q96FZ5; Q4VAQ0; Q8N6G5; Q07325; O43169; P78329; P56851; Q9BV81; P54852; O75355-2; Q9UKR5; P01350; P39905-3; Q9Y3E0; Q9NPR9; Q9HCP6; O60725; Q9Y5U4; P11215; Q969L2; Q13021; Q9P0N8; Q6N075; P30301; Q96S97; O95167; Q99519; Q92982; Q9NZG7; Q16617; Q8N912; Q8NH19; Q6TCH4; P26678; P60201-2; Q8IY26; P54315; Q59EV6; P30405; Q96AA3; Q02161-2; Q8TAC9; Q9Y6D0; Q8N6R1; P11686; Q8IWU4; Q969S0; Q6ICL7; Q9NVC3; Q9NRQ5; B2RUZ4; Q9NZ01; P07204; Q9BZW4; P17152; A0PK00; Q9BTD3; Q5BJH2-2; Q9BVK8; Q9Y6G1; Q9P0S9; Q14656; Q8NBD8; Q9BU79; Q8N2M4; Q8N661; Q5BJF2; Q9Y2Y6; O14763; Q8N609; Q5BVD1; Q53HI1; O95183; Q9BQB6; Q8IVQ6; P00519; P17676 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Cell membrane; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Lipoprotein; Membrane; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Tumor suppressor Protein physicochemical properties Chain ID B,C Molecular weight (Da) 25132.6 Length 230 Aromaticity 0.09 Instability index 44.8 Isoelectric point 7.12 Charge (pH=7) 0.18 2D Binding mode Binding energy (Kcal/mol) -6.46  Molscript Map 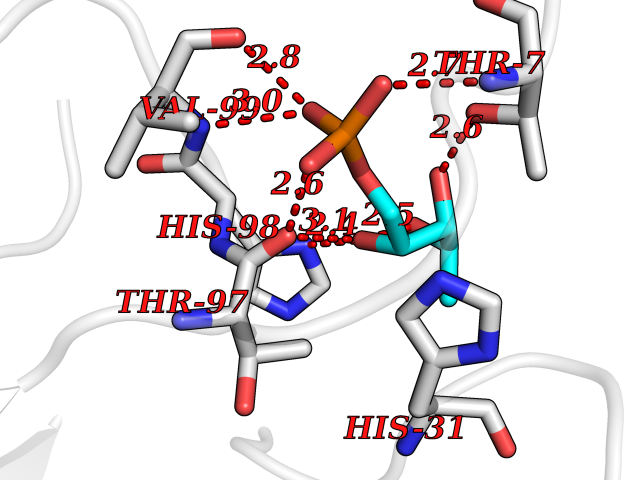 Pymol Map 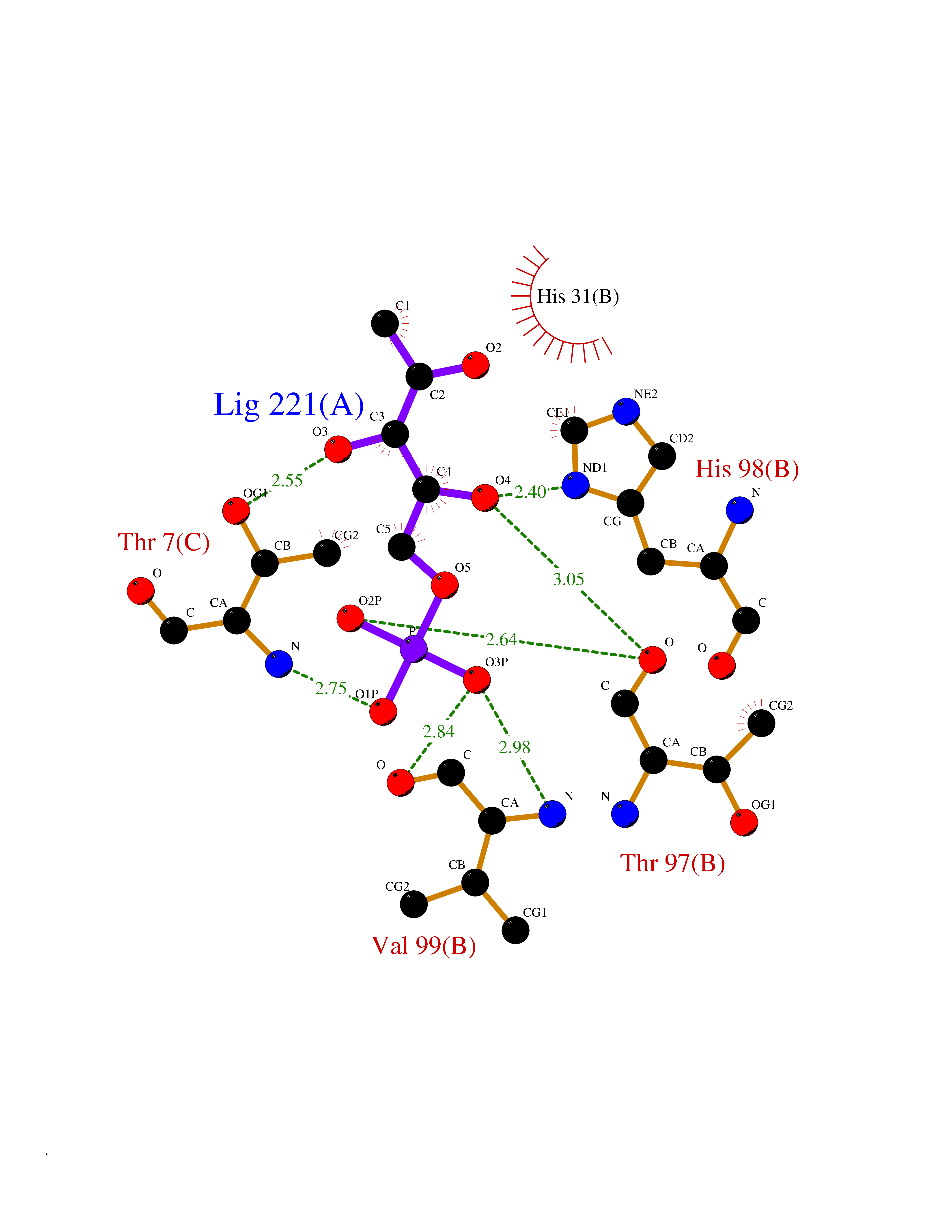 Ligplot Map 3D Binding mode Sequence DVVMTQTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLKISRVEAEDLGVYFCSQSTHVPPWTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNEXVTSAPDTRPA Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Peptidyl-prolyl cis-trans isomerase G | 2GW2 | 6.25 | |
Target general information Gen name PPIG Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Isomerase Function Cyclosporin A binding.Peptidyl-prolyl cis-trans isomerase activity.RNA binding. Related diseases Intellectual developmental disorder, autosomal dominant 6, with or without seizures (MRD6) [MIM:613970]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD6 additional features may include seizures, hypotonia, abnormal movements, such as dystonia, and autistic features. {ECO:0000269|PubMed:20890276, ECO:0000269|PubMed:23033978, ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24863970, ECO:0000269|PubMed:25356899, ECO:0000269|PubMed:27839871, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 27 (DEE27) [MIM:616139]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. {ECO:0000269|PubMed:24272827, ECO:0000269|PubMed:27839871, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberrations involving GRIN2B has been found in patients with intellectual disability. Translocations t(9;12)(p23;p13.1) and t(10;12)(q21.1;p13.1) with a common breakpoint in 12p13.1. Drugs (DrugBank ID) DB00172 Interacts with Q8N7W2-2; Q8NHQ1; O75553; Q9UI36-2; Q96C98; Q8NC69; P17931; Q6NVH9; Q15365; Q9UL42; Q96CD2; Q14498; Q16637; Q12800; Q9NVV9; PRO_0000037309 [P0C6X7] EC number 5.2.1.8 Uniprot keywords 3D-structure; Alternative splicing; Isomerase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Rotamase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 19125.4 Length 173 Aromaticity 0.1 Instability index 26.46 Isoelectric point 7.14 Charge (pH=7) 0.24 2D Binding mode Binding energy (Kcal/mol) -6.7  Molscript Map 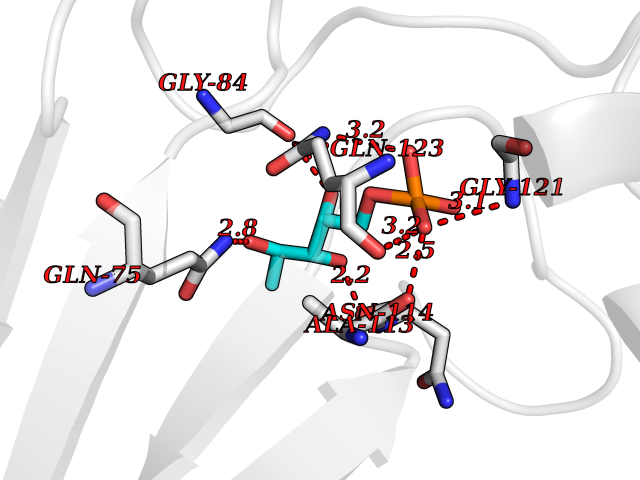 Pymol Map 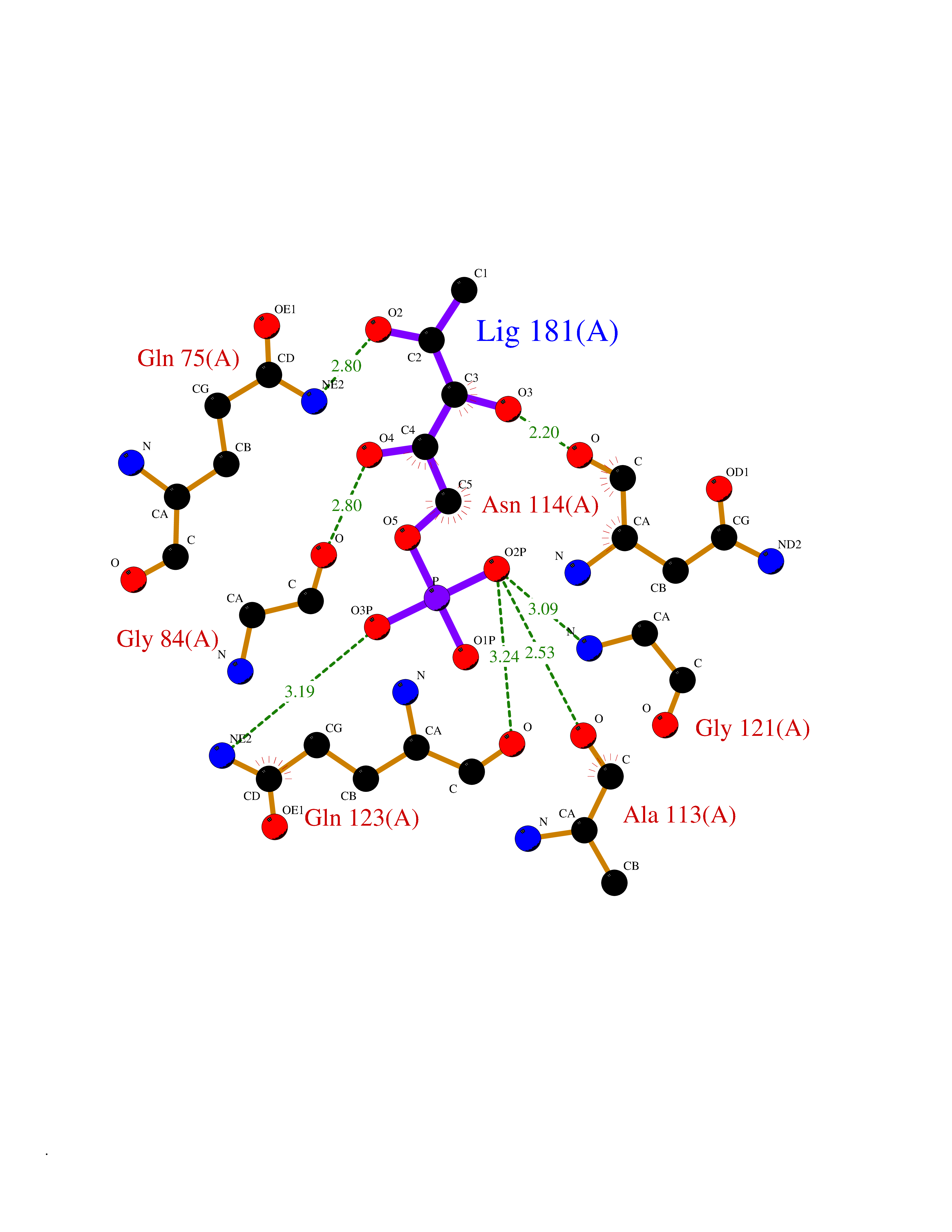 Ligplot Map 3D Binding mode Sequence RPRCFFDIAINNQPAGRVVFELFSDVCPKTCENFRCLCTGEKGTGKSTQKPLHYKSCLFHRVVKDFMVQGGDFSEGNGRGGESIYGGFFEDESFAVKHNAAFLLSMANRGKDTNGSQFFITTKPTPHLDGHHVVFGQVISGQEVVREIENQKTDAASKPFAEVRILSCGELIP Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 6.25 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -6.33 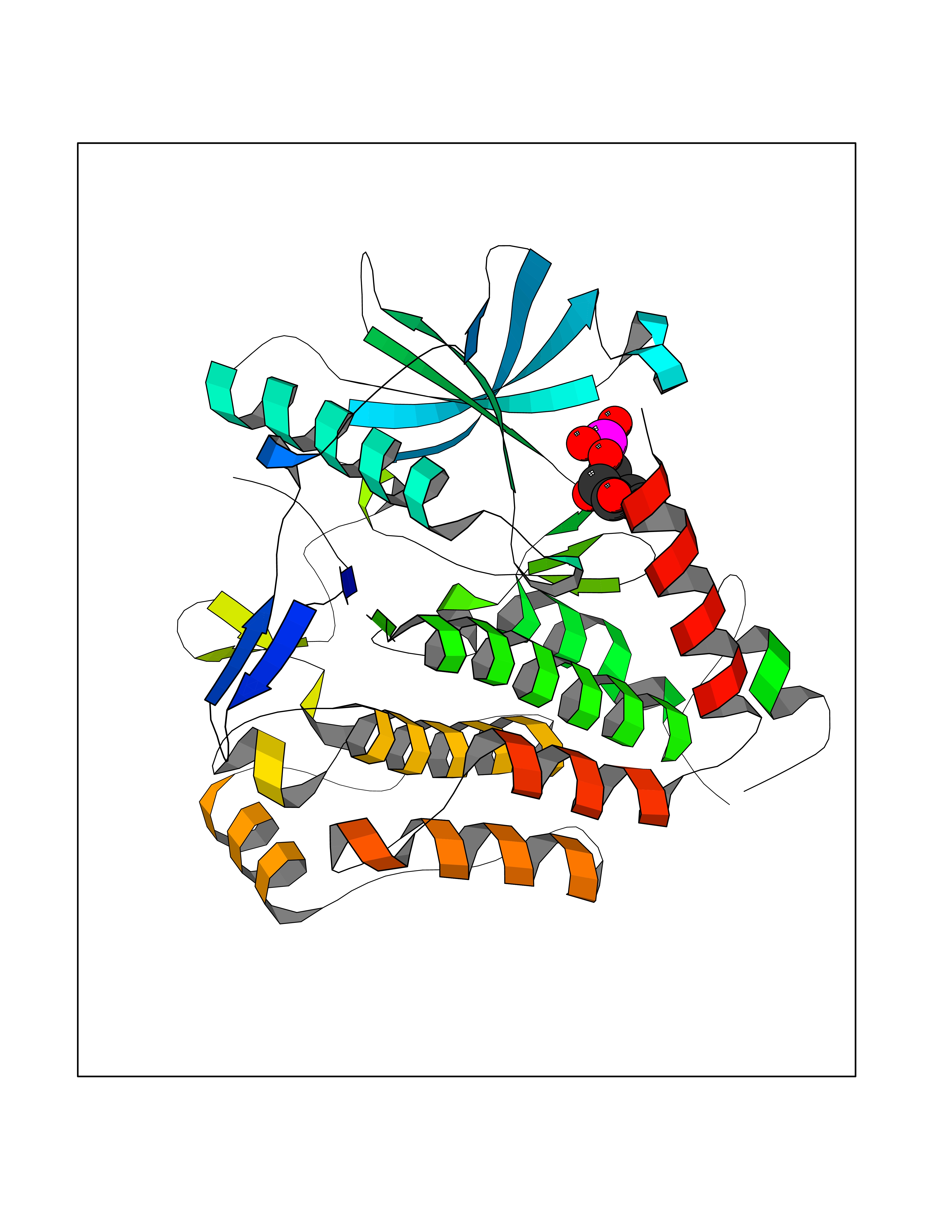 Molscript Map 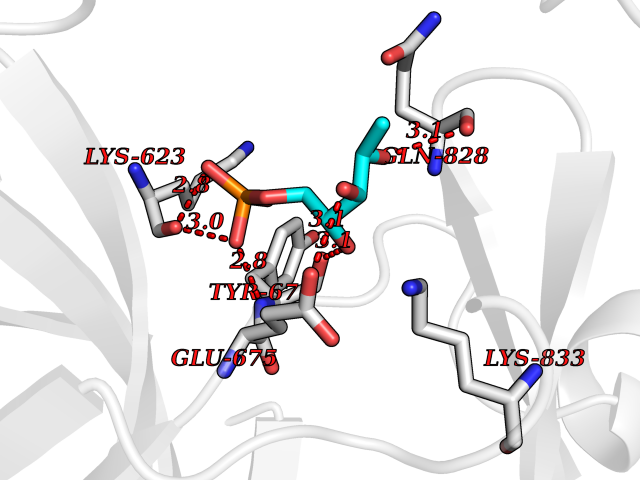 Pymol Map 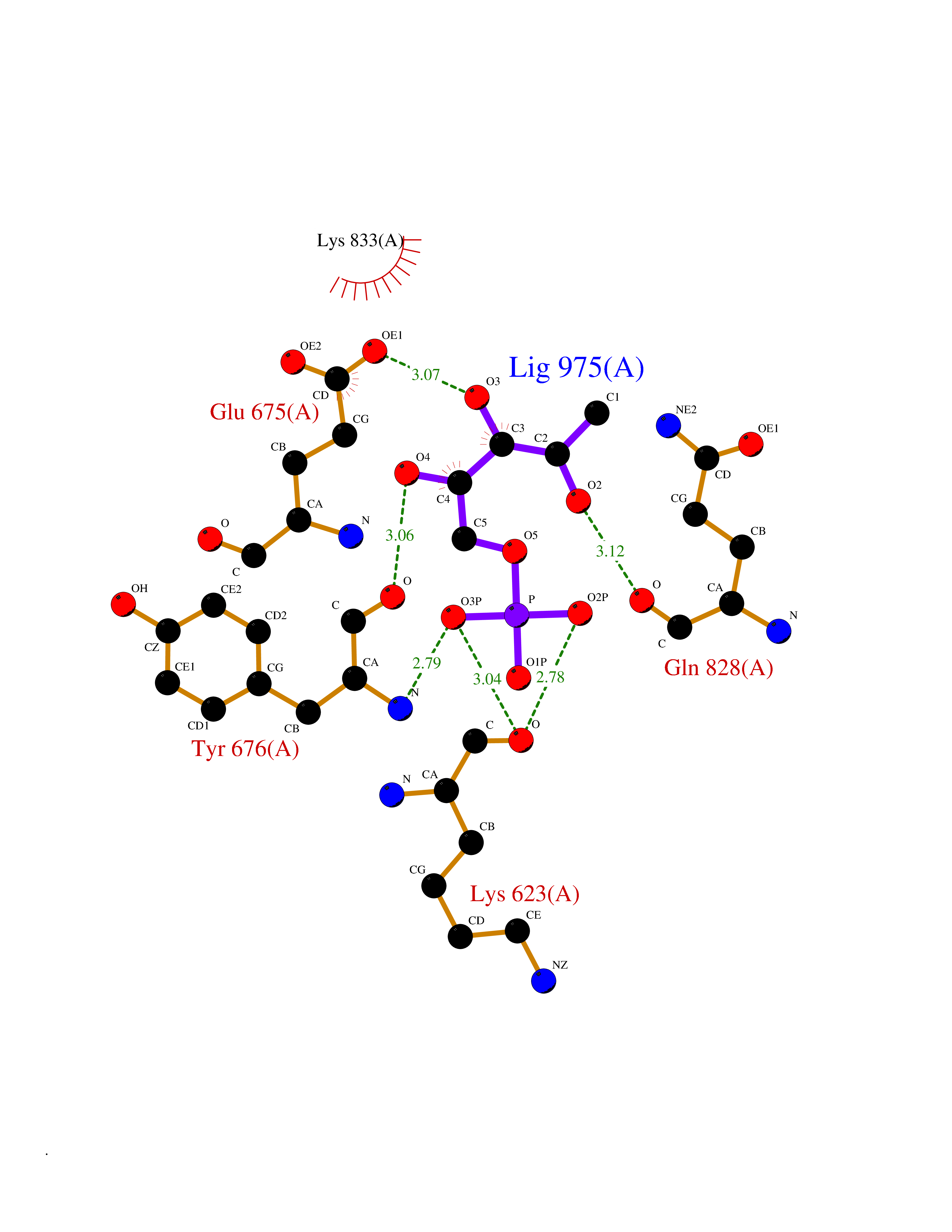 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||