Job Results:
Ligand
Structure
Job ID
0006599cba9dbe83f9144f4d36fa70dc
Job name
NA
Time
2024-05-29 06:52:44
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 21 | Aspartyl aminopeptidase (DNPEP) | 4DYO | 8.26 | |
Target general information Gen name DNPEP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DAP; ASPEP Protein family Peptidase M18 family Biochemical class Peptidase Function Likely to play an important role in intracellular protein and peptide metabolism. Aminopeptidase with specificity towards an acidic amino acid at the N-terminus. Related diseases Cohen-Gibson syndrome (COGIS) [MIM:617561]: An autosomal dominant overgrowth disorder characterized by accelerated osseous maturation, advanced bone age, skeletal abnormalities including flaring of the metaphyses of the long bones, large hands with long fingers and camptodactyly, scoliosis, cervical spine anomalies, dysmorphic facial features, and variable intellectual disability. {ECO:0000269|PubMed:25787343, ECO:0000269|PubMed:27193220, ECO:0000269|PubMed:27868325, ECO:0000269|PubMed:28229514, ECO:0000269|PubMed:28475857}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142 Interacts with Q9ULA0; Q8TBB1; Q00013; Q9UPN6 EC number EC 3.4.11.21 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Aminopeptidase; Cytoplasm; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 50574.6 Length 457 Aromaticity 0.07 Instability index 55.73 Isoelectric point 7.73 Charge (pH=7) 2.3 2D Binding mode Binding energy (Kcal/mol) -7.42 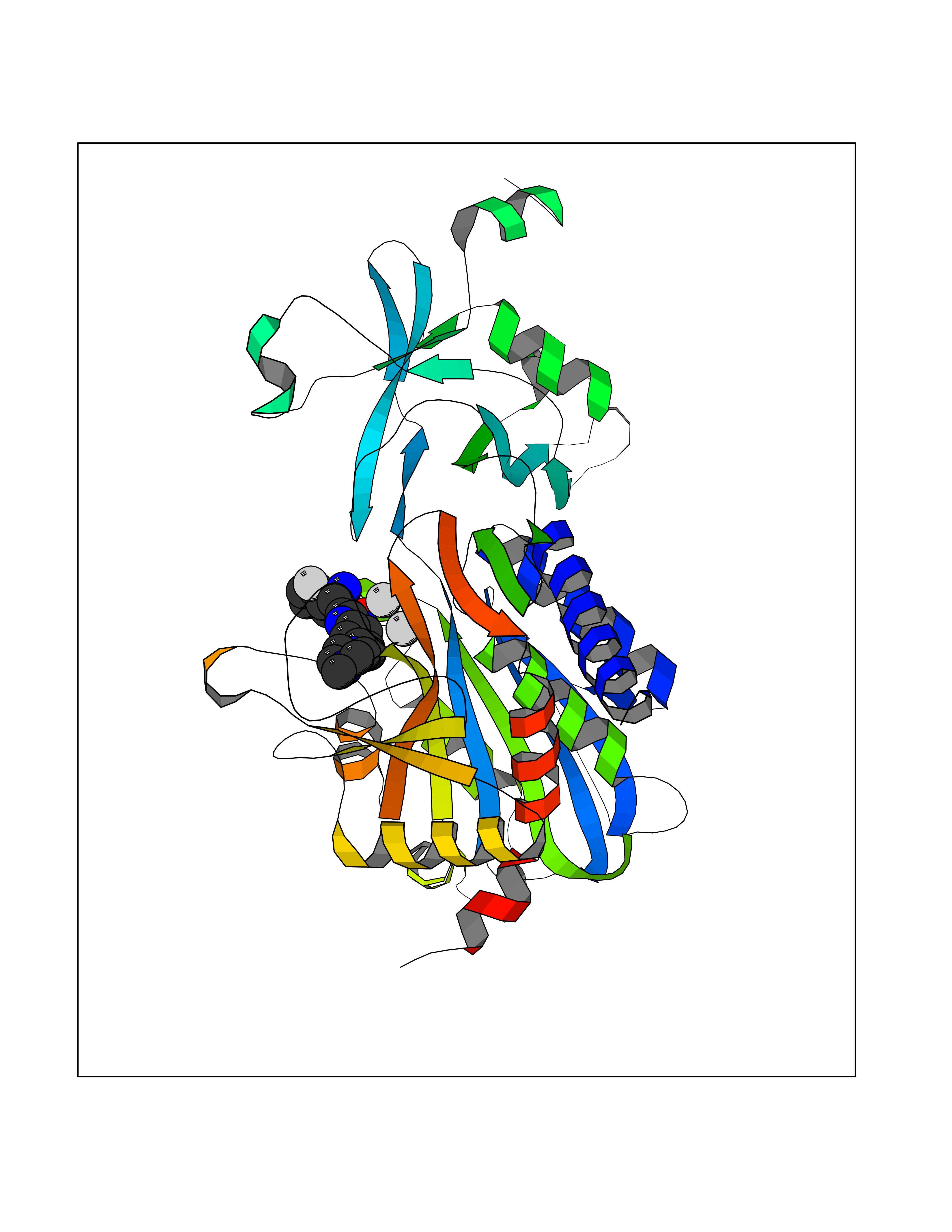 Molscript Map 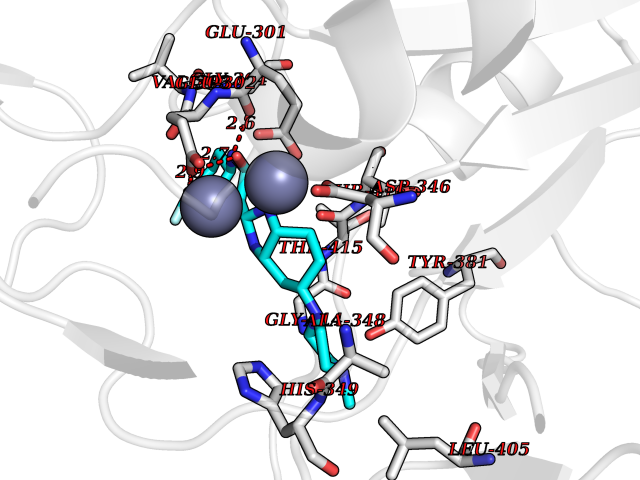 Pymol Map 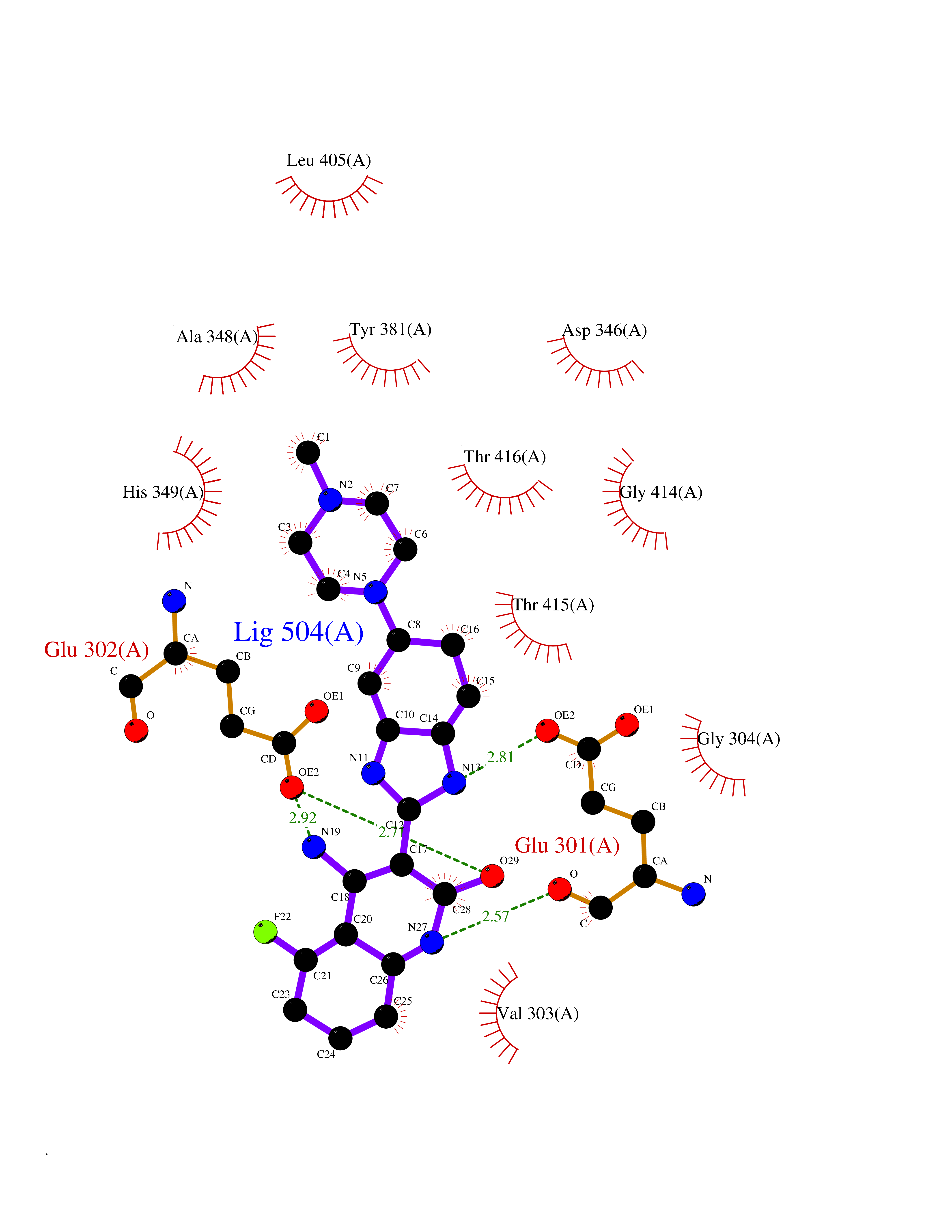 Ligplot Map 3D Binding mode Sequence GKARKEAVQTAAKELLKFVNRSPSPFHAVAECRNRLLQAGFSELKETEKWNIKPESKYFMTRNSSTIIAFAVGGQYVPGNGFSLIGAHTDSPCLRVKRRSRRSQVGFQQVGVETYGGGIWSTWFDRDLTLAGRVIVKCPTSGRLEQQLVHVERPILRIPHLAIHLQRNINENFGPNTEMHLVPILATAIQEELEKGTERHHSVLMSLLCAHLGLSPKDIVEMELCLADTQPAVLGGAYDEFIFAPRLDNLHSCFCALQALIDSCAGPGSLATEPHVRMVTLYDNEEVGSESAQGAQSLLTELVLRRISASCQHPTAFEEAIPKSFMISADMAHAVHPNYLDKHEENHRPLFHKGPVIKVNSKQRYASNAVSEALIREVANKVKVPLQDLMVRNDTPCGTTIGPILASRLGLRVLDLGSPQLAMHSIREMACTTGVLQTLTLFKGFFELFPSLAENLY Hydrogen bonds contact Hydrophobic contact | ||||
| 22 | Fatty acid synthase (FASN) | 3TJM | 8.26 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -9.21  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 23 | Glucagon-like peptide 1 receptor (GLP1R) | 6X1A | 8.25 | |
Target general information Gen name GLP1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GLP-1R; GLP-1-R; GLP-1 receptor Protein family G-protein coupled receptor 2 family Biochemical class GPCR secretin Function Ligand binding triggers activation of a signaling cascade that leads to the activation of adenylyl cyclase and increased intracellular cAMP levels. Plays a role in regulating insulin secretion in response to GLP-1. G-protein coupled receptor for glucagon-like peptide 1 (GLP-1). Related diseases Lynch syndrome 2 (LYNCH2) [MIM:609310]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10323887, ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10413423, ECO:0000269|PubMed:10480359, ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10627141, ECO:0000269|PubMed:10660333, ECO:0000269|PubMed:10671064, ECO:0000269|PubMed:10713887, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:11139242, ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11748856, ECO:0000269|PubMed:11754112, ECO:0000269|PubMed:11781295, ECO:0000269|PubMed:11793442, ECO:0000269|PubMed:11839723, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:12095971, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655562, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:14961575, ECO:0000269|PubMed:15064764, ECO:0000269|PubMed:15139004, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15365996, ECO:0000269|PubMed:16083711, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17301300, ECO:0000269|PubMed:17510385, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:20020535, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22753075, ECO:0000269|PubMed:7757073, ECO:0000269|PubMed:8566964, ECO:0000269|PubMed:8571956, ECO:0000269|PubMed:8574961, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:8993976, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9067757, ECO:0000269|PubMed:9218993, ECO:0000269|PubMed:9272156, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9326924, ECO:0000269|PubMed:9399661, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9833759, ECO:0000269|PubMed:9927034, ECO:0000269|Ref.5}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 1 (MMRCS1) [MIM:276300]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:17440981, ECO:0000269|PubMed:7661930}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:8751876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in MLH1 may contribute to lobular carcinoma in situ (LCIS), a non-invasive neoplastic disease of the breast.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Some epigenetic changes can be transmitted unchanged through the germline (termed 'epigenetic inheritance'). Evidence that this mechanism occurs in humans is provided by the identification of individuals in whom 1 allele of the MLH1 gene is epigenetically silenced throughout the soma (implying a germline event). These individuals are affected by Lynch syndrome but does not have identifiable mutations in MLH1, even though it is silenced, which demonstrates that an epimutation can phenocopy a genetic disease.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15184898, ECO:0000269|PubMed:18033691, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9032648, ECO:0000269|PubMed:9087566, ECO:0000269|PubMed:9611074}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09043; DB09045; DB15650; DB01276; DB00040; DB16697; DB06655; DB09265; DB13928; DB14027; DB15171 Interacts with A8MQ03; Q07627; Q8IUG1; P60409; P60410; P60411; Q9BYP8; P26371; Q7Z3S9; P0DPK4 EC number NA Uniprot keywords 3D-structure; ADP-ribosylation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 45579.6 Length 390 Aromaticity 0.16 Instability index 39.66 Isoelectric point 6.73 Charge (pH=7) -0.68 2D Binding mode Binding energy (Kcal/mol) -9.17  Molscript Map 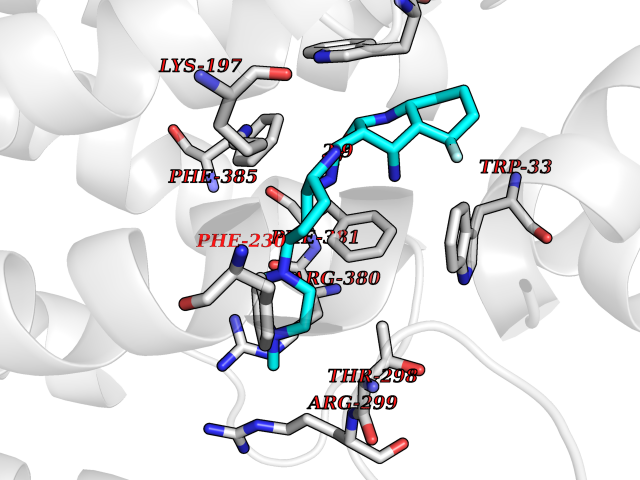 Pymol Map 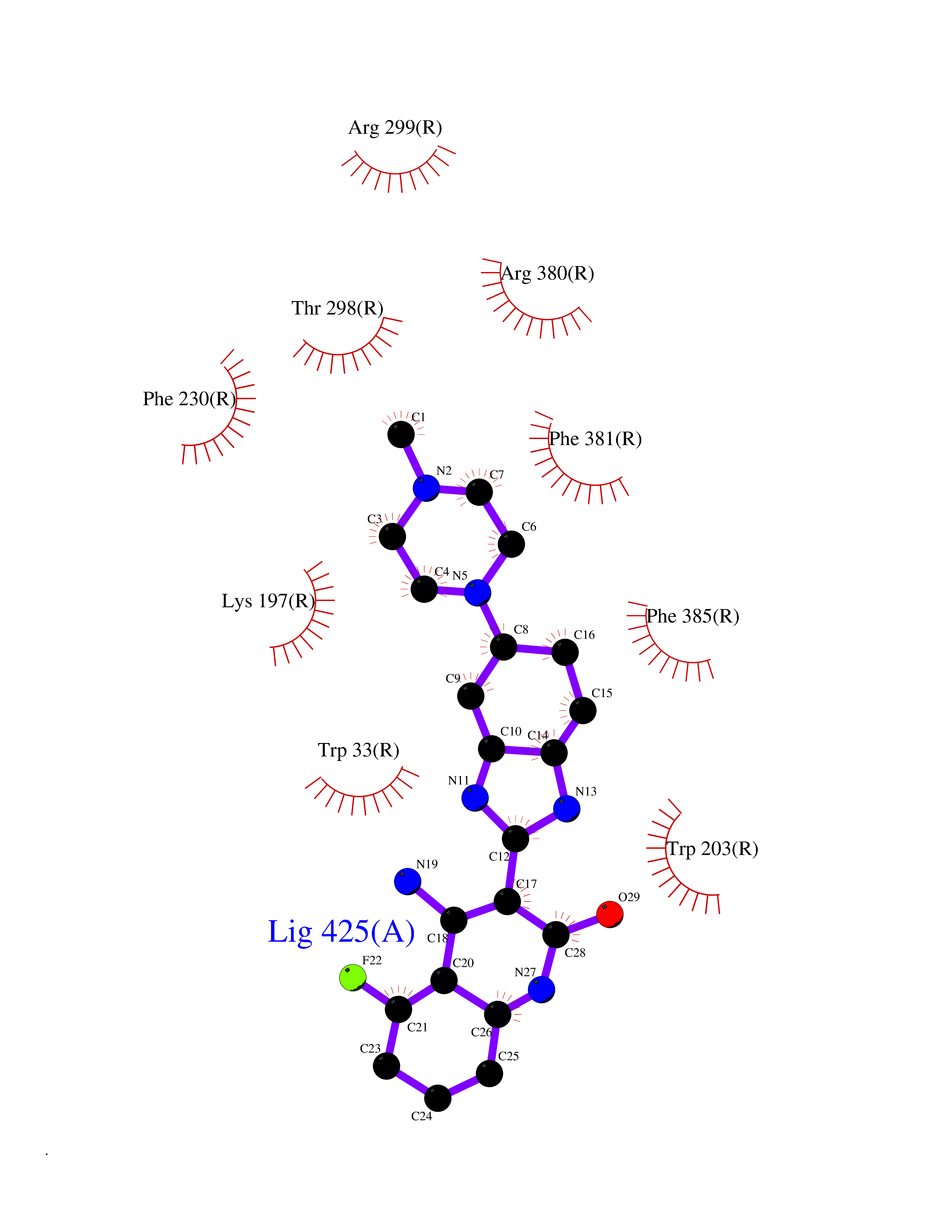 Ligplot Map 3D Binding mode Sequence ATVSLWETVQKWREYRRQCQRSLTEDPPPATDLFCNRTFDEYACWPDGEPGSFVNVSCPWYLPWASSVPQGHVYRFCTAEGLWLQKDNSSLPWRDLSECEESSPEEQLLFLYIIYTVGYALSFSALVIASAILLGFRHLHCTRNYIHLNLFASFILRALSVFIKDAALKWMYSTAAQQHQWDGLLSYQDSLSCRLVFLLMQYCVAANYYWLLVEGVYLYTLLAFSVFSEQWIFRLYVSIGWGVPLLFVVPWGIVKYLYEDEGCWTRNSNMNYWLIIRLPILFAIGVNFLIFVRVICIVVSKLKANLMCKTDIKCRLAKSTLTLIPLLGTHEVIFAFVMDEHARGTLRFIKLFTELSFTSFQGLMVAILYCFVNNEVQLEFRKSWERWRLE Hydrogen bonds contact Hydrophobic contact | ||||
| 24 | Multidrug resistance protein 3 (ABCB4) | 6S7P | 8.24 | |
Target general information Gen name ABCB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PGY3; MDR3; ATP-binding cassette sub-family B member 4; ABCB4 Protein family ABC transporter superfamily, ABCB family, Multidrug resistance exporter (TC 3.A.1.201) subfamily Biochemical class Acid anhydrides hydrolase Function Mediates ATP-dependent export of organic anions and drugs from the cytoplasm. Hydrolyzes ATP with low efficiency. Not capable of conferring drug resistance. Mediates the translocation of phosphatidylcholine across the canalicular membrane of the hepatocyte. Related diseases Cholestasis, progressive familial intrahepatic, 3 (PFIC3) [MIM:602347]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC3 inheritance is autosomal recessive. {ECO:0000269|PubMed:11313315, ECO:0000269|PubMed:12671900, ECO:0000269|PubMed:17726488, ECO:0000269|PubMed:21119540, ECO:0000269|PubMed:24045840, ECO:0000269|PubMed:24594635, ECO:0000269|PubMed:24806754, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:9419367}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cholestasis of pregnancy, intrahepatic 3 (ICP3) [MIM:614972]: A liver disorder of pregnancy. It presents during the second or, more commonly, the third trimester of pregnancy with intense pruritus which becomes more severe with advancing gestation and cholestasis. It causes fetal distress, spontaneous premature delivery and intrauterine death. Patients have spontaneous and progressive disappearance of cholestasis after delivery. Cholestasis results from abnormal biliary transport from the liver into the small intestine. {ECO:0000269|PubMed:10767346, ECO:0000269|PubMed:12746424, ECO:0000269|PubMed:15077010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gallbladder disease 1 (GBD1) [MIM:600803]: One of the major digestive diseases. Gallstones composed of cholesterol (cholelithiasis) are the common manifestations in western countries. Most people with gallstones, however, remain asymptomatic through their lifetimes. {ECO:0000269|PubMed:11313316, ECO:0000269|PubMed:12891548, ECO:0000269|PubMed:22331132, ECO:0000269|PubMed:23533021, ECO:0000269|PubMed:24723470, ECO:0000269|PubMed:28012258, ECO:0000269|PubMed:28587926, ECO:0000269|Ref.2}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06414; DB06207 Interacts with NA EC number EC 7.6.2.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Disease variant; Glycoprotein; Intrahepatic cholestasis; Lipid transport; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 123919 Length 1128 Aromaticity 0.1 Instability index 29.44 Isoelectric point 8.78 Charge (pH=7) 11.08 2D Binding mode Binding energy (Kcal/mol) -8.48  Molscript Map 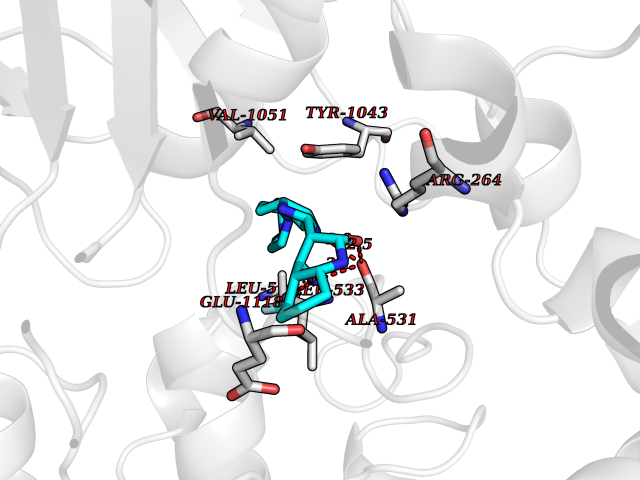 Pymol Map 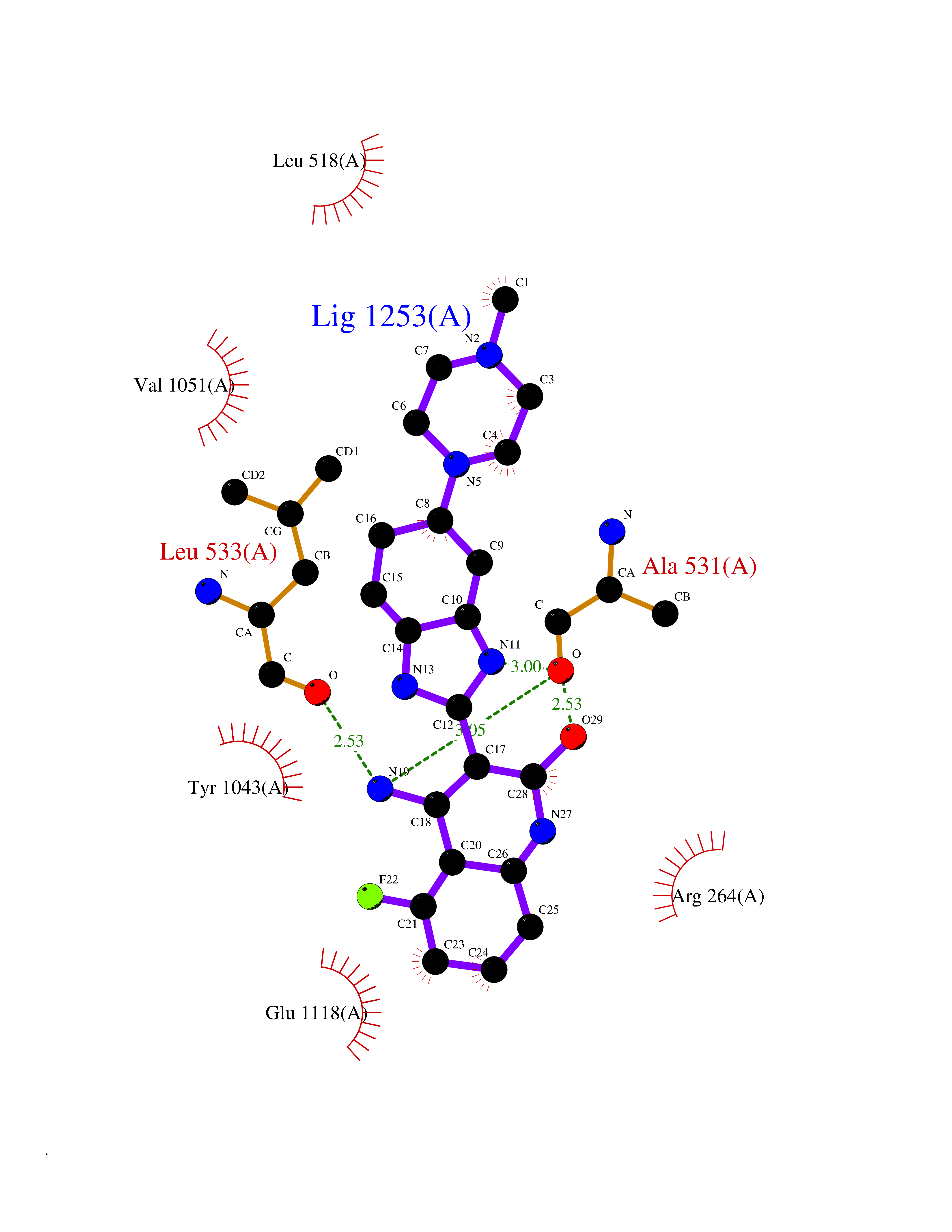 Ligplot Map 3D Binding mode Sequence VLTLFRYSDWQDKLFMSLGTIMAIAHGSGLPLMMIVFGEMTDKPGKILEEEMTRYAYYYSGLGAGVLVAAYIQVSFWTLAAGRQIRKIRQKFFHAILRQEIGWFDINDTTELNTRLTDDISKISEGIGDKVGMFFQAVATFFAGFIVGFIRGWKLTLVIMAISPILGLSAAVWAKILSAFSDKELAAYAKAGAVAEEALGAIRTVIAFGGQNKELERYQKHLENAKEIGIKKAISANISMGIAFLLIYASYALAFWYGSTLVISKEYTIGNAMTVFFSILIGAFSVGQAAPCIDAFANARGAAYVIFDIIDNNPKIDSFSERGHKPDSIKGNLEFNDVHFSYPSRANVKILKGLNLKVQSGQTVALVGSSGCGKSTTVQLIQRLYDPDEGTINIDGQDIRNFNVNYLREIIGVVSQEPVLFSTTIAENICYGRGNVTMDEIKKAVKEANAYEFIMKLPQKFDTLVGERGAQLSGGQKQRIAIARALVRNPKILLLDQATSALDTESEAEVQAALDKAREGRTTIVIAHRLSTVRNADVIAGFEDGVIVEQGSHSELMKKEGVYFKLVNVPPVSFLKVLKLNKTEWPYFVVGTVCAIANGGLQPAFSVIFSEIIAIFGPGDDAVKQQKCNIFSLIFLFLGIISFFTFFLQGFTFGKAGEILTRRLRSMAFKAMLRQDMSWFDDHKNSTGALSTRLATDAAQVQGATGTRLALIAQNIANLGTGIIISFIYGWQLTLLLLAVVPIIAVSGIVEMKLLAGNAKRDKKELEAAGKIATEAIENIRTVVSLTQERKFESMYVEKLYGPYRNSVQKAHIYGITFSISQAFMYFSYAGCFRFGAYLIVNGHMRFRDVILVFSAIVFGAVALGHASSFAPDYAKAKLSAAHLFMLFERQPLIDSYSEEGLKPDKFEGNITFNEVVFNYPTRANVPVLQGLSLEVKKGQTLALVGSSGCGKSTVVQLLERFYDPLAGTVLLDGQEAKKLNVQWLRAQLGIVSQEPILFDCSIAENIAYGDNSRVVSQDEIVSAAKAANIHPFIETLPHKYETRVGDKGTQLSGGQKQRIAIARALIRQPQILLLDQATSALDTESEKVVQEALDKAREGRTCIVIAHRLSTIQNADLIVVFQNGRVK Hydrogen bonds contact Hydrophobic contact | ||||
| 25 | MLK-related kinase (MLTK) | 5HES | 8.23 | |
Target general information Gen name MAP3K20 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ZAK; Sterile alpha motif- and leucine zipper-containing kinase AZK; Mixed lineage kinase-related kinase; Mitogen-activated protein kinase kinase kinase MLT; Mitogen-activated protein kinase kinase kin Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class NA Function Stress-activated component of a protein kinase signal transduction cascade. Regulates the JNK and p38 pathways. Part of a signaling cascade that begins with the activation of the adrenergic receptor ADRA1B and leads to the activation of MAPK14. Pro-apoptotic. Role in regulation of S and G2 cell cycle checkpoint by direct phosphorylation of CHEK2. Involved in limb development. Related diseases Split-foot malformation with mesoaxial polydactyly (SFMMP) [MIM:616890]: An autosomal recessive disorder characterized by a split-foot defect, mesoaxial polydactyly, nail abnormalities of the hands, and sensorineural hearing loss. {ECO:0000269|PubMed:26755636, ECO:0000269|PubMed:32266845}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, centronuclear, 6, with fiber-type disproportion (CNM6) [MIM:617760]: A form of centronuclear myopathy, a congenital muscle disorder characterized by progressive muscular weakness and wasting involving mainly limb girdle, trunk, and neck muscles. It may also affect distal muscles. Weakness may be present during childhood or adolescence or may not become evident until the third decade of life. Ptosis is a frequent clinical feature. The most prominent histopathologic features include high frequency of centrally located nuclei in muscle fibers not secondary to regeneration, radial arrangement of sarcoplasmic strands around the central nuclei, and predominance and hypotrophy of type 1 fibers. CNM6 is an autosomal recessive, slowly progressive form with onset in infancy or early childhood. {ECO:0000269|PubMed:27816943, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01254; DB12010 Interacts with O75582; P31947; P63104; Q8N184; Q16512; Q6P2D0; Q6ZN57; P13682; Q8N184; Q6AZW8; Q9NQZ8 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cytoplasm; Disease variant; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding; rRNA-binding; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 64591.5 Length 566 Aromaticity 0.1 Instability index 52.08 Isoelectric point 5.69 Charge (pH=7) -12.83 2D Binding mode Binding energy (Kcal/mol) -9.46 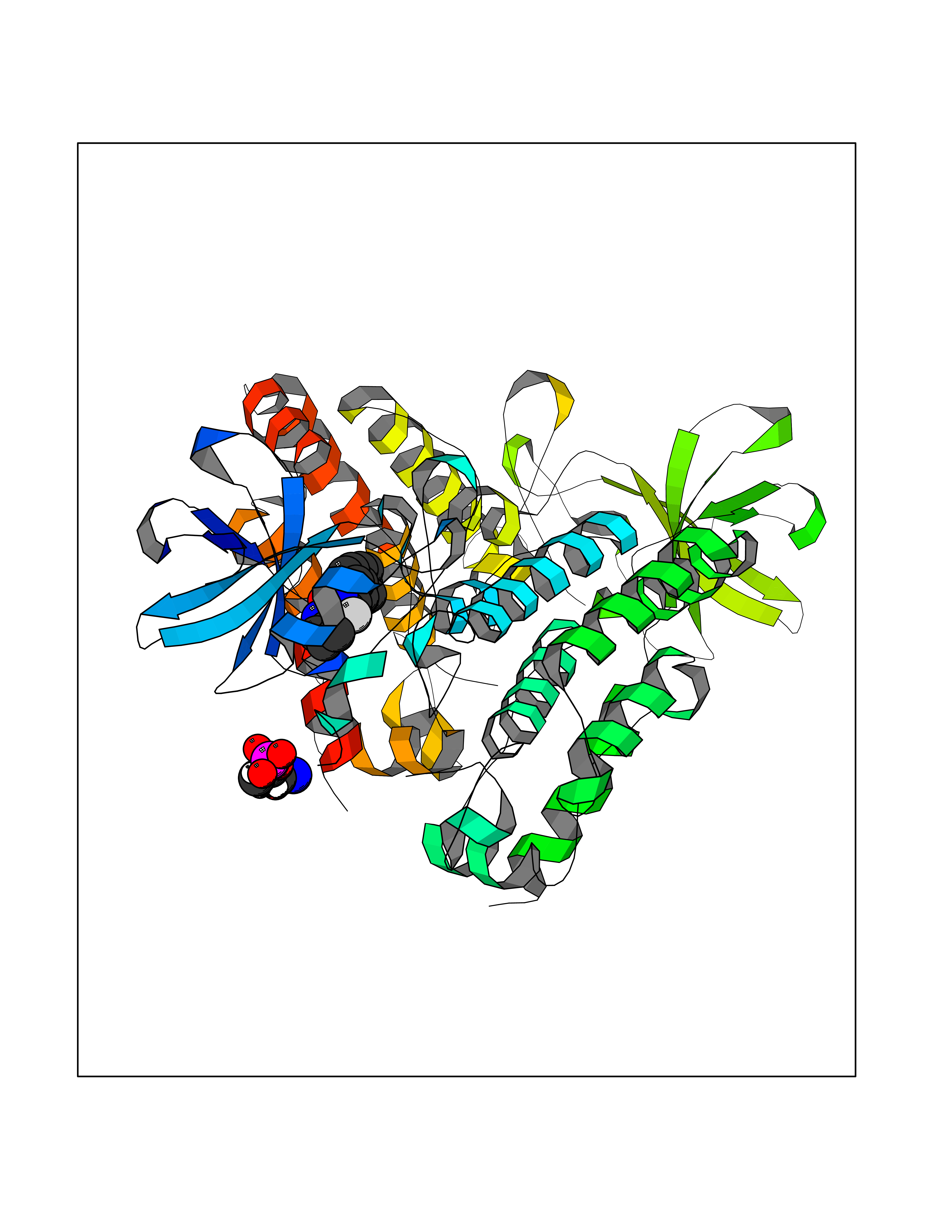 Molscript Map 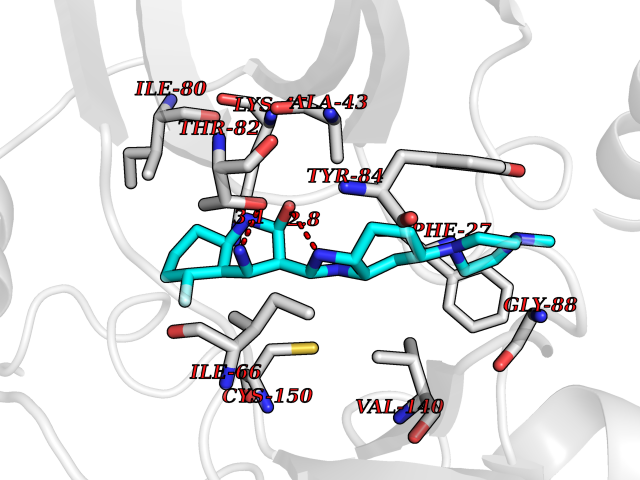 Pymol Map 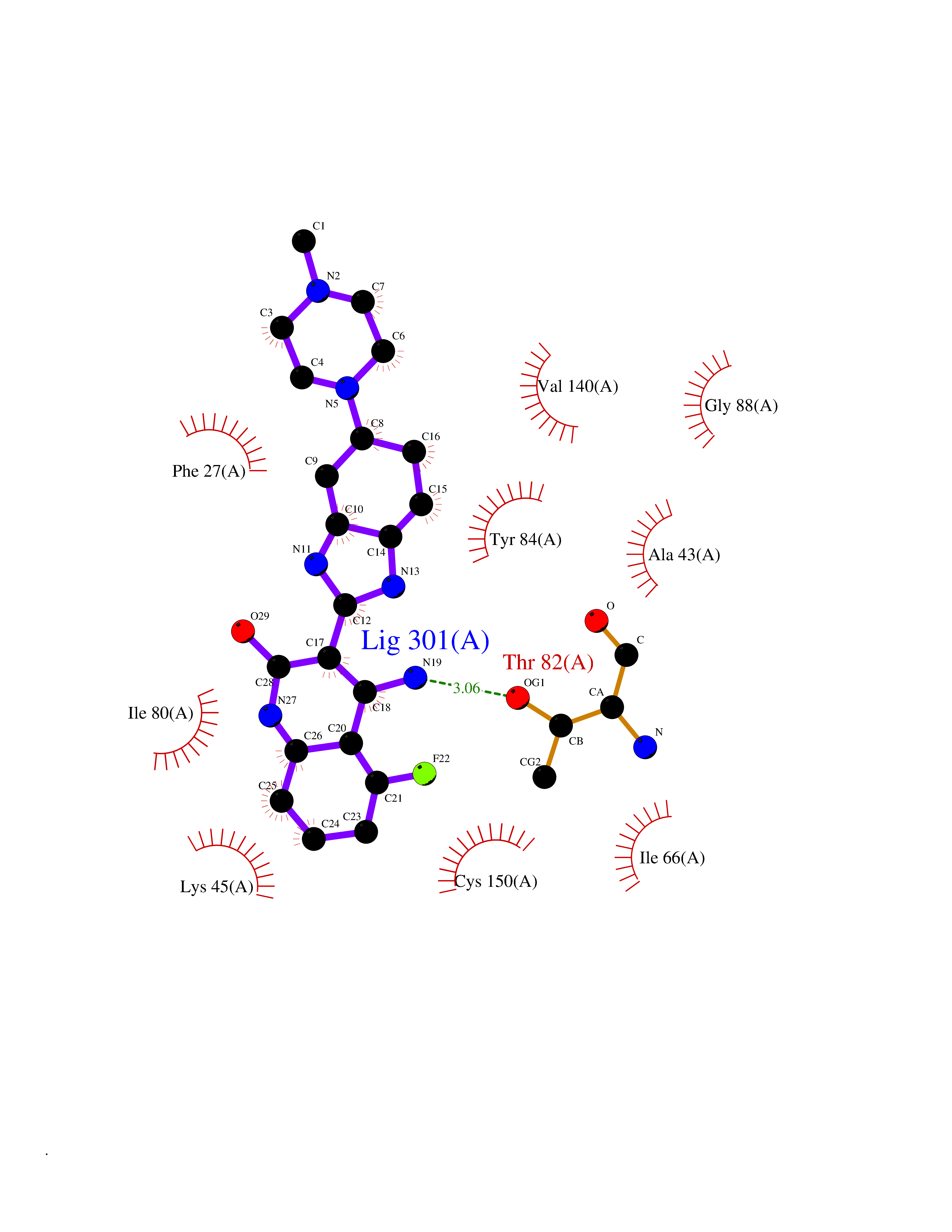 Ligplot Map 3D Binding mode Sequence ASFVQIKFDDLQFFENCGGGSFGSVYRAKWISQDKEVAVKKLLKIEKEAEILSVLSHRNIIQFYGVILEPPNYGIVTEYASLGSLYDYINSNRSEEMDMDHIMTWATDVAKGMHYLHMEAPVKVIHRDLKSRNVVIAADGVLKICDFGASRFHNHXGTFPWMAPEVIQSLPVSETCDTYSYGVVLWEMLTREVPFKGLEGLQVAWLVVEKNERLTIPSSCPRSFAELLHQCWEADAKKRPSFKQIISILESMSNDTSLPDKCNSFLHNKAEWRCEIEATLERLKKLERSFVQIKFDDLQFFENCGGGSFGSVYRAKWISQDKEVAVKKLLKIEKEAEILSVLSHRNIIQFYGVILEPPNYGIVTEYASLGSLYDYINSNRSEEMDMDHIMTWATDVAKGMHYLHMEAPVKVIHRDLKSRNVVIAADGVLKICDFGGTFPWMAPEVIQSLPVSETCDTYSYGVVLWEMLTREVPFKGLEGLQVAWLVVEKNERLTIPSSCPRSFAELLHQCWEADAKKRPSFKQIISILESMSNDTSLPDKCNSFLHNKAEWRCEIEATLERLKKLE Hydrogen bonds contact Hydrophobic contact | ||||
| 26 | Thymidylate synthase (TYMS) | 1HVY | 8.23 | |
Target general information Gen name TYMS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TSase; TS Protein family Thymidylate synthase family Biochemical class Methyltransferase Function Contributes to the de novo mitochondrial thymidylate biosynthesis pathway. Related diseases Dyskeratosis congenita, digenic (DKCD) [MIM:620040]: A form of dyskeratosis congenita, a rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy. DKCD transmission pattern is consistent with digenic inheritance. {ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. TYMS germline variants in the presence of a common ENOSF1 haplotype (defined by rs699517, rs2790 and rs1512643) result in severe thymidylate synthase deficiency and disease. The pathogenic mechanism involves increased expression of ENOSF1 relative to TYMS, and post-transcriptional inhibition of TYMS translation through ENOSF1-TYMS RNA-RNA interactions. {ECO:0000269|PubMed:35931051}. Drugs (DrugBank ID) DB03541; DB07577; DB08734; DB05308; DB01101; DB03800; DB00322; DB00544; DB00441; DB00563; DB08479; DB08478; DB05457; DB00642; DB06813; DB00293; DB04530; DB09256; DB09327; DB05116; DB01643; DB00432 Interacts with NA EC number EC 2.1.1.45 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Dyskeratosis congenita; Isopeptide bond; Membrane; Methyltransferase; Mitochondrion; Mitochondrion inner membrane; Nucleotide biosynthesis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 32889.3 Length 287 Aromaticity 0.1 Instability index 40.61 Isoelectric point 6.57 Charge (pH=7) -1.26 2D Binding mode Binding energy (Kcal/mol) -8.04 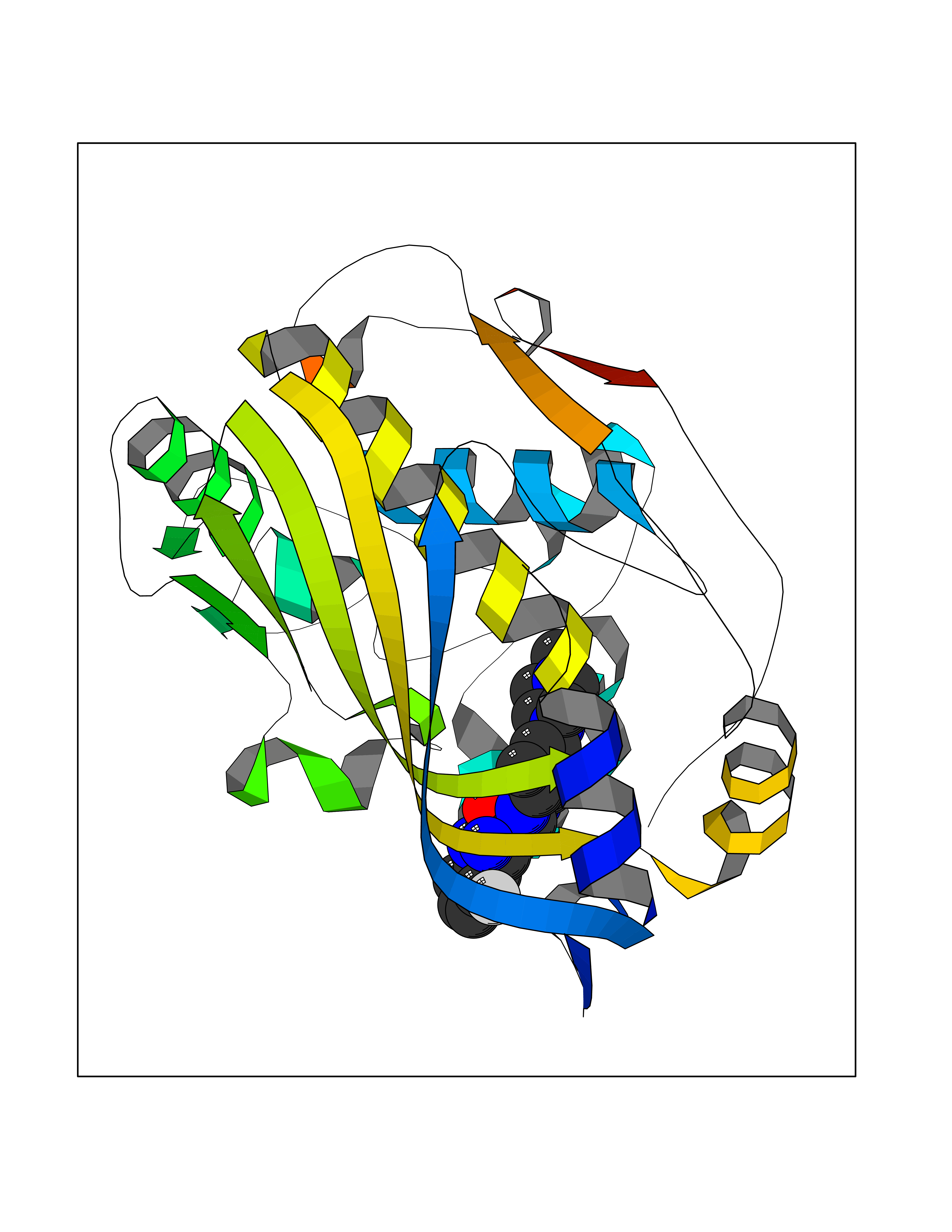 Molscript Map 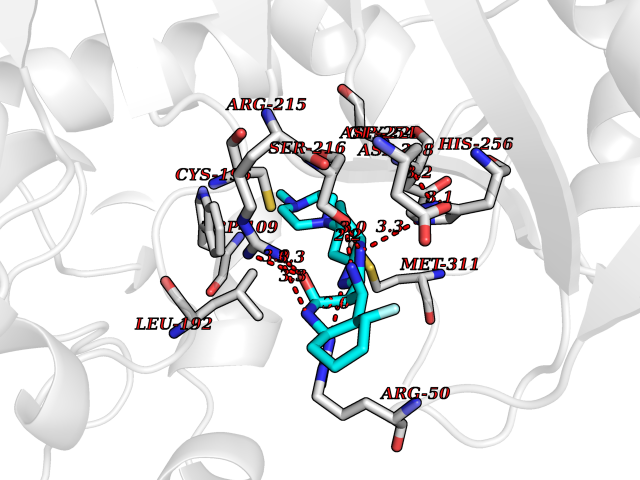 Pymol Map 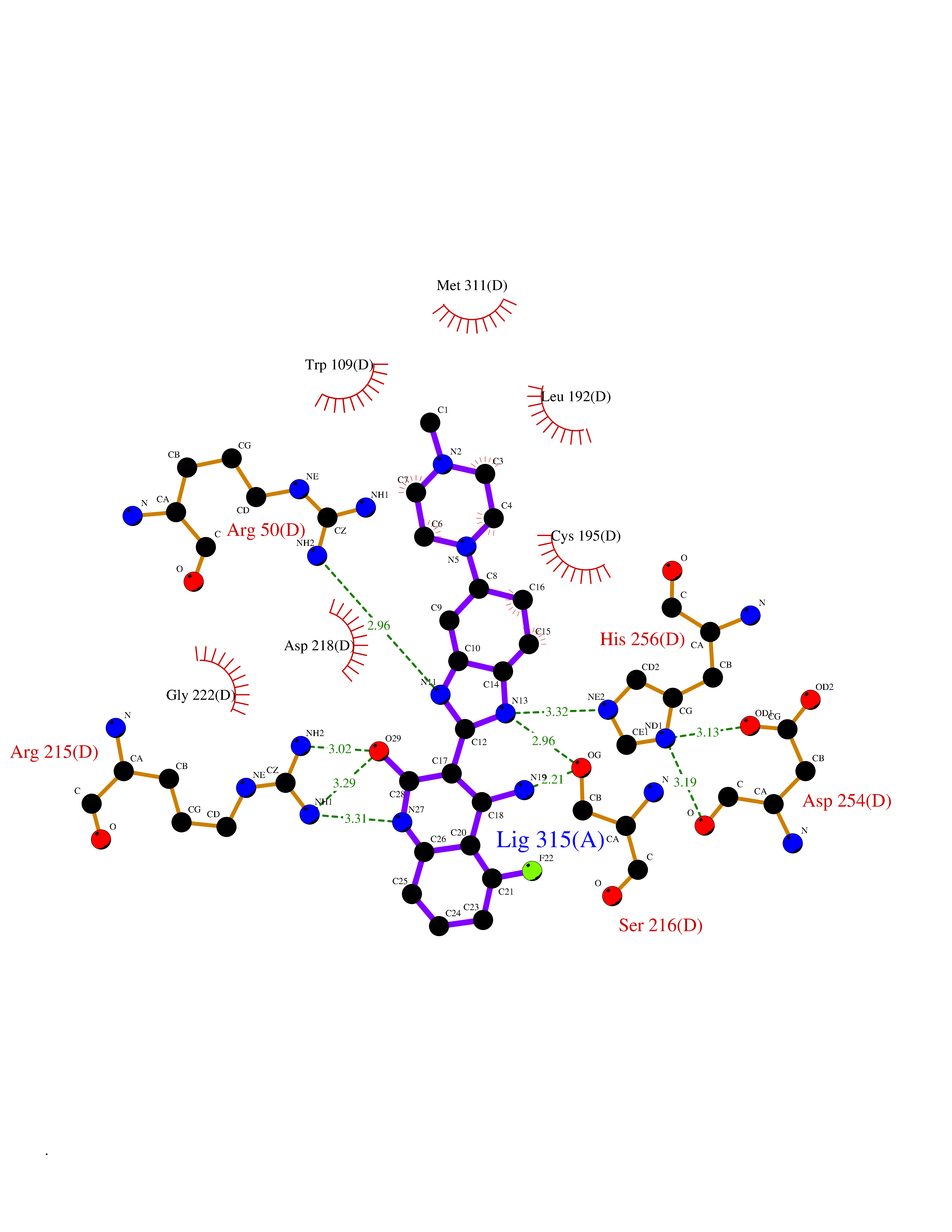 Ligplot Map 3D Binding mode Sequence PPHGELQYLGQIQHILRGVRKDDRTGTGTLSVFGMQARYSLRDEFPLLTTKRVFWKGVLEELLWFIKGSTNAKELSSKGVKIWDANGSRDFLDSLGFSTREEGDLGPVYGFQWRHFGAEYRDMESDYSGQGVDQLQRVIDTIKTNPDDRRIIMCAWNPRDLPLMALPPCHALCQFYVVNSELSCQLYQRSGDMGLGVPFNIASYALLTYMIAHITGLKPGDFIHTLGDAHIYLNHIEPLKIQLQREPRPFPKLRILRKVEKIDDFKAEDFQIEGYNPHPTIKMEMAV Hydrogen bonds contact Hydrophobic contact | ||||
| 27 | Voltage-gated sodium channel alpha Nav1.7 (SCN9A) | 7W9M | 8.22 | |
Target general information Gen name SCN9A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hNE-Na; Voltage-gated sodium channel subunit alpha Nav1.7; Sodium channel proteintype IX subunit alpha; Sodium channel proteintype 9 subunit alpha; Sodium channel protein type IX subunit alpha; Sodium Protein family Sodium channel (TC 1.A.1.10) family, Nav1.7/SCN9A subfamily Biochemical class Voltage-gated ion channel Function Assuming opened or closed conformations in response to the voltage difference across the membrane, the protein forms a sodium-selective channel through which Na(+) ions may pass in accordance with their electrochemical gradient. It is a tetrodotoxin-sensitive Na(+) channel isoform. Plays a role in pain mechanisms, especially in the development of inflammatory pain. Mediates the voltage-dependent sodium ion permeability of excitable membranes. Related diseases Primary erythermalgia (PERYTHM) [MIM:133020]: Autosomal dominant disease characterized by recurrent episodes of severe pain associated with redness and warmth in the feet or hands. {ECO:0000269|PubMed:14985375, ECO:0000269|PubMed:15385606, ECO:0000269|PubMed:15955112, ECO:0000269|PubMed:15958509, ECO:0000269|PubMed:16216943, ECO:0000269|PubMed:16392115, ECO:0000269|PubMed:16702558, ECO:0000269|PubMed:16988069, ECO:0000269|PubMed:18945915, ECO:0000269|PubMed:19369487, ECO:0000269|PubMed:24311784}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Indifference to pain, congenital, autosomal recessive (CIP) [MIM:243000]: A disorder characterized by congenital inability to perceive any form of pain, in any part of the body. All other sensory modalities are preserved and the peripheral and central nervous systems are apparently intact. Patients perceive the sensations of touch, warm and cold temperature, proprioception, tickle and pressure, but not painful stimuli. There is no evidence of a motor or sensory neuropathy, either axonal or demyelinating. {ECO:0000269|PubMed:20635406}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Paroxysmal extreme pain disorder (PEXPD) [MIM:167400]: An autosomal dominant paroxysmal disorder of pain and autonomic dysfunction. The distinctive features are paroxysmal episodes of burning pain in the rectal, ocular, and mandibular areas accompanied by autonomic manifestations such as skin flushing. {ECO:0000269|PubMed:17145499, ECO:0000269|PubMed:18945915, ECO:0000269|PubMed:25285947}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09088; DB13746; DB05541; DB00564; DB01161; DB00907; DB13269; DB13961; DB06218; DB00555; DB00281; DB00776; DB11186; DB09345; DB01069; DB09342; DB00243; DB06201; DB09085; DB00273; DB00313; DB00909 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Ubl conjugation; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 162402 Length 1418 Aromaticity 0.14 Instability index 35.66 Isoelectric point 6.72 Charge (pH=7) -1.33 2D Binding mode Binding energy (Kcal/mol) -8.13 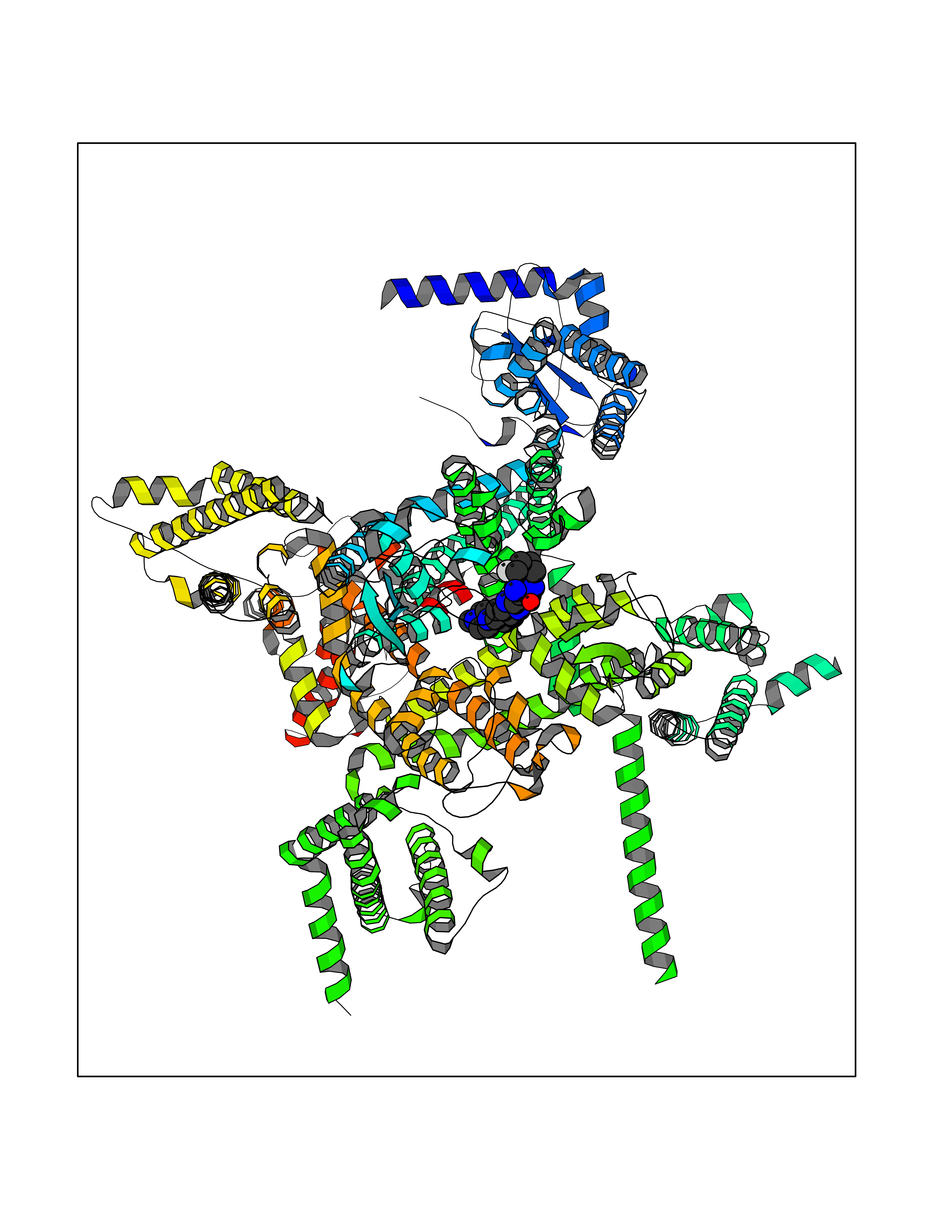 Molscript Map 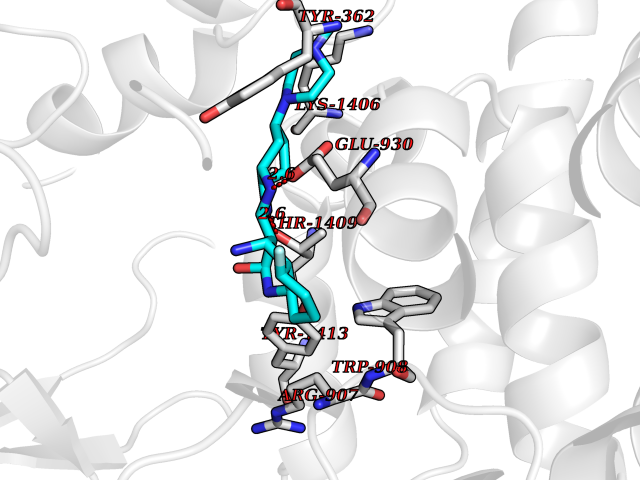 Pymol Map 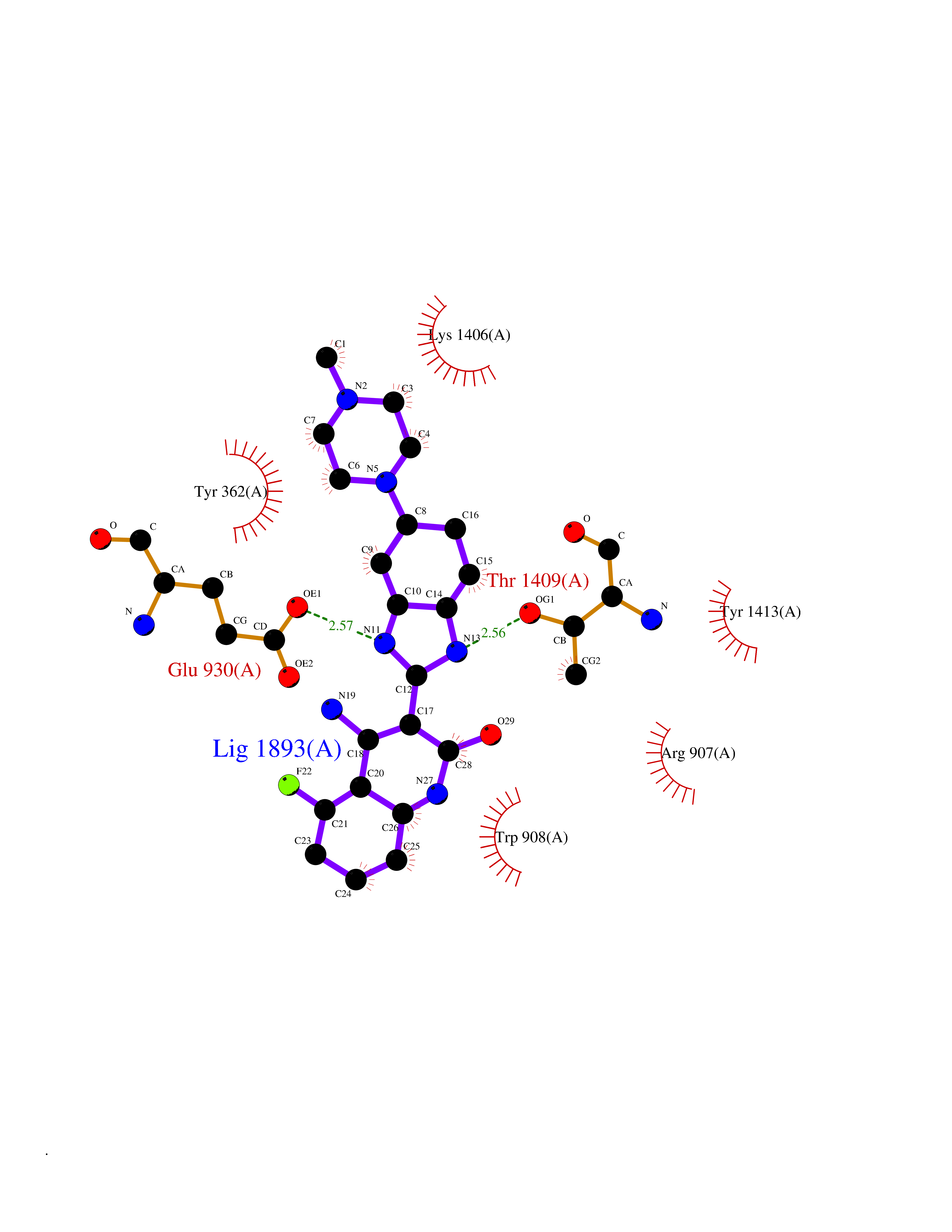 Ligplot Map 3D Binding mode Sequence GPQSFVHFTKQSLALIEQRIAERKSKEPKPSSDLEAGKQLPFIYGDIPPGMVSEPLEDLDPYYADKKTFIVLNKGKTIFRFNATPALYMLSPFSPLRRISIKILVHSLFSMLIMCTILTNCIFMTMNNPPDWTKNVEYTFTGIYTFESLVKILARGFCVGEFTFLRDPWNWLDFVVIVFAYLTEFVNNVSALRTFRVLRALKTISVIPGLKTIVGALIQSVKKLSDVMILTVFCLSVFALIGLQLFMGNLKHKCFRNSLENNETLESIMNTLESEEDFRKYFYYLEGSKDALLCGFSTDSGQCPEGYTCVKIGRNPDYGYTSFDTFSWAFLALFRLMTQDYWENLYQQTLRAAGKTYMIFFVVVIFLGSFYLINLILAVVAMAYKEQNQANIEEAKQKELEFQQMLDRLKKEQEPYWIKFKKCIYFIVMDPFVDLAITICIVLNTLFMAMEHHPMTEEFKNVLAIGNLVFTGIFAAEMVLKLIAMDPYEYFQVGWNIFDSLIVTLSLVELFLADVEGLSVLRSFRLLRVFKLAKSWPTLNMLIKIIGNSVGALGNLTLVLAIIVFIFAVVGMQLFGKSYKECVCKINDDCTLPRWHMNDFFHSFLIVFRVLCGEWIETMWDCMEVAGQAMCLIVYMMVMVIGNLVVLNLFLALLLSSFSSDNLTAIEEDPDANNLQIAVTRIKKGINYVKQTLREFILKAFGKIWWNIRKTCYKIVEHSWFESFIVLMILLSSGALAFEDIYIERKKTIKIILEYADKIFTYIFILEMLLKWIAYGYKTYFTNAWCWLDFLIVDVSLVTLVANTLGYSDLGPIKSLRTLRALRPLRALSRFEGMRVVVNALIGAIPSIMNVLLVCLIFWLIFSIMGVNLFAGKFYECINTTDGSRFPASQVPNRSECFALMNVSQNVRWKNLKVNFDNVGLGYLSLLQVATFKGWTIIMYAAVDSVNVDKQPKYEYSLYMYIYFVVFIIFGSFFTLNLFIGVIIDNFNQQKKKLGGQDIFMTEEQKKYYNAMKKLGSKKPQKPIPRPGNKIQGCIFDLVTNQAFDISIMVLICLNMVTMMVEKEGQSQHMTEVLYWINVVFIILFTGECVLKLISLRHYYFTVGWNIFDFVVVIISIVGMFLADLIETYFVSPTLFRVIRLARIGRILRLVKGAKGIRTLLFALMMSLPALFNIGLLLFLVMFIYAIFGMSNFAYVKKEDGINDMFNFETFGNSMICLFQITTSAGWDGLLAPILNSKPPDCDPKKVHPGSSVEGDCGNPSVGIFYFVSYIIISFLVVVNMYIAVILENFSVATEESTEPLSEDDFEMFYEVWEKFDPDATQFIEFSKLSDFAAALDPPLLIAKPNKVQLIAMDLPMVSGDRIHCLDILFAFTKRVLGESGEMDSLRSQMEERFMSANPSKVSYEPITTTLKRKQEDV Hydrogen bonds contact Hydrophobic contact | ||||
| 28 | Ecdysone receptor (20-hydroxy-ecdysone receptor) (EcRH) (Ecdysteroid receptor) (Nuclear receptor subfamily 1 group H member 1) | 3IXP | 8.22 | |
Target general information Gen name NA Organism Helicoverpa armigera (Cotton bollworm) (Heliothis armigera) Uniprot ID TTD ID NA Synonyms NA Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class NA Function NA Related diseases NA Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; DNA-binding; Metal-binding; Nucleus; Receptor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,D Molecular weight (Da) 52799.1 Length 462 Aromaticity 0.09 Instability index 51.97 Isoelectric point 8.09 Charge (pH=7) 2.37 2D Binding mode Binding energy (Kcal/mol) -8.93 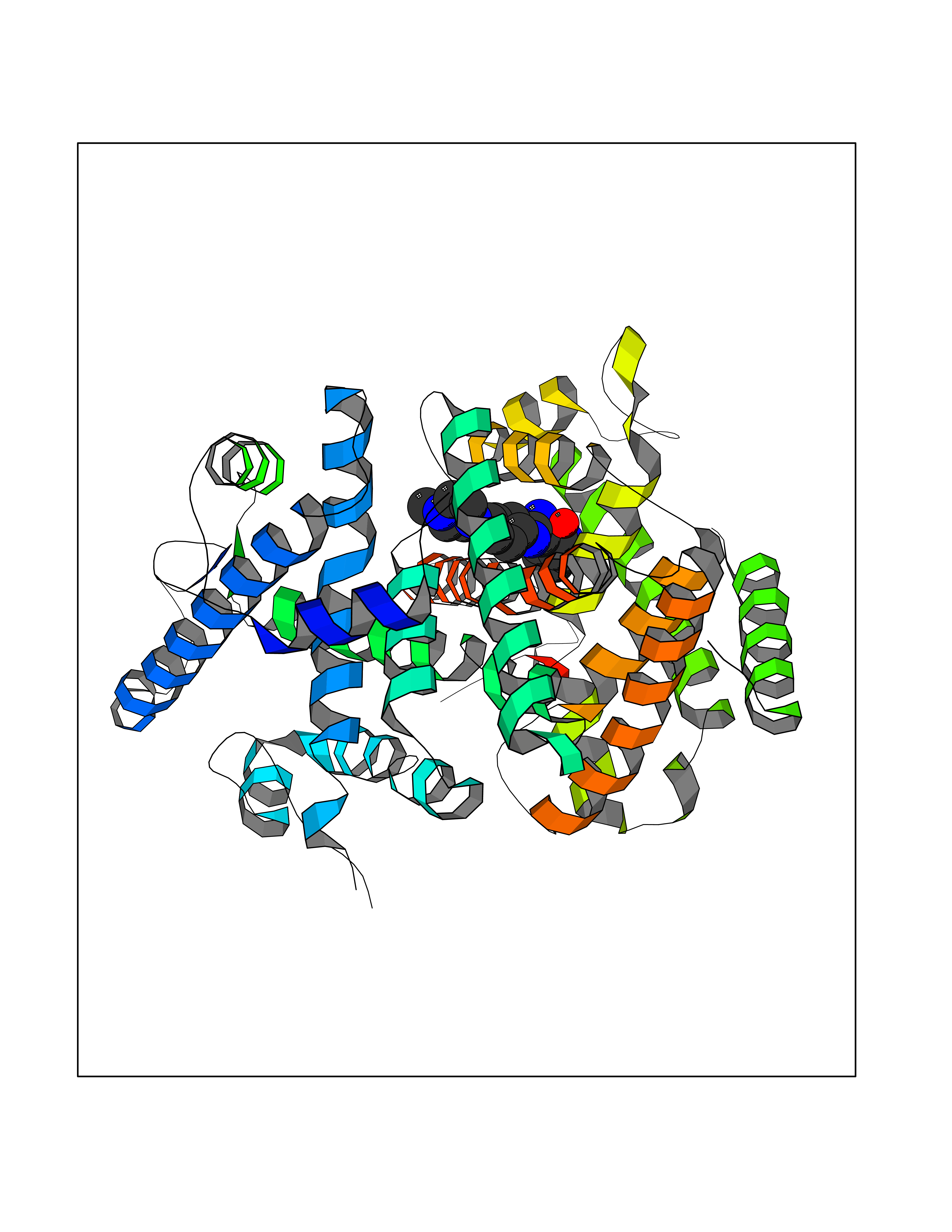 Molscript Map 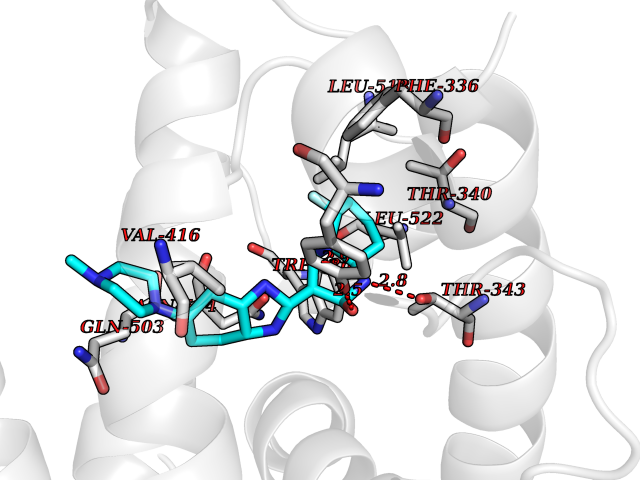 Pymol Map 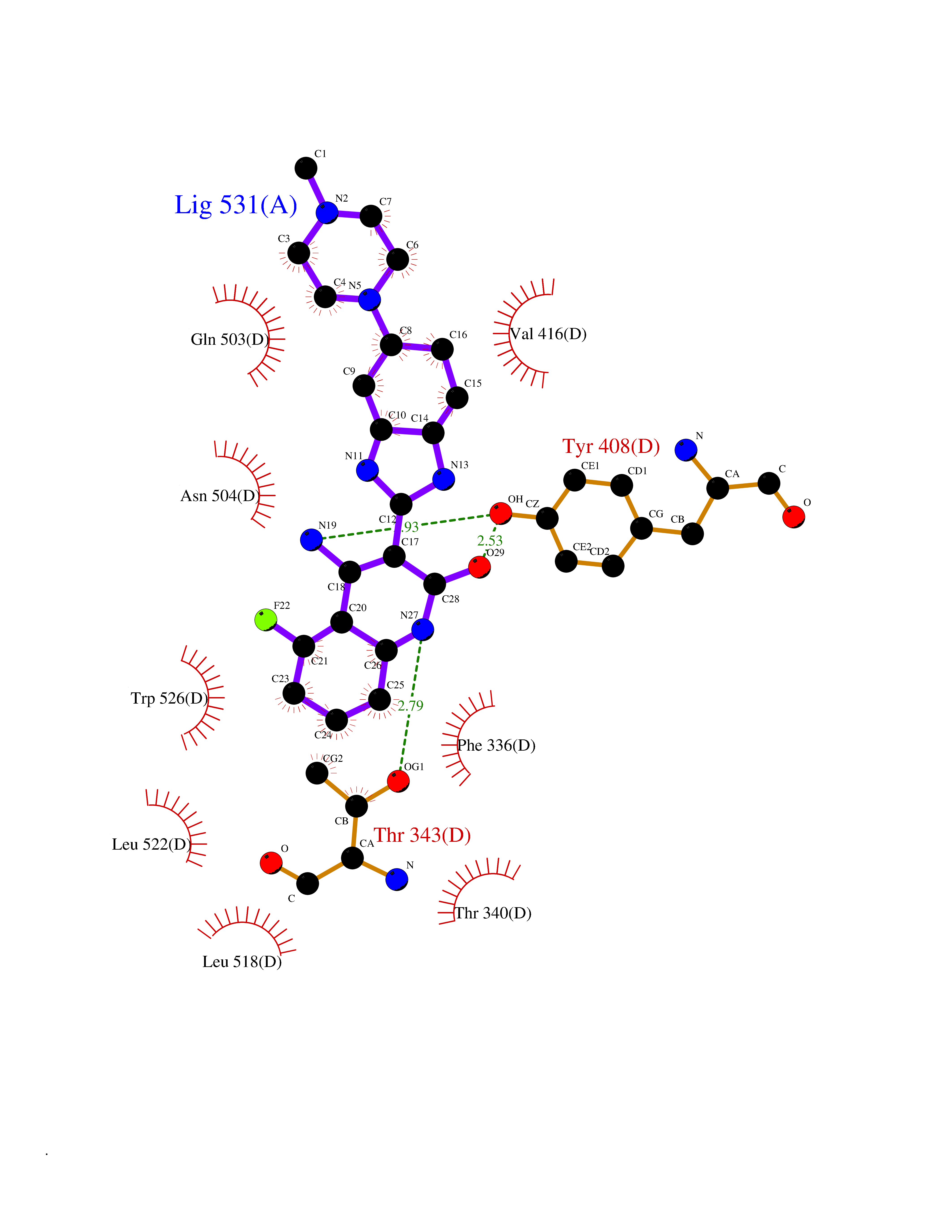 Ligplot Map 3D Binding mode Sequence QELSIERLLEMESLVADPSEEFQFLRVGPDSNVPPKFRAPVSSLCQIGNKQIAALVVWARDIPHFSQLEMEDQILLIKGSWNELLLFAIAWRSMEFLTSPPQLMCLMPGMTLHRNSALQAGVGQIFDRVLSELSLKMRTLRVDQAEYVALKAIILLNPDVKGLKNRQEVEVLREKMFLCLDEYCRRSRSSEEGRFAALLLRLPALRSISLKSFEHLFFFHLVADTSIAGYIRDALRNHAPPIVPPLTANQKSLIARLVYYQEGYEQMPFRQITEMTILTVQLIVEFAKGLPGFSKISQSDQITLLKACSSEVMMLRVARRYDAATDSVLFANNQAYTRDNYRKAGMAYVIEDLLHFCRCMYSMMMDNVHYALLTAIVIFSDRPGLEQPSLVEEIQRYYLNTLRVYILNQNSASPRSAVIFGKILGILTEIRTLGMQNSNMCISLKLKNRKLPPFLEEIWDVA Hydrogen bonds contact Hydrophobic contact | ||||
| 29 | Choline O-acetyltransferase | 2FY3 | 8.22 | |
Target general information Gen name CHAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Carnitine/choline acetyltransferase family Biochemical class Transferase Function Choline O-acetyltransferase activity. Related diseases Myasthenic syndrome, congenital, 6, presynaptic (CMS6) [MIM:254210]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS6 affected individuals have myasthenic symptoms since birth or early infancy, negative tests for anti-AChR antibodies, and abrupt episodic crises with increased weakness, bulbar paralysis, and apnea precipitated by undue exertion, fever, or excitement. CMS6 inheritance is autosomal recessive. {ECO:0000269|PubMed:11172068, ECO:0000269|PubMed:12756141}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006; DB00184 Interacts with Q6H8Q1-8; Q8N302-2; Q9NXL2-1; Q6XD76; Q9UII2; Q8TBE0; Q9UQB8-6; Q9ULD4-2; Q9NSI6-4; Q6P5X5; Q96LL4; P20807-4; O00257-3; Q6ZP82-1; O95674; Q9H3R5; Q8WUX9; Q9H2A9; Q3SX64; Q92782-2; Q14117; O14641; Q658K8; Q6UXG2-3; O00472; Q6NXG1; Q15910-2; Q8IZU1; P15407; P55318; Q06547-3; P23769-2; P23771; Q15486; Q8IV36; Q4VB01; Q53GQ0; P10809; P41134; Q9NZH6; Q8NA54; Q86U28; P17275; Q8N5Z5; Q6P597; P08727; Q14525; Q8IUC2; Q6IAA8; Q14847-2; P27338; Q9GZQ8; Q53S70; Q5JXC2; A0A0A0MR05; Q8NEH6; Q8TCY5; Q6IN84-2; Q96H12; P01106; P41271-2; P14598; Q9GZM8; Q5BJF6-2; Q9H8K7; Q9NR21-5; Q5VU43-8; Q13956; Q5SXH7-1; Q96T60; Q96I34; Q86UA1; Q15311; Q8TBY0; Q04206; P47804-3; Q9H0X6; P62899; Q66K80; Q9BY12-3; Q86SQ7-2; Q7Z6I5; Q496A3; Q7Z698; Q9C004; Q92783-2; Q8N4C7; O75528; Q15814; O15273; Q96A09; Q8WTV1; Q53NU3; Q71RG4-4; Q86WT6-2; Q9Y3Q8; Q99598; P49459; P11441; Q9H270; P19544-6; Q53FD0-2; Q3KNS6-3 EC number 2.3.1.6 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Neurotransmitter biosynthesis; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 66365.9 Length 595 Aromaticity 0.08 Instability index 53.36 Isoelectric point 8.16 Charge (pH=7) 4.64 2D Binding mode Binding energy (Kcal/mol) -9.21  Molscript Map 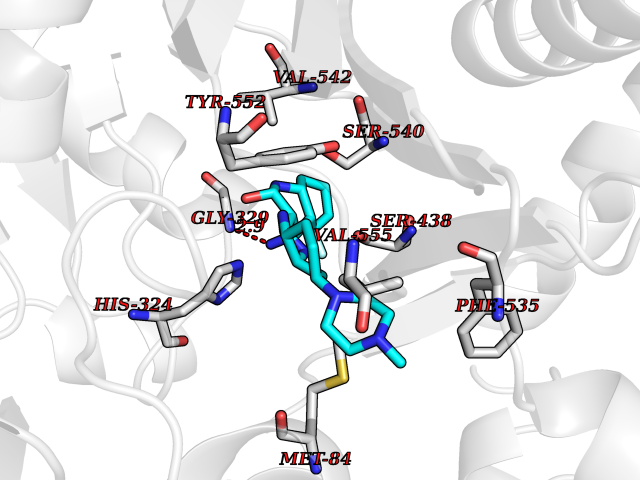 Pymol Map 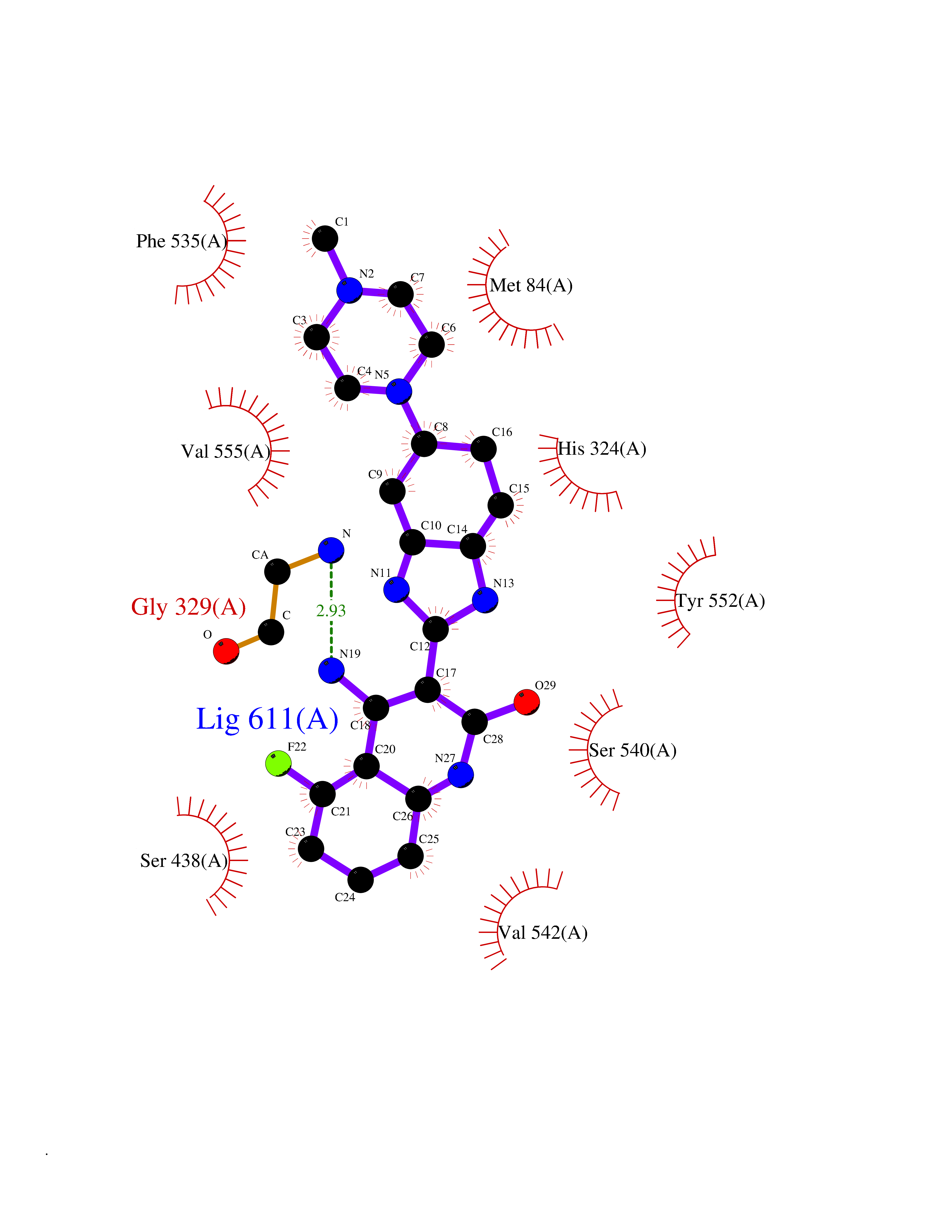 Ligplot Map 3D Binding mode Sequence SEESGLPKLPVPPLQQTLATYLQCMRHLVSEEQFRKSQAIVQQFGAPGGLGETLQQKLLERQEKTANWVSEYWLNDMYLNNRLALPVNSSPAVIFARQHFPGTDDQLRFAASLISGVLSYKALLDSHSIPTDCAKGQPLCMKQYYGLFSSYRLPGHTQDTLVAQNSSIMPEPEHVIVACCNQFFVLDVVINFRRLSEGDLFTQLRKIVKMASNAAARLPPIGLLTSDGRSEWAEARTVLVKDSTNRDSLDMIERCICLVCLDAPGGVELSDTHRALQLLHGGGYSKNGANRWYDKSLQFVVGRDGTCGVVCEHSPFDGIVLVQCTEHLLKHMTQPELVRSPMVPLPAPRRLRWKCSPEIQGHLASSAEKLQRIVKNLDFIVYKFDNYGKTFIKKQKCSPDAFIQVALQLAFYRLHRRLVPTYESASIRRFQEGRVDNIRSATPEALAFVRAVTDHKAAVPASEKLLLLKDAIRAQTAYTVMAITGMAIDNHLLALRELARAMCAALPEMFMDETYLMSNRFVLSTSQVPTTTEMFCCYGPVVPNGYGACYNPQPETILFCISSFHSCAATSSSKFAKAVEESLIDMRDLCSLLPP Hydrogen bonds contact Hydrophobic contact | ||||
| 30 | Non-heme chloroperoxidase | 1A8U | 8.21 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map 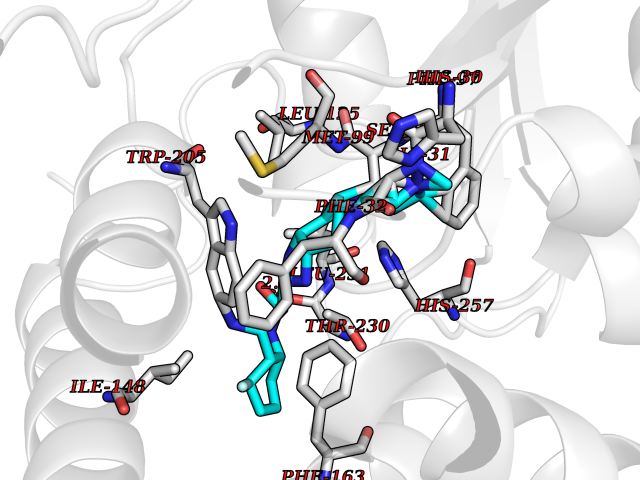 Pymol Map 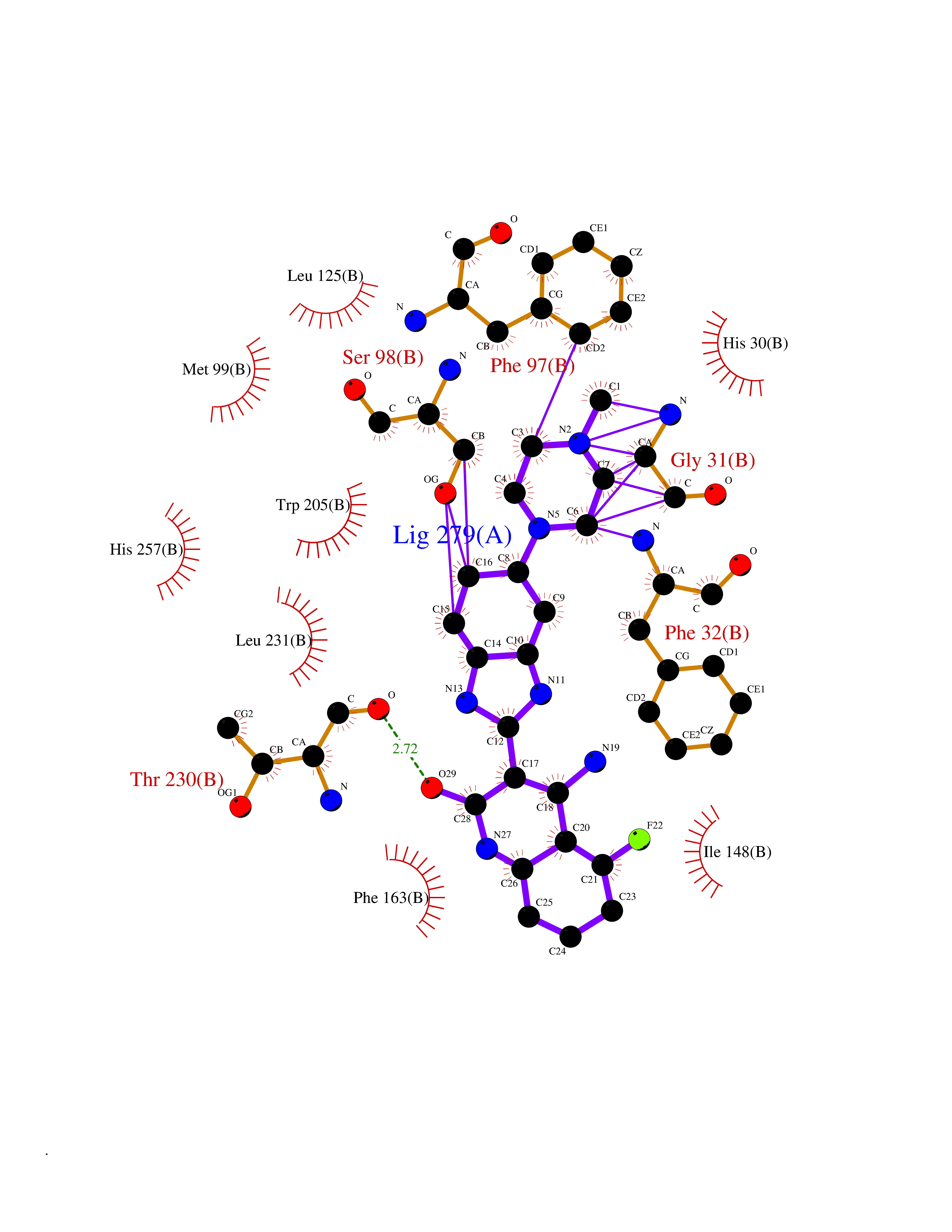 Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 31 | SET domain containing 8 (KMT5A) | 5TEG | 8.20 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -8.99  Molscript Map 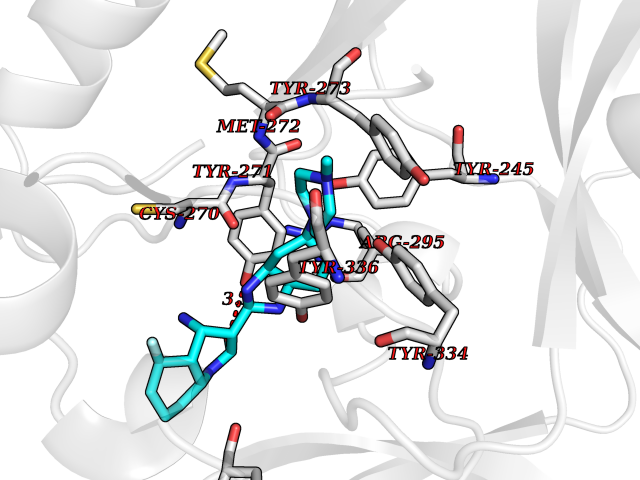 Pymol Map 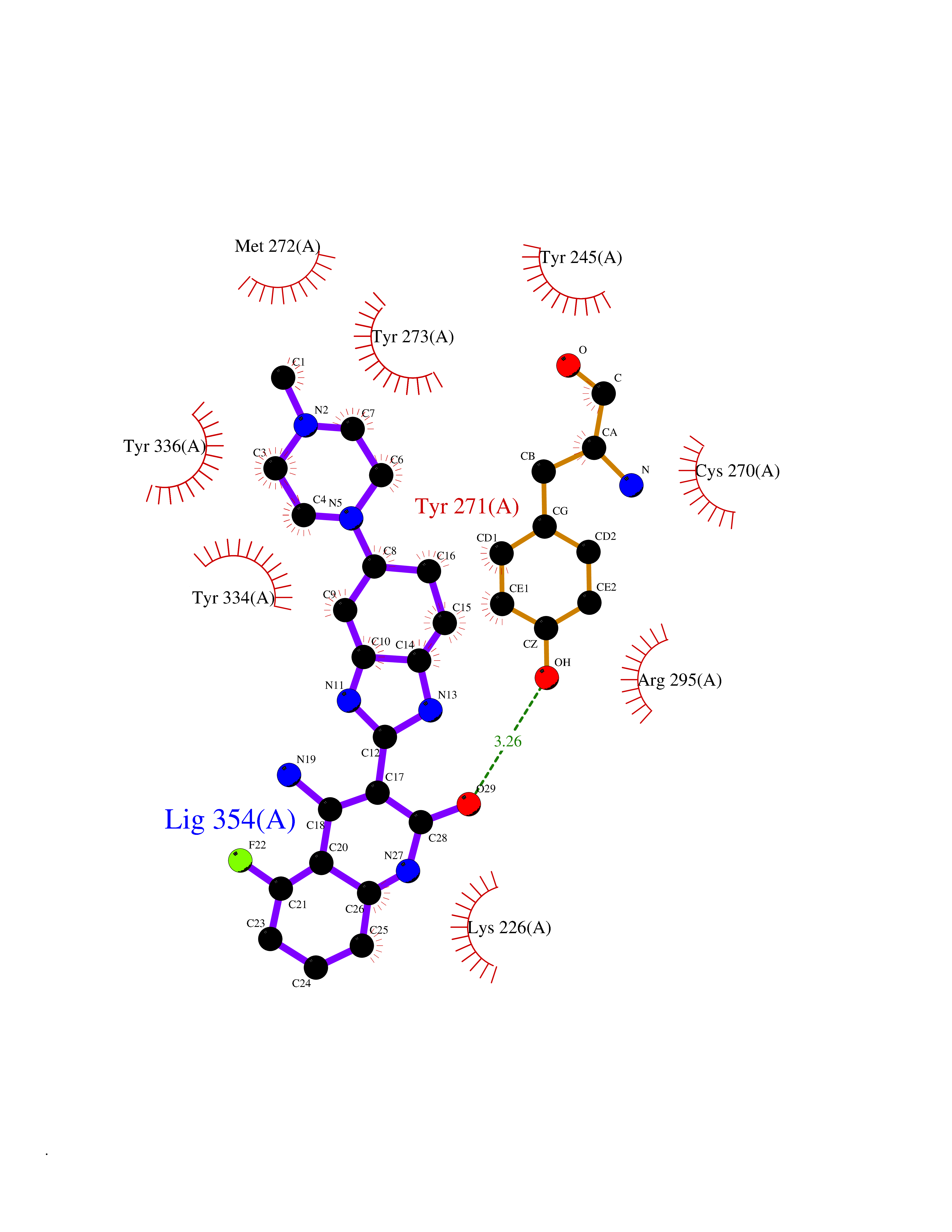 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 32 | Zinc finger protein Helios (IKZF2) | 7LPS | 8.19 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -9.49  Molscript Map 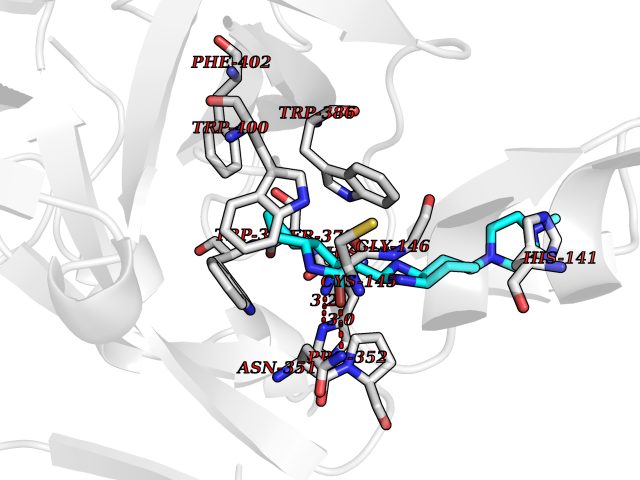 Pymol Map 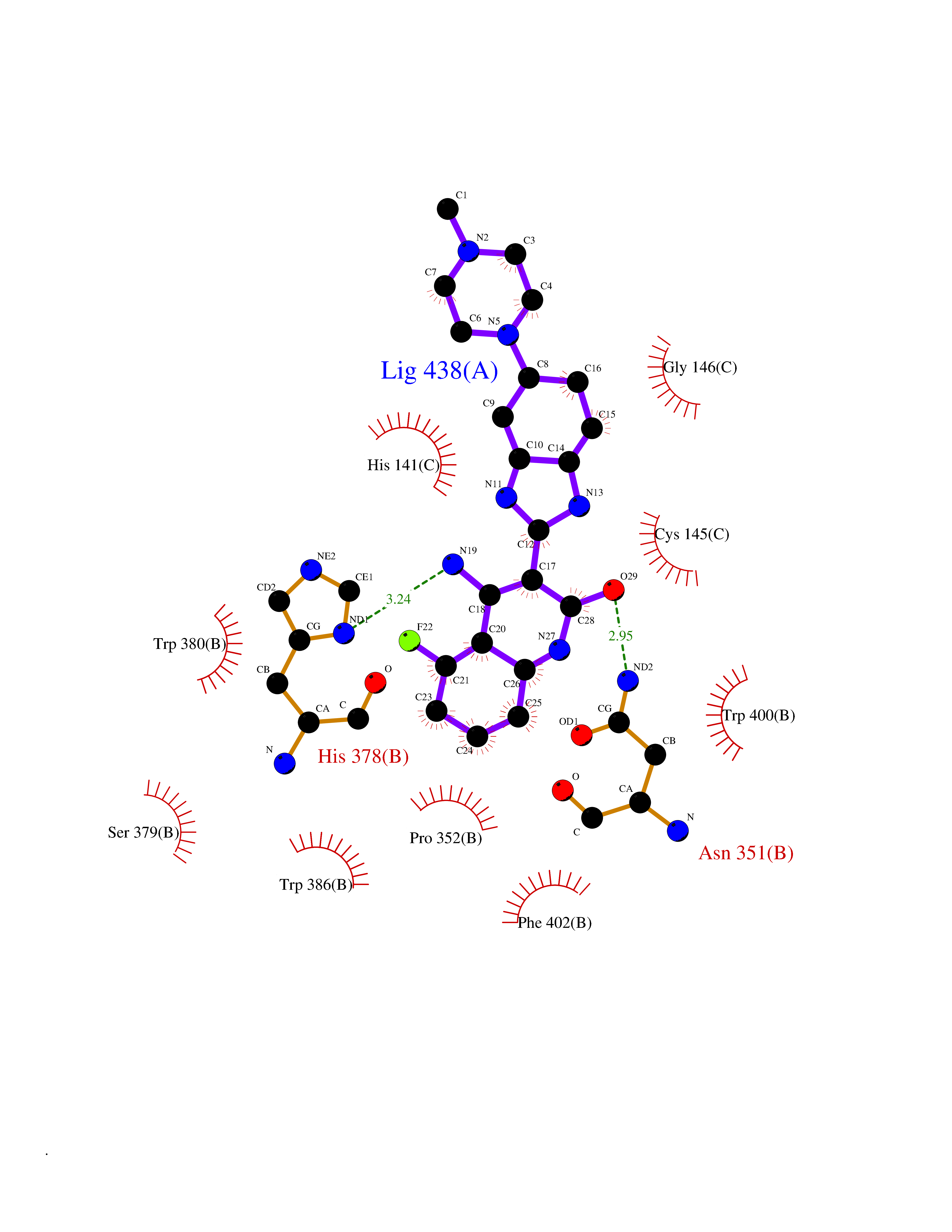 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 33 | ERK activator kinase 2 (MEK2) | 1S9I | 8.19 | |
Target general information Gen name MAP2K2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKMK2; MKK2; MEK 2; MAPKK 2; MAPK/ERK kinase 2; MAP kinase kinase 2; Dual specificity mitogenactivated protein kinase kinase 2; Dual specificity mitogen-activated protein kinase kinase 2 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Kinase Function Activates the ERK1 and ERK2 MAP kinases. Catalyzes the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in MAP kinases. Related diseases Cardiofaciocutaneous syndrome 4 (CFC4) [MIM:615280]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262, ECO:0000269|PubMed:20358587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11967; DB06616; DB12010; DB14904; DB11689; DB08911 Interacts with P05067; P10398; Q96II5; P15056; O95273; Q12959; P61978-2; Q8IVT5; P00540; P04049 EC number EC 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cardiomyopathy; Cytoplasm; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 33960.9 Length 303 Aromaticity 0.07 Instability index 45.61 Isoelectric point 6.29 Charge (pH=7) -2.53 2D Binding mode Binding energy (Kcal/mol) -9.35  Molscript Map 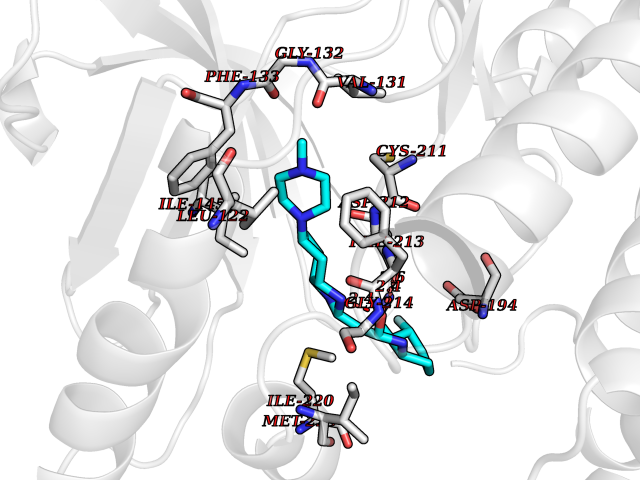 Pymol Map 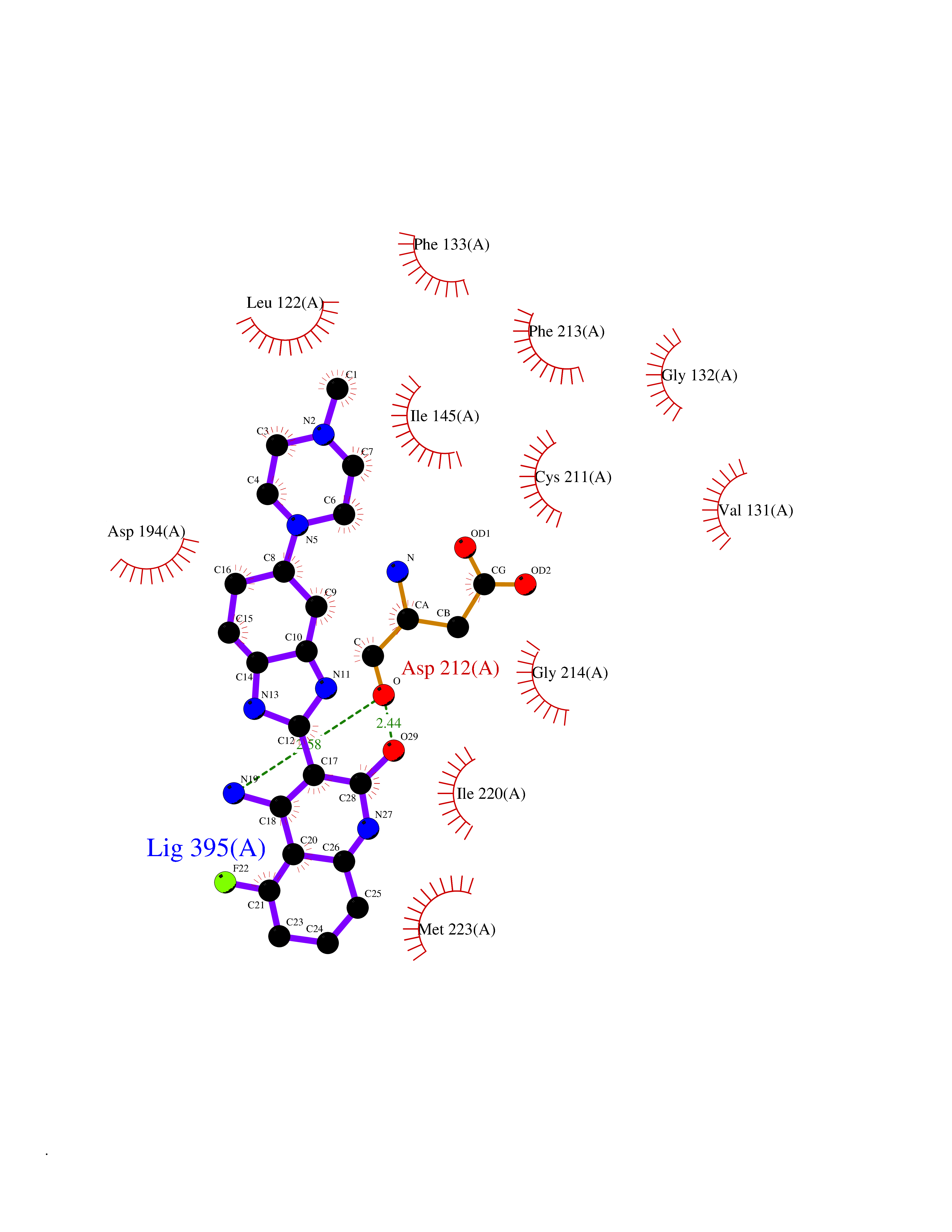 Ligplot Map 3D Binding mode Sequence QKAKVGELKDDDFERISELGAGNGGVVTKVQHRPSGLIMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKEAKRIPEEILGKVSIAVLRGLAYLREKHQIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDSMVGTRSYMAPERLQGTHYSVQSDIWSMGLSLVELAVGRYPIPPPDAKELEAIFGRPVVDRPAMAIFELLDYIVNEPPPKLPNGVFTPDFQEFVNKCLIKNPAERADLKMLTNHTFIKRSEVEEVDFAGWLCKTLRLNQPG Hydrogen bonds contact Hydrophobic contact | ||||
| 34 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 8.17 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -9.9  Molscript Map 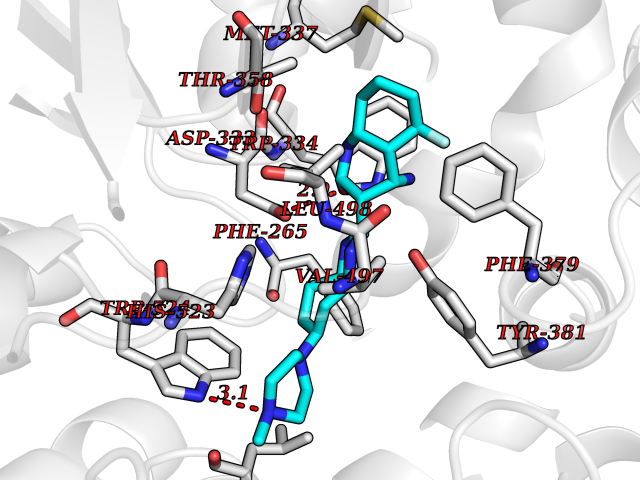 Pymol Map  Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 35 | Mutated oxalosuccinate decarboxylase (mIDH1) | 6ADG | 8.16 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD (mutated); Oxalosuccinate decarboxylase (mutated); NADP(+)-specific ICDH (mutated); Isocitrate dehydrogenase [NADP] cytoplasmic (mutated); IDP (mutated); IDH (mutated); Cytosolic NADP-isocitrate Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -9.04  Molscript Map 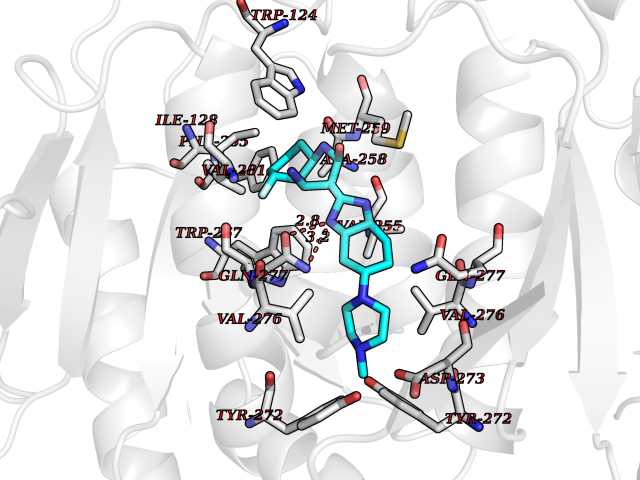 Pymol Map  Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 36 | Short transient receptor potential channel 5 (TRPC5) | 7WDB | 8.16 | |
Target general information Gen name TRPC5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hTRP5; hTRP-5; TrpC5; Transient receptor protein 5; TRP-5 Protein family Transient receptor (TC 1.A.4) family, STrpC subfamily, TRPC5 sub-subfamily Biochemical class Transient receptor potential catioin channel Function Thought to form a receptor-activated non-selective calcium permeant cation channel. Probably is operated by a phosphatidylinositol second messenger system activated by receptor tyrosine kinases or G-protein coupled receptors. Has also been shown to be calcium-selective. May also be activated by intracellular calcium store depletion. Related diseases Loss-of-function variants in TRPC5 may be involved in a mental disorder characterized by maladaptive behavior, anxiety, autism, postpartum depression, extreme food-seeking and hoarding behavior, hyperphagia and obesity. {ECO:0000269|PubMed:38959890}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ANK repeat; Calcium; Calcium channel; Calcium transport; Cell membrane; Disease variant; Glycoprotein; Ion channel; Ion transport; Membrane; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 76850.6 Length 665 Aromaticity 0.12 Instability index 40.05 Isoelectric point 6.16 Charge (pH=7) -5.94 2D Binding mode Binding energy (Kcal/mol) -8.55  Molscript Map  Pymol Map 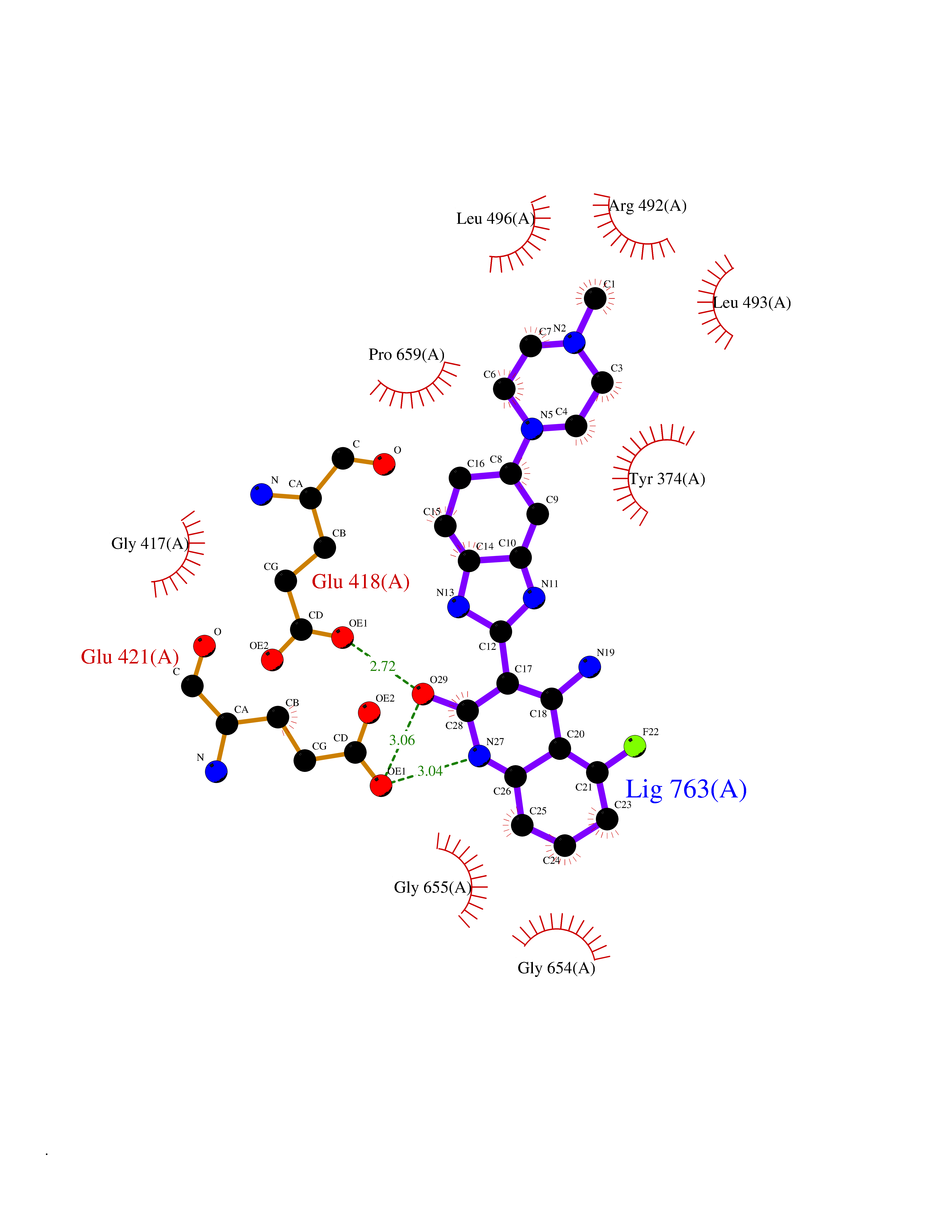 Ligplot Map 3D Binding mode Sequence RIPLQIVRAETELSAEEKAFLNAVEKGDYATVKQALQEAEIYYNVNINCMDPLGRSALLIAIENENLEIMELLLNHSVYVGDALLYAIRKEVVGAVELLLSYQFSEFTPDITPIMLAAHTNNYEIIKLLVQKRVTIPRPHQIRCNCVECVSSSEVDSLRHSRSRLNIYKALASPSLIALSSEDPILTAFRLGWELKELSKVENEFKAEYEELSQQCKLFAKDLLDQARSSRELEIILNHRDDLAKLKVAIKYHQKEFVAQPNCQQLLATLWYDGFPGWRRKHWVVKLLTCMTIGFLFPMLSIAYLISPRSNLGLFIKKPFIKFICHTASYLTFLFMLLLASQHVQGPPPTVVEWMILPWVLGFIWGEIKEMWDGGFTEYIHDWWNLMDFAMNSLYLATISLKIVAYVKYNGSRPREEWEMWHPTLIAEALFAISNILSSLRLISLFTANSHLGPLQISLGRMLLDILKFLFIYCLVLLAFANGLNQLYFYYETRAIDEPNNCKGIRCEKQNNAFSTLFETLQSLFWSVFGLLNLYVTNVKARHEFTEFVGATMFGTYNVISLVVLLNMLIAMMNNSYQLIADHADIEWKFARTKLWMSYFDEGGTLPPPFNIISLIQNQHYQEVIRNLVKRYVAAMIRNSKTTEENFKELKQDISSFRYEVLDLL Hydrogen bonds contact Hydrophobic contact | ||||
| 37 | Geranylgeranyl pyrophosphate synthase | 2Q80 | 8.15 | |
Target general information Gen name GGPS1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family FPP/GGPP synthase family Biochemical class Transferase Function Dimethylallyltranstransferase activity.Farnesyltranstransferase activity.Geranyltranstransferase activity.Identical protein binding.Metal ion binding. Related diseases Muscular dystrophy, congenital hearing loss, and ovarian insufficiency syndrome (MDHLO) [MIM:619518]: An autosomal recessive disorder characterized by early-onset progressive muscle weakness, sensorineural hearing loss, and primary amenorrhea due to ovarian insufficiency. Some patients become wheelchair-bound by the second decade, whereas others have a milder phenotype and maintain independent ambulation into adulthood. Most patients have respiratory insufficiency. {ECO:0000269|PubMed:32403198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06830; DB06931; DB07221; DB08529; DB07410; DB07780; DB04695; DB02552; DB07841; DB00710; DB04714; DB07873; DB06548; DB00282; DB00399 Interacts with O00244; O95749; O00560 EC number 2.5.1.-; 2.5.1.1; 2.5.1.10; 2.5.1.29 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Congenital muscular dystrophy; Cytoplasm; Deafness; Disease variant; Isoprene biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 32872.4 Length 284 Aromaticity 0.11 Instability index 41.09 Isoelectric point 6.17 Charge (pH=7) -3.37 2D Binding mode Binding energy (Kcal/mol) -8.51  Molscript Map 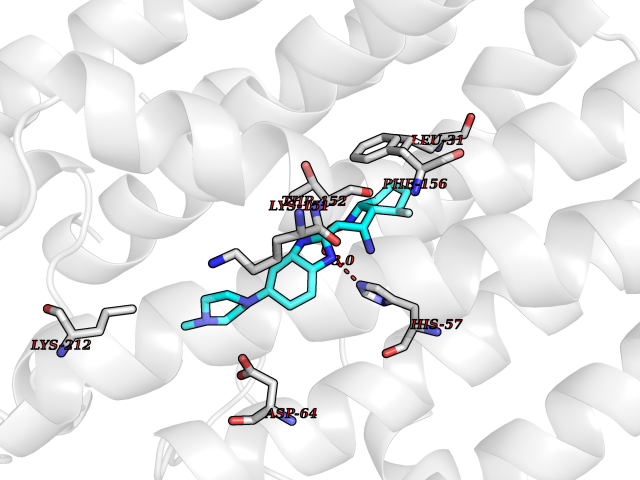 Pymol Map  Ligplot Map 3D Binding mode Sequence ETVQRILLEPYKYLLQLPGKQVRTKLSQAFNHWLKVPEDKLQIIIEVTEMLHNASLLIDDIEDNSKLRRGFPVAHSIYGIPSVINSANYVYFLGLEKVLTLDHPDAVKLFTRQLLELHQGQGLDIYWRDNYTCPTEEEYKAMVLQKTGGLFGLAVGLMQLFSDYKEDLKPLLNTLGLFFQIRDDYANLHSKSFCEDLTEGKFSFPTIHAIWSRPESTQVQNILRQRTENIDIKKYCVHYLEDVGSFEYTRNTLKELEAKAYKQIDARGGNPELVALVKHLSKMF Hydrogen bonds contact Hydrophobic contact | ||||
| 38 | Metabotropic glutamate receptor 2 (mGluR2) | 7E9G | 8.14 | |
Target general information Gen name GRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR2; Group II metabotropic glutamate receptor; Glutamate receptor mGLU2; GPRC1B Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling inhibits adenylate cyclase activity. May mediate suppression of neurotransmission or may be involved in synaptogenesis or synaptic stabilization. G-protein coupled receptor for glutamate. Related diseases Oocyte/zygote/embryo maturation arrest 21 (OZEMA21) [MIM:620610]: An autosomal dominant, female infertility disorder characterized by zygote development arrest due to failure of pronuclei fusion. {ECO:0000269|PubMed:33948904, ECO:0000269|PubMed:33953335}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with Q5T8D3-2; Q9BYF1; Q13520; Q13323; Q8WV48; P57739; O95484; Q7Z7G2; P00387; P27487; P28223-1; Q5SR56; O14880; Q8N4V1; Q58DX5; Q13113; Q9NR31; Q8IWU4; Q9H2H9; P27105; Q8N3G9; Q96Q45-2; Q9NWD8; Q8WUV1; Q9UMX0-2; P0DTC2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 85146.2 Length 769 Aromaticity 0.11 Instability index 36.84 Isoelectric point 8.48 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -9.08  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KKVLTLEGDLVLGGLFPVHQKGGPAEDCGPVNEHRGIQRLEAMLFALDRINRDPHLLPGVRLGAHILDSCSKDTHALEQALDFVRASLITGVIGGSYSDVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFFQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFELEARARNICVATSEKVGRAMSRAAFEGVVRALLQKPSARVAVLFTRSEDARELLAASQRLNASFTWVASDGWGALESVVAGSEGAAEGAITIELASYPISDFASYFQSLDPWNNSRNPWFREFWEQRFRCSFRQRDCAAHSLRAVPFEQESKIMFVVNAVYAMAHALHNMHRALCPNTTRLCDAMRPVNGRRLYKDFVLNVKFDAPFRPADTHNEVRFDRFGDGIGRYNIFTYLRAGSGRYRYQKVGYWAEGLTLDTSLIPWASPSAGPLPASRCSEPCLQNEVKSVQPGEVCCWLCIPCQPYEYRLDEFTCADCGLGYWPNASLTGCFELPQEYIRWGDAWAVGPVTIACLGALATLFVLGVFVRHNATPVVKAAGRELCYILLGGVFLCYCMTFIFIAKPSTAVCTLRRLGLGTAFSVCYSALLTKTNRIARIFGGAREGAQRPRFISPASQVAICLALISGQLLIVVAWLVVEAPGTGKETAPERREVVTLRCNHRDASMLGSLAYNVLLIALCTLYAFKTRKCPENFNEAKFIGFTMYTTCIIWLAFLPIFYVTSSDYRVQTTTMCVSVSLSGSVVLGCLFAPKLHI Hydrogen bonds contact Hydrophobic contact | ||||
| 39 | Extracellular calcium-sensing receptor (CASR) | 5FBK | 8.14 | |
Target general information Gen name CASR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCasR; Parathyroid cell calciumreceptor; Parathyroid cell calcium-sensing receptor 1; Parathyroid calcium receptor; Parathyroid Cell calcium-sensing receptor; PCaR1; GPRC2A; CaSR Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Senses fluctuations in the circulating calcium concentration and modulates the production of parathyroid hormone (PTH) in parathyroid glands. The activity of this receptor is mediated by a G-protein that activates a phosphatidylinositol-calcium second messenger system. The G-protein-coupled receptor activity is activated by a co-agonist mechanism: aromatic amino acids, such as Trp or Phe, act concertedly with divalent cations, such as calcium or magnesium, to achieve full receptor activation. G-protein-coupled receptor that senses changes in the extracellular concentration of calcium ions and plays a key role in maintaining calcium homeostasis. Related diseases Hypocalciuric hypercalcemia, familial 1 (HHC1) [MIM:145980]: A form of hypocalciuric hypercalcemia, a disorder of mineral homeostasis that is transmitted as an autosomal dominant trait with a high degree of penetrance. It is characterized biochemically by lifelong elevation of serum calcium concentrations and is associated with inappropriately low urinary calcium excretion and a normal or mildly elevated circulating parathyroid hormone level. Hypermagnesemia is typically present. Affected individuals are usually asymptomatic and the disorder is considered benign. However, chondrocalcinosis and pancreatitis occur in some adults. {ECO:0000269|PubMed:11762699, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:15579740, ECO:0000269|PubMed:15879434, ECO:0000269|PubMed:16598859, ECO:0000269|PubMed:16740594, ECO:0000269|PubMed:17473068, ECO:0000269|PubMed:17698911, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:19789209, ECO:0000269|PubMed:21566075, ECO:0000269|PubMed:21643651, ECO:0000269|PubMed:22114145, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25104082, ECO:0000269|PubMed:25292184, ECO:0000269|PubMed:26386835, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:7673400, ECO:0000269|PubMed:7726161, ECO:0000269|PubMed:7916660, ECO:0000269|PubMed:8636323, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9298824}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperparathyroidism, neonatal severe (NSHPT) [MIM:239200]: A disorder characterized by severe hypercalcemia, bone demineralization, and failure to thrive usually manifesting in the first 6 months of life. If untreated, NSHPT can be a devastating neurodevelopmental disorder, which in some cases is lethal without parathyroidectomy. {ECO:0000269|PubMed:14985373, ECO:0000269|PubMed:15572418, ECO:0000269|PubMed:17555508, ECO:0000269|PubMed:27434672, ECO:0000269|PubMed:8675635, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypocalcemia, autosomal dominant 1 (HYPOC1) [MIM:601198]: A disorder of mineral homeostasis characterized by blood calcium levels below normal, and low or normal serum parathyroid hormone concentrations. Disease manifestations include mild or asymptomatic hypocalcemia, paresthesias, carpopedal spasm, seizures, hypercalciuria with nephrocalcinosis or kidney stones, and ectopic and basal ganglia calcifications. Few patients manifest hypocalcemia and features of Bartter syndrome, including hypomagnesemia, hypokalemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia. {ECO:0000269|PubMed:10487661, ECO:0000269|PubMed:12050233, ECO:0000269|PubMed:12107202, ECO:0000269|PubMed:12241879, ECO:0000269|PubMed:12574188, ECO:0000269|PubMed:12915654, ECO:0000269|PubMed:15551332, ECO:0000269|PubMed:16608894, ECO:0000269|PubMed:19179454, ECO:0000269|PubMed:22789683, ECO:0000269|PubMed:23169696, ECO:0000269|PubMed:23966241, ECO:0000269|PubMed:25766501, ECO:0000269|PubMed:7874174, ECO:0000269|PubMed:8702647, ECO:0000269|PubMed:8733126, ECO:0000269|PubMed:8813042, ECO:0000269|PubMed:8878438, ECO:0000269|PubMed:9253358, ECO:0000269|PubMed:9661634, ECO:0000269|PubMed:9920108}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 8 (EIG8) [MIM:612899]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Seizure types are variable, but include myoclonic seizures, absence seizures, febrile seizures, complex partial seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:18756473}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11093; DB11348; DB14481; DB01012; DB12865; DB00994; DB05695; DB05255; DB00127 Interacts with Q15363; P41180-1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 52438.9 Length 467 Aromaticity 0.12 Instability index 39.62 Isoelectric point 5.63 Charge (pH=7) -10.18 2D Binding mode Binding energy (Kcal/mol) -8.74 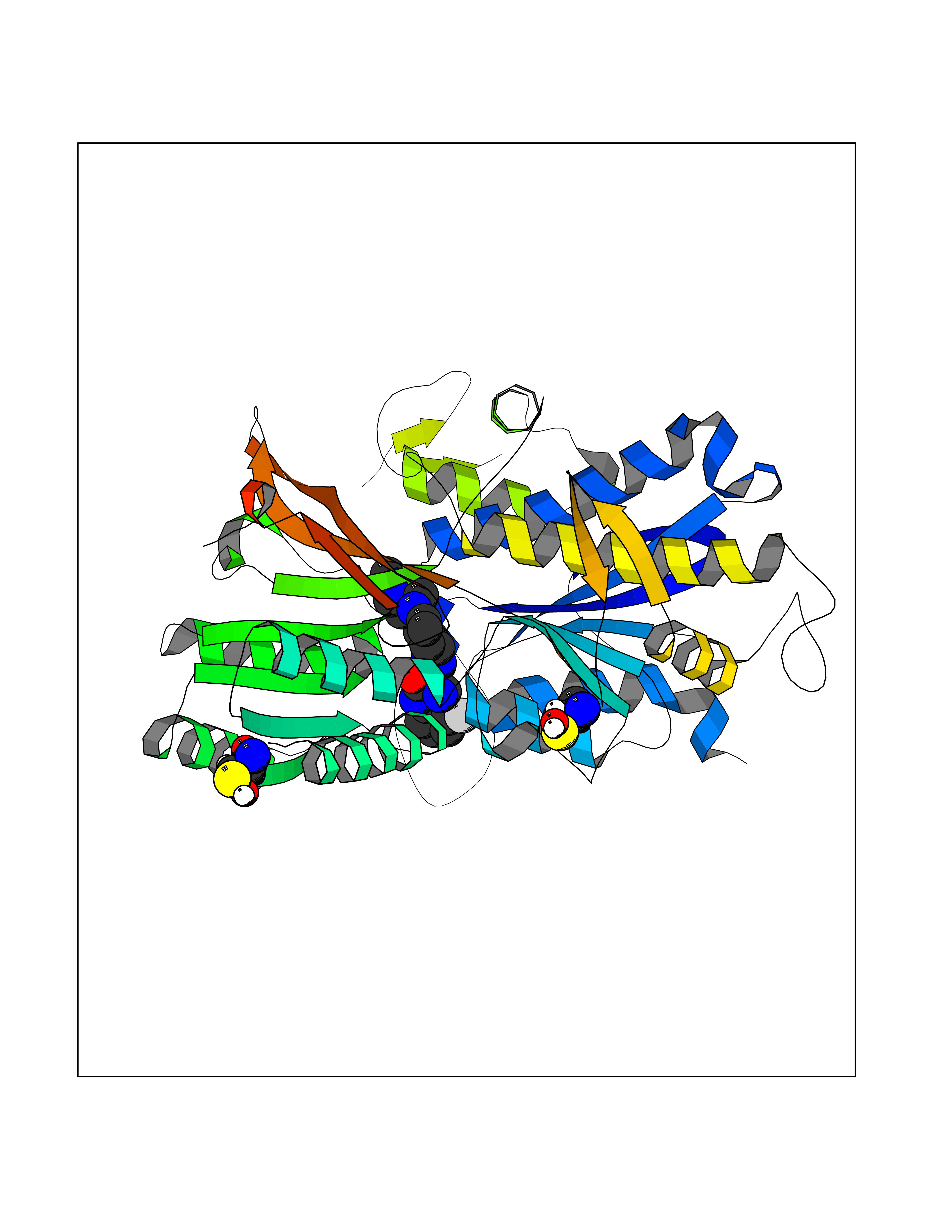 Molscript Map 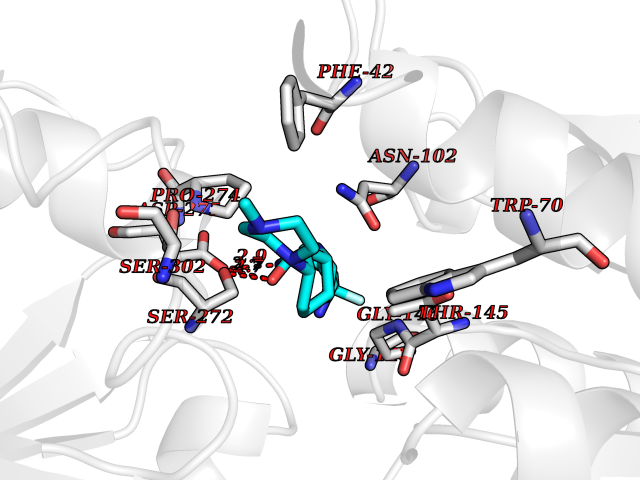 Pymol Map 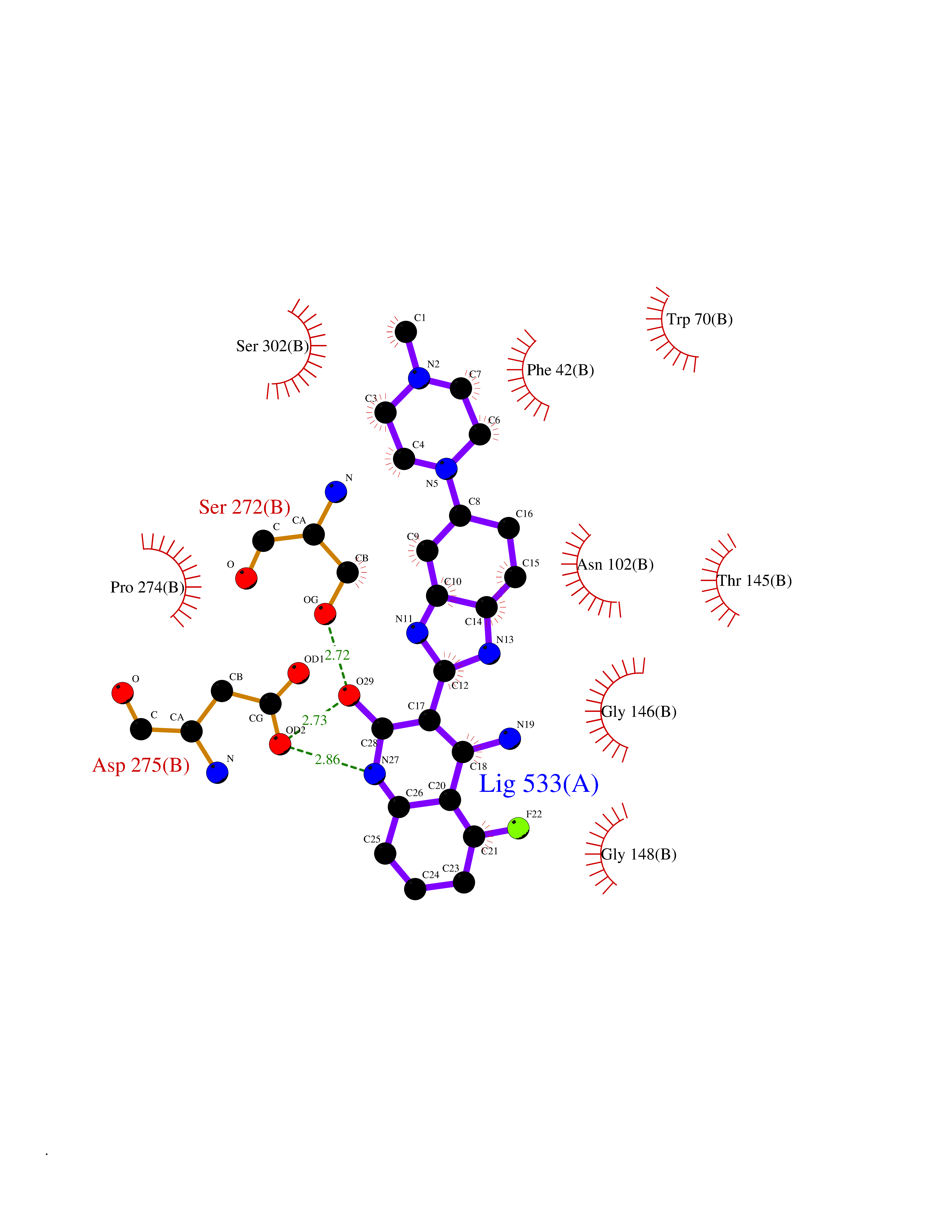 Ligplot Map 3D Binding mode Sequence GPDQRAQKKGDIILGGLFPIHFGVAAKDQDLKSRPESVECIRYNFRGFRWLQAMIFAIEEINSSPALLPNLTLGYRIFDTCNTVSKALEATLSFVAQNKIDSTIAVVGATGSGVSTAVANLLGLFYIPQVSYASSSRLLSNKNQFKSFLRTIPNDEHQATAMADIIEYFRWNWVGTIAADDDYGRPGIEKFREEAEERDIXIDFSELISQYSDEEEIQHVVEVIQNSTAKVIVVFSSGPDLEPLIKEIVRRNITGKIWLASEAWASSSLIAMPQYFHVVGGTIGFALKAGQIPGFREFLKKVHPRKSVHNGFAKEFWEETFNCHLQFRPLCTGDENISSVETPYIDYTHLRISYNVYLAVYSIAHALQDIYTCLPGRGLFTNGSCADIKKVEAWQVLKHLRHLNFTNNMGEQVTFDEXGDLVGNYSIINWHLSPEDGSIVFKEVGYYNVYAKKGERLFINEEKILWS Hydrogen bonds contact Hydrophobic contact | ||||
| 40 | Beta-adrenergic receptor kinase 1 (ADRBK1) | 3V5W | 8.14 | |
Target general information Gen name GRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GRK2; G-protein coupled receptor kinase 2; BetaARK1; Beta-ARK-1; Beta ARK1 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, GPRK subfamily Biochemical class Kinase Function Specifically phosphorylates the agonist-occupied form of the beta-adrenergic and closely related receptors, probably inducing a desensitization of them. Key regulator of LPAR1 signaling. Competes with RALA for binding to LPAR1 thus affecting the signaling properties of the receptor. Desensitizes LPAR1 and LPAR2 in a phosphorylation-independent manner. Related diseases 3-ketothiolase deficiency (3KTD) [MIM:203750]: An autosomal recessive inborn error of isoleucine catabolism characterized by intermittent ketoacidotic attacks associated with unconsciousness. Some patients die during an attack or are mentally retarded. Urinary excretion of 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid, triglylglycine, butanone is increased. It seems likely that the severity of this disease correlates better with the environmental or acquired factors than with the ACAT1 genotype. {ECO:0000269|PubMed:1346617, ECO:0000269|PubMed:1715688, ECO:0000269|PubMed:7728148, ECO:0000269|PubMed:9744475}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00171 Interacts with P05067; P48730-2; P21860; P21462; Q9Y2X7; P35626; Q00987; P13591; P25963; Q13635; P0CG48 EC number EC 2.7.11.15 Uniprot keywords 3D-structure; ATP-binding; Cell membrane; Cell projection; Cytoplasm; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Synapse; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 38433.9 Length 337 Aromaticity 0.09 Instability index 52.17 Isoelectric point 7.36 Charge (pH=7) 1.05 2D Binding mode Binding energy (Kcal/mol) -8.88 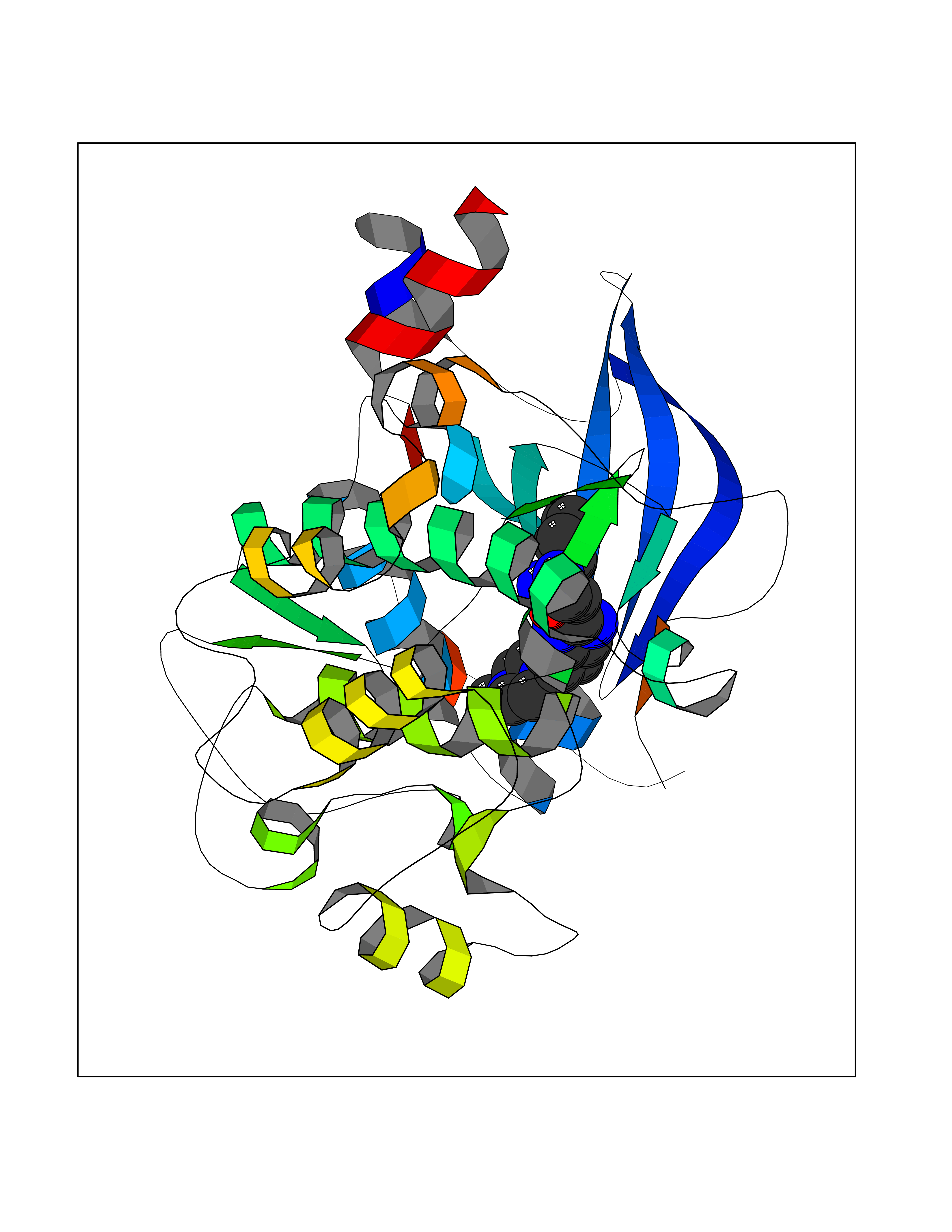 Molscript Map 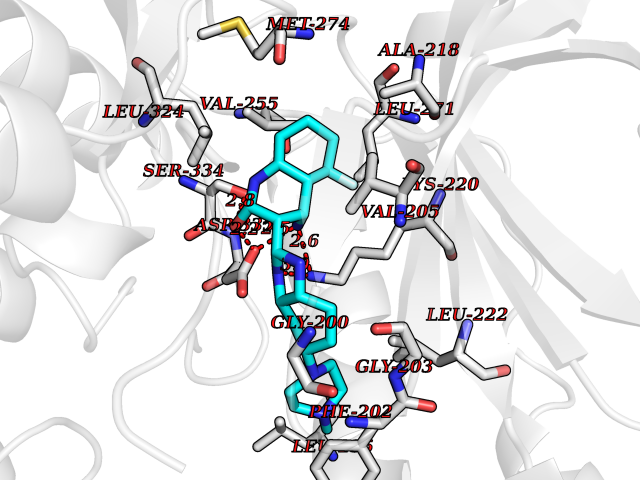 Pymol Map 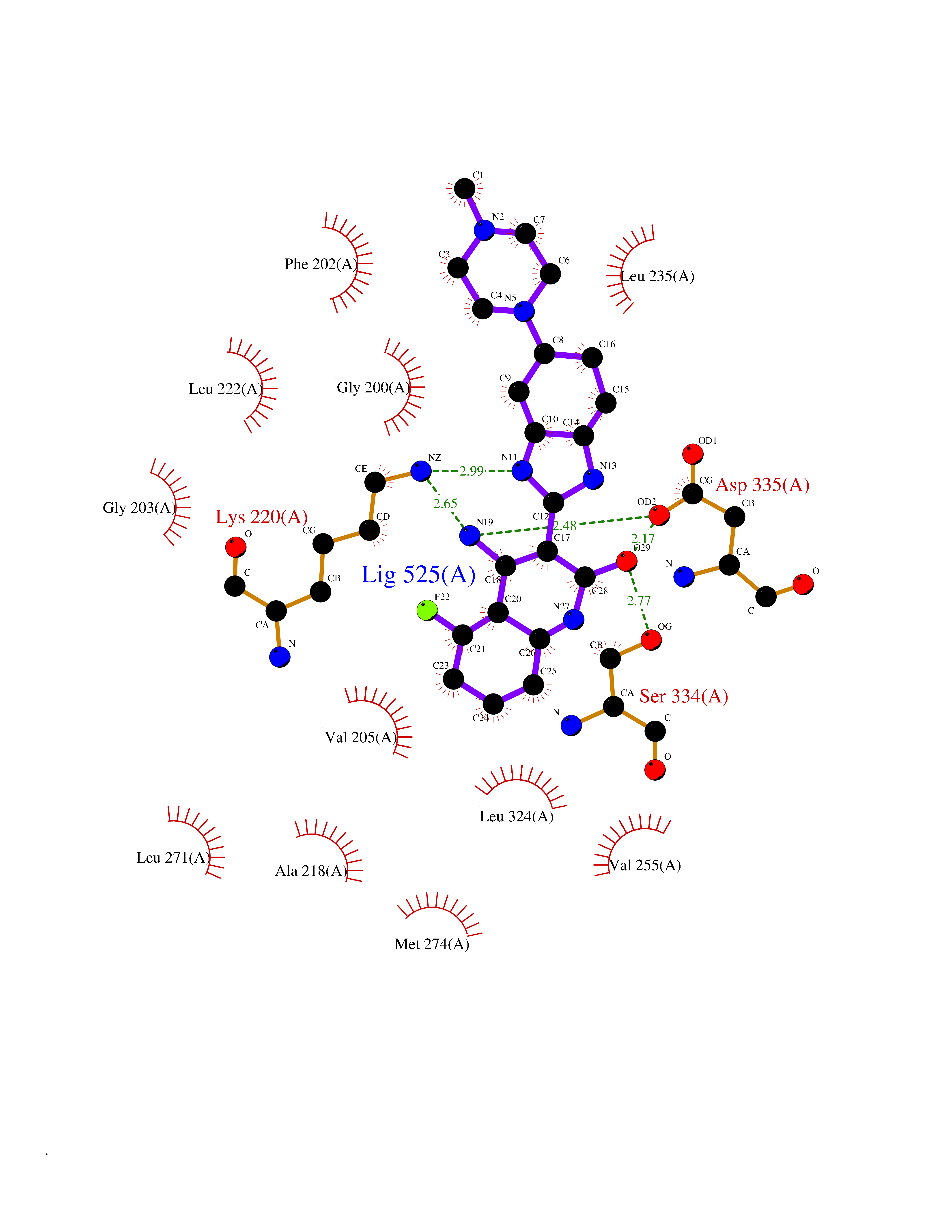 Ligplot Map 3D Binding mode Sequence KNVELNIHLTMNDFSVHRIIGRGGFGEVYGCRKADTGKMYAMKCLDKKRIKMKQGETLALNERIMLSLVSTGDCPFIVCMSYAFHTPDKLSFILDLMNGGDLHYHLSQHGVFSEADMRFYAAEIILGLEHMHNRFVVYRDLKPANILLDEHGHVRISDLGLACDFSKKKPHASVGTHGYMAPEVLQKGVAYDSSADWFSLGCMLFKLLRGHSPFRQHKTKDKHEIDRMTLTMAVELPDSFSPELRSLLEGLLQRDVNRRLGCLGRGAQEVKESPFFRSLDWQMVFLQKYPPPLIPPRGEVNAADAFDKGIKLLDSDQELYRNFPLTISERWQQEVAE Hydrogen bonds contact Hydrophobic contact | ||||